

Abstract
Biomineralization is a highly regulated process that plays a major role during the development of skeletal tissues. Despite its obvious importance, little is known about its regulation. Previously, it has been demonstrated that retinoic acid (RA) stimulates terminal differentiation and mineralization of growth plate chondrocytes (Iwamoto, M., I.M. Shapiro, K. Yagumi, A.L. Boskey, P.S. Leboy, S.L. Adams, and M. Pacifici. 1993. Exp. Cell Res. 207:413–420). In this study, we provide evidence that RA treatment of growth plate chondrocytes caused a series of events eventually leading to mineralization of these cultures: increase in cytosolic calcium concentration, followed by up-regulation of annexin II, V, and VI gene expression, and release of annexin II–, V–, VI– and alkaline phosphatase–containing matrix vesicles. Cotreatment of growth plate chondrocytes with RA and BAPTA-AM, a cell permeable Ca2+ chelator, inhibited the up-regulation of annexin gene expression and mineralization of these cultures. Interestingly, only matrix vesicles isolated from RA-treated cells that contained annexins, were able to take up Ca2+ and mineralize, whereas vesicles isolated from untreated or RA/BAPTA-treated cells, that contained no or only little annexins were not able to take up Ca2+ and mineralize. Cotreatment of chondrocytes with RA and EDTA revealed that increases in the cytosolic calcium concentration were due to influx of extracellular calcium. Interestingly, the novel 1,4-benzothiazepine derivative K-201, a specific annexin Ca2+ channel blocker, or antibodies specific for annexin II, V, or VI inhibited the increases in cytosolic calcium concentration in RA-treated chondrocytes. These findings indicate that annexins II, V, and VI form Ca2+ channels in the plasma membrane of terminally differentiated growth plate chondrocytes and mediate Ca2+ influx into these cells. The resulting increased cytosolic calcium concentration leads to a further up-regulation of annexin II, V, and VI gene expression, the release of annexin II–, V–, VI– and alkaline phosphatase–containing matrix vesicles, and the initiation of mineralization by these vesicles.
Keywords: annexin; calcium; matrix vesicles; mineralization; retinoic acid
Introduction
Mineralization plays a crucial role during normal development and replacement of cartilaginous skeleton via endochondral ossification. Mineralization of growth plate cartilage is restricted to terminally differentiated chondrocytes, and thus the mineralization process must be under cellular control (Kirsch and von der Mark, 1992; Kirsch et al., 1997b). In general, mineralization is restricted to skeletal tissues and teeth, where it is required for the proper function of these tissues. However, uncontrolled (pathological) mineralization can occur in all soft tissues. For example, excessive mineral deposition accompanies osteoarthritis. Crystal formation in articular cartilage may play a major role in the onset of inflammation and cartilage destruction (Doyle, 1982; Mitchell et al., 1992; Kirsch et al., 2000c). Calcification of cardiovascular tissues including arteries, heart valves, and cardiac muscle contributes to morbidity and mortality (Giachelli, 1999).
Annexins II, V, and VI are highly expressed in hypertrophic and mineralizing growth plate cartilage and in bone. These molecules belong to the annexin protein family, which have in common that they bind to acidic phospholipids in the presence of calcium (Crompton et al., 1988; Genge et al., 1989; Kirsch et al., 2000c, 2001). Annexin II and V each contain four 70–80 amino acid repeats with an annexin consensus sequence, whereas annexin VI contains eight such repeats. These four or eight repeats form the conserved core region, which is responsible for the Ca2+-dependent binding of the proteins to phospholipids. In contrast, the NH2-terminal regions of the annexins are highly variable and may contribute to the specific functions of the various annexins (Geisow et al., 1988). Previous studies have shown that annexins II, V, and VI form Ca2+ channels when inserted into artificial phospholipid bilayers or mediate Ca2+ influx into liposomes (Berendes et al., 1993; Chen et al., 1993; Benz et al., 1996; Burger et al., 1996; Matsuda et al., 1997; Kirsch et al., 2000b). Recently, we have demonstrated that annexins II, V, and VI form Ca2+ channels in matrix vesicles (Kirsch et al., 2000b). Matrix vesicles are particles that are released from the plasma membrane of mineralizing cells. These vesicles have the critical role of initiating the mineralization process (Anderson, 1995; Kirsch et al., 1997b). The first mineral phase forms inside the vesicles in a protected environment. Once these intralumenal crystals have reached a certain size they rupture the vesicle membrane and grow out into the extracellular matrix (Anderson, 1995). For the formation of the first intralumenal crystals, phase channel or transporter systems are required to mediate influx of mineral ions into the vesicles. Annexins II, V, and VI enable the influx of Ca2+ into matrix vesicles and the formation of the first intralumenal mineral phase. The inhibition of annexin Ca2+ channel activities led to a loss of the ability of these vesicles to mineralize (Kirsch et al., 2000b). Other studies have shown that matrix vesicles contain a Na+-coupled Pi symport system that mediates the influx of inorganic phosphate into the vesicles (Montessuit et al., 1991, 1995). The total amounts of Ca2+ and Pi in matrix vesicles are significantly higher than those in adjacent cells or in the extracellular fluid. The vast majority of Ca2+ and about half of Pi are in insoluble, protein/lipid-bound form in the vesicles, which allows permanent annexin-mediated Ca2+ influx into the vesicles.
Ultrastructural studies have shown a high concentration of matrix vesicles in the reserve zone of growth plate cartilage and another peak concentration in the hypertrophic zone just before the onset of matrix mineralization (Reinholt et al., 1982; Buckwalter et al., 1987). Furthermore, we have described a qualitative difference between vesicles released by the various growth plate chondrocytes, and that only mineralization-competent growth plate chondrocytes release specialized matrix vesicles that are capable of initiating the mineralization process, whereas nonmineralizing growth plate chondrocytes release vesicles that do not mineralize (Kirsch et al., 1997b, 2000a). Thus, it is reasonable to hypothesize that chondrocytes regulate the mineralization process by controlling the release of specialized matrix vesicles and that only mineralization-competent chondrocytes release matrix vesicles that initiate the mineralization process. However, very little is known about the mechanisms involved in regulating the release of these mineralization-competent matrix vesicles from the plasma membrane of growth plate chondrocytes.
Calcium is recognized as an important regulatory element for many cellular processes. Increase in the cytosolic calcium concentration, [Ca2+]i, which occurs in many cell types after stimulation by hormones, controls a diverse range of cell functions including adhesion, motility, gene expression, and proliferation. Several studies have provided evidences that growth plate chondrocytes accumulate large amounts of cytosolic calcium just before the initiation of mineralization (Gunter et al., 1990; Kirsch et al., 1992; Iannotti et al., 1994). Nevertheless, little is known about the mechanisms involved in alterations of Ca2+ homeostasis in growth plate chondrocytes and the role of altered Ca2+ homeostasis in chondrocyte differentiation and mineralization. Interestingly, vesiculation or ectocytosis of the plasma membrane has been shown to be dependent on an influx of calcium in the cytoplasm of many cell types. For example, Ca2+ ionophores can cause microvesiculation and membrane shedding in a variety of cell types including chondrocytes, fibroblasts, neutrophils, and platelets (Wiedmer et al., 1990; Iannotti et al., 1994; Bucki et al., 1998). Thus, it is possible that annexins II, V, and VI form Ca2+ channels in hypertrophic growth plate chondrocytes leading to influx of Ca2+ and alterations of Ca2+ homeostasis. The altered Ca2+ homeostasis leads to the release of annexin-containing, mineralization-competent matrix vesicles.
Previously we and others have shown that retinoic acid (RA)* affects the release of annexin-containing matrix vesicles and stimulates mineralization of hypertrophic growth plate chondrocytes (Iwamoto et al., 1993; Kirsch et al., 2000a). To test our hypothesis that annexin-mediated alterations in Ca2+ homeostasis of growth plate chondrocytes might regulate the mineralization process of growth plate chondrocytes, we cultured hypertrophic growth plate chondrocytes in the presence of RA and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), a cell permeable Ca2+ chelator, K-201, a specific annexin Ca2+ channel blocker (Kaneko, 1994; Kaneko et al., 1997; Kirsch et al., 2000b), or antibodies specific for annexin II, V, or VI. We measured changes in [Ca2+]i, and characterized matrix vesicles isolated from these cultures and the degree of mineralization in these cultures.
Results
Alteration in Ca2+ homeostasis and mineralization of growth plate chondrocytes
We and others have shown that RA treatment stimulates mineralization of hypertrophic growth plate chondrocytes (Iwamoto et al., 1993; Kirsch et al., 2000a). In addition, hypertrophic differentiation of chondrocytes is accompanied by an increase in [Ca2+]i (Iannotti and Brighton, 1989; Gunter et al., 1990; Kirsch et al., 1992). Since alterations in Ca2+ homeostasis affect differentiation processes in many cell types, we first determined whether RA treatment alters Ca2+ homeostasis of growth plate chondrocytes. As shown in Fig. 1, 1-d treatment of chondrocytes isolated from the hypertrophic zone of 19-d embryonic chick tibia growth plate cartilage with RA led to an approximately threefold increase in [Ca2+]i ([Ca2+]i of untreated chondrocytes, 887 nM; [Ca2+]i of RA-treated chondrocytes, 2,867 nM). Cotreatment of growth plate chondrocytes with RA and EDTA abolished the increase in [Ca2+]i, indicating that the increase in [Ca2+]i was due to the influx of extracellular Ca2+ into the cells.
Figure 1.
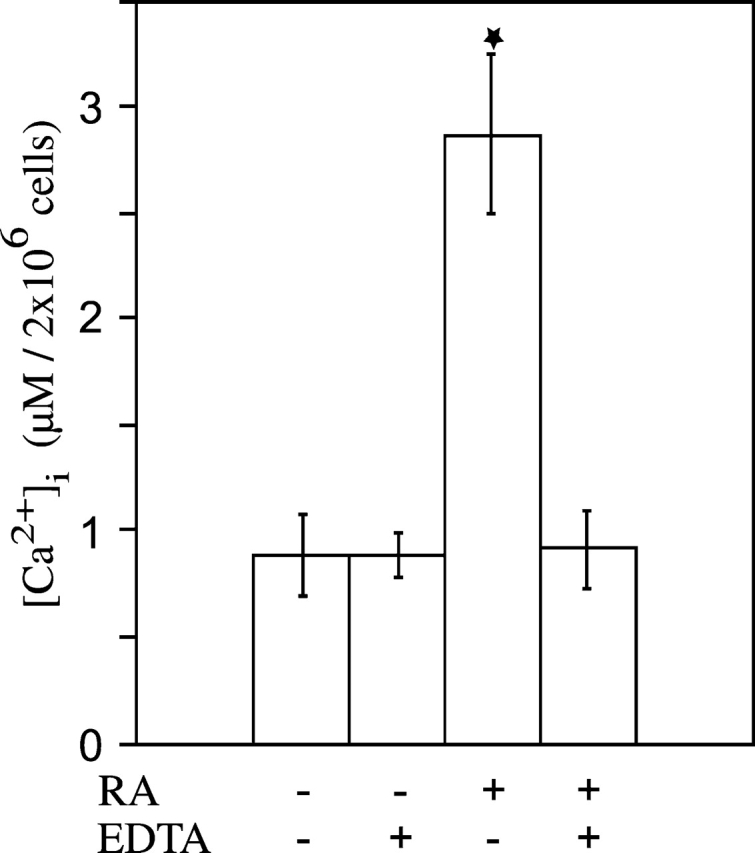
Cytosolic calcium concentration of untreated, RA- and RA/EDTA-treated growth plate chondrocytes. After growth plate chondrocytes isolated from 19-d chick embryonic tibia reached confluency, the cells were cultured in the absence or presence of 35 nM RA, or RA and 5 mM EDTA for 1 d. The cytosolic calcium concentration, [Ca2+]i, of chondrocytes was measured using fura-2AM as described in Materials and methods. Data were obtained from four different experiments and values are mean ± SD. (★, P ≤ 0.01 vs. untreated cultures.)
To determine whether the increase in [Ca2+]i affects mineralization of growth plate chondrocytes, we treated cells with RA and BAPTA-AM. After 6-d treatment with RA, cultures were heavily calcified as indicated by the intense alizarin red S staining (Fig. 2). In contrast untreated or RA/BAPTA-treated cultures showed significantly less alizarin red S staining (Fig. 2). Alkaline phosphatase (APase) activity, an enzyme whose activity is up-regulated just before the onset of mineralization (Bonucci et al., 1992), was also significantly increased in RA-treated cells compared with the APase activities in untreated or RA/BAPTA-treated cells (Fig. 3).
Figure 2.
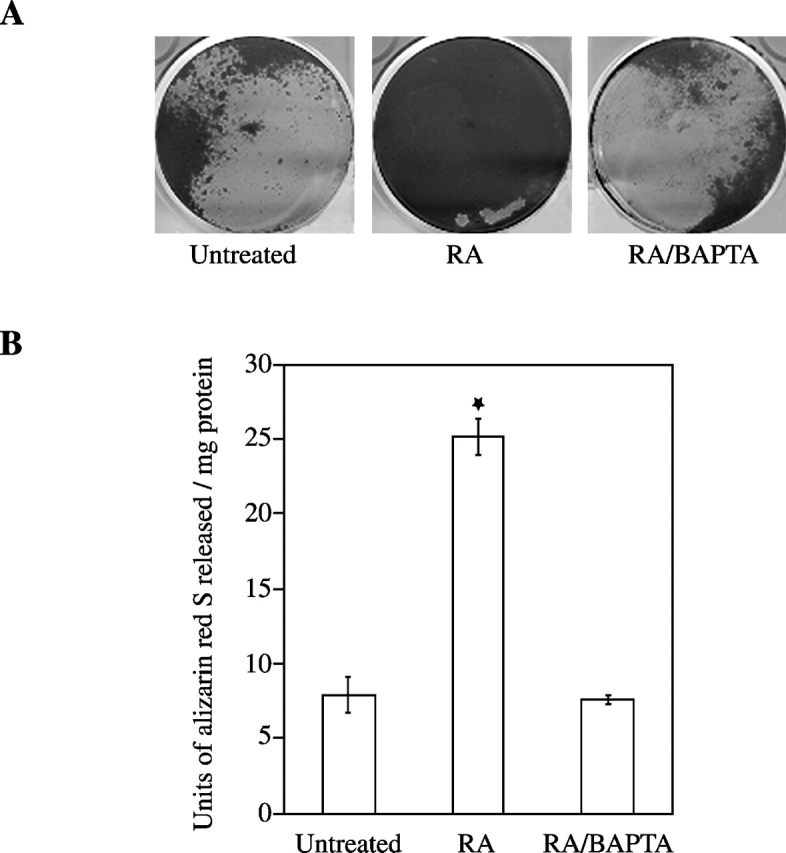
Extent of matrix mineralization in chondrocyte cultures treated with RA or RA/BAPTA. Growth plate chondrocytes were treated with RA or RA and BAPTA for 6 d. (A) Note the intense alizarin red S staining in cultures treated with RA. In contrast, less staining was detected in untreated or RA/BAPTA-treated cultures. (B) To quantitate the alizarin red S stain, each dish was incubated with 100 mM cetylpyridium chloride for 1 h. The alizarin red stain released into solution was collected, diluted when necessary, and read as units of alizarin red released (1 unit is equivalent to 1 unit optical density at 570 nm) per mg of protein. Data were obtained from four different experiments and values are mean ± SD. (★, P ≤ 0.01 vs. untreated cultures.)
Figure 3.
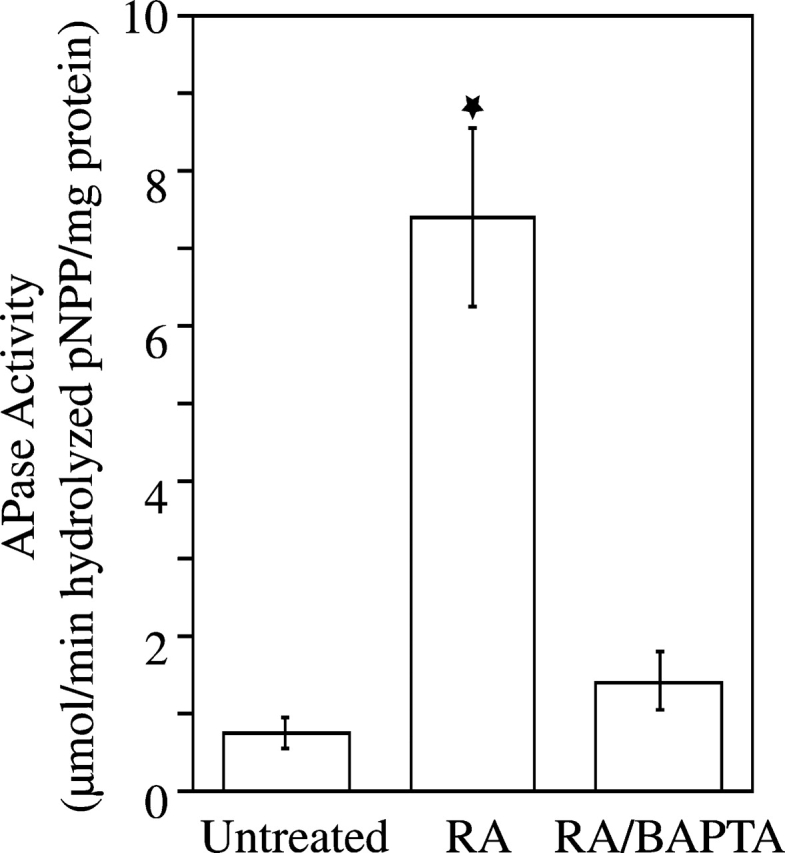
Alkaline phosphatase (APase) activity in untreated, RA-, and RA/BAPTA-treated chondrocyte cultures. After 6-d treatment, APase activity in the cell layer of RA-treated was significantly higher than APase activities in untreated or RA/BAPTA-treated cultures. Data were obtained from four different experiments; values are mean ± SD. (★, P ≤ 0.01 vs. untreated cultures.)
Previous studies have shown that matrix vesicles, which are released from the plasma membrane of mineralizing chondrocytes, initiate the mineralization process (Anderson, 1995; Kirsch et al., 1997b). In addition, we have demonstrated that only matrix vesicles that contain annexins II, V, and VI, and APase were able to initiate mineralization (Kirsch et al., 1997b). To test if alterations of Ca2+ homeostasis affect matrix vesicle release and/or composition, we isolated matrix vesicles from untreated, RA-treated, and RA/BAPTA-treated cultures and compared their composition and functions. APase activity (Fig. 4) and the amount of annexins II, V, and VI (Fig. 5) were significantly increased in matrix vesicles isolated from RA-treated cultures compared with vesicles isolated from untreated cultures. Matrix vesicles isolated from RA-treated chondrocytes were able to take up significant amounts of Ca2+ when incubated in synthetic cartilage lymph for 24 h. In contrast, vesicles isolated from untreated cultures were not able to take up significant amounts of Ca2+ (Fig. 6), confirming our previous findings that only vesicles containing Ca2+ channels formed by annexin II, V, and VI are able to take up Ca2+ (Kirsch et al., 1997b, 2000b). Interestingly, matrix vesicles isolated from RA/BAPTA-treated cultures showed similar properties as vesicles isolated from untreated cultures. These vesicles contained little APase activity, annexins II, V, and VI, and showed no significant Ca2+ uptake (Figs. 4–6). These findings indicate that alterations of Ca2+ homeostasis in growth plate chondrocytes regulate the release of mineralization-competent matrix vesicles and subsequent mineralization.
Figure 4.

Alkaline phosphatase (APase) activity in matrix vesicles isolated from untreated, RA- and RA/BAPTA-treated growth plate chondrocytes. After 3 d, matrix vesicles were isolated from the cell layer of untreated, RA-, and RA/BAPTA-treated chondrocytes as described in Materials and methods. APase activity was >10-fold increased in matrix vesicles isolated from RA-treated cultures compared with the activity in vesicles isolated from untreated or RA/BAPTA-treated cultures. Data were obtained from four different experiments; values are mean ± SD. (★, P ≤ 0.01 vs. APase activity of vesicles isolated from untreated cultures.)
Figure 5.
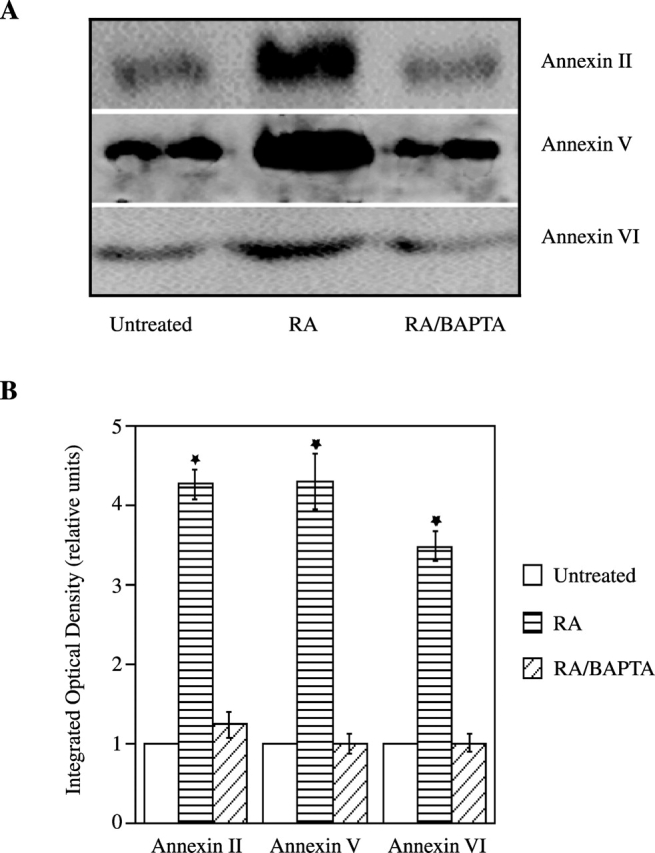
Amount of annexins II, V, and VI in matrix vesicles isolated from untreated, RA- or RA/BAPTA-treated chondrocytes. Matrix vesicle fractions (50 μg of total protein) isolated from 3-d untreated, RA-, or RA/BAPTA-treated cultures were subjected to SDS-PAGE and immunoblotting using antibodies specific for annexin II, V, or VI (A). The optical densities of the annexin bands were quantitated by densitometry. The optical densities obtained for annexin bands in matrix vesicle fractions isolated from untreated cultures were set as 1. Data were obtained from four different experiments; values are mean ± SD. (★, P ≤ 0.01 vs. vesicles isolated from untreated cultures.)
Figure 6.

Ca 2+ uptake by matrix vesicles isolated from untreated, RA-, and RA/BAPTA-treated chondrocytes. Matrix vesicles were isolated from 3-d untreated, RA- or RA/BAPTA-treated chondrocytes. The vesicle fractions (100 μg of total protein) were incubated in synthetic cartilage lymph for 24 h at 37°C and Ca2+ uptake of these vesicle fractions was measured as described in Materials and methods. Note that only vesicles isolated from RA-treated cultures showed significant Ca2+ uptake, whereas vesicles isolated from untreated or RA/BAPTA-treated cultures showed no significant Ca2+ uptake. Ca2+ uptake by matrix vesicles isolated from RA-treated cultures was set as 1. Data were obtained from four different experiments; values are mean ± SD.
Annexins mediate Ca2+ influx into growth plate chondrocytes
Based on our previous findings showing that annexins II, V, and VI form Ca2+ channels in matrix vesicles enabling Ca2+ influx into these particles (Kirsch et al., 2000b), we hypothesized that these annexins might also form Ca2+ channels in the plasma membrane of growth plate chondrocytes, thereby regulating Ca2+ homeostasis and ultimately mineralization. First, we tested whether the amount of membrane-bound annexins increased in RA-treated cultures. Indeed, 3-d treatment of chondrocytes with RA led to a significant relocation of annexins II, V, and VI from the cytoplasm to the plasma membrane. Cotreatment with BAPTA reduced the amount of plasma membrane–bound annexin II, V, and VI to levels similar as in untreated cultures (Fig. 7). Next, we tested whether RA treatment not only leads to a relocation of annexins to the plasma membrane, but also to Ca2+ channel formation. Therefore, we cotreated growth plate chondrocytes with RA and the specific annexin Ca2+ channel blocker K-201 and measured [Ca2+]i after 1-d treatment. As shown above, RA treatment led to more than threefold increase in [Ca2+]i compared with the levels in untreated cultures. Interestingly, K-201 significantly reduced this increase in RA-treated cultures. [Ca2+]i was only slightly higher in RA/K-201-treated cultures compared with untreated cultures, indicating that the majority of the increased [Ca2+]i in growth plate chondrocytes is due to the influx of Ca2+ through annexin channels (Fig. 8). Finally, to test whether all three annexins form Ca2+ channels in the plasma membrane, we treated chondrocyte cultures with RA and antibodies specific for annexin II, V, or VI. The anti–annexin V IgG fraction decreased RA-mediated increase in [Ca2+]i by 75%, the anti–annexin VI IgG fraction by 65%, and the anti–annexin II IgG fraction by 57%, indicating that all three annexins form Ca2+ channels in the plasma membrane of growth plate chondrocytes and mediate Ca2+ influx into these cells (Fig. 8).
Figure 7.

Amount of annexin II, V, or VI bound to the plasma membrane of untreated, RA-, and RA/BAPTA-treated growth plate chondrocytes. The membrane and cytosolic fractions were isolated from 3-d untreated, RA-, or RA/BAPTA-treated chondrocytes and analyzed by SDS-PAGE and immunoblotting using antibodies specific for annexin II, V, or VI. The optical densities of the annexin bands in membrane and cytosolic fractions were quantitated by densitometry. Data were obtained from four different experiments; values are mean ± SD. (★, P ≤ 0.01 vs. untreated cultures.)
Figure 8.
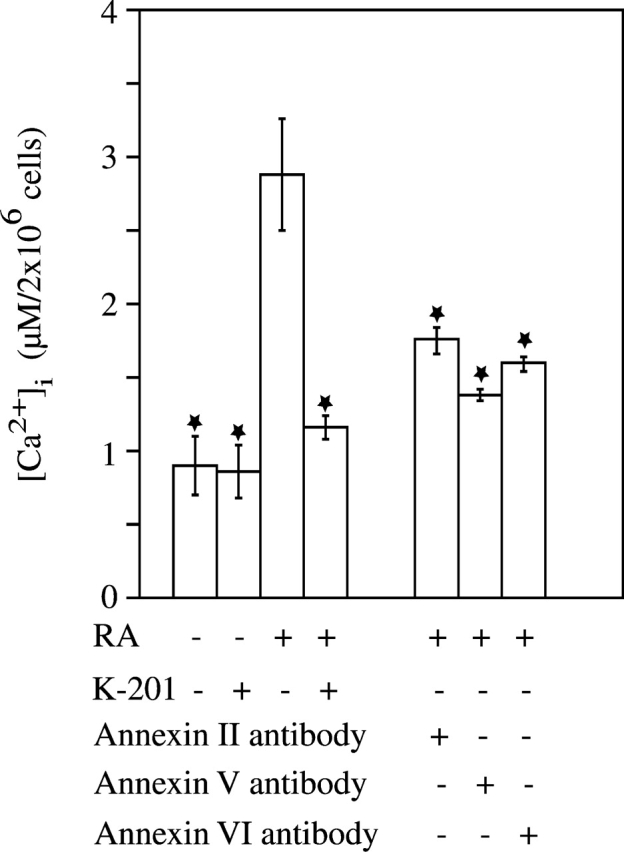
Cytosolic calcium concentration in untreated, RA-, RA/K-201-, RA/anti–annexin II IgG, RA/anti–annexin V IgG, RA/anti–annexin VI IgG-treated chondrocytes. [Ca2+]i in chondrocytes was measured after 1-d treatment with RA, RA/K-201, RA/anti–annexin II IgG, RA/anti–annexin V IgG, and RA/anti–annexin VI IgG as described in Materials and methods. Data were obtained from four different experiments and values are mean ± SD. (★, P ≤ 0.01 vs. RA-treated cultures.)
Interestingly, annexin-mediated Ca2+ influx into growth plate chondrocytes led to the up-regulation of their own gene expression. Real-time PCR analysis revealed a three- to sevenfold up-regulation of annexin II, V, and VI gene expression after 3- and 4-d treatment with RA. Chelation of cytosolic Ca2+ with BAPTA decreased up-regulation of annexin gene expression (Fig. 9, A–C). In this study chondrocytes were isolated from the hypertrophic zone of tibia growth plate cartilage, and thus these cells showed high expression levels of type X collagen. RA or RA/BAPTA treatment did not further increase these already high type X collagen expression levels (Fig. 9 D). The fact that RA and RA/BAPTA treatment does not alter the high expression of type X collagen in growth plate chondrocytes reveals that these treatments do not change the chondrocytic phenotype.
Figure 9.
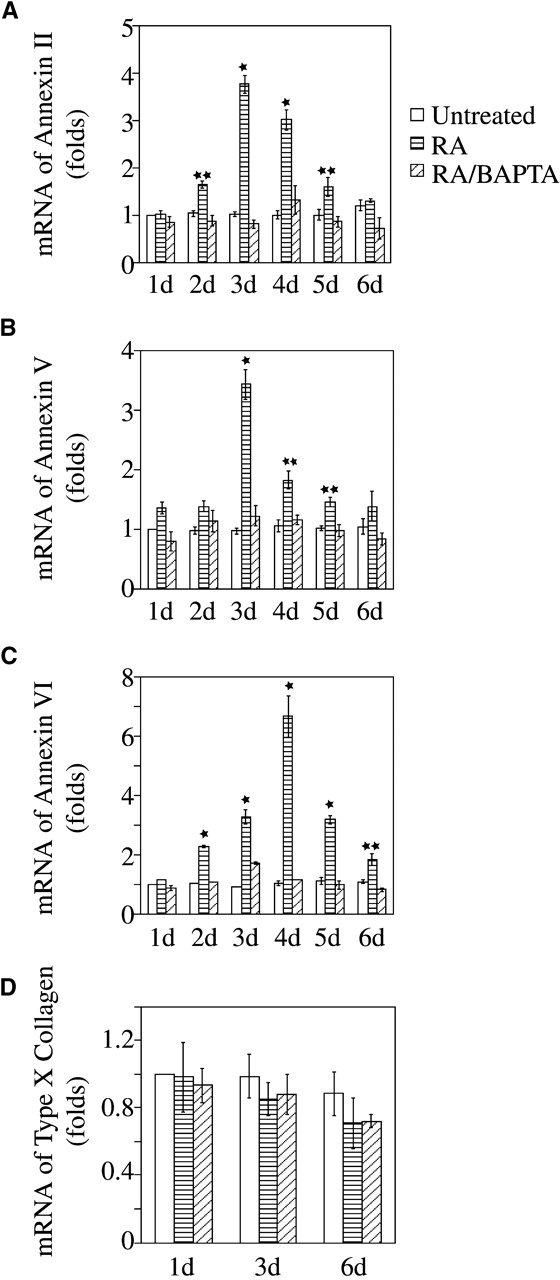
Quantitative real time PCR analysis of annexin II, V, and VI, and type X collagen gene expression in chondrocytes treated with RA or RA/BAPTA. Total RNA was isolated from 1- to 6-d untreated, RA-, and RA/BAPTA-treated chondrocytes. Annexin II, V, and VI, and type X collagen gene expression were detected by quantitative real-time PCR using SYBR Green. PCR reactions were run in triplicates. Data were obtained from triplicated PCR reactions of three different cultures and values are mean ± SD (★, P ≤ 0.01, ★★, P ≤ 0.05 vs. 1-d untreated cultures.)
Discussion
Ca2+ homeostasis controls a diverse range of cellular functions including adhesion, motility, gene expression, proliferation, and differentiation. Interestingly, many studies have demonstrated that hypertrophic growth plate chondrocytes can accumulate large amounts of cytosolic calcium, suggesting that these increases might regulate chondrocyte differentiation and mineralization. Here we provide direct evidence that RA treatment, which has been shown to induce terminal differentiation and mineralization of growth plate chondrocytes (Iwamoto et al., 1993; Kirsch et al., 2000a), leads to an increase in [Ca2+]i. This increase regulates a whole series of events eventually leading to matrix mineralization of growth plate chondrocytes: up-regulation of annexin II, V, and VI gene expression, release of APase- and annexin II, V, and VI–containing matrix vesicles, and initiation of mineralization by these vesicles. Moreover, the increase in [Ca2+]i during terminal differentiation of growth plate chondrocytes is to a large degree mediated by annexins II, V, and VI. These annexins form Ca2+ channels in the plasma membrane of RA-treated chondrocytes, enabling Ca2+ influx into these cells. Thus, through alteration of Ca2+ homeostasis in growth plate chondrocytes, annexins II, V, and VI regulate their own gene expression and subsequent release of annexin-containing, mineralization-competent matrix vesicles.
Our results reveal that under certain physiological conditions, annexins are able to form Ca2+ channels. These findings present a new concept to regulate Ca2+ homeostasis by annexin Ca2+ channels. Annexins are cytosolic proteins, which in the presence of Ca2+, can bind to acidic phospholipids. However, channel formation not only requires membrane binding, but also membrane insertion. Previous studies have provided evidence that annexins V, VI, and XII can insert into the membrane and form Ca2+ channels at low pH in a Ca2+-independent manner (Isas et al., 2000; Golczak et al., 2001a,b). For example, annexin VI undergoes major conformational changes under low pH that leads to an increase of hydrophobicity (Golczak et al., 2001a,b). Furthermore, a biophysical study has demonstrated that the secondary structure of annexin V is affected by the combined interactions with Ca2+ and membrane components (Wu et al., 1999). Interestingly, a recent study has shown that annexin V mediates peroxide-induced Ca2+ influx into B cells under physiological pH (Kubista et al., 1999). These and our findings indicate that the cell not only can regulate gene expression of annexins, but also their channel formation, allowing a precise regulation of the biological consequences of annexin channel formation. It is not clear which factors regulate physiological channel formation by annexins. It is possible that a certain Ca2+ concentration is required for membrane binding and channel formation. Other possibilities are that intracellular acidification or, as we have shown previously, the lipid composition of the membrane, may play regulatory roles in annexin channel formation (Kirsch et al., 1997a). Furthermore, a previous study has shown that vitamin D caused an increase in fluidity of the plasma membrane (Swain et al., 1993). Thus, it is possible that RA has a similar effect on the plasma membrane that could favor annexin channel formation.
Our results and the findings showing that annexin V mediated peroxide-induced Ca2+ influx into B cells (Kubista et al., 1999) establish that annexin Ca2+ channels have important physiological functions in altering cell behavior and differentiation. In case of growth plate chondrocytes, annexin channel formation regulates Ca2+ homeostasis in these cells, and thereby the mineralization process including the release of mineralization-competent matrix vesicles. These vesicles contain annexins II, V, and VI as major components (Genge et al., 1989). We have demonstrated that these annexins also form Ca2+ channels in matrix vesicles, allowing Ca2+ influx into these particles and the formation of the first mineral phase inside the vesicles (Kirsch et al., 2000b). Thus, these annexins not only regulate Ca2+ homeostasis in growth plate chondrocytes, but they also play a direct role in the initiation of mineralization. Interestingly, our data indicate that annexin-mediated Ca2+ influx into growth plate chondrocytes up-regulates annexin gene expression. Thus, these annexins regulate their own gene expression, thereby possibly enabling sufficient amounts of annexins II, V, and VI to be available for the release of mineralization-competent matrix vesicles.
[Ca2+]i in hypertrophic chondrocytes is higher than the concentration in immature chondrocytes (Iannotti and Brighton, 1989; Gunter et al., 1990). As indicated by previous studies, this increase is most likely mediated by other Ca2+ channels than the annexins (Zimmermann et al., 1994; Zuscik et al., 1997). Initial increased [Ca2+]i might lead to annexin Ca2+ channel formation and subsequent further increase in [Ca2+]i. K-201 treatment, which inhibits Ca2+ channel activities of annexin II, V, and VI (Kirsch et al., 2000b), did not completely reduce RA-mediated increase in [Ca2+]i to levels in untreated cultures. However, K-201 reduced the increase by ∼87%, indicating that the majority of Ca2+ influx into growth plate chondrocytes is mediated by annexins. There are several reasons why K-201 did not completely inhibit RA-mediated increase in [Ca2+]i. First, we have shown that the concentration of K-201 required to completely block annexin II–, V–, and VI–mediated Ca2+ influx into artificial liposomes or matrix vesicles is 100 μM (Kirsch et al., 2000b). However, 100 μM K-201 is toxic to chondrocytes (unpublished data). Thus, it is possible that 20 μM K-201, which was not toxic to the cells, might not completely block annexin channels. Another possibility is that growth plate chondrocytes contain other Ca2+ channels that might contribute to the Ca2+ influx into RA-treated chondrocytes. Previous characterizations of Ca2+ channels in growth plate chondrocytes are controversial. One study provided evidence that growth plate chondrocytes contain N-type Ca2+ channels, whereas another study suggested that growth plate chondrocytes contain L-type Ca2+ channels (Zimmermann et al., 1994; Zuscik et al., 1997). Each antibody specific for annexin II, V, or VI decreased RA-mediated increase in [Ca2+]i by between 57 and 75%, revealing that all three annexins contribute to Ca2+ influx into growth plate chondrocytes and blocking the channel activities of one annexin only partially reduces RA-mediated increase in [Ca2+]i. Thus, our findings indicate that the annexins contribute the majority to the alteration of Ca2+ homeostasis in RA-treated growth plate chondrocytes and thereby regulate the mineralization process.
Annexin-mediated Ca2+ influx into growth plate chondrocytes may be directly involved in the release of mineralization-competent matrix vesicles. Vesiculation or ectocytosis of the plasma membrane has been shown to be dependent on an influx of Ca2+ into the cytoplasm of many cell types. Ca2+ ionophores can cause microvesiculation and membrane shedding in a variety of cell types, including chondrocytes, fibroblasts, neutrophils, and platelets (Wiedmer et al., 1990; Iannotti et al., 1994; Bucki et al., 1998). In contrast, when platelets were suspended in a solution containing EDTA, microparticle formation was inhibited (Wiedmer et al., 1990). Furthermore, we and others have provided evidence that matrix vesicles contain Ca2+-Pi-phospholipid or nucleational core complex. This core complex is required for the formation of the first mineral phase inside the vesicles (Kirsch et al., 1994; Wu et al., 1997). There is some evidence to suggest that this complex might already be assembled at the plasma membrane of the cell before matrix vesicles are released (Wuthier, 1982). Thus, it is possible that high [Ca2+]i is required for the assembly of this complex.
Our study indicates that RA action is required for completion of the chondrocyte maturation process, and it provides the first important insight into the underlying mechanisms. Interestingly, a previous study demonstrated that the perichondral tissue surrounding the cartilage contained large amounts of retinoids and that implantation of beads containing RA antagonist near the humeral anlagen severely affected humerus development (Koyama et al., 1999). Thus, it is likely that RA plays an important role in regulating mineralization and terminal differentiation of growth plate chondrocytes in vivo.
In conclusion, our findings provide evidence that RA treatment of growth plate chondrocytes leads to Ca2+ channel formation by annexins II, V, and VI and influx of Ca2+ into these cells. The annexin-mediated alteration in Ca2+ homeostasis regulates a whole sequence of events eventually leading to matrix mineralization, including up-regulation of annexin II, V, and VI gene expression, relocation of these annexins to the plasma membrane, and release of APase- and annexin-containing matrix vesicles that initiate the mineralization process.
Materials and methods
Chondrocyte culture
Chondrocytes were isolated from the hypertrophic zone of day 19 embryonic chick tibia growth plate cartilage as described previously (Kirsch et al., 1997b). In brief, sliced hypertrophic zone of growth plate cartilage was digested with 0.25% trypsin and 0.05% collagenase for 5 h at 37°C. Cells were plated at a density of 3 × 106 into 10-cm tissue culture dishes and grown in monolayer cultures in DME (GIBCO BRL) containing 5% FCS (Hyclone), 2 mM l-glutamine (GIBCO BRL), and 50 U/ml penicillin and streptomycin (complete medium; GIBCO BRL). After cultures reached confluency, chondrocytes were cultured in the presence of 1.5 mM phosphate, and in the absence or presence of (a) 35 nM RA (Sigma-Aldrich), (b) 35 nM RA + 10 μM BAPTA-AM (Molecular Probes, Inc.), (c) 35 nM RA + 5 mM EDTA, (d) 35 nM RA + 20 μM 1,4-benzothiazepine derivative K-201 (JTV519) (provided by Drs. Noboro Kareko [Dokkyo University, Tochigo, Japan] and Toshizo Tanaka [Japan Tobcco Inc., Osaka, Japan]), or (e) 35 nM RA + 20 μg/ml IgG fractions specific for annexin II, V, or VI. [Ca2+]i of chondrocytes was measured after 1-d treatment, matrix vesicles were isolated after 3-d treatment, and the degree of mineralization in these cultures was determined after 6-d treatment.
Measurement of [Ca2+]i
Chondrocytes were trypsinized first, and then 2 × 106 cells were incubated with 4 μM of fura-2AM (Molecular Probes, Inc.) at 37°C for 15 min in complete medium. [Ca2+]i was measured as described previously (Kirsch et al., 1992). Briefly, labeled cells were resuspended in measuring buffer ([mM] 140 NaCl, 5 KCl, 1 CaCl2, 20 Hepes, 1 NaH2PO4, 5.5 glucose, pH 7.4), transferred to a cuvette (magnetically stirred), and fluorescence was measured in a fluorimeter (Photon Technology Instruments) using the excitation wavelength of 340 nm (Ca2+-bound form of fura-2AM) and the emission wavelength of 510 nm. The fluorescence maximum (Fmax) was determined by addition of 2 pM ionomycin (Calbiochem-Novabiochem), and the fluorescence minimum (Fmin) was determined in the presence of 1 mM EDTA/10 mM Tris. [Ca2+]i was calculated according to the following equation: ([Ca2+]i = K d × [(F − Fmin)/(Fmax − F)] with K d = 224 nM (Grynkiewicz et al., 1985).
Isolation of total RNA and real-time PCR
Total RNA was isolated from chondrocyte cultures after 1-, 2-, 3-, 4-, 5-, and 6-d treatments using RNeasy Mini Kit (QIAGEN). 1 μg RNA was reverse transcribed using Ominiscript RT Kit (QIAGEN). Then a 1:100 dilution of the resulting cDNA was used as the template to quantitate the relative content of mRNA by real-time PCR (ABI PRISM 7700 sequence detection system) using respective primers and SYBR Green. The following primers for real-time PCR were designed using Primers Express software: annexin II, forward primer 5′-CATGCCTATCTGCTCTTCGTT-3′, reverse primer 5′-AGCCACCACACCGTCCATAA-3′; annexin V, forward primer 5′-AGAGACATCAGGCCATTTTCAGA-3′, reverse primer 5′-CTGCCATCAGGATCTCTATTTGC-3′; annexin VI, forward primer 5′-GCGGCTGATTGTAAGCTTGAT-3′, reverse primer 5′-GTCGGTGGTCCAGCACTTA-3′; and type X collagen, forward primer 5′-AGTGCTGTCATTGATCTCATGGA-3′, reverse primer 5′-TCAGAGGAATAGAGACCATTGGATT-3′. PCR reactions were performed with TaqMan PCR master mix kit (Applied Biosystems). The 18 S RNA was amplified at the same time and used as an internal control. The cycle threshold values for 18 S RNA and that of the samples were measured and calculated by computer software. Relative transcript levels were calculated as x = 2−ΔΔCt, in which ΔΔCt = ΔE − ΔC, and ΔE = Ctexp − Ct18S; ΔC = Ctctl − Ct18S.
Isolation of matrix vesicles
Matrix vesicles were isolated from chondrocyte cultures by enzymatic digestion as described previously (Kirsch et al., 1997b). Briefly, adherent chondrocyte layers were washed with PBS and incubated with crude collagenase (500 U/ml; type IA; Sigma Chemical Co.) in HBSS at 37°C for 3 h. Matrix vesicles were harvested by different ultracentrifugation.
Ca2+ uptake by matrix vesicles
Ca2+ uptake by matrix vesicles was assayed by incubating matrix vesicle aliquots (100 μg of protein) in 400 μl of synthetic cartilage lymph solution at 37°C for 24 h. After incubation, matrix vesicles were pelleted, washed twice in TNE buffer (TES pH 7.4, NaCl 1.5 mM and EDTA 2 μM), resuspended in TNE buffer containing 1 μM fura-2, transferred to a cuvette, and fluorescence was measured in fluorimeter using the excitation wavelength of 340 nm and the emission wavelength of 510 nm. Matrix vesicles were lysed with 1% Triton X-100 to release intravesicular calcium.
Recombinant annexin proteins and antibodies
Recombinant annexins II, V, and VI were prepared using the pGEX expression vector (Amersham Pharmacia Biotech) as described previously (Kirsch et al., 1997a). The preparation of antibodies specific for annexins II, V, and VI were also described previously (Kirsch et al., 2000c, 2001).
SDS-PAGE and immunoblotting
Samples were dissolved in 3% SDS sample buffer with DTT, denatured at 100°C for 3 min, and analyzed by electrophoresis in 10 or 12% (wt/vol) SDS–polyacrylamide gels. Samples were electroblotted onto nitrocellulose filters after electrophoresis. After blocking with a solution of low-fat milk protein, blotted proteins were immunostained with primary antibodies followed by peroxidase-conjugated secondary antibody, and the signal was detected by enhanced chemiluminescene (Pierce Chemical Co.).
Measurement of APase activity and protein content
The measurements of APase activity and protein content were described previously (Kirsch et al., 1997b). Cells or vesicles were washed in PBS and suspended in 10 mM Tris-HCl, pH 7.5, 0.1% Triton X-100, and 0.5 mM MgCl2. Protein content was analyzed by BCA assay (Pierce Chemical Co.). APase activity was determined using p-nitrophenyl phosphate as a substrate (Tietz et al., 1983).
Alizarin red S staining
To determine the degree of mineralization in chondrocyte cultures, cultures were stained with alizarin red S. After washing, chondrocyte cultures were fixed with 70% ethanol for 10 min, and then stained with 0.5% alizarin red S in H2O, pH 4.0, for 5 min at room temperature. After staining, cultures were washed three times with H2O followed by 70% ethanol. To quantify matrix mineralization, the alizarin red S–stained cultures were incubated with 100 mM cetylpyridinium chloride for 1 h to solubilize and release calcium-bound alizarin red into solution (Johnson et al., 2001). The absorbance of the released alizarin red S was measured at 570 nm using a spectrophotometer. Data are expressed as units of alizarin red S released per milligram of protein in each culture.
Statistical analysis
Numerical data are presented as mean + SD (n > 4) and statistical significance between groups was identified using the two-tailed t test (P values are reported in the figure legends).
Acknowledgments
This work was supported by grants from the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases, AR 43732 and AR 46245 to T. Kirsch).
Footnotes
Abbreviations used in this paper: APase, alkaline phosphatase; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester); RA, retinoic acid.
References
- Anderson, H.C. 1995. Molecular biology of matrix vesicles. Clin. Orthop. 314:266–280. [PubMed] [Google Scholar]
- Benz, J., A. Bergner, A. Hofmann, P. Demange, P. Gottig, S. Liemann, R. Huber, and D. Voges. 1996. The structure of recombinant human annexin VI in crystals and membrane-bound. J. Mol. Biol. 260:638–643. [DOI] [PubMed] [Google Scholar]
- Berendes, R., D. Voges, P. Demange, R. Huber, and A. Burger. 1993. Structure-function analysis of the ion channel selectivity filter in human annexin V. Science. 262:427–430. [DOI] [PubMed] [Google Scholar]
- Bonucci, E., G. Silvestrini, and P. Bianco. 1992. Extracellular alkaline phosphatase activity in mineralizing matrices of cartilage and bone: ultrastructural localization using a cerium-based method. Histochemistry. 97:323–327. [DOI] [PubMed] [Google Scholar]
- Bucki, R., C. Bachelot-Loza, A. Zachowski, F. Giraud, and J.C. Sulpice. 1998. Calcium induces phospholipid redistribution and microvesicle release in human erythrocyte membranes by independent pathways. Biochemistry. 37:15383–15391. [DOI] [PubMed] [Google Scholar]
- Buckwalter, J.A., D. Mower, and J. Schaeffer. 1987. Differences in matrix vesicle concentration among growth plate zones. J. Orthop. Res. 5:157–163. [DOI] [PubMed] [Google Scholar]
- Burger, A., R. Berendes, S. Liemann, J. Benz, A. Hofmann, P. Gottig, R. Huber, V. Gerke, C. Thiel, J. Romisch, and K. Weber. 1996. The crystal structure and ion channel activity of human annexin II, a peripheral membrane protein. J. Mol. Biol. 257:839–847. [DOI] [PubMed] [Google Scholar]
- Chen, J.M., A. Sheldon, and M.R. Pincus. 1993. Structure-function correlations of calcium binding and calcium channel activities based on 3-dimensional models of human annexins I, II, III, V and VII. J. Biomol. Struct. Dyn. 10:1067–1089. [DOI] [PubMed] [Google Scholar]
- Crompton, M.R., S.E. Moss, and M.J. Crumpton. 1988. Diversity in the lipocortin/calpactin family. Cell. 55:1–3. [DOI] [PubMed] [Google Scholar]
- Doyle, D.V. 1982. Tissue calcification and inflammation in osteoarthritis. J. Pathol. 136:199–216. [DOI] [PubMed] [Google Scholar]
- Geisow, M.J., J.H. Walker, C. Boustead, and W. Taylor. 1988. Annexins: a new family of Ca2+ regulated phospholipid-binding proteins. Molecular Mechanisms in Secretion. N.A. Thorn, M. Traiman, and O.H. Peterson, editors. Munksgaard, Copenhagen. 598–608.
- Genge, B.R., L.N.Y. Wu, and R.E. Wuthier. 1989. Identification of phospholipid-dependent calcium-binding proteins as constituents of matrix vesicles. J. Biol. Chem. 264:10917–10921. [PubMed] [Google Scholar]
- Giachelli, C.M. 1999. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am. J. Pathol. 154:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak, M., A. Kirilenko, J. Bandorowicz-Pikula, and S. Pikula. 2001. a. Conformational states of annexin VI in solution induced by acidic pH. FEBS Lett. 496:49–54. [DOI] [PubMed] [Google Scholar]
- Golczak, M., A. Kirilenko, J. Bandorowicz-Pikula, and S. Pikula. 2001. b. N- and C-terminal halves of human annexin VI differ in ability to form low pH-induced ion channels. Biochem. Biophys. Res. Commun. 284:785–791. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R.Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Gunter, T.E., M.J. Zuscik, J.E. Puzas, K.K. Gunter, and R.N. Rosier. 1990. Cytosolic free calcium concentrations in avian growth plate chondrocytes. Cell Calcium. 11:445–457. [DOI] [PubMed] [Google Scholar]
- Iannotti, J.P., and C.T. Brighton. 1989. Cytosolic ionized calcium concentration in isolated chondrocytes from each zone of the growth plate. J. Orthop. Res. 7:511–518. [DOI] [PubMed] [Google Scholar]
- Iannotti, J.P., S. Naidu, Y. Noguchi, R.M. Hunt, and C.T. Brighton. 1994. Growth-plate matrix vesicle biogenesis. The role of intracellular calcium. Clin. Orthop. 306:222–229. [PubMed] [Google Scholar]
- Isas, J.M., J.-P. Cartailler, Y. Sokolov, D.R. Patel, R. Langen, H. Luecke, J.E. Hall, and H.T. Haigler. 2000. Annexins V and XII insert into bilayers at mildly acidic pH and form ion channels. Biochemistry. 39:3015–3022. [DOI] [PubMed] [Google Scholar]
- Iwamoto, M., I.M. Shapiro, K. Yagami, A.L. Boskey, P.S. Leboy, S.L. Adams, and M. Pacifici. 1993. Retinoic acid induces rapid mineralization and expression of mineralization-related genes in chondrocytes. Exp. Cell Res. 207:413–420. [DOI] [PubMed] [Google Scholar]
- Johnson, K., S. Hashimoto, M. Lotz, K. Pritzker, J. Goding, and R. Terkeltaub. 2001. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 44:1071–1081. [DOI] [PubMed] [Google Scholar]
- Kaneko, N. 1994. New 1,4-benzothiazepine derivative, K201, demonstrates cardioprotective effects against sudden cardiac cell death and intracellular blocking action. Drug Dev. Res. 33:429–438. [Google Scholar]
- Kaneko, N., H. Ago, R. Matsuda, E. Inagaki, and M. Miyano. 1997. Crystal structure of annexin v with its ligand K-201 as a calcium channel activity inhibitor. J. Mol. Biol. 274:16–20. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., and K. von der Mark. 1992. Remodelling of collagen types I, II and X and calcification of human fetal cartilage. Bone Miner. 18:107–117. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., B. Swoboda, and K. von der Mark. 1992. Ascorbate independent differentiation of human chondrocytes in vitro: simultaneous expression of types I and X collagen and matrix mineralization. Differentiation. 52:89–100. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., Y. Ishikawa, F. Mwale, and R.E. Wuthier. 1994. Roles of the nucleational core complex and collagens (types II and X) in calcification of growth plate cartilage matrix vesicles. J. Biol. Chem. 269:20103–20109. [PubMed] [Google Scholar]
- Kirsch, T., H.-D. Nah, D.R. Demuth, G. Harrison, E.E. Golub, S.L. Adams, and M. Pacifici. 1997. a. Annexin V-mediated calcium flux across membranes is dependent on the lipid composition. Implications for cartilage mineralization. Biochemistry. 36:3359–3367. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., H.D. Nah, I.M. Shapiro, and M. Pacifici. 1997. b. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J. Cell Biol. 137:1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, T., G. Harrison, and E.E. Golub. 2000. a. Regulatory roles of zinc in matrix vesicle-mediated mineralization of growth plate cartilage. J. Bone Miner. Res. 15:261–270. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., G. Harrison, E.E. Golub, and H.-D. Nah. 2000. b. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J. Biol. Chem. 275:35577–35583. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., B. Swoboda, and H.-D. Nah. 2000. c. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthritis Cartilage. 8:294–302. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., B. Swoboda, and H.-D. Nah. 2001. Expression of early and late differentiation markers (proliferating cell nuclear antigen, syndecan-3, annexin VI, and alkaline phosphatase) by human osteoarthritic chondrocytes. Am. J. Pathol. 159:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama, E., E.B. Golden, T. Kirsch, S.L. Adams, R.A.S. Chandraratna, J.-J. Michaille, and M. Pacifici. 1999. Retinoid signaling is required for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Dev. Biol. 208:375–391. [DOI] [PubMed] [Google Scholar]
- Kubista, H., T.E. Hawkins, D.R. Patel, H.T. Haigler, and S.E. Moss. 1999. Annexin V mediates a peroxidase-induced Ca2+ influx in B cells. Curr. Biol. 9:1403–1406. [DOI] [PubMed] [Google Scholar]
- Matsuda, R., N. Kaneko, and Y. Horikawa. 1997. Presence and comparison of Ca2+ transport activity of annexins I, II, V, and VI in large unilamellar vesicles. Biochem. Biophys. Res. Commun. 237:499–503. [DOI] [PubMed] [Google Scholar]
- Mitchell, P.G., J.A. Struve, G.M. McCarthy, and H.S. Cheung. 1992. Basic calcium phosphate crystals stimulate cell proliferation and collagenase message accumulation in cultured adult articular chondrocytes. Arthritis Rheum. 35:343–350. [DOI] [PubMed] [Google Scholar]
- Montessuit, C., J. Caverzasio, and J.P. Bonjour. 1991. Characterization of a Pi transport system in cartilage matrix vesicles. Potential role in the clarification process. J. Biol Chem. 266:17791–17797. [PubMed] [Google Scholar]
- Montessuit, C., J.P. Bonjour, and J. Caverzasio. 1995. Expression and regulation of Na-dependent P(i) transport in matrix vesicles produced by osteoblast-like cells. J. Bone Miner. Res. 10:625–631. [DOI] [PubMed] [Google Scholar]
- Reinholt, F.P., B. Engfeldt, A. Hjerpe, and K. Jansson. 1982. Stereological studies on the epiphyseal growth plate with special reference to the distribution of matrix vesicles. J. Ultrastruct. Res. 80:270–279. [DOI] [PubMed] [Google Scholar]
- Swain, L.D., Z. Schwartz, K. Caulfield, B.P. Brooks, and B.D. Boyan. 1993. Nongenomic regulation of chondrocyte membrane fluidity by 1,25-(OH)2d3 and 24,25-(OH)2d3 is dependent on cell maturation. Bone. 14:609–617. [DOI] [PubMed] [Google Scholar]
- Tietz, N.W., C.A. Burtis, P. Duncan, K. Ervin, C.J. Petitclerd, A.D. Rinker, D. Shney, and E.R. Zygowicz. 1983. A reference method for measurement of alkaline phosphatase activity in human serum. Clin. Chem. 29:751–761. [PubMed] [Google Scholar]
- Wiedmer, T., S.J. Shattil, M. Cunningham, and P.J. Sims. 1990. Role of calcium and calpain in complement-induced vesiculation of the platelet plasma membrane and its exposure of the platelet factor Va receptor. Biochemistry. 29:623–632. [DOI] [PubMed] [Google Scholar]
- Wu, F., C.R. Flach, B.A. Seaton, T.R. Mealy, and R. Mendelsohn. 1999. Stability of annexin V in ternary complexes with Ca2+ and anionic phospholipids: IR studies of monolayer and bulk phases. Biochemistry. 38:792–799. [DOI] [PubMed] [Google Scholar]
- Wu, L.N.Y., B.R. Genge, D.G. Dunkelberger, R.Z. Legeros, B. Concannon, and R.E. Wuthier. 1997. Physicochemical characterization of the nucleational core of matrix vesicles. J. Biol. Chem. 272:4404–4411. [DOI] [PubMed] [Google Scholar]
- Wuthier, R.E. 1982. A review of the primary mechanism of endochondral calcification with special emphasis on the role of cells, mitochondria and matrix vesicles. Clin. Orthop. 169:219–242. [PubMed] [Google Scholar]
- Zimmermann, B., K. Lange, P. Mertens, and J. Bernimoulin. 1994. Inhibition of chondrogenesis and endochondral mineralization in-vitro by different calcium-channel blockers. Eur. J. Cell Biol. 63:114–121. [PubMed] [Google Scholar]
- Zuscik, M.J., T.E. Gunter, J.E. Puzas, and R.N. Rosier. 1997. Characterization of voltage-sensitive calcium channels in growth plate chondrocytes. Biochem. Biophys. Res. Commun. 234:432–438. [DOI] [PubMed] [Google Scholar]