

Abstract
We have selectively inhibited Notch1 signaling in oligodendrocyte precursors (OPCs) using the Cre/loxP system in transgenic mice to investigate the role of Notch1 in oligodendrocyte (OL) development and differentiation. Early development of OPCs appeared normal in the spinal cord. However, at embryonic day 17.5, premature OL differentiation was observed and ectopic immature OLs were present in the gray matter. At birth, OL apoptosis was strongly increased in Notch1 mutant animals. Premature OL differentiation was also observed in the cerebrum, indicating that Notch1 is required for the correct spatial and temporal regulation of OL differentiation in various regions of the central nervous system. These findings establish a widespread function of Notch1 in the late steps of mammalian OPC development in vivo.
Keywords: ectopic expression; brain; Cre-lox P; newborn; apoptosis
Introduction
Signaling through the Notch pathway is involved in various cell fate decisions, in particular in maintenance of precursor cells in an undifferentiated state until they are competent to respond to inductive cues (for reviews see Artavanis-Tsakonas et al., 1995; Robey, 1997; Weinmaster, 1998). In this context, Notch signaling plays a key role in tissue-autonomous fate determination in central nervous system (CNS)* development, in a process referred to as a lateral inhibition (Artavanis-Tsakonas et al., 1999), and also regulates mammalian neurogenesis (Lütolf et al., 2002). A role of Notch signaling in the regulation of terminal neural differentiation also has been found in the development and remodeling of dendrites (Berezovska et al., 1999; Franklin et al., 1999; Sestan et al., 1999; Redmond et al., 2000).
In vertebrate glial cell development, the Notch pathway has been implicated in a number of crucial events. Activated Notch promotes the formation of radial glia in the fetal forebrain (Gaiano et al., 2000; Chambers et al., 2001), Schwann cells in dorsal root ganglia (Wakamatsu et al., 2000), and Müller glia in the retina (Furukawa et al., 2000). In cell culture, even transient activation of Notch strongly promotes the differentiation of adult hippocampus-derived multipotent progenitors into astroglia (Tanigaki et al., 2001), and of neural crest stem cells into the Schwann cell lineage (Morrison et al., 2000).
However, the oligodendrocyte (OL) lineage appears to respond somewhat differently. Activation of Notch signaling suppresses rather than promotes the differentiation of OLs from multipotent progenitor cells (Gaiano et al., 2000; Tanigaki et al., 2001). Furthermore, a potential regulatory role of the Notch pathway in mammalian oligodendrocyte precursor cell (OPC) differentiation has been suggested by cell culture studies showing that OPCs derived from the postnatal rat optic nerve could be inhibited in their differentiation by incubation with Notch ligands (Wang et al., 1998).
How proliferation and differentiation are regulated in the OL lineage is only partially understood (Barres et al., 1992; Barres and Raff, 1994; Raff et al., 1998; Kondo and Raff, 2000a,b; Qi et al., 2001; Wang et al., 2001; Zhou et al., 2001). On the basis of morphology and molecular phenotype, cells of the OL lineage can be grouped into OPCs, immature OLs (premyelinating), and mature OLs (myelinating cells; Gard and Pfeiffer, 1990; Hardy and Reynolds, 1991; Armstrong et al., 1992; Miller, 1996). OPCs of the developing spinal cord are generated along the length of the neural tube from a narrow zone in the ventral region of the neuroepithelium (Noll and Miller, 1993; Pringle and Richardson, 1993; Yu et al., 1994). Determination of the OL lineage is promoted by the transcription factors Olig1, Olig2, and Nkx2.2 (Lu et al., 2001; Zhou et al., 2001; Fu et al., 2002), but the exact nature of the interplay among them is not clear. OPCs arising in the ventral spinal cord subsequently proliferate and disperse throughout the developing CNS (for reviews see Miller, 1996; Richardson et al., 1997; Spassky et al., 2000).
Postmitotic OLs appear first in the ventral funiculus and later in the dorsal and lateral funiculi, the large axonal bundles that become the white matter tracts of the spinal cord (Jordan et al., 1989; Yu et al., 1994). However, it remains an open question how OPCs are able to migrate through an already established neuronal system around, over, and along the axons they may ultimately myelinate, before differentiating at the appropriate time point (Blaschuk and ffrench-Constant, 1998). Progression past the OPC stage is strongly delayed and reduced in Nkx2.2−/ − mice, suggesting a possible role for this transcription factor in later development of OLs (Qi et al., 2001). Ectopic OPCs arising on overexpression of Notch1 together with Olig2 in chick spinal cord do not mature, which is consistent with a negative role for Notch1 in late stages of differentiation (Zhou et al., 2001). In contrast, Givogri et al. (2002) have reported a transient increase of myelination in some parts of the CNS during the early postnatal weeks in heterozygous-null Notch1 mutants. In vitro studies and correlative evidence based on the regulation of Notch1 and its ligand Jagged1 in the developing optic nerve have also implicated the Notch pathway in the control of the timing of OPC differentiation (Wang et al., 1998). This is a tantalizing idea supporting a model in which loss of responsiveness to growth factors may permit OPC differentiation, but local cues (e.g., the Notch pathway) regulate the timing of final maturation in different myelinated tracts (Blaschuk and ffrench-Constant, 1998). Unfortunately, further support for this model has not been particularly forthcoming over the last years and several crucial questions remain open: is the observed function of Notch signaling in the control of rat OPC differentiation restricted to the optic nerve or is it a general phenomenon throughout the rodent CNS? Is the observed effect specifically Notch1 dependent or may other members of the Notch family be involved (Notch 2–4; Lindsell et al., 1996; Irvin et al., 2001)? Are the findings with isolated OPCs in vitro directly transferable to the in vivo situation, in particular given the recent reappraisal that OL development is under stringent axonal control and the proposed interactions of OPCs with mature OLs via the Notch pathway in tissue (Wang et al., 1998; Barres and Raff, 1999; Casaccia-Bonnefil, 2000)? The “acid test” to answer these questions requires an analysis in a physiologically accurate system in which specifically Notch1 has been reduced or eliminated. Transgenic mice provide such a proper setting. Notch1 knockout mice were not informative, however, due to early embryonic lethality (Swiatek et al., 1994; Conlon et al., 1995). Thus, in this study we have selectively inhibited Notch1 signaling using a conditional Notch1 knockout mouse strain (Radtke et al., 1999). Our data demonstrate a crucial function of Notch1 in late steps of OL differentiation in the spinal cord and suggest a similar function in the brain.
Results
We have used a “floxed” allele of Notch1, in which the exon encoding the signal sequence (Fig. 1 A) is flanked by loxP sites (Radtke et al., 1999) for conditional gene ablation. Recombination mediated by the Cre recombinase excises the region between the lox sites, thereby inactivating the gene. Cre was expressed under the control of regulatory elements known to be active in the OL lineage, either those of the Cnp gene (with the Cre coding region inserted into the endogenous gene; unpublished data) or of the proteolipid (Plp) gene (as a transgene driving Cre; unpublished data) (Fig. 1 A). Newborn mice homozygous for the floxed Notch1 allele and carrying either the Cnp-Cre (Cnp-Cre Δ/Δ) or the Plp-Cre (Plp-Cre Δ/Δ) allele were outwardly normal, nursed, moved, breathed, and responded to mechanical stimulation, but usually survived only a few hours after birth. Recombination outside the CNS was assessed in Cnp-Cre Δ/Δ mice by reporter gene expression (for method, see next paragraph) and was observed in various organs whose development is known to be affected by Notch signaling, including kidney (McLaughlin et al., 2000), liver (Nijjar et al., 2001), lung (Ito et al., 2000), and pancreas (Apelqvist et al., 1999). These findings could explain the early death of the Cnp-Cre Δ/Δ animals. A small number of Cnp-Cre Δ/Δ individuals survived longer, exhibited only modest defects (smaller in size, poor sense of balance, and partially closed eyes), and could be kept until adulthood. In contrast, Plp-Cre Δ/Δ mice that survived after birth had to be killed because of severe growth retardation and motor defects. Some Cnp-Cre Δ/Δ and Plp-Cre Δ/Δ mice had strikingly enlarged lateral ventricles at birth (unpublished data).
Figure 1.
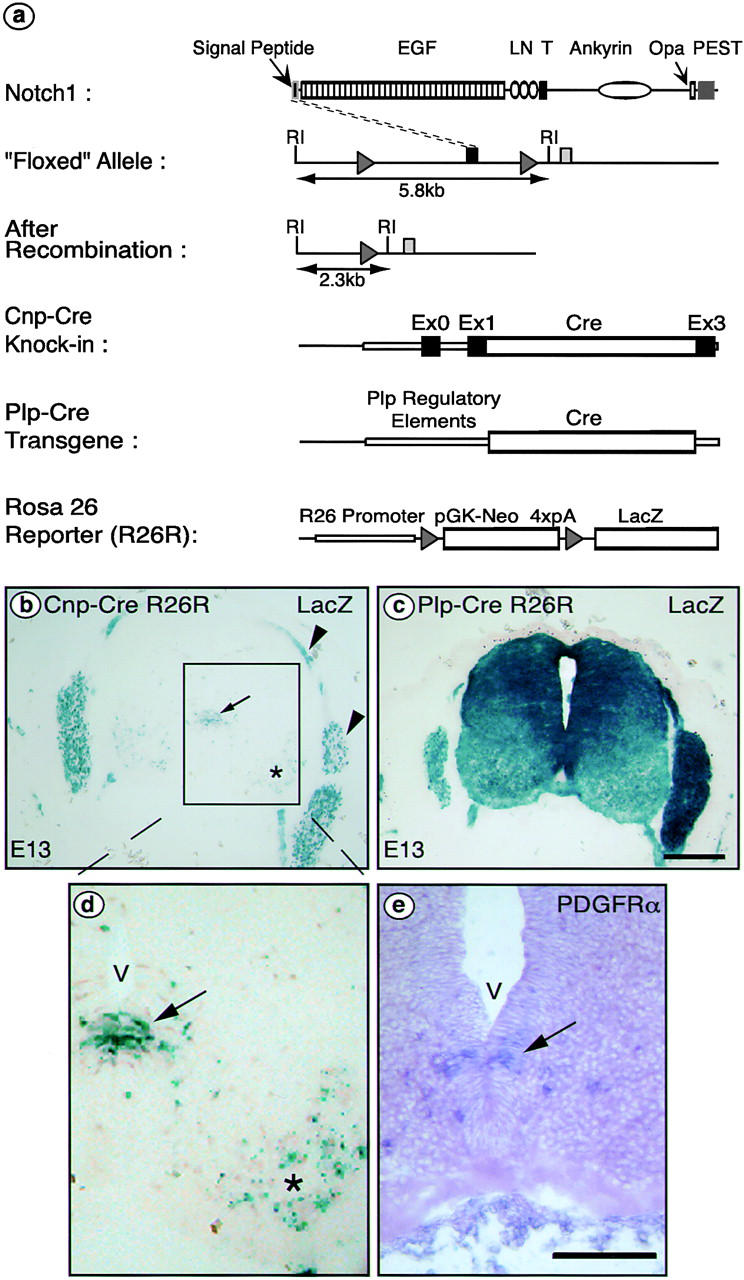
Experimental strategy and Cre-mediated recombination in OPCs at E13. (a) Schematic representation of the murine Notch1 protein and LoxP/Cre-mediated deletion strategy. The Notch protein contains 2,531 amino acid residues that encompass a signal peptide, 36 EGF repeats (EGF), 3 Lin/Notch domains (LN), a transmembrane domain (T), cytoplasmic ankyrin repeats, a polyglutamine stretch (Opa), and a PEST sequence. In the floxed Notch1 allele (Radtke et al., 1999), the first coding exon is flanked by LoxP sequences (gray triangles). After Cre-mediated recombination, a null allele is generated. Arrows indicate EcoRI fragments that differ in size between the floxed locus compared with the locus after deletion of the 3.5-kb segment flanked by loxP sites. The Cre recombinase gene was inserted into the Cnp locus (Cnp-Cre) or is driven from Plp/DM20 regulatory elements (Spassky et al., 1998) (Plp-Cre). The ROSA26 reporter mouse (Soriano, 1999) (R26R) was used to follow recombination events: Cre-mediated recombination deletes the neomycin phosphotransferase gene (PGK-neo) plus four polyadenylation sites (4xpA), activating the production of β-gal encoded by LacZ. RI indicates EcoRI restriction sites. (b–d) Cre-induced β-gal expression patterns. Cnp-Cre or Plp-cre mice were crossed with R26R reporter mice. Cells in which Cre had been expressed were detected by X-gal staining for β-galactosidase activity in sections of E13 embryos. The Cnp-Cre line shows expression in the ventral ventricular zone (b, arrow) and motoneurons (b, asterisk); these regions are enlarged in panel d. The area indicated by an arrow (b and d) is the site of origin of the OPCs consistent with the expected endogenous CNP expression pattern and confirmed by PDGFR-α in situ hybridization (e). (b) Arrowheads indicate the nerve roots and dorsal root ganglia that are part of the PNS. Expression in the Plp-Cre line used is much broader, affecting most cells of the spinal cord (c). V, ventricle. Bars: 10 μm.
We focused our studies on the spinal cord because OL development has been extensively studied in this structure. To determine where and when Cre recombinase was produced in the spinal cord in our Cre-transgenic mice, ROSA26 reporter (R26R) mice (Soriano, 1999) were crossed with mice carrying Cnp-Cre (yielding Cnp-Cre R26R) or Plp-Cre (yielding Plp-Cre R26R). This approach allowed lineage tracing via detection of the LacZ product β-galactosidase (β-gal; Fig. 1 a). Sections were cut at the forelimb level of doubly transgenic mice at embryonic day 13 (E13), and β-gal was detected by histological X-gal staining. In Cnp-Cre R26R mice (Fig. 1 b, enlarged in d), X-gal–positive cells were observed within the spinal cord in the ventral ventricular zone, in a pattern consistent with recombination in OPCs as indicated by PDGF receptor-α (PDGFR-α) in situ hybridization (Fig. 1 e). This restricted expression in OPCs was expected based on the reported endogenous 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) expression in the rat (Yu et al., 1994) and the analysis of transgenic mice under the control of CNP regulatory elements (Gravel et al., 1998; Chandross et al., 1999). Additional recombination was observed in the ventral horns of the neural tube, presumably in developing motoneurons. Furthermore, X-gal–positive cells were already found in the ventral ventricular zone as early as E11.5 (unpublished data). By E17.5, the number of X-gal–positive cells was increased substantially and the cells were dispersed throughout the spinal cord (see Fig. 2 b), which is consistent with the expected proliferation and migration of OPCs (Miller et al., 1997). In contrast to the Cnp-Cre R26R embryos, developing animals with a Plp-Cre R26R genotype showed a much broader pattern of β-gal expression at E13 (Fig. 1 c), involving most of the cells in the spinal cord. This is most likely an artifact of the transgene insertion because expression of the endogenous Plp gene is more restricted (Yu et al., 1994; Timsit et al., 1995). Therefore, further analysis was performed mainly with the Cnp-Cre mice.
Figure 2.
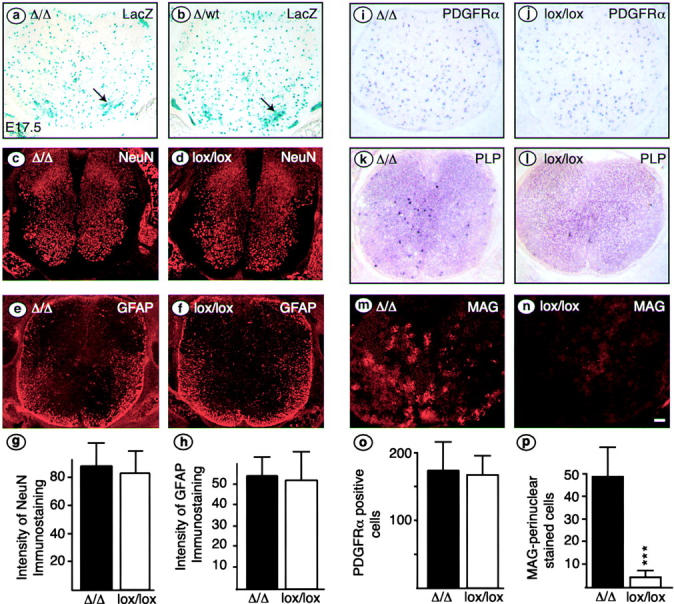
Precocious differentiation of immature OLs in E17.5 spinal cord of Cnp-CreΔ/Δ mice. (a and b) Sections were cut from triple transgenic animals carrying Cnp-Cre, R26R, and one (b) or two copies (a) of the floxed Notch1 allele. X-gal stainings indicate recombination with a similar pattern in homozygous mutant (a, Δ/Δ) or heterozygous mutant (b, Δ/wt) spinal cords; motoneurons are indicated by arrows. Neurons, astrocytes, OPCs, and immature OLs were compared in Cnp-Cre Δ/Δ and control (lox/lox) spinal cords by immunohistochemistry (c–f, m, and n) or in situ hybridization (i–l). Neurons, astrocytes, and OPCs, assessed by the markers NeuN (c and d), GFAP (e and f), and PDGFR-α (i and j), respectively, appear normal in conditional mutant animals (Δ/Δ) compared with control (lox/lox) littermates. Total image intensity was determined for 10 sections of each genotype. For NeuN (g), we integrated only in the region of the gray matter, and for GFAP (h) intensity was averaged over the whole spinal cord section. Intensities are in arbitrary units. The number of cells positive for PDGFR-α mRNA was similar in mutant and control animals (o). By contrast, the number of immature OLs, assessed by counting cells positive for perinuclear MAG immunoreactivity (m and n) is significantly increased in mutant compared with control spinal cord (p, mean ± SD; ***, P < 0.001, t test). The precocious differentiation was confirmed by in situ hybridization for Plp/DM20 (k and l). Bar: (a–f and i–n) 10 μm.
Precocious appearance of OLs in the spinal cord at E17.5
Next, we examined spinal cords at E17.5, shortly before OLs start to accumulate in substantial numbers in the ventral fiber tracts of the mouse thoracic spinal cord. Initial X-gal staining in Cnp-Cre Δ/Δ R26R (Fig. 2 a) and Cnp-Cre Δ/wt R26R (Fig. 2 b) embryos showed no major differences in the pattern of recombined cells in the homozygous (Δ/Δ) compared with heterozygous (Δ/wt) mice. We examined the cellular composition of the neural tube in Cnp-Cre Δ/Δ versus control (lox/lox) mice using immunostaining and in situ hybridization for markers characteristic of neurons, astrocytes, OPCs, and immature OLs. We used antibodies against neuronal nuclear antigen (NeuN; Fig. 2, c and d), neurofilament (unpublished data), and β-tubulin III (unpublished data) for neurons, glial fibrillary acidic protein (GFAP) for astrocytes (Fig. 2, e and f), and detected PDGFR-α by in situ hybridization for OPCs (Fig. 2, i and j). None of these markers showed significant differences between mutant and control mice. The number of neurons and astrocytes was indirectly quantitated by determining the average overall intensity of NeuN and GFAP immunostaining of 10 representative sections from three embryos of each genotype. Staining and image processing were performed identically for sections of mutant and control animals. No significant differences were obtained, when intensity was calculated either per whole spinal cord cross section (Fig. 2, g and h) or per unit area (unpublished data). The size of the white matter area (i.e., that part of the spinal cord outside the area stained by NeuN) also did not differ significantly (38 ± 7.4 relative units for Cnp-Cre Δ/Δ vs. 42 ± 6.0 for control, P = 0.27, t test). However, for immature OLs, the situation was quite different. The number of cells expressing high levels of PLP/DM20 mRNA (Fig. 2, k and l) was strongly increased in the spinal cord of mutants compared with control animals, both in the gray matter and in the future white matter (PLP/DM-20 has been described to be expressed at earlier times in the ventral ventricular zone, but expression outside this region is characteristic of differentiated OLs; Richardson et al., 2000). An increased number of differentiating OLs was also observed by immunostaining for myelin-associated glycoprotein (MAG; Fig. 2, m and n). We found ∼10-fold more perinuclear MAG-positive cells per section compared with controls (Fig. 2 p). Interestingly, this increased number of immature OLs was not the result of an increased number of OPCs (PDGFR-α mRNA–positive cells; Fig. 2 o). Furthermore, no significant increase in proliferating cells was observed using phosphorylated histone H3 as a marker for cells in the S phase of the cell cycle (11.1 ± 5.2 positive cells per section in mutant animals vs. 10.1 ± 3.9 in control animals).
Ectopic appearance of immature OLs in the gray matter at birth
Dramatic changes in the number and spatial distribution of OLs were also evident in spinal cords of newborn (P0) Cnp-Cre Δ/Δ compared with control (lox/lox) mice (Fig. 3 , compare c and e with d and f). Many immature OLs were abnormally located in the gray matter. This altered distribution was seen along the entire length of the spinal cord in mutant mice (unpublished data). We quantified the MAG-positive cells showing perinuclear staining (immature OLs; Fig. 3 i), using their perinuclear staining to distinguish them from myelinating mature OLs (Trapp et al., 1997). After excluding the mature OLs at the ventral margin of both mutant and control spinal cords (Fig. 3, e and f; see also MBP staining for mature OLs in Fig. 3, g and h for comparison), there were approximately sixfold more immature OLs present in mutants as compared with controls (Fig. 3 l). Similar observations were made in Plp-Cre Δ/Δ mice (53 ± 19 vs. 5 ± 4 in mutant and control, respectively). At E17.5, we found no difference between the numbers of cells positive for PDGFR-α mRNA in Cnp-Cre Δ/Δ and control mice (Fig. 3, a, b, and k).
Figure 3.

OPCs and immature OLs in P0 Cnp-CreΔ/Δ spinal cord. Sections of thoracic spinal cord of Cnp-Cre Δ/Δ or control (lox/lox) mice were labeled with PDGFR-α cRNA or PLP cRNA (a–d), with anti-MAG (e, f, and i) or anti-MBP (g and h) antibodies, or with MAG antibodies combined with TUNEL staining (j). The number and location of PDGFR-α–positive OPCs was similar in mutant and control animals (a, b, and k). Also, the number and location of mature MBP-positive OLs in the developing white matter of the cervical spinal cord was not altered (g and h). In contrast, in the gray matter, there was a significant increase in ectopic immature OLs with extensive perinuclear MAG staining (i, quantified in panel l, mean ± SD; ***, P < 0.001). A subpopulation of these cells were TUNEL-positive (j, arrow). The percentage of apoptotic cells among perinuclear MAG-positive cells was significantly higher in mutant than in control spinal cord (m, 1,077 cells counted in mutant mice and 303 cells in control mice; [χ2 distribution] χ2= 86.29; **, P < 0.005). Bars: (a–h) 100 μm; (i and j) 10 μm.
Increased apoptosis of prematurely differentiated OLs
Although we observed ectopic perinuclear MAG-positive OLs in Cnp-Cre Δ/Δ mice, staining for MBP (a late marker for myelinating OLs) was absent from the gray matter of both thoracic (unpublished data) and cervical spinal cord (Fig. 3, g and h), even though the latter is at a more advanced stage of differentiation than the thoracic segment. However, some staining was observed in the future white matter of both mutant and control animals (Fig. 3, g and h). These observations raised the possibility that the ectopic immature OLs might be cleared by programmed cell death. To test this hypothesis, we double stained sections from Cnp-Cre Δ/Δ spinal cords at P0 for TUNEL (Gavrieli et al., 1992) and MAG (Fig. 3 j). Significantly more TUNEL-positive dying cells were found in mutant spinal cords: 14.9 ± 3.9 per section in mutants versus 7.8 ± 2.3 per section in controls (P < 0.001, n = 17, t test). Further, a higher percentage of MAG-positive cells were TUNEL-positive in mutant mice (Fig. 3 m). Note that most, but not all, of the dying MAG-positive immature OLs were localized in the gray matter. Together, these results suggest that the ectopic and precociously appearing immature OLs are eliminated before they fully differentiate into MBP-positive mature OLs.
Efficiency of Notch1 ablation
Because our multiple efforts to directly show elimination of the Notch1 protein failed due to a lack of reliable reagents, we demonstrated that Cre recombinase had been active in the differentiating OLs. We prepared short-term cultures of cells isolated from the spinal cord of single Cnp-Cre R26R mice homozygous for the floxed Notch1 allele (Δ/Δ). Heterozygous littermates (Δ/wt) were used as controls. The cells were first stained with X-gal (Fig. 4 , a and b) and then for O4 (Fig. 4, c and d), a marker for both immature and mature OLs (Hardy and Reynolds, 1991). 89 out of 100 and 71 out of 100 O4-positive cells derived from homozygous and heterozygous animals, respectively, were also positive for β-gal, demonstrating expression of Cre recombinase and efficient recombination of the R26R allele within the O4-positive cell population. To directly demonstrate Notch1 recombination, DNA was isolated from spinal cords of newborn mice and analyzed by Southern blotting (Fig. 4 e). Quantification by PhosphorImager analysis revealed ∼20–25% recombination in Cnp-Cre Δ/Δ whole spinal cord, after normalization to DNA isolated from control lox/0 mice.
Figure 4.
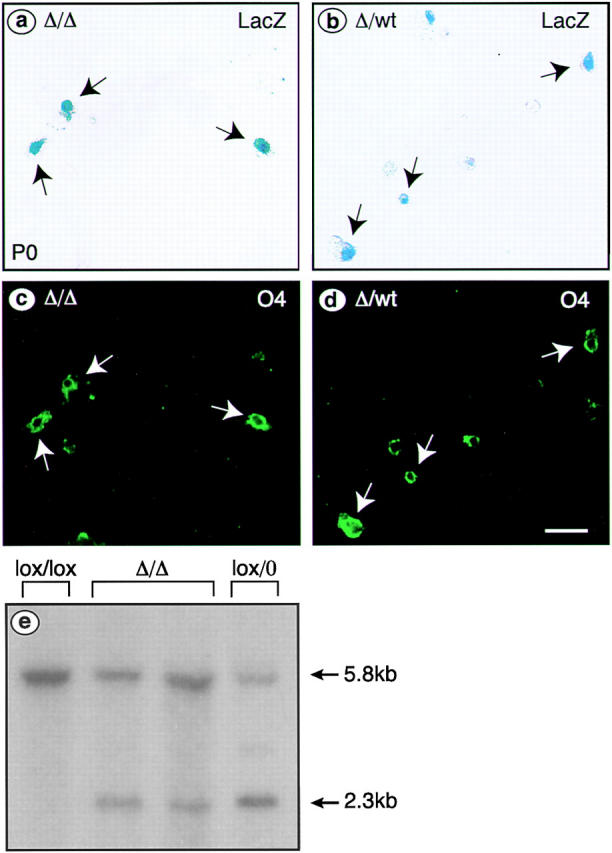
Recombination in cultured OL and efficiency of ablation of Notch1 in P0 Cnp-CreΔ/Δ spinal cord. Acutely dissociated cells isolated from individual P0 Cnp-Cre R26R mice homozygous for the floxed Notch1 allele (a and c, Δ/Δ) or from Cnp-Cre R26R heterozygous littermates (b and d, Δ/wt) were stained for X-gal (a and b) and O4 (c and d). Arrows mark double-labeled cells. (e) Southern blot analysis of DNA isolated from P0 spinal cord using a probe derived from the 5′ upstream region of the Notch1 locus revealed a 5.8-kb fragment from the wild-type allele after digestion with EcoRI. After recombination, a 2.3-kb fragment can be detected. Genotypes of the animals used are indicated above the lanes.
Early stages of OPC development are normal in Notch1 mutant embryos
Because expression of Cre recombinase starts as early as E11.5 in Cnp-Cre R26R mice (unpublished data), we examined the early development of OPCs in Cnp-Cre Δ/Δ mice. PDGFR-α mRNA–positive OPCs were found at E12.5 in the neural tube in homozygous (Δ/Δ) as well as in heterozygous (Δ/wt) mice (unpublished data). At E14.5, the PDGFR-α mRNA-positive OPCs were dispersed throughout the cross section (Fig. 5 , a and b), and their number was similar in mutants and controls (Fig. 5 g). MAG-positive cells were not detected at this time point (Fig. 5, c and d). Thus, early development of the OL lineage appeared normal in Cnp-Cre Δ/Δ mice. The distribution of motoneurons showed no major alterations in Cnp-Cre Δ/Δ mice at E12.5 (unpublished data), E14.5 (Fig. 5, e and f, arrows), and E17.5 (Fig. 2, a and b, arrows), as judged by immunostaining for the markers Isl1 and Isl2 (Tsuchida et al., 1994; Pfaff et al., 1996) and X-gal staining in Cnp-Cre Δ/Δ R26R mice.
Figure 5.
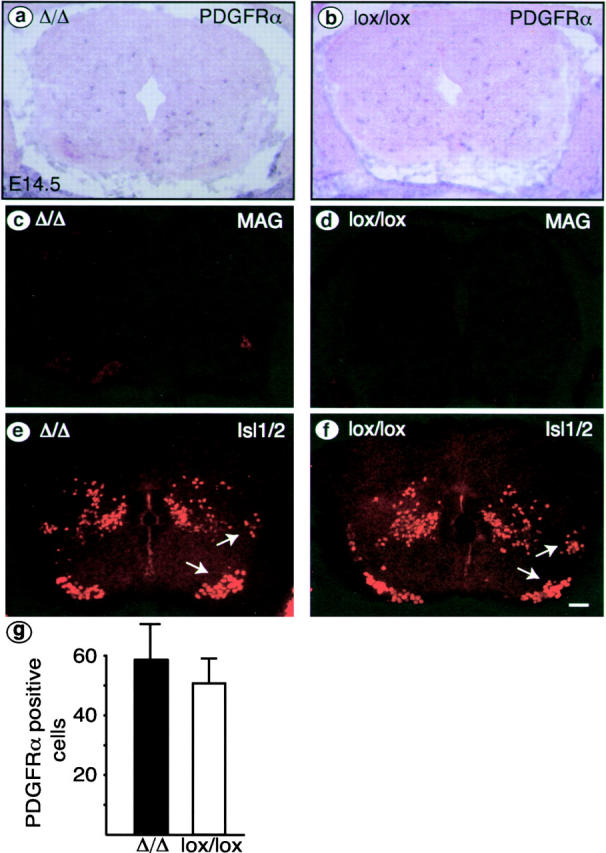
E14.5 spinal cord appears normal in Cnp-CreΔ/Δ mice. Transverse sections of E14.5 Cnp-Cre Δ/Δ and control (lox/lox) spinal cords following in situ hybridization (a and b) or immunohistochemistry (c–f). OPCs, OLs, and motoneurons were analyzed by the expression of PDGFR-α mRNA (a and b), MAG (c and d), and Isl1/2, respectively. Comparable numbers of OPCs (g) and no MAG- positive cells were observed. Motoneurons (e and f, arrows) appear normal; some interneurons and dorsal root ganglia are also marked by Isl1/2. Bar: 10 μm.
Precocious differentiation of OLs in the forebrain
We asked next whether precocious OL differentiation is confined exclusively to the spinal cord. Thus, we examined the forebrain, where OL differentiation starts slightly later compared with the spinal cord. As in the spinal cord, ablation of Notch1 in Cnp-Cre Δ/Δ mice led to a strong increase in the number of perinuclear MAG-positive immature OLs compared with controls, with a large fraction (∼50%) found in the gray matter (Fig. 6 , a–c). However, we did not observe precocious differentiation using antibodies against MAG in the optic nerve of P0 mutant animals (unpublished data), likely because OL differentiation starts later in the optic nerve.
Figure 6.

Precocious development of immature OLs in the cerebrum of P0 Cnp-CreΔ/Δ mice. Transverse sections of P0 cerebrum were immunostained for MAG. (b) At P0, there were many immature (perinuclear MAG-positive) OLs present in the mutant brain (Δ/Δ), both in white matter (WM) and in gray matter (GM, here cortex, Co), whereas these were rare in the control (lox/lox) brain (c). These results are quantitated in panel a, which shows the mean number of cells (±SD) with perinuclear MAG staining. Bar: 20 μm.
Discussion
In the present paper, we used defined conditional gene ablation to address the question of how the OL lineage is affected when Notch1 function is specifically eliminated in vivo. We provide direct evidence that Notch1 is critically important for correct temporal and spatial differentiation of OLs from the precursor to the immature stage in the mouse spinal cord by demonstrating that ablation of Notch1 in OPCs leads to ectopic production of prematurely differentiated immature OLs. Most of the prematurely differentiated OLs were found in the gray matter at P0 where immature OLs are usually very scarce in control animals. Apoptosis was strongly increased among the precociously differentiated cells. However, the number of OPCs was not altered at the ages examined, and early OL development appeared normal. Precocious differentiation was also observed in the anterior forebrain, suggesting that Notch1 signaling may play an important role in OL development throughout the CNS.
An important issue is whether the observed effects of Notch1 deletion on OPC differentiation depend directly on deletion within the OPCs themselves or are an indirect consequence of deletion in some other cell type. We used Cre-mediated recombination driven by Cre recombinase inserted into the Cnp locus. In agreement with published reports on CNP regulation and according to our own data, in the developing spinal cord Cre recombinase was expressed exclusively by early oligodendrocyte progenitors and motoneurons. Given the pattern of X-gal and immunostaining at E17.5, Cre is likely not to be expressed in astrocytes (Fig. 2, compare b with f). Development of neurons appears normal in the spinal cords of mutant mice (Fig. 2, c, d, and g). However, we cannot exclude subtle effects such as altered trophic support or abnormal electrical signaling from the periphery. Electrical activity of neurons has been shown to influence proliferation of OPCs (Barres and Raff, 1993) and the process of myelination (Demerens et al., 1996), but we are not aware of evidence that such electrical activity influences differentiation of OPCs into immature OLs (for review see Barres and Raff, 1999). OLs develop normally in explant cultures from spinal cord of Isl1 − / − mice (Sun et al., 1998), even though no motoneurons or V1 interneurons are produced (Pfaff et al., 1996). OLs also develop in hindbrain of Olig2 − / − mutants (Lu et al., 2002), which lack somatic motoneurons. The most likely interpretation of our results is therefore that they reflect a direct effect of Notch1 deletion in OPCs.
Although β-gal expression was seen as early as E11.5, the first immature OLs were not found until E17.5. Several possibilities may be envisaged to explain this delay. Persistence of Notch1 protein might render the cells effectively Notch1-positive for some time after recombination has occurred. This appears unlikely because, although we were not able to directly assess the lifetime of the Notch1 protein, the presence of a PEST sequence near the COOH terminus suggests that Notch1 turns over rapidly (Rechsteiner and Rogers, 1996). Alternatively, ablation of Notch1 might have an immediate effect, but progress through the developmental program might require several days (Durand and Raff, 2000). This would be consistent with the clonal analyses of purified precursor cells isolated from P7–8 (Barres et al., 1994) and E18 rat optic nerve (Gao et al., 1998) suggesting that a cell intrinsic program plays an important role in determining when OPCs stop dividing and differentiate. Finally, Notch1 may inhibit the transition from OPCs to immature OLs only relatively late in development, shortly before E17.5 in the mouse spinal cord.
Why do many of the prematurely differentiated OLs in Notch1 mutants die? Immunostaining with late differentiation markers and assessment of apoptosis suggest that precociously differentiated OLs cannot terminally differentiate and are unable to survive in the long term if they arise at the wrong place and time, most likely due to missing appropriate survival cues. Similar conclusions were reached by Calver et al. (1998). Overexpression of PDGF led to the appearance of an excess of immature OLs at ectopic sites in the gray matter of transgenic spinal cords, due to an excess production of OPCs, but the OLs survived in large numbers only in white matter. In our study, the number of precursors was normal, and it was precocious and uncontrolled differentiation that led to a high number of immature OLs in the gray matter. However, in both cases, these cells were eliminated by apoptosis, and mature OLs accumulated only in the white matter. It has been suggested that OPCs differentiate preferentially in fiber tracts (Hardy and Friedrich, 1996). However, our results and those of Calver et al. (1998) indicate that progenitor cells can differentiate both in gray and white matter, but survive in large numbers only in white matter.
The increase in immature OLs was not accompanied by a decrease in the number of OPCs. It has been proposed that expansion of OPCs is regulated by a density-dependent mechanism (Zhang and Miller, 1996), which could have served to prevent depletion of the OPC pool in our experiments. We did not observe a significant increase in the number of proliferating cells. However, if the OLs live considerably longer than the cell cycle time of the OPCs (which ranges from ≈30 h at E13 to ≈100 h at E17; van Heyningen et al., 2001), the expected increase in proliferation rate might have escaped detection. We also considered the possibility that Notch ablation might have produced MAG-positive OLs that were also still positive for early markers. We tested this by coimmunostaining for MAG and the early marker NG2, but found that the MAG-positive cells in the gray matter were NG2 negative (unpublished data).
Our analysis demonstrates that Notch1 is required for the regulation of the differentiation of OPCs to OLs, and thus contributes to the timing of oligodendrocyte differentiation in vivo. It has been suggested that this timing might be controlled by an intrinsic mechanism that allows a single OPC to undergo controlled cell cycle arrest and to differentiate in vitro without neurons (Temple and Raff, 1986). Our data and the observations by Wang et al. (1998) show that additional regulatory mechanisms that are mediated by direct cell–cell interactions via the Notch1 receptor and its ligands throughout the CNS are functional in this process. This most probably involves interactions between the axon and OPCs but there might also be a contribution by interactions between mature OLs and OPCs (Wang et al., 1998). The intracellular mechanisms that underlie precocious OL differentiation in the Notch conditional knockout animals remain unknown at this stage. The finding that maturation of OLs is strongly inhibited in the absence of the transcription factor Nkx2.2 (Qi et al., 2001) suggests the possibility that this transcription factor or one of its regulatory targets is antagonized by the Notch signaling pathway.
Very recently, examination of mutants heterozygous for a Notch1-null allele has provided evidence for an effect of Notch1 on myelination (Givogri et al., 2002). Several changes in the pattern of myelination were observed, including premature myelination in the cortex, increased myelination of small caliber axons in the optic nerve, and ectopic myelination in the molecular layer of the cerebellum. Some of these changes may reflect premature differentiation of OPCs, such as we describe here but in the heterozygous-null setting, there also seem to be effects on survival of OLs and on the choice of axons to be myelinated.
Cell–cell interactions mediated by Notch signaling are likely to play a pivotal role in development as part of a local regulatory circuit that forms the basis for the asynchronous myelination of the CNS and its different fiber tracts. The final trigger for OL differentiation is likely to be a down-regulation of the axonal component of the Notch1–ligand interaction, possibly linked to electrical activity (Rogister et al., 1999). Likewise, OPCs in the adult nervous system might be held in the undifferentiated state via this regulatory system. If correct, inhibition of Notch1 might be an effective way of activating these quiescent cells for repair in demyelinating diseases like multiple sclerosis. This is particularly warranted by the fact that lesions in multiple sclerosis are not starved of OPCs as previously thought, but rather that other factors, possibly including activated Notch signaling, are responsible for the failure of remyelination (Solanky et al., 2001).
Materials and methods
Mice
Mice carrying LoxP-flanked Notch1 alleles have been described previously (Radtke et al., 1999). Recombination between the LoxP sites reduces expression of Notch1 protein to undetectable levels, and no shorter fragments are seen (Wolfer et al., 2001). Mice carrying the Cre recombinase under the transcriptional control of the Plp (line PLCD) and Cnp regulatory elements will be described elsewhere. The ROSA26 mouse reporter line was provided by Dr. P. Soriano (Soriano, 1999). Embryos were generated by timed mating, counting the morning after pairing as E0.5. Procedures for animal experiments were approved by the Veterinary Office of the Canton of Zurich (permit 113/99).
For most experiments, we compared Cnp-Cre Δ/Δ or Plp-Cre Δ/Δ animals with lox/lox littermates as a control. Findings were reproduced from at least three animals of each genotype and from at least two different litters. Spinal cord sections were cut at the forelimb level in embryos and at the thoracic level in newborns, unless otherwise indicated. The lateral ventricles were used as landmarks for the brain sections.
X-gal histochemistry, in situ hybridization, immunofluorescence, and TUNEL staining
Timed pregnant mothers were killed, and embryos were isolated, fixed for 1–2 h in 4% paraformaldehyde, incubated for a few hours to overnight in 30% sucrose, embedded in OCT (TissueTech), and immediately frozen on dry ice. Newborns were anesthetized by intraperitoneal injection of a pentobarbital solution, and then perfused with a 0.9% saline solution followed by 4% ice-cold paraformaldehyde. Brains and spinal cords were isolated, fixed for 24–72 h in the same fixative at 4°C, incubated in 30% sucrose overnight, and embedded in OCT. 20-μm (embryos) or 5-μm (newborn)–frozen sections were thaw-mounted onto Superfrost slides (Mettler) and air dried.
For the X-gal staining, the sections were incubated overnight in a PBS solution containing 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2 mM magnesium chloride, and 2 mM X-gal (Calbiochem). In situ hybridizations were performed with digoxigenin-labeled RNA probes overnight at 72°C in buffer containing 50% formamide and detected using an anti–DIG-AP antibody according to the manufacturer's instructions (Roche Diagnostics). The PDGFR-α and PLP/DM20 plasmids were gifts from Drs. W.D. Richardson and B. Zalc, respectively. Immunohistochemistry was performed overnight at 4°C with antibodies against NeuN (1:100; Chemicon Inc.), GFAP (1:500; Accurate), β-tubulin III (1:300; Sigma-Aldrich), neurofilament 160 (1:20; Sigma-Aldrich) and phosphorylated histone H3 (1:100; Upstate Biotechnology). Antibodies against MBP (1:500), PLP/DM20 (1:500), Isl1/2 (1:500), and MAG (1:500) were provided by Drs. N. Baumann, I.R. Griffiths, T.M. Jessell, and J.L. Salzer, respectively. Secondary antibody incubations (1:300; Jackson Immunochemicals) were performed for 1 h at room temperature.
Apoptotic cell death was analyzed by TUNEL staining using biotin-labeled UTP and an FITC-conjugated streptavidin complex according to the manufacturer's instructions (Roche Diagnostics).
Images were collected using an Axiophot microscope (Carl Zeiss MicroImaging, Inc.) in conjunction with a ProgRes 3008 (Jenoptik) or Hamamatsu CCD camera. Image processing was performed with Adobe Photoshop® 5.0 software. Staining intensity was measured with NIH Image 1.62 software. The number of cells expressing PDGFR-α mRNA or showing perinuclear MAG staining was determined by counting all cells in at least 10 sections.
Primary cell dissociation
Spinal cords were individually isolated from newborn animals by incubation for 20 min at 37°C in 200 μl DME medium (Life Technologies) containing 2 mg/ml collagenase type IV (Worthington Biochemical Corp.), 1.2 mg/ml hyaluronidase type IV-S (Sigma-Aldrich) and 0.3 mg/ml trypsin inhibitor (Sigma-Aldrich). After trituration, dissociated cells were plated onto poly-d-lysine (Sigma-Aldrich)–coated dishes (35 mm; Corning) in Eagle's medium with 10% FCS (Sera-Tech) and maintained at 37°C in 5% CO2 overnight. These cells were used for X-gal and O4 staining (Fig. 4, a–d). The carcasses were genotyped by PCR.
Southern blot and quantification
Spinal cord was isolated from newborn animals and cells were mechanically dissociated as previously described (Milner and Ffrench-Constant, 1994). Southern blotting (5 μg EcoRI-digested genomic DNA) to a Hybond™-N+ membrane (Amersham Biosciences) was performed essentially as described by Radtke et al. (1999), using as probe a 750-bp BamHI-EcoRI fragment derived from the 5′ upstream region of the Notch1 locus. The probe was hybridized at 42°C, and the signals quantified using a PhosphorImager (Storm 820; Molecular Dynamics).
Acknowledgments
We thank Drs L. Sommer and V. Taylor for many helpful discussions and valuable advice, as well as for critically reading the manuscript. We are grateful to Drs. N. Baumann, I.R. Griffiths, T.M. Jessell, W.D. Richardson, J.L. Salzer, and B. Zalc for supplying us with plasmids and antibodies. We thank Kathrin Mannigel and Roger Staub for animal care.
This work was supported by the Swiss National Science Foundation and the Fifth European Community Framework Programme (Control of specification and migration of oligodendrocytes).
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; CNP, 2′,3′-cyclic nucleotide 3′-phosphodiesterase; CNS, central nervous system; GFAP, glial fibrillary acidic protein; MAG, myelin-associated glycoprotein; MBP, myelin basic protein; NeuN, neuronal nuclear antigen; OPC, oligodendrocyte precursor cell; OL, oligodendrocyte; PDGFR-α, PDGF receptor α; Plp, proteolipid protein.
References
- Apelqvist, A., H. Li, L. Sommer, P. Beatus, D.J. Anderson, T. Honjo, M. Hrabe de Angelis, U. Lendahl, and H. Edlund. 1999. Notch signalling controls pancreatic cell differentiation. Nature. 400:877–881. [DOI] [PubMed] [Google Scholar]
- Armstrong, R.C., H.H. Dorn, C.V. Kufta, E. Friedman, and M.E. Dubois-Dalcq. 1992. Pre-oligodendrocytes from adult human CNS. J. Neurosci. 12:1538–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas, S., K. Matsuno, and M.E. Fortini. 1995. Notch signaling. Science. 268:225–232. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas, S., M.D. Rand, and R.J. Lake. 1999. Notch signaling: cell fate control and signal integration in development. Science. 284:770–776. [DOI] [PubMed] [Google Scholar]
- Barres, B.A., and M.C. Raff. 1993. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 361:258–260. [DOI] [PubMed] [Google Scholar]
- Barres, B.A., and M.C. Raff. 1994. Control of oligodendrocyte number in the developing rat optic nerve. Neuron. 12:935–942. [DOI] [PubMed] [Google Scholar]
- Barres, B.A., and M.C. Raff. 1999. Axonal control of oligodendrocyte development. J. Cell Biol. 147:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres, B.A., I.K. Hart, H.S. Coles, J.F. Burne, J.T. Voyvodic, W.D. Richardson, and M.C. Raff. 1992. Cell death in the oligodendrocyte lineage. J. Neurobiol. 23:1221–1230. [DOI] [PubMed] [Google Scholar]
- Barres, B.A., M.A. Lazar, and M.C. Raff. 1994. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 120:1097–1108. [DOI] [PubMed] [Google Scholar]
- Berezovska, O., P. McLean, R. Knowles, M. Frosh, F.M. Lu, S.E. Lux, and B.T. Hyman. 1999. Notch1 inhibits neurite outgrowth in postmitotic primary neurons. Neuroscience. 93:433–439. [DOI] [PubMed] [Google Scholar]
- Blaschuk, K.L., and C. ffrench-Constant. 1998. Developmental neurobiology: notch is tops in the developing brain. Curr. Biol. 8:R334–R337. [DOI] [PubMed] [Google Scholar]
- Calver, A.R., A.C. Hall, W.P. Yu, F.S. Walsh, J.K. Heath, C. Betsholtz, and W.D. Richardson. 1998. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 20:869–882. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil, P. 2000. Cell death in the oligodendrocyte lineage: a molecular perspective of life/death decisions in development and disease. Glia. 29:124–135. [DOI] [PubMed] [Google Scholar]
- Chambers, C.B., Y. Peng, H. Nguyen, N. Gaiano, G. Fishell, and J.S. Nye. 2001. Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development. 128:689–702. [DOI] [PubMed] [Google Scholar]
- Chandross, K.J., R.I. Cohen, P. Paras, Jr., M. Gravel, P.E. Braun, and L.D. Hudson. 1999. Identification and characterization of early glial progenitors using a transgenic selection strategy. J. Neurosci. 19:759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon, R.A., A.G. Reaume, and J. Rossant. 1995. Notch1 is required for the coordinate segmentation of somites. Development. 121:1533–1545. [DOI] [PubMed] [Google Scholar]
- Demerens, C., B. Stankoff, M. Logak, P. Anglade, B. Allinquant, F. Couraud, B. Zalc, and C. Lubetzki. 1996. Induction of myelination in the central nervous system by electrical activity. Proc. Natl. Acad. Sci. USA. 93:9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, B., and M. Raff. 2000. A cell-intrinsic timer that operates during oligodendrocyte development. Bioessays. 22:64–71. [DOI] [PubMed] [Google Scholar]
- Franklin, J.L., B.E. Berechid, F.B. Cutting, A. Presente, C.B. Chambers, D.R. Foltz, A. Ferreira, and J.S. Nye. 1999. Autonomous and non-autonomous regulation of mammalian neurite development by Notch1 and Delta1. Curr. Biol. 9:1448–1457. [DOI] [PubMed] [Google Scholar]
- Fu, H., Y. Qi, M. Tan, J. Cai, H. Takebayashi, M. Nakafuku, W. Richardson, and M. Qiu. 2002. Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development. 129:681–693. [DOI] [PubMed] [Google Scholar]
- Furukawa, T., S. Mukherjee, Z.Z. Bao, E.M. Morrow, and C.L. Cepko. 2000. rax, Hes1, and notch1 promote the formation of Müller glia by postnatal retinal progenitor cells. Neuron. 26:383–394. [DOI] [PubMed] [Google Scholar]
- Gaiano, N., J.S. Nye, and G. Fishell. 2000. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 26:395–404. [DOI] [PubMed] [Google Scholar]
- Gao, F.B., J. Apperly, and M. Raff. 1998. Cell-intrinsic timers and thyroid hormone regulate the probability of cell-cycle withdrawal and differentiation of oligodendrocyte precursor cells. Dev. Biol. 197:54–66. [DOI] [PubMed] [Google Scholar]
- Gard, A.L., and S.E. Pfeiffer. 1990. Two proliferative stages of the oligodendrocyte lineage (A2B5+O4− and O4+GalC−) under different mitogenic control. Neuron. 5:615–625. [DOI] [PubMed]
- Gavrieli, Y., Y. Sherman, and S.A. Ben-Sasson. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givogri, M.I., R.M. Costa, V. Schonmann, A.J. Silva, A.T. Campagnoni, and E.R. Bongarzone. 2002. Central nervous system myelination in mice with deficient expression of Notch1 receptor. J. Neurosci. Res. 67:309–320. [DOI] [PubMed] [Google Scholar]
- Gravel, M., A. Di Polo, P.B. Valera, and P.E. Braun. 1998. Four-kilobase sequence of the mouse CNP gene directs spatial and temporal expression of lacZ in transgenic mice. J. Neurosci. Res. 53:393–404. [DOI] [PubMed] [Google Scholar]
- Hardy, R., and R. Reynolds. 1991. Proliferation and differentiation potential of rat forebrain oligodendroglial progenitors both in vitro and in vivo. Development. 111:1061–1080. [DOI] [PubMed] [Google Scholar]
- Hardy, R.J., and V.L. Friedrich, Jr. 1996. Oligodendrocyte progenitors are generated throughout the embryonic mouse brain, but differentiate in restricted foci. Development. 122:2059–2069. [DOI] [PubMed] [Google Scholar]
- Irvin, D.K., S.D. Zurcher, T. Nguyen, G. Weinmaster, and H.I. Kornblum. 2001. Expression patterns of Notch1, Notch2, and Notch3 suggest multiple functional roles for the Notch-DSL signaling system during brain development. J. Comp. Neurol. 436:167–181. [PubMed] [Google Scholar]
- Ito, T., N. Udaka, T. Yazawa, K. Okudela, H. Hayashi, T. Sudo, F. Guillemot, R. Kageyama, and H. Kitamura. 2000. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development. 127:3913–3921. [DOI] [PubMed] [Google Scholar]
- Jordan, C., V. Friedrich, Jr., and M. Dubois-Dalcq. 1989. In situ hybridization analysis of myelin gene transcripts in developing mouse spinal cord. J. Neurosci. 9:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, T., and M. Raff. 2000. a. Basic helix-loop-helix proteins and the timing of oligodendrocyte differentiation. Development. 127:2989–2998. [DOI] [PubMed] [Google Scholar]
- Kondo, T., and M. Raff. 2000. b. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 19:1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsell, C.E., J. Boulter, G. diSibio, A. Gossler, and G. Weinmaster. 1996. Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol. Cell. Neurosci. 8:14–27. [DOI] [PubMed] [Google Scholar]
- Lu, Q.R., L. Cai, D. Rowitch, C.L. Cepko, and C.D. Stiles. 2001. Ectopic expression of Olig1 promotes oligodendrocyte formation and reduces neuronal survival in developing mouse cortex. Nat. Neurosci. 4:973–974. [DOI] [PubMed] [Google Scholar]
- Lu, Q.R., T. Sun, Z. Zhu, N. Ma, M. Garcia, C.D. Stiles, and D.H. Rowitch. 2002. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 109:75–86. [DOI] [PubMed] [Google Scholar]
- Lütolf, S., F. Radtke, M. Aguet, U. Suter, and V. Taylor. 2002. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development. 129:373–385. [DOI] [PubMed] [Google Scholar]
- McLaughlin, K.A., M.S. Rones, and M. Mercola. 2000. Notch regulates cell fate in the developing pronephros. Dev. Biol. 227:567–580. [DOI] [PubMed] [Google Scholar]
- Miller, R.H. 1996. Oligodendrocyte origins. Trends Neurosci. 19:92–96. [DOI] [PubMed] [Google Scholar]
- Miller, R.H., J. Payne, L. Milner, H. Zhang, and D.M. Orentas. 1997. Spinal cord oligodendrocytes develop from a limited number of migratory highly proliferative precursors. J. Neurosci. Res. 50:157–168. [DOI] [PubMed] [Google Scholar]
- Milner, R., and C. Ffrench-Constant. 1994. A developmental analysis of oligodendroglial integrins in primary cells: changes in alpha v-associated beta subunits during differentiation. Development. 120:3497–3506. [DOI] [PubMed] [Google Scholar]
- Morrison, S.J., S.E. Perez, Z. Qiao, J.M. Verdi, C. Hicks, G. Weinmaster, and D.J. Anderson. 2000. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 101:499–510. [DOI] [PubMed] [Google Scholar]
- Nijjar, S.S., H.A. Crosby, L. Wallace, S.G. Hubscher, and A.J. Strain. 2001. Notch receptor expression in adult human liver: a possible role in bile duct formation and hepatic neovascularization. Hepatology. 34:1184–1192. [DOI] [PubMed] [Google Scholar]
- Noll, E., and R.H. Miller. 1993. Oligodendrocyte precursors originate at the ventral ventricular zone dorsal to the ventral midline region in the embryonic rat spinal cord. Development. 118:563–573. [DOI] [PubMed] [Google Scholar]
- Pfaff, S.L., M. Mendelsohn, C.L. Stewart, T. Edlund, and T.M. Jessell. 1996. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 84:309–320. [DOI] [PubMed] [Google Scholar]
- Pringle, N.P., and W.D. Richardson. 1993. A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development. 117:525–533. [DOI] [PubMed] [Google Scholar]
- Qi, Y., J. Cai, Y. Wu, R. Wu, J. Lee, H. Fu, M. Rao, L. Sussel, J. Rubenstein, and M. Qiu. 2001. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development. 128:2723–2733. [DOI] [PubMed] [Google Scholar]
- Radtke, F., A. Wilson, G. Stark, M. Bauer, J. van Meerwijk, H.R. MacDonald, and M. Aguet. 1999. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 10:547–558. [DOI] [PubMed] [Google Scholar]
- Raff, M.C., B. Durand, and F.B. Gao. 1998. Cell number control and timing in animal development: the oligodendrocyte cell lineage. Int. J. Dev. Biol. 42:263–267. [PubMed] [Google Scholar]
- Rechsteiner, M., and S.W. Rogers. 1996. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21:267–271. [PubMed] [Google Scholar]
- Redmond, L., S.R. Oh, C. Hicks, G. Weinmaster, and A. Ghosh. 2000. Nuclear Notch1 signaling and the regulation of dendritic development. Nat. Neurosci. 3:30–40. [DOI] [PubMed] [Google Scholar]
- Richardson, W.D., N.P. Pringle, W.P. Yu, and A.C. Hall. 1997. Origins of spinal cord oligodendrocytes: possible developmental and evolutionary relationships with motor neurons. Dev. Neurosci. 19:58–68. [DOI] [PubMed] [Google Scholar]
- Richardson, W.D., H.K. Smith, T. Sun, N.P. Pringle, A. Hall, and R. Woodruff. 2000. Oligodendrocyte lineage and the motor neuron connection. Glia. 29:136–142. [DOI] [PubMed] [Google Scholar]
- Robey, E. 1997. Notch in vertebrates. Curr. Opin. Genet. Dev. 7:551–557. [DOI] [PubMed] [Google Scholar]
- Rogister, B., T. Ben-Hur, and M. Dubois-Dalcq. 1999. From neural stem cells to myelinating oligodendrocytes. Mol. Cell. Neurosci. 14:287–300. [DOI] [PubMed] [Google Scholar]
- Sestan, N., S. Artavanis-Tsakonas, and P. Rakic. 1999. Contact-dependent inhibition of cortical neurite growth mediated by notch signaling. Science. 286:741–746. [DOI] [PubMed] [Google Scholar]
- Solanky, M., Y. Maeda, X. Ming, W. Husar, W. Li, S. Cook, and P. Dowling. 2001. Proliferating oligodendrocytes are present in both active and chronic inactive multiple sclerosis plaques. J. Neurosci. Res. 65:308–317. [DOI] [PubMed] [Google Scholar]
- Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- Spassky, N., C. Goujet-Zalc, E. Parmantier, C. Olivier, S. Martinez, A. Ivanova, K. Ikenaka, W. Macklin, I. Cerruti, B. Zalc, and J.L. Thomas. 1998. Multiple restricted origin of oligodendrocytes. J. Neurosci. 18:8331–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky, N., C. Olivier, E. Perez-Villegas, C. Goujet-Zalc, S. Martinez, J. Thomas, and B. Zalc. 2000. Single or multiple oligodendroglial lineages: a controversy. Glia. 29:143–148. [PubMed] [Google Scholar]
- Sun, T., N.P. Pringle, A.P. Hardy, W.D. Richardson, and H.K. Smith. 1998. Pax6 influences the time and site of origin of glial precursors in the ventral neural tube. Mol. Cell. Neurosci. 12:228–239. [DOI] [PubMed] [Google Scholar]
- Swiatek, P.J., C.E. Lindsell, F.F. del Amo, G. Weinmaster, and T. Gridley. 1994. Notch1 is essential for postimplantation development in mice. Genes Dev. 8:707–719. [DOI] [PubMed] [Google Scholar]
- Tanigaki, K., F. Nogaki, J. Takahashi, K. Tashiro, H. Kurooka, and T. Honjo. 2001. Notch1 and Notch3 instructively restrict bFGF-responsive multipotent neural progenitor cells to an astroglial fate. Neuron. 29:45–55. [DOI] [PubMed] [Google Scholar]
- Temple, S., and M.C. Raff. 1986. Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell. 44:773–779. [DOI] [PubMed] [Google Scholar]
- Timsit, S., S. Martinez, B. Allinquant, F. Peyron, L. Puelles, and B. Zalc. 1995. Oligodendrocytes originate in a restricted zone of the embryonic ventral neural tube defined by DM-20 mRNA expression. J. Neurosci. 15:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, B.D., A. Nishiyama, D. Cheng, and W. Macklin. 1997. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol. 137:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida, T., M. Ensini, S.B. Morton, M. Baldassare, T. Edlund, T.M. Jessell, and S.L. Pfaff. 1994. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 79:957–970. [DOI] [PubMed] [Google Scholar]
- van Heyningen, P., A.R. Calver, and W.D. Richardson. 2001. Control of progenitor cell number by mitogen supply and demand. Curr. Biol. 11:232–241. [DOI] [PubMed] [Google Scholar]
- Wakamatsu, Y., T.M. Maynard, and J.A. Weston. 2000. Fate determination of neural crest cells by NOTCH-mediated lateral inhibition and asymmetrical cell division during gangliogenesis. Development. 127:2811–2821. [DOI] [PubMed] [Google Scholar]
- Wang, S., A.D. Sdrulla, G. diSibio, G. Bush, D. Nofziger, C. Hicks, G. Weinmaster, and B.A. Barres. 1998. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 21:63–75. [DOI] [PubMed] [Google Scholar]
- Wang, S., A. Sdrulla, J.E. Johnson, Y. Yokota, and B.A. Barres. 2001. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron. 29:603–614. [DOI] [PubMed] [Google Scholar]
- Weinmaster, G. 1998. Notch signaling: direct or what? Curr. Opin. Genet. Dev. 8:436–442. [DOI] [PubMed] [Google Scholar]
- Wolfer, A., T. Bakker, A. Wilson, M. Nicolas, V. Ioannidis, D.R. Littman, P.P. Lee, C.B. Wilson, W. Held, H.R. MacDonald, and F. Radtke. 2001. Inactivation of Notch 1 in immature thymocytes does not perturb CD4 or CD8T cell development. Nat. Immunol. 2:235–241. [DOI] [PubMed] [Google Scholar]
- Yu, W.P., E.J. Collarini, N.P. Pringle, and W.D. Richardson. 1994. Embryonic expression of myelin genes: evidence for a focal source of oligodendrocyte precursors in the ventricular zone of the neural tube. Neuron. 12:1353–1362. [DOI] [PubMed] [Google Scholar]
- Zhang, H., and R.H. Miller. 1996. Density-dependent feedback inhibition of oligodendrocyte precursor expansion. J. Neurosci. 16:6886–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q., G. Choi, and D.J. Anderson. 2001. The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron. 31:791–807. [DOI] [PubMed] [Google Scholar]