

Abstract
The effects of bromocriptine (10 mg/kg), known to inhibit prolactin secretion and lower autoimmune processes, were studied on glucose homeostasis in non-fasted non-obese diabetic mice, a spontaneous model of type 1 diabetes. Hyperglycemia was observed 120 and 240 min after i.p. but not s.c. injection. Bromocriptine administration i.p. led to rapid and marked hyperglycemia characterized by sexual dimorphism with males having higher glycemia than females. Bromocriptine induced a rapid but transient decrease in insulinemia in males only and biphasic increases in glucagon levels and a sustained stimulatory effect on circulating corticosterone in both sexes. Bromocriptine-induced hyperglycemia involved D2-dopaminergic receptors, as demonstrated by the inhibitory effect of the D2-dopamine antagonist, metoclopramide (10 mg/kg). Simultaneous injection of bromocriptine and metoclopramide also blocked the rise in blood corticosterone. In conclusion, by inducing hyperglycemia, i.p. bromocriptine administration to prediabetic autoimmune mice may counteract its beneficial anti-immunostimulatory effects.
Keywords: type 1 diabetes, NOD mouse, bromocriptine, insulin, glucagon, corticosterone, dopamine, L-Dopa
Introduction
A beneficial effect of bromocriptine, a D2-dopaminergic agonist, has been described in various rodent models of autoimmune diseases, such as arthritis and uveitis, which do not involve alterations of glucose homeostasis [1]. Attenuation of these diseases reflected the ability of the dopaminergic agonist to suppress pituitary secretion of the immunostimulatory hormone, prolactin. Similarly, in a spontaneous model of autoimmune type 1 diabetes, the non-obese diabetic (NOD) mouse, diabetes incidence was lower in NOD females receiving chronic s.c. injections of bromocriptine [2]. Intriguingly, we were unable to reproduce those data in NOD mice with chronic i.p. injections [3].
Type 1 diabetes is both an autoimmune and a metabolic disease, and we thought that the hyperglycemic effect of dopamine or its analogs, which has been described in control rodents [4-6], might explain the contrast in the reported effects of bromocriptine. The hyperglycemic effect observed in rats involves a specific interaction between dopamine or dopaminergic agonists and D2-subtype receptors [7]. We previously showed that acute or chronic i.p. bromocriptine injection induced hyperglycemia in NOD mice [3], but it remained to be investigated whether or not subcutaneous injections had the same effect.
Moreover, little information was available on the mechanisms of the in vivo hyperglycemic effect of D2-dopaminergic agonists. Glucose homeostasis primarily depends on the opposing metabolic actions of two pancreatic hormones, insulin, which decreases glycemia, and glucagon, which together with other non-pancreatic counterregulatory hormones, increases it [8]. In fasted male rats, sacrificed one hour after drug injection, bromocriptine markedly reduced insulinemia [4], whereas lergotrile, another D2-dopaminergic agonist, did not significantly alter glucagon levels [6].
The main non-pancreatic counterregulatory hormones released during hypoglycemia and other stress conditions are glucocorticoids, catecholamines and growth hormone [9]. Like glucagon, these hormones have insulin-antagonistic effects both in the liver and peripheral tissues [10]. Bromocriptine-induced hyperglycemia in rodents appears to be independent of any variation in growth hormone release [11]. With regard to catecholamines, the stimulatory effect of the D2-dopaminergic agonist, quinpirol, on epinephrine release from rat adrenals [12], strongly suggests that they are involved in inducing hyperglycemia. Finally, measurement of circulating glucocorticoids has been used routinely in rats to evaluate D2-dopaminergic activity and dose-dependent increases in serum corticosterone following the injection of various D2-dopaminergic agonists [13-15].
In this study, we analyzed in greater depth the mechanisms of bromocriptine-induced hyperglycemia, which, to the best of our knowledge, have yet to be elucidated in mice. We investigated, in 8-week old female and male NOD mice, i.e. in pre-diabetic animals, the effect of bromocriptine on: 1) glycemia, after i.p. versus s.c. injection; 2) the kinetics of serum concentrations of glucose, insulin, glucagon and corticosterone after i.p. injection and 3) these parameters in the presence of a D2-dopaminergic antagonist, metoclopramide.
Material and methods
Animals
NOD mice (originally provided by Clea-Japan Inc., Tokyo, Japan) were bred in our own facilities under pathogen-free conditions. NOD mice were anti-virus antibody-free for thirteen strains including diabetogenic viruses. All the mice were maintained on a 12-h light/12-h dark cycle with lights on from 7:00 a.m. to 7:00 p.m. at 22 ± 1°C under laminar flow ventilation, and were given standard food pellets and water ad libitum. The animal facilities and care respected the norms stipulated by the European Community. In the experiments, female and male nondiabetic NOD mice (with basal non-fasting glycemia < 11 mmol/l as assessed by Glukotest, Boehringer-Mannheim, Mannheim, Germany) were used at eight weeks of age. In our NOD colony, during the investigation period, overt diabetes appeared at twelve and sixteen weeks of age for females and males respectively, and 80% of females and 40% of males were diabetic at six months of age.
Treatments and blood sampling
Drugs were obtained from Sigma-Coger (Paris, France). Saline:water:ethanol (60:20:20) served as the vehicle for bromocriptine and metoclopramide, a D2-dopaminergic antagonist, was dissolved in 0.9% NaCl. Bromocriptine (10 mg/kg) and metoclopramide (10 mg/kg) were injected i.p. unless otherwise stated. All experiments started at 9:00 am and unanesthetized animals were bled in less than two minutes by retro-orbital puncture in a way that avoided stress-induced metabolic changes, at least during the sampling time [16]. In kinetic studies, different groups of animals were bled at different times to avoid the hyperglycemic effect of repeated orbital puncture routinely observed in mice [16]. For circulating glucagon, blood samples were supplemented with recombinant aprotinin, 3000 kallikrein IU/ml of blood (Bayer Inc., Kankakee, IL, USA). Mice were sacrificed immediately after being bled. Blood samples were kept on ice, centrifuged at 13000 × g for two min at 4°C, and stored at -20°C.
Glucose, insulin, glucagon and corticosterone determinations
Glucose concentrations were measured using the glucose-oxidase method (Biotrol glucose enzymatic color, Biotrol, Paris, France). Standard radioimmunoassay kits were used for insulin (Insulin-CT, CIS Biointernational, Gif-sur-Yvette, France), glucagon (Biodata, Pharmacia, St-Quentin-en-Yvelines, France) and corticosterone (ICN Biomedicals Inc., Sorin Biomedica, Antony, France) determinations.
Mean (± SE) non-fasting, non-vehicle treated serum values for two-month old NOD females and males respectively were: 6.6 ± 0.2 and 6.9 ± 0.1 mmol/l for glucose, 588 ± 102 and 354 ± 30 pmol/l for insulin, 252 ± 7 and 210 ± 3 pg/ml for glucagon and 69 ± 8 and 25 ± 3 ng/ml for corticosterone (n = 12-15 mice/sex).
Statistical analyses
All data are expressed as means ± SE. The differences between mean glucose, insulin, glucagon and corticosterone values were compared using two, three or four p-factor analysis of variance (ANOVA). Post-hoc analysis using the Newman-Keuls test was performed when the main effect and interaction were significant (p < 0.05) as assessed by analysis of variance.
Results
Dependence of the hyperglycemic effect of bromocriptine on the administration route
Blood glucose levels were determined in NOD mice 120 and 240 min following either a single s.c. or i.p. injection of bromocriptine (10 mg/kg). As shown in Table 1, in both sexes of non-fasted NOD mice, bromocriptine had no effect on glycemia when injected s.c., regardless of the time of investigation. In constrast, glycemia was significantly higher in females and males 120 min (p = 0.0001 in both cases) and 240 min (p = 0.002 and p = 0.0001 respectively) after i.p. injection. Moreover, a significant sex effect could be noted: hyperglycemia was higher in males than females 120 and 240 min after injection (p = 0.001 and p = 0.0003 respectively).
Table 1. Effect of the bromocriptine administration routes on glycemia in 8-week old NOD female and male mice.
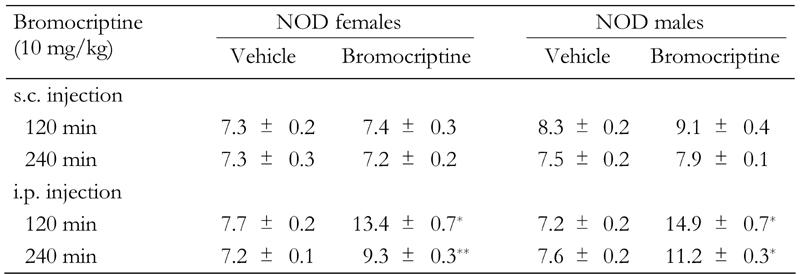
Data are blood glucose levels in mmol/l (mean ± SE) 120 min and 240 min after injection. s.c.: subcutaneous. i.p.: intraperitoneal. n = 6 mice/group. * p = 0.0001, ** p = 0.002.
Kinetics of bromocriptine effects on glycemia in NOD mice
Bromocriptine effects on non-fasting blood glucose levels in mice of both sexes were compared from 5 to 360 min after i.p. administration (Figure 1). In females, glycemia was significantly increased as early as 10 min following bromocriptine injection (p = 0.00003). The maximum drug effect (an approximately 1.7-fold increase) was observed 15 min after injection and persisted for 180 min (p range: 0.0001 to 0.0004), before returning to basal values 360 min post administration. In males, bromocriptine also induced significant hyperglycemia as early as 10 min after injection (p = 0.00002). The maximum drug effects (an approximately 2.5-fold increase) were obtained between 30 min and 60 min (p = 0.00004 in both cases). Thereafter, blood glucose levels gradually declined but remained significantly higher than basal levels up to 360 min following bromocriptine administration (p range: 0.0002 to 0.00004). Finally, at any time other than 180 min, bromocriptine-induced hyperglycemia was significantly higher in males than females (p range: 0.04 to 0.00002).
Figure 1. Kinetics of the bromocriptine effect on glycemia in NOD females and males.
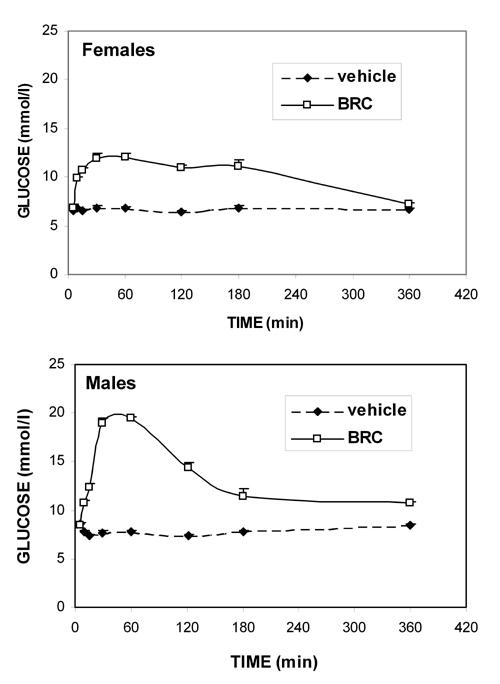
Means ± SE, n = 6 animals per sex/time group. From 10 to 180 min following bromocriptine injection (10 mg/kg, i.p.), glycemia was significantly higher in bromocriptine-treated mice than in vehicle-treated mice of both sexes (p range: 0.00004 to 0.0002). Significant bromocriptine-induced hyperglycemia persisted 360 min after injection only in males (p = 0.00002).
Kinetics of bromocriptine effects on NOD mouse insulinemia
We then studied the kinetics of i.p. injected bromocriptine or its vehicle on blood insulin, glucagon and corticosterone, which are the major hormones that regulate glucose homeostasis. Bromocriptine effects on insulin (Figure 2) differed according to sex: in females, while apparently decreased at the earliest time point, insulin concentrations were not significantly altered, regardless of the time after administration; in males, a significant decrease was observed 15 min and 30 min after injection (p = 0.04 and p = 0.02, respectively). Thereafter, insulin values from bromocriptine-treated animals of both sexes were comparable to those of vehicle-treated animals.
Figure 2. Kinetics of the bromocriptine effect on insulinemia in NOD females and males.
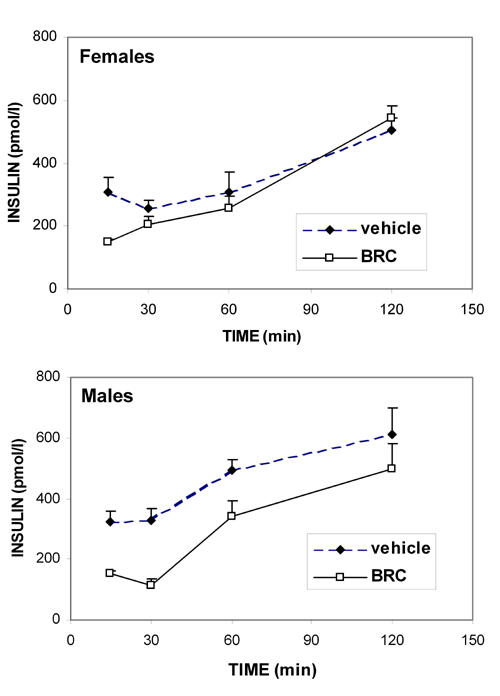
Means ± SE, n = 6 animals per sex/time group. Bromocriptine (10 mg/kg, i.p.) significantly lowered insulinemia in NOD males only 15 min (p = 0.04) and 30 min (p = 0.02) after injection. Moreover, 120 min after administration, insulin levels were significantly increased in vehicle-injected NOD males (p = 0.001), and bromocriptine-injected female and male NOD mice (p = 0.0001 and p = 0.0002 mice respectively).
Interestingly, blood insulin concentrations progressively rose with time after administration of the vehicle, which was significant in males at 120 min (p = 0.001), while it only tended towards significance in females (p = 0.06).
Kinetics of bromocriptine effects on NOD mouse glucagon levels
Blood glucagon concentrations (Figure 3), determined at different times after vehicle injection, did not change significantly for females or males. Moreover, bromocriptine action was similar in both sexes throughout the kinetic study: it was characterized by biphasic increases in glucagon levels. First, a sharp and significant rise was observed between 5 and 15 min after i.p. bromocriptine injection (p range: 0.002 to 0.0001 in both sexes), followed by a drop to vehicle-treated levels at 30 min. Then, 60 and 120 min after bromocriptine injection, a second, mild but significant increase of glucagon was detected in both sexes (p range: 0.04 to 0.0007).
Figure 3. Kinetics of the bromocriptine effect on glucagon levels in NOD females and males.
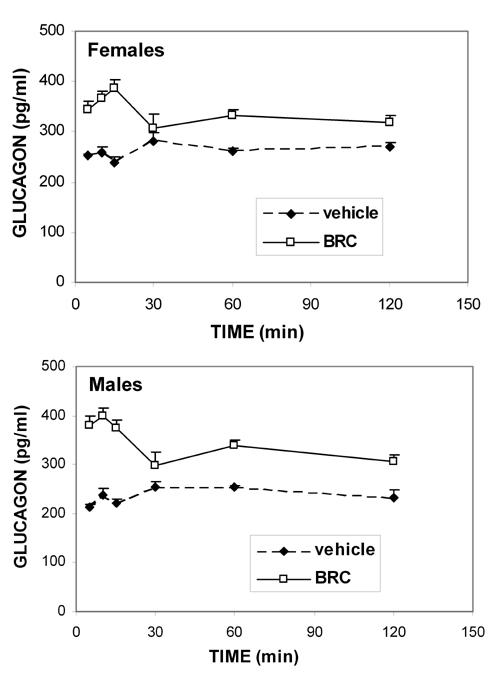
Means ± SE, n = 6 animals per sex/time group. The stimulatory bromocriptine effect (10 mg/kg, i.p.) on glucagon levels is biphasic, being significant in both sexes at all times investigated, except 30 min (p range: 0.04 to 0.0001 for both sexes). When no SE bar is visible, the value is smaller than the data symbol.
Kinetics of bromocriptine effects on NOD mouse corticosterone levels
During the first 30 min after vehicle injection, blood corticosterone concentrations (Figure 4) were markedly enhanced in both sexes, because of the stress of the injection alone. Corticosterone levels comparable to basal non-injected levels were recovered around 60 min post-injection in both sexes.
Figure 4. Kinetics of the bromocriptine effect on circulating corticosterone in NOD females and males.
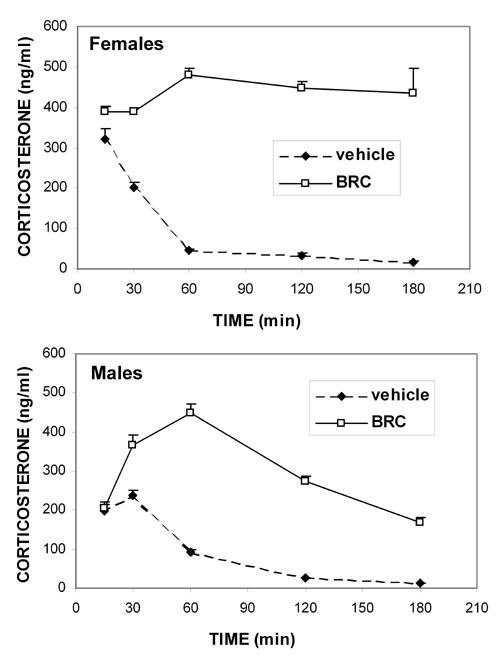
Means ± SE, n = 6 in each sex/time group. Note the significant stress effect of i.p. injection alone (p = 0.001 in both sexes) 15 and 30 min after vehicle injection. From 30 to 180 min following bromocriptine injection (10 mg/kg, i.p.), blood corticosterone values were significantly higher in NOD females and males than in sex-matched vehicle-treated mice (p = 0.0001 for females and p range 0.0009 to 0.0001 for males). A significant sexual dimorphism was observed, with females having higher corticosterone levels than males (p range: 0.005 to 0.0001). When no SE bar is visible, the value is smaller than the data symbol.
Blood corticosterone concentrations were similar in bromocriptine- and vehicle-injected mice 15 min after i.p. injection in both sexes of NOD mice, because of the stress induced by injection alone, as stated above. Then, in females, a sustained elevated corticosterone concentration was observed at all times investigated (p = 0.0001). In males, however, although bromocriptine induced significantly higher than vehicle blood corticosterone levels from 30 to 180 min after injection (p range: 0.0009 to 0.0001), this effect was not sustained as it was in females. Furthermore, a sex effect was observed, with bromocriptine-treated females having significantly higher blood corticosterone concentrations than males at all times except at 60 min (p range: 0.005 to 0.0001).
Effects of the D2-dopaminergic antagonist, metoclopramide, on glycemia and some of its regulatory hormones
We first studied the effects of metoclopramide and bromocriptine (10 mg/kg, each) administered i.p., alone or concomitantly, on the glycemia of NOD females and males, 120 min after injection, i.e. a time at which bromocriptine still had a significant hyperglycemic effect in both sexes but was late enough to avoid the stress effect of injection and to allow metoclopramide to exert its antagonistic action. In both sexes, metoclopramide alone had no effect on glycemia, while it significantly inhibited hyperglycemic response to bromocriptine (p = 0.004 and p = 0.0001 for females and males respectively, Figure 5).
Figure 5. Specificity of the bromocriptine effect on glycemia in NOD females and males.
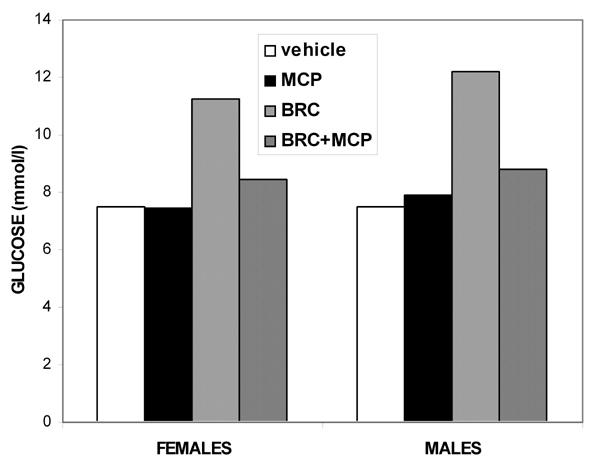
Means ± SE, n = 6 animals per sex/drug group, 120 min after administration of 10 mg/kg bromocriptine (BRC) alone or concomitantly with 10 mg/kg i.p. metoclopramide (MCP), a D2-dopaminergic antagonist. Metoclopramide significantly inhibited bromocriptine-induced glycemia in females and males respectively. Bromocriptine vs. vehicle: p = 0.0002 and p = 0.0001. Bromocriptine vs. metoclopramide: p = 0.004 and p = 0.0001.
We then assessed the possible antagonistic action of metoclopramide on bromocriptine modulation of the various hormones involved in glucose homeostasis, the kinetics of which are presented above. However, because of the diversity of kinetic responses to bromocriptine, i.e. rapid and non-sustained (insulin), more or less sustained (glucose and glucagon) or involving a transient early stress effect from the injection alone (corticosterone), two different protocols were used: 1) metoclopramide injection, followed 30 min later by bromocriptine, with regulatory hormone determinations at 60 min or 2) concomitant injection of both compounds with measurements made 120 min later. These two approaches were assessed only in NOD males. In the separate or concomitant administration experiments, metoclopramide significantly counteracted the hyperglycemic effect of bromocriptine (p = 0.0002 and p = 0.0001 respectively, Figure 6), reproducing the data presented in Figure 5 for both sexes. However, no significant antagonistic action on bromocriptine-induced modulation of hormones could be demonstrated after separate drug injections, possibly because of the high corticosterone levels present in each group after two successive injections (Figure 6).
Figure 6. Specificity of the bromocriptine effect on glycemia, insulinemia, glucagon levels and circulating corticosterone in NOD males.
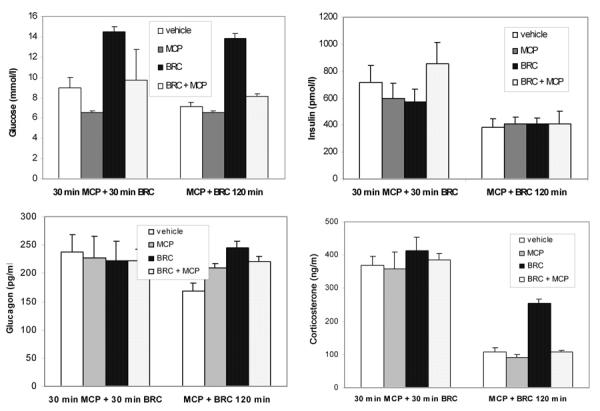
Means ± SE, n = 6 in each group, for the 2 different protocols: either first 10 mg/kg i.p. metoclopramide (MCP), a D2-dopaminergic antagonist, at time 0. This was followed 30 min later by 10 mg/kg i.p. bromocriptine (BRC) injection, designated as a "separate injection", with determinations at 60 min or 120 min after concomitant administration of the 2 agents, designated as a "concomitant injection". Metoclopramide significantly inhibited the bromocriptine-induced effects on glycemia in both protocols (p = 0.0002 and p = 0.0001 respectively) and the effects on blood corticosterone after simultaneous injection only (p = 0.0001).
However, as also shown in Figure 6, 120 min after concomitant injection of metoclopramide and bromocriptine into NOD males, the bromocriptine-induced increase of corticosterone concentrations was significantly inhibited (p = 0.0001), while insulin levels were (as expected) unaffected, regardless of the agent administered. Metoclopramide induced a slight but significant increase in glucagon levels, as did bromocriptine (p = 0.03 and 0.04, respectively), but had no antagonistic activity.
Discussion
We first showed that the bromocriptine-induced hyperglycemic effect in NOD mice is clearly dependent on the route of administration. Therefore, bromocriptine differs from other dopaminergic agonists, such as lergotrile or quinpirole, which are hyperglycemic even after s.c. injection [7, 17]. The absence of a hyperglycemic action on the part of s.c. injected bromocriptine remains unexplained, but it is possible to hypothesize that i.p. administration leads to faster or more complete absorption and/or increased conversion into active metabolite(s), as suggested by others [7]. However, a rapid action mediated through central nervous system dopamine receptors together with faster absorption may also be causative for the velocity of the bromocriptine-induced hyperglycemic effect [7, 24]. The differential effect obtained according to the route of administration might explain why NOD females were protected from diabetes, when s.c. injected but not i.p. [2, 3]. Bromocriptine administered s.c. inhibits prolactin secretion only, thus lowering its immunostimulatory effects and dampening the autoimmune reaction [1, 2], while i.p. bromocriptine induces a hyperglycemic effect that counteracts its beneficial action on the immune system.
Kinetic studies revealed that bromocriptine-induced hyperglycemia occurs rapidly, is significant in both sexes 10 min after i.p. injection, and persists up to 180 and 360 min in non-fasted NOD females and males respectively. Previously reported kinetic data concerned only the injection of dopaminergic agonists into fasted male rodents: a significant hyperglycemic effect of apomorphine in rats 15 min after s.c. injection [7] and marked hyperglycemia lasting up to 5 h after i.p. bromocriptine administration to mice [4]. Our data underline a marked sex difference in the bromocriptine-induced hyperglycemic effect in non-fasted mice, with males having higher blood glucose levels than females, as expected from the well known androgen-induced insulin resistance [18]. Because metoclopramide injection is able to abolish the hyperglycemic effect of bromocriptine, in NOD mice the latter appears to be due to D2-dopaminergic receptor stimulation, as previously demonstrated for various D2-dopaminergic agonists [4, 7].
Regarding the bromocriptine effect on non-fasted NOD mouse insulinemia, kinetic studies reveal an early significant decline 15 min and 30 min after i.p. injection only in males. These data corroborate those showing an in vivo diminution of serum insulin one hour after i.p. bromocriptine injection into fasted male rats [4]. Moreover, in vitro, various dopaminergic agonists, including bromocriptine, inhibit insulin release from isolated rodent islets [19, 20]. In mouse islets, this effect is blocked by pimozide, a D2-receptor-subtype antagonist, suggesting that insulin release is inhibited, at least partially, through bromocriptine activation of D2-dopaminergic receptors [20]. It is worth noting here: 1) the presence of non-negligible quantities of dopamine in the islets of mouse, hamster and guinea pig [21], and 2) that i.v. L-Dopa injection into mice rapidly increases β-cell dopamine and consequently lowers insulin response to different secretagogues [22]. Thereafter, insulinemia gradually rose during the rest of the kinetic study. In bromocriptine-treated animals, the latter effect is mainly the consequence of increased glycemia and glucagon levels. Indeed, glucagon is known to stimulate insulin release directly through specific receptors on β-cells [23]. In vehicle-treated animals, late insulin increase might reflect recovery from the stress-induced inhibitory effects of injection on insulin secretion, NOD β-cell hyperactivity and/or insulin resistance [16, 18].
Bromocriptine inhibition of insulin secretion may also result from indirect pathways involving the release of catecholamines and/or glucocorticoids, both known for their ability to block insulin secretion. In fact, various D2-dopaminergic agonists induce rapid and marked increases in circulating epinephrine [12, 24, 25]. In this context, the partial inhibition of a quinpirole effect by domperidone, a D2-dopaminergic antagonist unable to cross the blood-brain barrier, suggests that dopamine analogs stimulate epinephrine release from the adrenal medulla via peripheral D2-dopaminergic receptors [12]. Moreover, dopaminergic agonists also activate the sympathoadrenal system through a primary action on dopamine receptors in the central nervous system [24]. Finally, catecholamines present in islet innervation [26-28] might also directly modulate insulin release [21]. Regardless of their origin, catecholamines inhibit insulin secretion through specific α2-adrenergic receptors present on β-cells [29]. Still at the β-cell level, corticosteroids, the secretion of which is strongly enhanced by bromocriptine (present data and [14, 15]), might potentially be responsible for part of the inhibitory effect observed, because of the presence of specific glucocorticoid receptors in β-cells and their rapid inhibitory effect on pancreatic insulin release [30-33]. Corticosteroids may also have more long-term inhibitory effects on insulin synthesis.
Regarding glucagon, the α-cell counterregulatory hormone, a rapid, marked and sustained increase is observed after i.p. bromocrip-tine injection, with similar kinetics in both sexes. Studies conducted on rodents after dopamine or dopaminergic-agonist administration showed either an increase [34] or no change [6, 17, 24]. However, differences in the methodologies applied make comparison difficult and the biphasic pattern, observed here, may explain these discrepancies. Since pancreatic α-cells do not appear to contain dopamine receptors, only indirect effects mediated by catecholamines and/or corticosterone release appear to be involved in bromocriptine-induced increased glucagon levels in mice. As mentioned above, dopamine agonists can stimulate catecholamine release from the adrenal medulla either directly [12] or indirectly through central nervous system activation of the sympathoadrenal system [24]. In addition, islets are, particularly at their periphery where α-cells are located, richly innervated by sympathetic nerves, which synthesize catecholamines locally [26, 28, 35]. Therefore, the elevated circulating glucagon concentration observed after dopaminergic-agonist injection might, at least in part, indirectly result from the interaction of catecholamines with specific α2- and β2-adrenergic receptors present on pancreatic α-cells [36]. Moreover, bromocriptine sharply increases corticosterone levels in NOD mice, an effect that could contribute, via the presence of specific glucocorticoid receptors in α-cells [33], to stimulation of glucagon release, as described in the isolated perfused rat pancreas [30]. Such an effect might be predominant in the second phase of kinetic response.
Finally, a significant increase of corticosterone, another counterregulatory hormone, can be observed only 30 min after i.p. bromocriptine injection, because of the stress of the injection alone. Thereafter, bromocriptine effects on blood corticosterone are comparable in both sexes and, after 60 min, vary according to sex, with more sustained levels in females than males. This difference reflects the sexual dimorphism that exists in rodent hypothalamo-pituitary-adrenal axis activity, because of a stimulatory action on the part of estrogens [37]. The D2-dopaminergic antagonist, metoclopramide, completely inhibits late bromocriptine effects on NOD corticosterone levels, suggesting an action mediated by specific D2-dopaminergic-receptor stimulation. In rats, the observation that the pergolide-induced increase in corticosterone is blocked by haloperidol, a dopaminergic antagonist that acts both centrally and peripherally, but not by the peripheral antagonist, domperidone, is indicative of central D2-dopaminergic-receptor involvement [15]. These receptors are probably located in the hypothalamus, since direct quinpirole administration into the third ventricle increases serum adrenocorticotrophic hormone (ACTH) levels in rats [13]. A rapid rise in blood ACTH is also observed after i.p. injection of haloperidol or bromocriptine into adult male rats or mice [38, 39]. Considered as a whole, these data suggest that the stimulatory effect of bromocriptine on corticosterone results from enhanced hypothalamo-pituitary-adrenal axis activity mediated by hypothalamic dopaminergic receptors and pituitary ACTH release.
In conclusion, in pre-diabetic NOD mice, complex neuroendocrine interactions are involved in the bromocriptine-induced hyperglycemic effect. The latter appears to result from successive and synergistic interactions, involving decreased insulin and increased anti-insulin hormones, such as glucagon, catecholamines and corticosterone, as previously shown in various models of stress-induced hyperglycemia [40, 41]. Some of these hormones are short-term regulators of glucose homeostasis (catecholamines and glucagon), others are long-term regulators (corticosterone) [42]. Moreover, from a therapeutic point of view, it should be mentioned that D2-dopaminergic agonists might have the opposite effects on glucose homeostasis. For example, in obese and/or diabetic (type 2) humans or rodents, chronic bromocriptine administration significantly lowers glycemia and therefore has a potentially beneficial effect [43, 44], in contrast to that observed in normal rodents and type 1 pre-diabetic, i.p. injected NOD mice [3, 7, 19, 38]. The mechanisms underlying these differences remain to be investigated in the light of an increasing incidence of intermediary forms of diabetes, e.g. latent autoimmune diabetes in adults, also called type 1 ½ diabetes [45].
Acknowledgments
The authors thank Mrs. Cisse for technical assistance. This research was funded by grants from CNRS, Universite Paris V, Fondation de France, Alfediam (associated with Lilly Laboratories), INSERM-NWO, BIOMED "Betimmune" and 5th PCRD "Monodiab".
References
- 1.Berczi I. The role of prolactin in the pathogenesis of autoimmune disease. Endocr Pathol. 1993;4:178–195. doi: 10.1007/BF02915460. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins TA, Gala RR, Dunbar JC. Prolactin modulates the incidence of diabetes in male and female NOD mice. Autoimmunity. 1994;18:155–162. doi: 10.3109/08916939409007991. [DOI] [PubMed] [Google Scholar]
- 3.Durant S, Alves V, Coulaud J, El Hasnaoui A, Dardenne M, Homo-Delarche F. Attempts to pharmacologically modulate prolactin levels and type 1 autoimmune diabetes in the non-obese diabetic (NOD) mouse. J Autoimmun. 1995;8:875–885. doi: 10.1016/s0896-8411(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 4.Ageel AM, El-Denshary ES, Abu-Jayyab AR. Effect of bromocriptine on sugar level in mice. IRCS Med Sci. 1984;12:475–476. [Google Scholar]
- 5.Hakanson R, Lundquist I, Rerup C. On the hyperglycaemic effect of DOPA and dopamine. Eur J Pharmacol. 1967;1(2):114–119. doi: 10.1016/0014-2999(67)90047-7. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt MJ, Root MA, Hall JL. Dopamine agonist-induced hyperglycemia in rats: structure-activity relationships and mechanisms of action. Eur J Pharmacol. 1983;90:169–177. doi: 10.1016/0014-2999(83)90234-0. [DOI] [PubMed] [Google Scholar]
- 7.Arneric SP, Chow SA, Long JP, Fischer LJ. Dopamine analog-induced hyperglycemia in rats: involvement of the adrenal medulla and the endocrine pancreas. J Pharmacol Exp Ther. 1984;228:551–559. [PubMed] [Google Scholar]
- 8.Gerich JE. Lillie lecture 1988. Glucose couterregulation and its impact on diabetes mellitus. Diabetes. 1988;37:1608–1617. doi: 10.2337/diab.37.12.1608. [DOI] [PubMed] [Google Scholar]
- 9.Lager I. The insulin-antagonistic effect of the counterregulatory hormones. J Intern Med. 1991;735:41–47. [PubMed] [Google Scholar]
- 10.Gerich JE. Control of glycaemia. Baillieres Clin Endocrinol Metab. 1993;7(3):551–586. doi: 10.1016/s0950-351x(05)80207-1. [DOI] [PubMed] [Google Scholar]
- 11.Sinha YN, Salocks CB, VanderLaan WP. A comparison of the effects of CB-154 and lergotrile mesylate on prolactin and growth hormone secretion in mice. Horm Metab Res. 1976;8(5):332–336. doi: 10.1055/s-0028-1093627. [DOI] [PubMed] [Google Scholar]
- 12.Kujacic M, Hansson LO, Carlsson A. Acute dopaminergic influence on plasma adrenaline levels in the rat. Eur J Pharmacol. 1995;273:247–257. doi: 10.1016/0014-2999(94)00699-8. [DOI] [PubMed] [Google Scholar]
- 13.Borowsky B, Kuhn CM. D1 and D2 dopamine receptors stimulate hypothalamo-pituitary-adrenal activity in rats. Neuropharmacology. 1992;31(7):671–678. doi: 10.1016/0028-3908(92)90145-f. [DOI] [PubMed] [Google Scholar]
- 14.Foreman MM, Fuller RW, Hynes MD, Gidda JS, Nichols CL, Schaus JM, Kornfeld EC, Clemens JA. Preclinical studies on quinelorane, a potent and highly selective D2-dopaminergic agonist. J Pharmacol Exp Ther. 1989;250:227–235. [PubMed] [Google Scholar]
- 15.Fuller RW, Snoddy HD. Elevation of serum corticosterone concentrations in rats by pergolide and other dopamine agonists. Endocrinology. 1981;109:1026–1032. doi: 10.1210/endo-109-4-1026. [DOI] [PubMed] [Google Scholar]
- 16.Amrani A, Jafarian-Tehrani M, Mormede P, Durant S, Pleau JM, Haour F, Dardenne M, Homo-Delarche F. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J Endocrinol. 1996;148:139–148. doi: 10.1677/joe.0.1480139. [DOI] [PubMed] [Google Scholar]
- 17.Uvnas-Moberg K, Ahlenius S, Alster P, Hillegaart V. Effects of selective serotonin and dopamine agonists on plasma levels of glucose, insulin and glucagon in the rat. Neuroendocrinology. 1996;63:269–274. doi: 10.1159/000126970. [DOI] [PubMed] [Google Scholar]
- 18.Amrani A, Durant S, Throsby M, Coulaud J, Dardenne M, Homo-Delarche F. Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology. 1998;139:1115–1124. doi: 10.1210/endo.139.3.5823. [DOI] [PubMed] [Google Scholar]
- 19.Arneric SP, Chow SA, Long JP, Fischer LJ. Inhibition of insulin release from rat pancreatic islets by drugs that are analogues of dopamine. Diabetes. 1984;33:888–893. doi: 10.2337/diab.33.9.888. [DOI] [PubMed] [Google Scholar]
- 20.El-Denshary EE, Ismail NA, Gagerman E, Sehlin J, Taljedal IB. Bromocriptine and insulin secretion. Biosci rep. 1982;2(2):115–116. doi: 10.1007/BF01116177. [DOI] [PubMed] [Google Scholar]
- 21.Lundquist I, Ahren B, Hansson C, Hakanson R. Monoamines in pancreatic islets of guinea pig, hamster, rat, and mouse determined by high performance liquid chromatography. Pancreas. 1989;4:662–667. doi: 10.1097/00006676-198912000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Ericson LE, Hakanson R, Lundquist I. Accumulation of dopamine in mouse pancreatic B-cells following injection of L-DOPA. Localization to secretory granules and inhibition of insulin secretion. Diabetologia. 1977;13:117–124. doi: 10.1007/BF00745138. [DOI] [PubMed] [Google Scholar]
- 23.Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia. 2000;43:1012–1019. doi: 10.1007/s001250051484. [DOI] [PubMed] [Google Scholar]
- 24.Arneric SP, Chow SA, Bhatnagar RK, Webb RL, Fischer LJ, Long JP. Evidence that central dopamine receptors modulate sympathetic neuronal activity to the adrenal medulla to alter glucoregulatory mechanisms. Neuropharmacology. 1984;23:137–147. [PubMed] [Google Scholar]
- 25.Nagahama S, Chen YF, Oparil S. Mechanism of the depressor effect of bromocriptine in the spontaneously hypertensive rat. J Pharmacol Exp Ther. 1984;228:370–375. [PubMed] [Google Scholar]
- 26.Ahren B. Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia. 2000;43:393–410. doi: 10.1007/s001250051322. [DOI] [PubMed] [Google Scholar]
- 27.Borelli MI, Gagliardino JJ. Possible modulatory effect of endogenous islet catecholamines on insulin secretion. BMC Endocr Disord. 2001;1:1–5. doi: 10.1186/1472-6823-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundquist I, Ericson LE. Beta-Adrenergic insulin release and adrenergic innervation of mouse pancreatic islets. Cell Tissue Res. 1978;193:73–85. doi: 10.1007/BF00221602. [DOI] [PubMed] [Google Scholar]
- 29.Ostenson CG, Cattaneo AG, Doxey JC, Efendic S. Alpha-adrenoceptors and insulin release from pancreatic islets of normal and diabetic rats. Am J Physiol. 1989;257:E439–E443. doi: 10.1152/ajpendo.1989.257.3.E439. [DOI] [PubMed] [Google Scholar]
- 30.Barseghian G, Levine R. Effect of corticosterone on insulin and glucagon secretion by the isolated perfused rat pancreas. Endocrinology. 1980;106:547–552. doi: 10.1210/endo-106-2-547. [DOI] [PubMed] [Google Scholar]
- 31.Billaudel B, Mathias PC, Sutter BC, Malaisse WJ. Inhibition by corticosterone of calcium inflow and insulin release in rat pancreatic islets. J Endocrinol. 1984;100:227–233. doi: 10.1677/joe.0.1000227. [DOI] [PubMed] [Google Scholar]
- 32.Delaunay F, Khan A, Cintra A, Davani B, Ling ZC, Andersson A, Ostenson CG, Gustafsson J, Efendic S, Okret S. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest. 1997 doi: 10.1172/JCI119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthes H, Kaiser A, Stier U, Riecken EO, Rosewicz S. Glucocorticoid receptor gene expression in the exocrine and endocrine rat pancreas. Endocrinology. 1994;135:476–479. doi: 10.1210/endo.135.1.8013388. [DOI] [PubMed] [Google Scholar]
- 34.George D, Bailey P. The effect of adrenergic and ganglionic blockers upon the L-dopa-stimulated release of glucagon in the rat. Proc Soc Exp Biol Med. 1978;157(1):1–4. doi: 10.3181/00379727-157-39978. [DOI] [PubMed] [Google Scholar]
- 35.Ushiki T, Watanabe S. Distribution and ultrastructure of the autonomic nerves in the mouse pancreas. Microsc Res Tech. 1997;37:399–406. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<399::AID-JEMT4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 36.Hirose H, Maruyama H, Ito K, Kido K, Koyama K, Saruta T. Effects of alpha 2- and beta-adrenergic agonism on glucagon secretion from perfused pancreata of normal and streptozocin-induced diabetic rats. Metabolism. 1993;42:1072–1076. doi: 10.1016/0026-0495(93)90025-j. [DOI] [PubMed] [Google Scholar]
- 37.Spinedi E, Suescun MO, Hadid R, Daneva T, Gaillard RC. Effects of gonadectomy and sex hormone therapy on the endotoxin-stimulated hypothalamo-pituitary-adrenal axis: evidence for a neuroendocrine-immunological sexual dimorphism. Endocrinology. 1992;131:2430–2436. doi: 10.1210/endo.131.5.1330501. [DOI] [PubMed] [Google Scholar]
- 38.El-Denshary ES, Ageel AM, el-Wakkad I, Abu-Jayyab AR. Effect of bromocriptine on lipid plasma levels in rats. Life Sci. 1987;40:1531–1535. doi: 10.1016/0024-3205(87)90386-9. [DOI] [PubMed] [Google Scholar]
- 39.Jezova D, Jurcovicova J, Vigas M, Murgas K, Labrie F. Increase in plasma ACTH after dopaminergic stimulation in rats. Psychopharmacology (Berl) 1985;85:201–203. doi: 10.1007/BF00428414. [DOI] [PubMed] [Google Scholar]
- 40.Shamoon H, Hendler R, Sherwin RS. Synergistic interactions among antiinsulin hormones in the pathogenesis of stress hyperglycemia in humans. J Clin Endocrinol Metab. 1981;52:1235–1241. doi: 10.1210/jcem-52-6-1235. [DOI] [PubMed] [Google Scholar]
- 41.Yamada F, Inoue S, Saitoh T, Tanaka K, Satoh S, Takamura Y. Glucoregulatory hormones in the immobilization stress-induced increase of plasma glucose in fasted and fed rats. Endocrinology. 1993;132:2199–2205. doi: 10.1210/endo.132.5.8477665. [DOI] [PubMed] [Google Scholar]
- 42.Porte D, Woods SC. Neural regulation of islet hormones and its role in energy balance and stress hyperglycemia. In: Ellenberg, Rifkins, editors. Series M. Medical Examination. Pub.Co; New Hyde Park: 1983. pp. 175–197. [Google Scholar]
- 43.Pijl H, Ohashi S, Matsuda M, Miyazaki Y, Mahankali A, Kumar V, Pipek R, Iozzo P, Lancaster JL, Cincotta AH, DeFronzo RA. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care. 2000;23:1154–1161. doi: 10.2337/diacare.23.8.1154. [DOI] [PubMed] [Google Scholar]
- 44.Scislowski PW, Tozzo E, Zhang Y, Phaneuf S, Prevelige R, Cincotta AH. Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int J Obes Relat Metab Disord. 1999;23:425–431. doi: 10.1038/sj.ijo.0800893. [DOI] [PubMed] [Google Scholar]
- 45.Juneja R, Hirsch IB, Naik RG, Brooks-Worrell BM, Greenbaum CJ, Palmer JP. Islet cell antibodies and glutamic acid decarboxylase antibodies, but not the clinical phenotype, help to identify type 1(1/2) diabetes in patients presenting with type 2 diabetes. Metabolism. 2001;50:1008–1013. doi: 10.1053/meta.2001.25654. [DOI] [PubMed] [Google Scholar]