

Abstract
The activity of serum response factor (SRF), an essential transcription factor in mouse gastrulation, is regulated by changes in actin dynamics. Using Srf(−/−) embryonic stem (ES) cells, we demonstrate that SRF deficiency causes impairments in ES cell spreading, adhesion, and migration. These defects correlate with defective formation of cytoskeletal structures, namely actin stress fibers and focal adhesion (FA) plaques. The FA proteins FA kinase (FAK), β1-integrin, talin, zyxin, and vinculin were downregulated and/or mislocalized in ES cells lacking SRF, leading to inefficient activation of the FA signaling kinase FAK. Reduced overall actin expression levels in Srf(−/−) ES cells were accompanied by an offset treadmilling equilibrium, resulting in lowered F-actin levels. Expression of active RhoA-V14 rescued F-actin synthesis but not stress fiber formation. Introduction of constitutively active SRF-VP16 into Srf(−/−) ES cells, on the other hand, strongly induced expression of FA components and F-actin synthesis, leading to a dramatic reorganization of actin filaments into stress fibers and lamellipodia. Thus, using ES cell genetics, we demonstrate for the first time the importance of SRF for the formation of actin-directed cytoskeletal structures that determine cell spreading, adhesion, and migration. Our findings suggest an involvement of SRF in cell migratory processes in multicellular organisms.
Keywords: SRF; ES cells; actin cytoskeleton; focal adhesion; cell migration
Introduction
The actin cytoskeleton directs many essential cellular processes, including cell–matrix and cell–cell adhesion, cell motility, cytokinesis, and endo-/exocytosis (Salmon, 1989; Gumbiner, 1996; Chen et al., 2000). F-actin and monomeric actin (G-actin) are balanced by an equilibrium reaction of actin incorporation at the barbed end of a filament and actin dissociation at its pointed end (“treadmilling”). The actin treadmilling cycle is temporally and spatially coordinated by signaling cascades that target actin binding proteins, such as ADF/cofilin and profilin (Beckerle, 1998).
Actin cytoskeletal structures include cortical actin, stress fibers, lamellipodia, and microspikes. Remodelling of preexisting actin filaments into such structures is mainly controlled by members of the Rho family of GTPases (Nobes and Hall, 1995). RhoA activation leads to the formation of actin stress fibers, consisting of long bundles of filaments traversing the cell (Ridley and Hall, 1992). Actin stress fibers are linked to integrins at the inner surface of the plasma membrane involving a multicomponent protein complex called focal adhesion (FA)* (Critchley, 2000). Components of FAs include α-actinin, FA kinase (FAK), talin, vinculin, and zyxin. Vinculin and zyxin are adaptors for other FA proteins, such as α-actinin, talin, F-actin, and VASP. Whereas the interaction between vinculin and talin/F-actin apparently stabilizes FA complexes (Gilmore and Burridge, 1996), binding of VASP to zyxin and/or vinculin appears to stimulate actin polymerization via recruitment of profilin/G-actin complexes (Huttelmaier et al., 1998; Drees et al., 2000). FAK links FAs to different signaling pathways involved in cell growth, migration, and survival (Schlaepfer et al., 1999). FA assembly has been studied using genetically modified cell lines. Embryonic stem (ES) cells deficient for vinculin are able to assemble FAs when plated on fibronectin, demonstrating that vinculin is dispensable for this process (Volberg et al., 1995; Xu et al., 1998). Fibroblasts lacking FAK also contain FAs, albeit with altered size and distribution (Ilic et al., 1995a,b). Null mutations of the Zyxin gene have not been described so far. However, peptides that block the interaction between zyxin and α-actinin cause reduced cell motility (Drees et al., 1999).
Serum response factor (SRF) is a transcription factor of the MADS box family (Norman et al., 1988; Shore and Sharrocks, 1995). DNA binding sites for SRF (serum response elements [SREs] or CArG boxes) have been found in the promoters of ∼50 different genes so far (unpublished data), including immediate early genes (IEGs) like c-fos and Egr-1 (Herschman, 1991) and muscle-specific genes (e.g., the Actin genes, Dystrophin) (for review see Sobue et al., 1999). SRF exhibits important functions in concert with accessory proteins, e.g., ternary complex factors (TCFs) (Shaw et al., 1989) or other partner proteins. TCF-independent activation of SRF target genes has also been described (Graham and Gilman, 1991; Hill et al., 1994; Johansen and Prywes, 1994) and seems to be particularly important for the regulation of muscle-specific genes (Carnac et al., 1998; Wei et al., 1998). One TCF-independent signaling pathway targeting SRF involves RhoA (Hill et al., 1995), whereby downstream signaling components may include Rho kinase (Chihara et al., 1997), and possibly the transcription factors NF-κB and C/EBPβ as potential SRF partner proteins (Montaner et al., 1999). Recently, actin dynamics were demonstrated to activate SRF in fibroblasts (Sotiropoulos et al., 1999), regulating the endogenous Vinculin, Actin, and Srf genes largely in a RhoA-dependent manner (Gineitis and Treisman, 2001). On the other hand, in cardiac myocytes, an organized actin cytoskeleton was found to be essential for RhoA-dependent activation of SRF (Wei et al., 2001). SRF itself controls the expression of genes encoding cytoskeletal proteins, including vinculin and different actins (Frederickson et al., 1989; Sartorelli et al., 1990; Lee et al., 1991; Moiseyeva et al., 1993; Mack and Owens, 1999).
Functions of SRF in multicellular organisms were revealed by the Drosophila melanogaster mutations pruned and blistered, two alleles of the Drosophila SRF homologue (DSRF) (Affolter et al., 1994; Guillemin et al., 1996). Both mutant phenotypes are, at least in part, due to cytoskeletal defects. In mammals, SRF fulfills an essential function in mesoderm formation. Mouse embryos lacking SRF do not form any detectable mesoderm cells, apparently do not express mesodermal marker genes, show a drastic reduction in IEG expression, and die during early gastrulation (Arsenian et al., 1998).
To explore a potential role of SRF in regulating cytoskeletal activities, we used murine ES cells homozygous for an Srf null allele (Srf[−/−]). We provide evidence that SRF-deficient ES cells have reduced adhesive and migratory capacities, accompanied by the inability to assemble actin stress fibers and FAs. This reveals a direct functional link between the transcription factor SRF, the formation and remodelling of actin-based cytoskeletal structures, and the generation of functional FA complexes.
Results
Srf(−/−) ES cells display impairments in cell spreading, substrate adhesion, and cell migration
ES cells represent a highly informative in vitro cell system to study the phenotypic effects of gene disruption at the cellular level. Srf(−/−), Srf(−/+), and Srf(−/−)rescue murine ES cell lines have been described previously (Weinhold et al., 2000; Schratt et al., 2001). Plating of undifferentiated wild-type or heterozygous Srf(−/+) ES cells on gelatine-coated tissue culture plates in the presence of leukemia inhibitory factor (LIF) resulted in cell spreading of ∼30–40% of the cells after 2 h (arrows in Fig. 1 A, top). In contrast, cells of two independent Srf(−/−) ES cell lines remained rounded under these conditions, without any signs of cell surface protrusions (Fig. 1 A, bottom). ES cell spreading on different matrices after 2 h is quantified in Fig. 1 B. In contrast to plating on gelatine, only little or no difference between ES cells of different Srf genotype was observed regarding cell spreading on laminin and fibronectin. However, individual Srf(−/−) ES cells were spread to a lesser extent on these two matrices (unpublished data).
Figure 1.

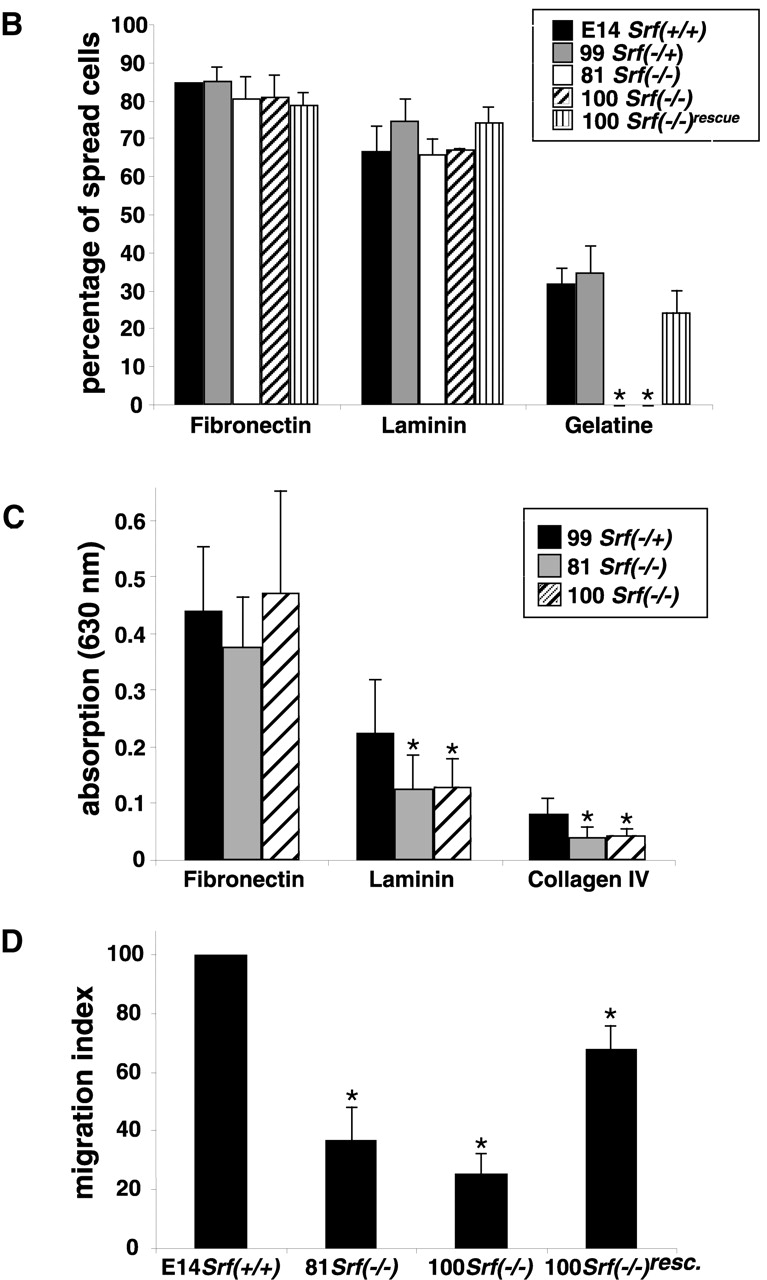
SRF-deficient ES cells are impaired in cell spreading, adhesion, and migration. (A) Srf(−/−) ES cells do not spread on gelatin. ES cells of the indicated Srf genotypes were allowed to settle on gelatine-coated tissue culture plates for 2 h and inspected by scanning electron microscopy. Arrows indicate cells engaged in spreading. (B) Quantitation of ES cell spreading on different matrices. ES cells of the indicated Srf genotype were allowed to settle on tissue culture plates coated with fibronectin (25 μg/ml), laminin (25 μg/ml), or gelatine (0.2%) for 2 h and inspected by light microscopy. Arbitrary fields containing at least 100 cells were counted. Cells that displayed any sign of protrusive activity after 2 h, such as filopodia or lamellipodia, were scored as “spread.” Values represent the percentage of spread cells counted in two independent experiments ± SD. (C) Srf(−/−) ES cells display adhesion defects on laminin and collagen IV. ES cells of the indicated Srf genotype were trypsinized, seeded onto 96-wells coated with either fibronectin, laminin, or collagen IV, and fixed. Adherent cells were stained with crystal violet, the dye extracted and absorption of the obtained solution measured at 630 nm. Absorption at 630 nm is a measure of adherent cells on the respective matrix. Background adhesion of ES cells on BSA was subtracted from each value. Data represent the mean of five independent experiments ± SD. Asterisk indicates that the value of the Srf(−/−) cell line is significantly different from the value of the Srf(−/+) cell line (P < 0.05). (D) Srf(−/−) ES cells are impaired in chemotactic migration. ES cells were allowed to migrate in a FBS-gradient for 22 h in a Boyden chamber assay (see Materials and methods). Cells that reached the lower surface of the chamber were fixed, and staining was performed as described in the legend to C. Obtained values were corrected for migration toward a BSA-coated lower surface, and wild-type was set to 100%. Data represent the mean of three independent experiments ± SD. Asterisk indicates that the value of the Srf(−/−) cell line is significantly different from the value of the Srf(+/+) cell line (P < 0.05). Bar, 25 μm.
The ability to form cell projections and associated cell–matrix contacts is a prerequisite for cellular activities like substrate adhesion and migration. Therefore, we monitored adhesion properties and cell migration behavior of SRF-deficient ES cell lines. ES cells were plated for 1 h on fibronectin, laminin, or collagen IV, and the number of stably adhered cells was measured (Fig. 1 C). No differences were observed between different ES cell lines regarding adhesion to fibronectin. In contrast, adhesion to laminin and collagen IV was significantly reduced by ∼50% in Srf(−/−) ES cells as compared with Srf(−/+) ES cells. These results indicate that a subset of adhesion receptors that confers specific binding to laminin and collagen IV, but not to fibronectin, is affected by SRF deficiency.
To assess the effect of SRF deficiency on the migration behavior of ES cells, we performed Boyden chamber assays. A FBS gradient served as chemo-attractant. The amount of cells that migrated through the membrane toward higher FBS concentration was significantly reduced in both Srf(−/−) ES cell lines tested, as compared with wild-type, Srf(−/+), or Srf(−/−) rescue ES cells (Fig. 1 D). This difference did not arise as a consequence of altered adhesion properties, since Srf(−/−) ES cells adhere to a fibronectin matrix as strongly as wild-type cells (Fig. 1 C). In summary, these results show that SRF deficiency is associated with impaired cell surface activities, namely cell spreading, adhesion, and migration, which all depend on the proper establishment of FAs.
Deregulated expression of FA proteins in Srf(−/−) ES cells
Integrins and FA complexes link the extracellular matrix to the actin cytoskeleton. Since Srf(−/−) ES cells display impaired spreading and adhesion properties when plated on gelatine, we measured by Western blotting whether SRF deficiency affects the expression of the FA proteins α-actinin, FAK, β1-integrin, talin, vinculin, and zyxin (Fig. 2 A). Neither the expression of α-actinin nor FAK was affected by the absence of SRF. The β1-integrin antiserum recognized two proteins in the range of 120 kD in SRF-containing cell extracts. These proteins most likely represent the precursor (p) and the mature (m) β1-integrin chain (Belkin et al., 1997). In the two independent Srf(−/−) ES cell lines tested, the expression of the mature β1-integrin subunit was significantly reduced. In addition, a smaller protein of ∼60 kD was recognized by the β1-integrin antibody in Srf(−/−) ES cell extracts (asterisk in Fig. 2 A). These data suggest that the maturation of β1-integrin is impaired in the absence of SRF. In agreement with previous findings (Priddle et al., 1998), two talin isoforms were present in ES cell extracts (Fig. 2 A). Protein levels of both talin isoforms were severely reduced in Srf(−/−) ES cells. Furthermore, protein levels of the known SRF target vinculin were reduced to ∼50% of wild-type levels in Srf(−/−) ES cells. Interestingly, the full-length zyxin protein was nearly undetectable in Western blots of Srf(−/−) ES cell extracts (Fig. 2 A). Constitutive expression of wild-type SRF in an Srf null background completely restored the expression of β1-integrin, talin, vinculin, and zyxin, demonstrating that the observed defects in FA protein expression are a direct consequence of SRF deficiency.
Figure 2.
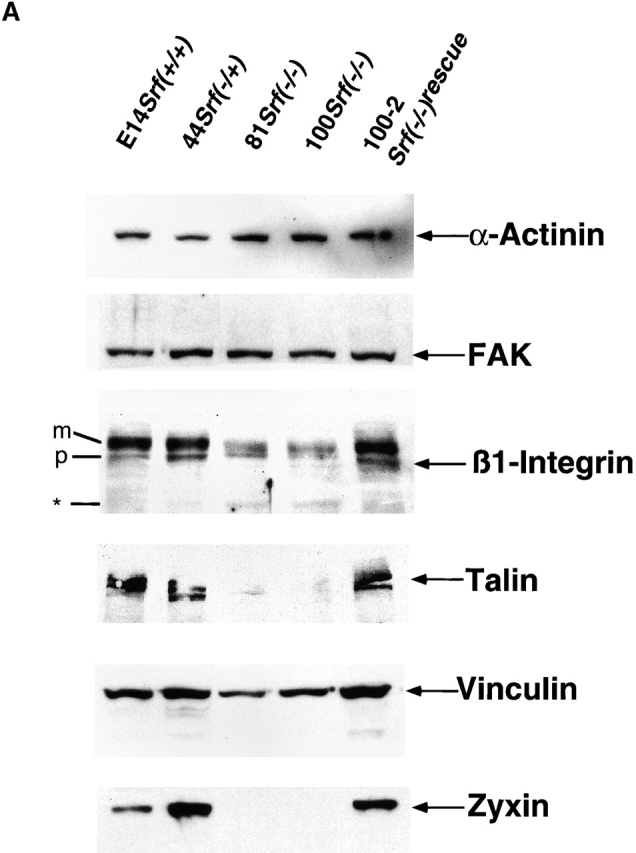

Srf( −/−) ES cells display an impaired expression of FA proteins. (A) Analysis of expression of FA components by Western blotting. ES cell extracts were resolved by SDS-PAGE, blotted, and the membranes probed with antibodies against α-actinin, FAK, β1-integrin, talin, vinculin, and zyxin. p, β1-integrin precursor; m, mature β1-integrin. Asterisk indicates truncated β1-integrin. (B) Measuring of transcript levels in ES cells by quantitative RT-PCR. Relative transcript levels were obtained as described in Materials and methods. Values are given as a percentage of the wild-type and represent the mean of four measurements ± SD, except for 100 Srf(−/−), β1-integrin, where five measurements were averaged. Asterisks denote mRNA levels that are significantly different from the respective wild-type levels (P < 0.005). Note that in Srf(−/−) rescue cells Zyxin mRNA levels did not reach wild-type levels. They were however significantly higher than the Zyxin mRNA levels in Srf(−/−) cells.
To quantitate FA protein expression at the transcript level, we performed quantitative reverse transcription (RT)-PCR analysis with primers specific for β-actin, β1-integrin, Talin, Vinculin, and Zyxin. β1-Integrin mRNA levels did not significantly differ between ES cell lines of different Srf genotype (Fig. 2 B). Therefore, in light of the corresponding protein data (Fig. 2 A), we infer that SRF deficiency influences β1-Integrin expression at the posttranscriptional level. Similarly, in the case of Talin, mRNA levels in Srf(−/−) ES cells were reduced to ∼60% of wild-type levels, whereas talin protein was almost undetectable in Srf(−/−) ES cells (Fig. 2 A). Apparently, SRF-dependent posttranscriptional mechanisms also contribute to the regulation of talin expression. In contrast, SRF deficiency affects β-actin, Vinculin, and Zyxin expression at both the transcript and protein levels to similar extents (compare Fig. 2, A and B). We are currently analyzing whether SRF acts directly on the promoters of the Talin and Zyxin genes, in addition to regulating transcription of β-actin (Frederickson et al., 1989) and Vinculin (Moiseyeva et al., 1993; Gineitis and Treisman, 2001). The data presented here show that loss of SRF is associated with a reduction in the expression levels of several FA proteins. This dysregulation is seen at both the transcriptional and posttranscriptional levels and suggests a mechanism by which loss of SRF function interferes with cytoskeletal organization and FA activity.
Mislocalization of FA proteins in individual Srf(−/−) ES cells
Next, we tested the distribution of FA proteins in individual ES cells. Srf(+/+) and Srf(−/−) ES cells were trypsinized and replated on gelatine for 1 h. Subsequently, adherent cells were fixed and processed for indirect immunofluorescence with antibodies against α-actinin, FAK, β1-integrin, talin, vinculin, and zyxin, together with phalloidin to visualize F-actin. The majority of wild-type ES cells is not yet well spread after 1 h of cultivation. The small fraction of wild-type cells already spread after 1 h displays a characteristic punctate staining of FA proteins at the tips of actin stress fibers (unpublished data).
We first monitored the distribution of FA proteins in representative, individual Srf(+/+) and Srf(−/−) ES cells that were not spread. The distribution of α-actinin in isolated Srf(−/−) ES cells is similar to that in wild-type ES cells (Fig. 3, A and H). Membrane localization of FAK, however, is dramatically reduced in Srf(−/−) ES cells (Fig. 3, B and I), although overall expression levels of FAK are unchanged (Fig. 2 A). The epitope recognized by the β1-integrin antibody is localized preferentially at the cell margin in both ES cell lines tested (Fig. 3, C and J). Since protein levels of the mature β1-integrin subunit are reduced in Srf(−/−) ES cells (Fig. 2 A), we speculate that the immunofluorescence signal is generated by a COOH-terminally truncated β1-integrin variant that is still able to localize to the membrane. Talin is predominantly found at the membrane of Srf(+/+) and Srf(−/−) ES cells (Fig. 3, D and K), although the overall protein level is greatly diminished by SRF deficiency (Fig. 3 K). Staining for Vinculin and Zyxin revealed that these two proteins do not exclusively localize to the membrane in Srf(−/−) ES cells (Fig. 3, E, F, L, and M). Surprisingly, staining of SRF-deficient cells with antizyxin antibody gave a relatively strong signal (Fig. 3 M), although full-length zyxin protein was not detectable by Western blotting in Srf(−/−) cell extracts (Fig. 2 A). This may be explained by the presence of additional zyxin variants in Srf(−/−) ES cells (Murthy et al., 1999).
Figure 3.

Delocalization of FA proteins in isolated Srf( −/−) ES cells. Srf(−/−) and Srf(+/+) ES cells were plated on gelatine for 1 h. The distribution of the indicated FA proteins (red) and F-actin (green) in both E14Srf(+/+) cells (A–G) and 81Srf(−/−) cells (H–N) was monitored by immunofluorescence and confocal microscopy. Only single, nonspread ES cells, which represent ∼70% of the whole Srf(+/+) population at the 1 h time point, were taken into account. Arrow in N denotes F-actin concentration in “membrane blebs” of Srf(−/−) ES cells. Bar, 10 μm.
The cells investigated above were simultaneously analyzed for the localization of F-actin by phalloidin staining (Fig. 3, G and N). In wild-type ES cells, F-actin is predominantly found in the cell cortex (Fig. 3 G). All FA proteins tested colocalize with F-actin at the cell margins of wild-type ES cells (unpublished data). In agreement with earlier findings (Schratt et al., 2001), F-actin staining in Srf(−/−) ES cells is severely reduced (Fig. 3 N), with a frequent local concentration at structures that resemble membrane blebs (arrow). In summary, isolated Srf(−/−) ES cells display both reduction and mislocalization of a variety of proteins that have important functions in the attachment of the cortical actin network to the membrane and the transmission of signals from the ECM.
Impaired FAK activation in Srf(−/−) ES cells
The nonreceptor tyrosine-kinase FAK is not essential for the formation of FAs (Ilic et al., 1995a), but plays a critical role in integrin-stimulated cell migration (Sieg et al., 1999). Activation of FAK involves its recruitment to the membrane upon integrin-clustering and subsequent autophosphorylation at critical tyrosine residues. Phosphorylation of FAK at Tyr-397 creates a binding site for other kinases with known functions in cytoskeletal dynamics, such as Src, Fyn, and PI-3 kinase. We therefore investigated whether the observed FAK delocalization in isolated Srf(−/−) ES cells (Fig. 3 I) correlated with changes in FAK activity. Plating of wild-type ES cells on fibronectin led to a strong induction of FAK-Tyr397 phosphorylation after 10 min, indicative of FAK activation. The amount of Tyr-397–phosphorylated FAK stayed fairly constant in a 60-min time course (Fig. 4 A, top). A similar kinetic profile of FAK activation was observed for Srf(−/−) ES cells. However, the amount of Tyr-397 phosphorylated FAK was substantially reduced to ∼50% of wild-type levels at each measured time point. Western blotting with pan-FAK antibody was used to normalize for overall FAK recovery in wild-type and Srf(−/−) ES cells (Fig. 4 A, bottom). Transient overexpression of SRF-VP16 in Srf(−/−) ES cells was used to demonstrate the ability of activated SRF to restore FAK Tyr-397 phosphorylation (Fig. 4 B). The relative amount of Tyr-397–phosphorylated FAK was 2–3-fold higher in SRF-VP16 transfected as compared with mock-transfected 100 Srf(−/−) ES cells. A rescue of FAK activation was not observed in the ES cell line 100-2 Srf(−/−) rescue (unpublished data). This unexpected finding may reflect the fact that this cell clone expresses 3.5-fold more SRF protein than wild-type cells (Weinhold et al., 2000). It is therefore conceivable that a large fraction of SRF in these cells is not activated and competes with activated SRF on cytoskeletal promoters.
Figure 4.
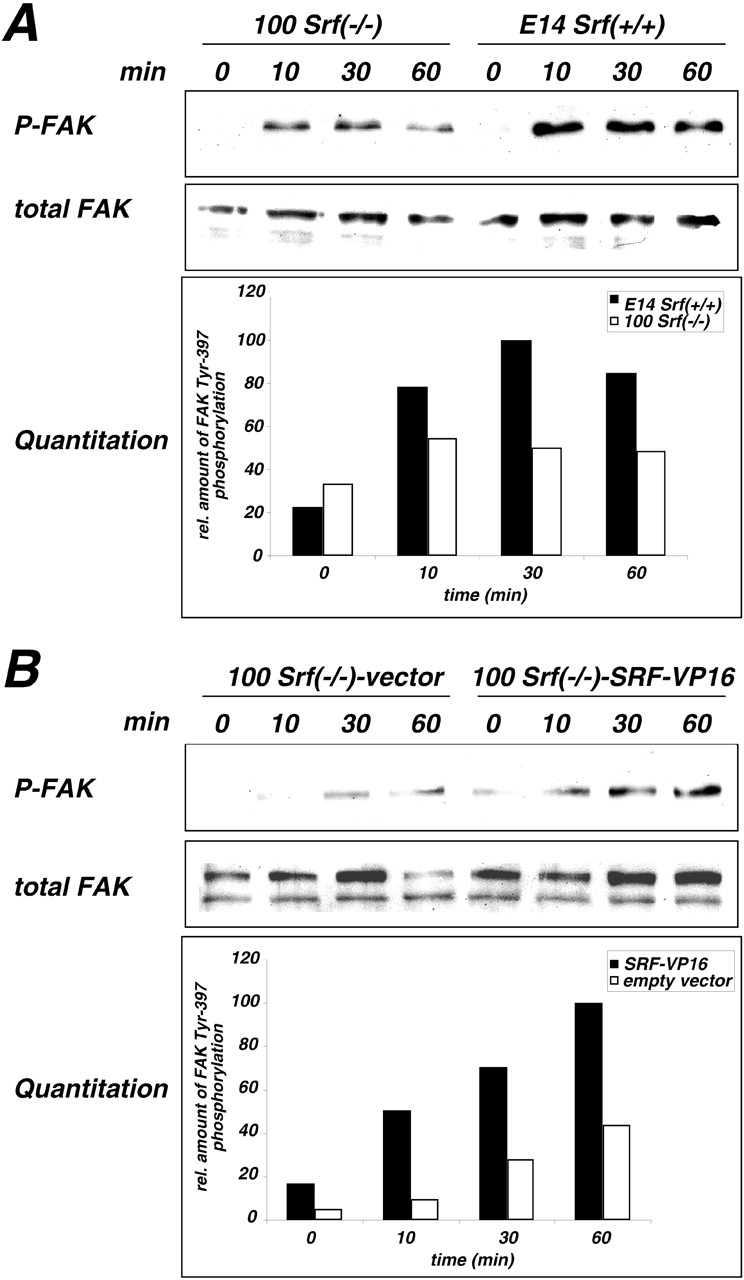
Impaired integrin-mediated FAK activation in Srf( −/−) ES cells. (A) Serum-starved 100 Srf(−/−) and E14Srf(+/+) ES cells were plated on fibronectin-coated dishes for a time course of 60 min, and whole protein extracts were prepared at the indicated time points. Proteins were resolved on 8.5% SDS-PAGE, transferred to PVDF membrane, and the blots were probed either with P-Tyr397-FAK specific antibody (top) or pan-FAK antibody (middle). For quantitation, signals from the P-Tyr397 blot were normalized to the respective signals from the Pan-FAK blot and the maximum value was arbitrarily set to 100. Representative results from one of six experiments are shown. (B) Experiment was performed as in A, except that 100 Srf(−/−) ES cells were transfected with an SRF-VP16 expression plasmid or with pCS2+ (empty vector) before plating. Quantitation of FAK Tyr397 phosphorylation was performed as described in the legend to A.
Our data indicate that impaired expression and mislocalization of FA components in Srf(−/−) ES cells correlate with defective transmission of signals from the ECM to important intracellular signal transducers like FAK. This finding offers a molecular link between the observed structural defects in FA complexes and reduced cell motility of Srf(−/−) ES cells.
Srf(−/−) ES cells are unable to assemble FAs and associated actin stress fibers
Actin-based cytoskeletal structures, such as stress fibers and lamellipodia, are characteristically found with spread cells. We therefore analyzed wild-type, Srf(−/−), and Srf(−/−) rescue ES cells for their abilities to form actin stress fibers, lamellipodia, and associated FAs. For this assay, cells were plated for 48 h on either fibronectin (unpublished data) or gelatine. Under these conditions, only wild-type, Srf(−/+), and Srf(−/−) rescue cells were able to form individual, well-spread cells. Srf(−/−) ES cells, in contrast, were always distinctly nonspread and part of large colonies after 48 h of cultivation. Wild-type and Srf(−/−) rescue ES cells assembled numerous actin stress fibers that terminated in FAs. These cells stained positive for the FA marker proteins α-actinin, FAK, vinculin, and zyxin (Fig. 5, left and right). Moreover, the formation of large lamellipodia could be observed in a subset of these cells (arrows in Fig. 5). In contrast, in Srf(−/−) ES cells, F-actin staining was limited to cell margins and was rather diffuse (Fig. 5, middle). Lamellipodia or stress fibers were never observed in Srf(−/−) ES cells. In addition, staining for α-actinin, FAK, and vinculin was limited to the cell margins and did not show the typical punctate pattern. In the case of vinculin, however, some punctate staining could be observed at the periphery of Srf(−/−) ES cell colonies. Zyxin staining was rather diffuse, and neither displayed a punctate pattern nor a preferential membrane localization. Fibronectin as substrate neither lead to an assembly of FAs nor of stress fibers in Srf(−/−) ES cells (unpublished data), as was previously seen with Talin(−/−) ES cells (Priddle et al., 1998). We conclude that undifferentiated Srf(−/−) ES cells are severely impaired in building up distinct cytoskeletal structures, such as actin stress fiber arrays, lamellipodia, and associated FA plaques. The defects in cytoskeletal organization are probably a major reason for altered spreading, adhesion, and migration of SRF-deficient ES cells.
Figure 5.
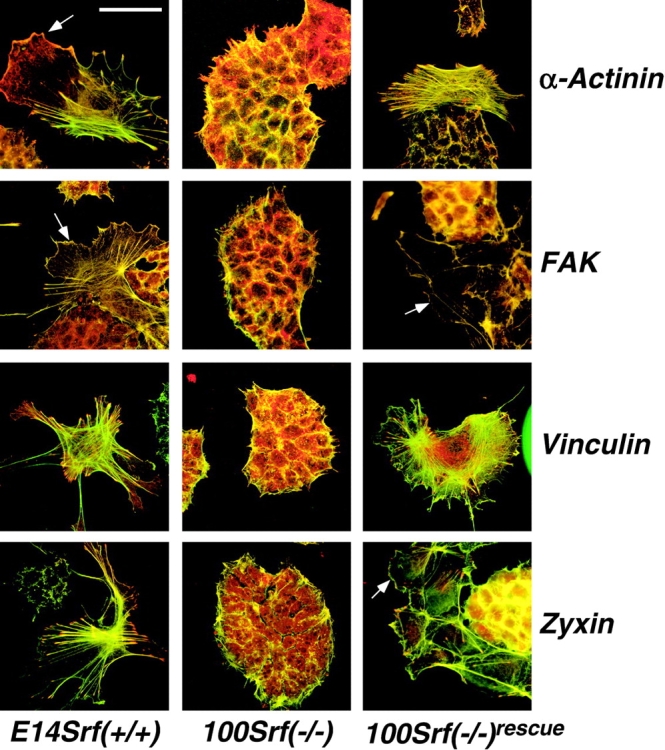
Srf( −/−) ES cells are unable to form FAs and actin stress fibers. ES cells were grown on gelatine-coated dishes for 48 h, fixed, and processed for indirect immunofluorescence analysis. Red channel represents staining with antibodies specified on the right of the figure. Green channel represents staining with Oregon green–conjugated phalloidin. Genotypes of the cell lines used are indicated. Arrows point to large lamellipodia that were exclusively observed in SRF-containing ES cells. The shown individual spread cells represent a subpopulation in our ES cell culture (20–30%). The number of these cells does not increase with passage; however, we cannot formally exclude that they represent spontaneously differentiated cells having lost stem cell character. Bar, 50 μm.
Srf(−/−) ES cells display reduced overall actin expression and an altered ratio of F- versus G-actin
The expression of genes encoding for several muscle- and nonmuscle actin isoforms is regulated by SRF (Chen et al., 1996; Frederickson et al., 1989; Sobue et al., 1999). In addition, we recently showed a two- to threefold reduction of overall actin protein levels in Srf(−/−) ES cells (Schratt et al., 2001). Given the lack of lamellipodia and actin stress fibers in Srf(−/−) ES cell colonies (Fig. 5), we investigated whether SRF deficiency also had an impact on the F/G-actin treadmilling equilibrium. Thus, we analyzed the actin polymerization state inside cells. First, we performed a sedimentation assay to monitor G- and F-actin levels in cell extracts. In agreement with previous results, total actin in cell extracts of two Srf(−/−) ES cell lines was reduced to ∼40% of wild-type levels (Fig. 6 A, lanes 1–4). The amount of nonpelletable G-actin in the supernatants of Srf(−/−) ES cell extracts after high-speed centrifugation was reduced by 20–40%, as compared with wild-type extracts (Fig. 6 A, lanes 5–8). In contrast, pelletable F-actin from Srf(−/−) ES cells was reduced by ∼80% (Fig. 6 A, lanes 9–12). SRF deficiency affecting F-actin levels more severely than G-actin levels was confirmed by FACS analysis. In this assay, a twofold reduction of G-actin levels was observed in Srf(−/−) ES cells as compared with Srf(−/+) cells (Fig. 6 B, top). In contrast, F-actin levels, as determined by phalloidin staining, were affected more than 3.5-fold (Fig. 6 B, bottom). Srf(−/−) rescue cells displayed a G-/F-actin ratio nearly identical to that of Srf(−/+) cells (unpublished data). Thus, both sedimentation and FACS analysis demonstrate that the F-actin pool is more severely affected by the absence of SRF than the G-actin pool. These results point to a role for SRF not only in the regulation of overall actin expression levels, but also in balancing F- versus G-actin. Reduced F-actin levels correlate with the impairment of Srf(−/−) ES cells in stress fiber assembly.
Figure 6.
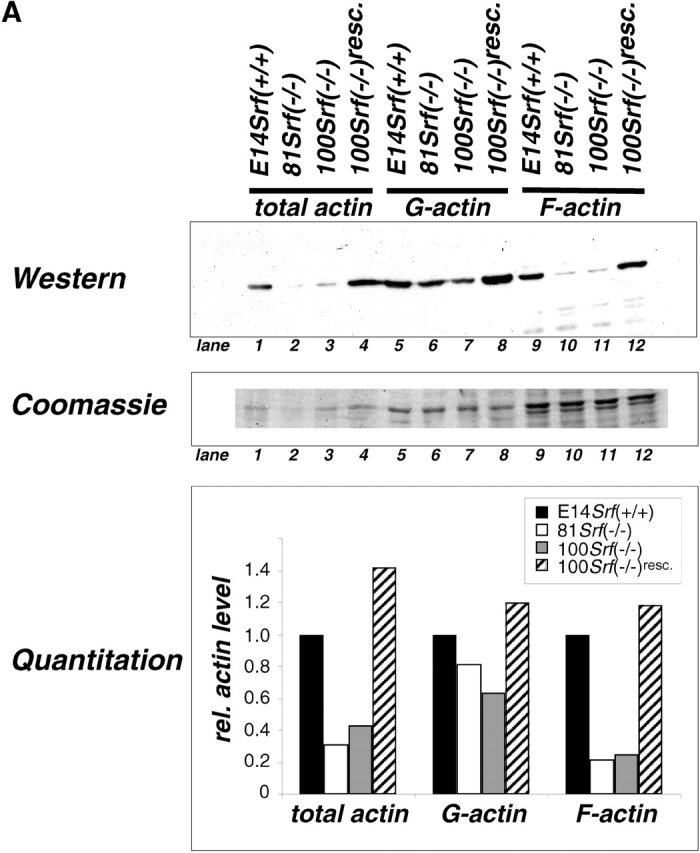
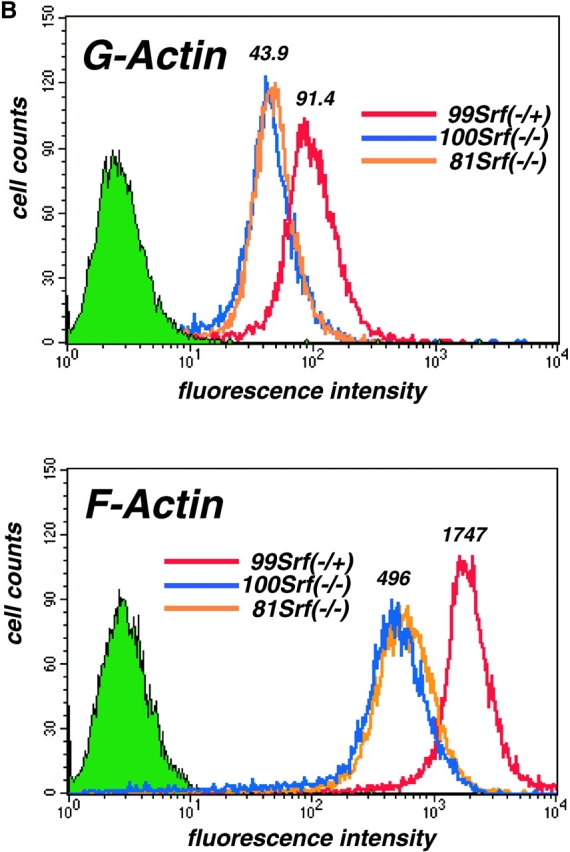
Impaired actin treadmilling in Srf( −/−) ES cells. (A) Quantitation of actin isoforms by sedimentation assay. Antiactin Western blot of extracts from four types of ES cell representing the pools of total cellular actin (lanes 1–4), G-actin (lanes 5–8), and F-actin (lanes 9–12) (top). Preparation of extracts was as described in Materials and methods. Coomassie staining of the gel after blotting confirms equal protein loading of samples within each actin pool (middle). Quantitation of relative actin levels (bottom) was as described in Materials and methods. Values for E14Srf(+/+) were arbitrarily set to one for each actin pool. (B) Quantitation of actin isoforms by flow cytometry. 99 Srf(−/+) ES cells (red), and Srf(−/−) ES lines 81 (orange) and 100 (blue), were stained with Texas red DNaseI conjugate (top) and Oregon green phalloidin (bottom) to specifically stain for G-Actin and F-Actin, respectively. Fluorescence intensity values are given on the abscissae.
Overexpression of constitutively active RhoA fails to induce stress fibers in Srf(−/−) ES cells
The function of RhoA in the induction of FAs and associated actin stress fibers has been extensively characterized (Chrzanowska-Wodnicka and Burridge, 1996). Furthermore, RhoA has been implicated in regulating the actin treadmilling cycle more directly, either through inactivation of the actin depolymerizing factor ADF/cofilin (Maekawa et al., 1999) or through activation of the polymerization-promoting factor profilin (Watanabe et al., 1997). Finally, activated RhoA is able to drive SRF-dependent transcription (Hill et al., 1995). We therefore determined the importance of functional SRF for RhoA-mediated stress fiber formation. Constitutively active RhoA-V14 induces characteristic stress fiber bundles in wild-type ES cells (arrows in Fig. 7 B). This effect is Rho-specific, since expression of the control protein red fluorescent protein (RFP) had no effect on the cytoskeleton (unpublished data). In contrast, RhoA-V14 is not able to induce stress fiber formation in SRF-deficient cells (Fig. 7 E). Instead, RhoA-V14–transfected cells frequently display a large number of bleb-like structures (Fig. 7, D–F). Phalloidin staining revealed that F-actin is concentrated within these blebs (arrow in Fig. 7 E). Again, expression of RFP did not lead to cytoskeletal changes (unpublished data). These results demonstrate that RhoA-mediated stress fiber formation requires the expression of SRF-dependent genes downstream of RhoA. If such genes are not properly expressed due to SRF-deficiency, increased acto-myosin contractility in response to RhoA activation leads to membrane blebbing.
Figure 7.

RhoA-mediated actin polymerization and stress fiber formation in ES cells. Wild-type (A–C) or 100 Srf(−/−) (D–F) ES cells were transfected with myc-tagged, constitutively active Rho (RhoA-V14-mt) and analyzed by indirect immunofluorescence. Red channel, anti-myc; green channel, phalloidin. Arrows in B indicate stress fiber bundles and in E indicate F-actin accumulation in bleb-like structures. Bar, 50 μm.
Constitutively active SRF (SRF-VP16) is sufficient to induce molecular and cell morphological changes in ES cells of Srf(−/−) genetic background
SRF activity is stimulated by effector kinases downstream of the RhoGTPase and MAP kinase pathways (Hill et al., 1995). SRF-VP16 fusion proteins were previously used to drive SRF-dependent transcription independent of the activity of upstream signaling cascades (Treisman and Ammerer, 1992; Johansen and Prywes, 1994; Hines et al., 1999). Such a constitutively active SRF variant, when introduced into Srf(−/−) ES cells, allowed us to test for a putative link between the SRF activation state and the establishment of cytoskeletal structures. To control for potential unspecific effects of the VP16 activation domain, we constructed SRFΔM-VP16, which lacks most of the MADS box. This mutant cannot dimerize nor bind to SREs, but still locates to nuclei (Fig. 8 A and unpublished data). Undifferentiated ES cells were transiently transfected with SRF-VP16 or SRFΔM-VP16 expression vectors, and RNA expression levels were measured by quantitative RT-PCR (Fig. 8 B). In an Srf(−/−) background, the mRNA levels for the IEGs Egr-1 (Fig. 8 B, top left) and c-fos (unpublished data) were increased ∼400-fold by the introduction of SRF-VP16, as compared with mock-transfected cells, confirming the capacity of SRF to drive expression of these genes. Importantly, introduction of SRFΔM-VP16 had no significant effect. Zyxin mRNA levels were specifically induced 3–6-fold by SRF-VP16 (Fig. 8 B, top right), similar to the known SRF-regulated genes Vinculin and β-Actin (Fig. 8 B, bottom). Therefore, SRF is identified to drive the expression of several cytoskeletal genes, including Zyxin. These results confirm previous findings that showed the ability of SRF expression in Srf(−/−) ES cells to restore vinculin and zyxin protein levels (Fig. 2 A). At the morphological level, introduction of SRF-VP16 into Srf(−/−) ES cells led to extensive cell flattening (arrows in Fig. 8 C, right) in ∼25% of cells, whereas cells transfected with the control construct looked unaltered (Fig. 8 C, left). Since the transfection efficiency was ∼20–30% in this experiment (unpublished data), flattened cells most likely represent cells that overexpress SRF-VP16. We speculated that SRF-VP16–induced cytoskeletal rearrangements are responsible for these changes in cell morphology. Indeed, staining for F-actin revealed the formation of massive arrays of stress fibers (Fig. 8 D, left) and frequent occurrence of lamellipodia (unpublished data) in each SRF-VP16–transfected Srf(−/−) ES cell. The cytoskeleton of SRFΔM-VP16–transfected Srf(−/−) ES cells did not undergo any detectable changes (Fig. 8 D, right). Both immunofluorescence staining with anti-VP16 antibody and Western blotting (unpublished data) confirmed that SRF-VP16 and SRFΔM-VP16 were expressed at comparable levels and that both proteins accumulated in the cell nuclei. Interestingly, the effects elicited by expression of constitutively active SRF on the expression of target genes (Fig. 8 B) and ES cell morphology (unpublished data) were much more pronounced in Srf(−/−) ES cells than in ES cells of wild-type or Srf(−/+) genetic background. Endogenous SRF protein present in the latter two cell lines, not being activated under these growth conditions, probably competes with SRF-VP16 for binding to cognate promoters. We note that transient overexpression of wild-type SRF in Srf(−/−) ES cells caused cell spreading only in a small fraction of transfected cells (unpublished data). In agreement with previous results (Schratt et al., 2001), introduction of SRF-VP16 into Srf(−/−) ES cells led to a strong increase in overall actin expression, accompanied by a strong elevation in F-actin and only a minor increase in G-actin, as judged by FACS analysis (Fig. 8 E). Clearly, a net shift of the actin equilibrium in favor of F-actin is seen. This effect was specific for SRF-VP16, since the expression of the control construct SRFΔM-VP16 had no significant influence on actin treadmilling. These data demonstrate conclusively that a constitutively active form of SRF is sufficient to regulate actin treadmilling and to elicit drastic cell morphological and molecular changes that are linked to remodelling of the actin cytoskeleton.
Figure 8.


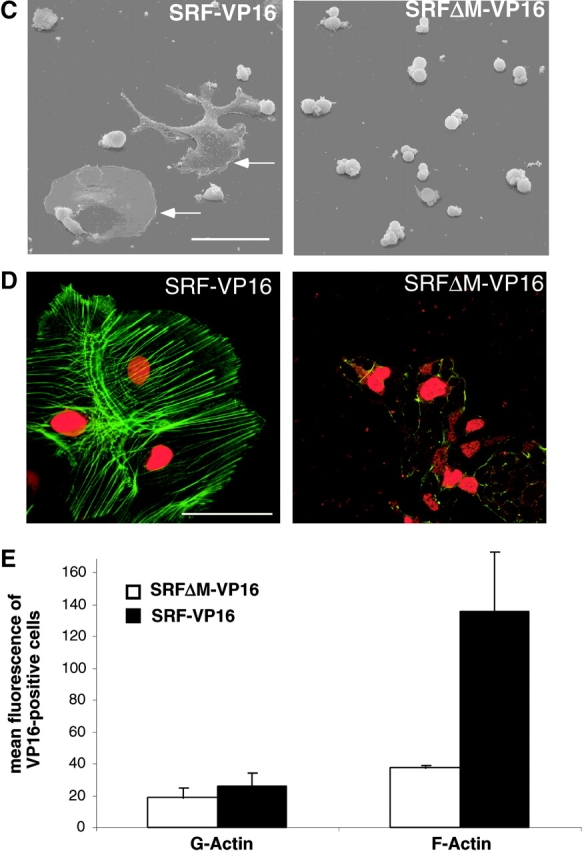
SRF-VP16 activates cytoskeletal genes. (A) Schematic representation of the various SRF coding sequences expressed upon transient transfection of ES cells. In SRF-VP16, part of the SRF transactivation domain (TAD) is replaced by the HSV VP16 activation domain. In the control construct SRFΔM-VP16, the major part of the MADS box is removed. NLS, nuclear localization signal. (B) Induction of Egr-1, Vinculin, Zyxin, and β-actin at the transcriptional level by transiently transfected SRF effector molecules. RT-PCR analysis was performed on ES cell extracts of the indicated genotypes 3d after transfection with either empty vector, SRFΔM-VP16, or SRF-VP16 encoding vector. Relative mRNA levels for each sample were calculated as described in Materials and methods. Values are given as fold induction over cells transfected with empty vector only. Comparable expression levels of the SRF effector proteins was confirmed by quantitative Western blotting. A representative dataset of two independent experiments is shown. (C) SRF-VP16 expression leads to extensive cell flattening. 100 Srf(−/−) ES cells were transiently transfected with SRF-VP16 (left) and the SRFΔM-VP16 control construct (right) and grown for 72 h. Subsequently, cells were replated on gelatine for 1 h, fixed, and inspected by scanning electron microscopy. Arrows denote large lamellipodia that occur in ∼25% of the cells. This population most likely represents SRF-VP16 transfectants. (D) SRF-VP16 expression leads to increased stress fiber formation. 100 Srf(−/−) ES cells were transiently transfected with SRF-VP16 (top) and the SRFΔM-VP16 control construct (bottom) and processed for immunofluorescence analysis 3 d later. Cells were simultaneously stained with phalloidin (green) and anti-VP16 (red). (E) SRF-VP16 induces F-Actin synthesis in Srf(−/−) ES cells. ES cells were transiently transfected with SRF-VP16 or SRFΔM-VP16 and analyzed for their actin polymerization state by FACS analysis. Cells were simultaneously stained with anti-VP16 antibody and either Texas red phalloidin (F-actin) or Texas red DNaseI (G-actin). Staining with IgG isotype served as a control (unpublished data). Given values represent the mean fluorescent G-actin (left) or F-actin (right) signal of VP16-positive cells (between 20–30% of all gated cells). Data represent the mean of two independent experiments ± SD. Bars, 50 μm.
Discussion
The targeted mutagenesis of ES cell genomes by homologous recombination offers a powerful tool to study gene function at the cellular level. We have used such ES cell genetics to study the physiological roles of the transcription factor SRF. In addition to generating null alleles at the Srf locus, we have used Srf(−/−) ES cells to reintroduce constitutively active variants of SRF and RhoA into these SRF-deficient cells. We uncovered a far reaching role of SRF in governing the structure and function of cytoskeletal structures, namely the actin cortex, actin stress fibers, and FA plaques. Consequently, cellular activities dependent upon these structures, such as cell spreading, adhesion, and migration, were found to be affected by SRF deficiency.
SRF regulates the actin treadmilling equilibrium
We have shown previously that expression of the SRE-containing β-actin gene was found to be significantly reduced in ES cells lacking SRF (Schratt et al., 2001). Furthermore, the promoters for the cell-type-specific α- and γ-Actins also contain functional SRF binding sites (Mohun et al., 1989; Kovacs and Zimmer, 1998). Interestingly, in addition to the reduced overall actin expression levels, the treadmilling equilibrium of G- versus F-actin is shifted in Srf(−/−) ES cells, thereby reducing F-actin levels disproportionately. It is conceivable that the altered G/F-actin ratio is simply due to mass action, i.e., due to reduced overall actin expression levels. However, increasing the pool of monomeric actin in Srf(−/−) ES cells by overexpressing HA-tagged actin did not result in a detectable increase in F-actin synthesis (unpublished observation). Overexpression of both actin and a constitutively active variant of RhoA (RhoA-V14) allowed some de novo actin polymerization (unpublished data). However, this treatment did not fully rescue stress fiber formation in Srf(−/−) ES cells, whereas SRF-VP16 elicited a very strong stress fiber response in these cells. Therefore, we postulate that SRF affects actin treadmilling by regulating the expression of additional proteins involved in actin turnover downstream of RhoA. Such candidates include members of the LIM kinase or mDia pathways, which have been described to influence actin polymerization via regulation of cofilin and profilin activation states, respectively (Watanabe et al., 1997; Maekawa et al., 1999). Overexpression of LIM kinase 2 alone or in conjunction with RhoA-V14, however, did not lead to enhanced F-actin levels (unpublished data). SRF-regulated zyxin/vinculin expression may also contribute to F-actin synthesis, since these proteins are able to recruit the actin polymerization machinery via binding to Ena/VASP family members (Laurent et al., 1999). Finally, talin, whose expression is partially dependent on SRF, has been reported to possess actin nucleation activity (Kaufmann et al., 1991). Treisman and coworkers recently demonstrated that, in fibroblasts, SRF itself is regulated by actin dynamics (Sotiropoulos et al., 1999). In that study, decreases in G-actin levels were argued to stimulate SRF activation. We describe here that SRF controls signaling components, yet to be identified (class Y in the model of Fig. 9), which act downstream of RhoA and influence the F-/G-actin equilibrium. Therefore, we propose that SRF is a central component of an autoregulatory feed-back loop controlling actin polymerization (Fig. 9). Such a mechanism may allow cells to adapt their F-actin synthesis to signal-induced cytoskeletal changes.
Figure 9.

Model depicting the influence of SRF on cytoskeletal structures and dynamics. RhoA activation by serum components (e.g., LPA), or in response to FA stimulation by the extracellular matrix (ECM), leads to actin organization into stress fibers/FAs (e.g., via Rho-kinase), enhanced actin polymerization (e.g., via mDia, PIP2) or SRF activation (via yet ill-defined signaling paths). Depletion of the G-actin pool upon actin polymerization (Sotiropoulos et al., 1999), and initial signaling by FAs (Wei et al., 2001) are able to activate SRF. Activated SRF in turn regulates the expression of proteins that either influence actin organization (protein group X, containing e.g., FA proteins) or the actin treadmilling equilibrium (protein group Y). This regulation establishes SRF-dependent autoregulatory feed-back loops that contribute to actin-based cell spreading, adhesion, and migration. For further discussion, see text.
SRF is required for the formation of FA plaques and stress fibers
Several lines of evidence suggest that actin polymerization and actin organization, the latter referring to the formation of both stress fibers and FAs, are mediated by distinct effectors of RhoA (for review see Van Aelst and D'Souza-Schorey, 1997). This distinction is mirrored by the ability of SRF not only to regulate components acting downstream of RhoA but also to control the expression of cytoskeletal components, like the myosin light chain (MLC) gene (Papadopoulos and Crow, 1993). A potential mechanism for Rho-induced stress fiber formation involves phosphorylation of MLC by activated MLC kinase (MLCK) and subsequent increase in acto-myosin contractility (Burridge and Chrzanowska-Wodnicka, 1996). It is therefore possible that impaired MLC expression may contribute to the defect in stress fiber and FA formation in Srf(−/−) ES cells. Introduction of RhoA-V14 in Srf(−/−) ES cells lead to increased membrane blebbing which was also observed in cells with defective actin-membrane coupling in response to MLC phosphorylation (Coleman et al., 2001; Sebbagh et al., 2001). This indicates that the Rho-kinase/MLCK pathway can be activated, at least to some extent, in Srf(−/−) ES cells.
Regarding FAs, the failure of Srf(−/−) ES cells to assemble functional FAs may be associated with inadequate expression of essential FA components. Indeed, we find misexpression and/or mislocalization of several structural FA proteins, namely FAK, β1-integrin, talin, vinculin, and zyxin. Integrin clustering is the initial event in FA formation (Gumbiner, 1996). Therefore, impaired integrin expression and/or activation could account for the adhesion and spreading defects of Srf(−/−) ES cells. Since spreading is most severely reduced on a gelatin matrix, the function of the collagen receptors α1β1 and α2β1 may be compromised. We observe a reduced protein expression of the full-length β1 integrin subunit in Srf(−/−) ES cells, whereas expression of the α1 and α2 subunit is not affected at the mRNA level (unpublished data). β1-integrin is also important for adhesion to fibronectin (Fassler et al., 1995), but the upregulation of other β subunits (e.g., β3) may compensate for reduced β1-integrin expression in this case (Priddle et al., 1998). The regulation of β1-integrin expression by SRF seems to occur at the post-transcriptional level, since (a) β1-integrin mRNA levels are unaffected by SRF deficiency and (b) a truncated β1-integrin protein is enriched in Srf(−/−) ES cells. Recently, calpain-mediated cleavage of β1-integrin has been reported (Pfaff et al., 1999), and we are currently testing a putative link between SRF and calpain activity in ES cells.
Since FAK is able to bind β1-integrin, FAK delocalization in Srf(−/−) ES cells may be a direct consequence of β1-integrin cleavage. Reduced FAK activation may affect migration, differentiation, and survival of cells. Fibroblasts expressing the FAK Y397A mutant have migration defects (Sieg et al., 2000), indicating that recruitment to FAK of tyrosine kinases, such as Fyn and Src, is critical for cell migration. Also, FAK activation enables recruitment and activation of PI-3 kinase. The FAK/PI-3-kinase pathway counteracts apoptosis initiated by serum deprivation or deadhesion (Schlaepfer et al., 1999). Indeed, differentiating Srf(−/−) ES cells tend to undergo increased apoptosis (unpublished data), possibly due to inadequate activation of the FAK/PI-3 kinase survival pathway. We also present evidence that the scaffolding protein talin is regulated by SRF at the transcriptional level. Undifferentiated talin-deficient ES cells display striking phenotypic similarities to Srf(−/−) ES cells, including defects in cell shape, adhesion, and β1-integrin expression (Priddle et al., 1998). However, talin is dispensable for cytoskeletal organization in differentiated ES cells. Since we do not observe a well-established cytoskeleton in differentiated Srf(−/−) ES cells (unpublished data), we believe that reduced talin expression alone is not sufficient to explain the abnormal actin cytoskeleton in the absence of SRF. The same is true for another SRF-regulated protein, vinculin, that has been shown to be dispensable for FA formation (Xu et al., 1998). Finally, the expression of zyxin is strongly dependent upon SRF. Inhibitory peptides that block zyxin/α-actinin interaction, and thereby displace zyxin from FAs, predict a role for zyxin in cell spreading and motility (Drees et al., 1999). We do not know how the loss of zyxin affects FA formation, but downregulation of zyxin may contribute to the reduced motility of Srf(−/−) ES cells.
Together, we demonstrate that SRF is important for the expression of a variety of proteins that have known functions in the formation and/or stabilization of FAs. Furthermore, others have implicated SRF in the transcriptional regulation of both myosin light and heavy chains (Papadopoulos and Crow, 1993; Catala et al., 1995; Zilberman et al., 1998), α1-integrin (Sobue et al., 1999), tropomyosin (Toutant et al., 1994), and dystrophin (Galvagni et al., 1997). This strongly argues that SRF is a central determinant of the formation and activity of FAs. In addition, a recent study reported that, in myocytes, FA signaling cooperates with RhoA in SRF activation requiring a properly organized actin cytoskeleton (Wei et al., 2001). In light of these results, SRF-dependent regulation of β1-integrin maturation and FAK activity might also be part of a positive feed-back loop (Fig. 9). Such a mechanism may be important in muscle cells, where initial Rho-induced cytoskeletal changes have to be reinforced by increased transcription of SRF-dependent cytoskeletal genes.
Cellular behavior under SRF control
We have demonstrated that the cellular activities of spreading, adhesion, and migration are severely affected by SRF deficiency. The structural basis for these defects in Srf(−/−) ES cell behavior may rest jointly on unbalanced actin dynamics and impaired formation of FA plaques and stress fibers. Cytoskeletal defects might also contribute to the inability of Srf(−/−) mouse embryos to form mesoderm (Arsenian et al., 1998). Inductive events during embryogenesis require precise cell adhesion mechanisms, either to neighboring cells or to the extracellular matrix (Hynes, 1999). Inappropriate cell adhesion displayed by cells of the early Srf(−/−) embryo could therefore impair mesoderm induction and migration of newly formed mesodermal germ layer cells (Tam and Behringer, 1997). In summary, the Srf(−/−) ES cell system revealed a hitherto unrecognized and physiologically important role of SRF in determining the formation of specific cytoskeletal structures and, consequently, in governing cellular activities that require dynamic changes of the cytoskeleton. Such SRF-directed activities include cell adhesion and migration, and are suggested to be of relevance for more complex biological processes, like directed cell movement during embryogenesis, wound healing, and metastasis.
Materials and methods
Reagents and antibodies
Laminin (mouse) and fibronectin (calf) were purchased by Harbor Bio Products and collagen IV was purchased by GIBCO BRL. Primary antisera were used which recognized the following antigens: FAK (Transduction Laboratories), phospho-FAK (BioSource International), vinculin, α-actinin, talin (all from Sigma-Aldrich), zyxin (gift of J. Wehland, GBF Braunschweig), β1-integrin (gift of R. Hynes, MIT), VP16, HA (both from Santa Cruz Biotechnology, Inc.), actin (gift of H. Knauth, MPI Tübingen), and myc (9E10 hybridoma supernatant).
Plasmids
To obtain pCS2+/hSRF-VP16, a 1.8-kb NcoI-XbaI fragment (NcoI site filled-in) was isolated from pKD24 (Dalton and Treisman, 1992), and subcloned into pCS2+ (gift of R. Rupp, MPI Tübingen) linearized with StuI-XbaI. SRFΔM-VP16 was generated from pCS2+/hSRF-VP16 by removing a fragment from the BglII to the BbsI site. The remaining vector sequences were religated with a double-stranded oligonucleotide of the following sequence: 5′-GG GGC GAA GAG ACA-3′ (top strand); 5′-G ATC TGT CTC TTC G-3′ (bottom strand). RhoA-V14 myc tag was excised from pUHD-RhoA-V14 myc-tag (a gift from M. Baehler, Muenster) and subcloned into pCS2+. Expression of the inserted cDNAs in pCS2+ is driven by the CMV enhancer/promoter.
ES cell culture conditions
General growth conditions.
The E14.1 ES cell line is derived from the 129/Ola mouse strain and has been described previously (Kuhn et al., 1991). ES cells were kept without feeders on gelatine-coated dishes in DME medium containing 4.5 g/liter glucose and 3.7 g/liter NaHCO3, supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 g/ml streptomycin, 450 μM monothioglycerol, 15% FBS, and 100 U/ml LIF (referred to as complete medium) at 37°C in a humidified atmosphere at 5% CO2, and split every 2 d.
Transient transfection.
ES cells were seeded the day before transfection at 4 × 105 cells/10 cm dish or 105 cells per 6-well and grown overnight. Transfection was performed with LipofectAmin (GIBCO BRL) according to the manufacturer's protocol. A 10-fold excess of LipofectAmin over total DNA was used throughout the transfections. 10 μg of the described SRF expression plasmids were used per 10-cm dish, or 1 μg of all other expression plasmids per 6-well. Total DNA amount was kept constant at 10 μg per 10-cm dish or 2 μg per 6-well with pCS2+mt or pCS2+. 48–72 h after transfection, cells were either harvested for RNA preparation or fixed for immunofluorescence analysis.
Adhesion assay
96-well plates were coated with 25 μg/ml fibronectin, 25 μg/ml laminin or 50 μg/ml collagen IV at 4°C overnight. To avoid nonspecific binding, wells were washed once with PBS and blocked with 1% BSA for 1 h at 37°C. ES cells grown for 48 h in the presence of LIF were trypsinized to obtain a single-cell suspension, washed once in serum-free medium, and plated on the matrix-coated wells for 1 h at 37°C in serum-free medium. Nonadherent cells were washed away and adherent cells were fixed for 30 min with 10% formaldehyde. After washing with PBS, fixed cells were stained with crystal violet for 15 min. Residual dye was removed by extensive washing with PBS. Stained cells were air-dried, and the dye was extracted with 50 μl 2% SDS per well. Absorption of the solution was measured at 630 nm. The absorption measured from wells coated with BSA only served as a reference.
Migration assay (Boyden chamber assay)
Cell culture filter inserts (Becton Dickinson; pore size 8 μm) for 6-well plates were coated on the lower surface overnight with 20 μg/ml fibronectin at 4°C. After washing with PBS, the inserts were put into 6-well plates to obtain a modified Boyden chamber. The lower reservoir of the chamber was filled with complete medium. ES cells were trypsinized to a single-cell suspension, washed in serum-free medium, resuspended in serum-free medium to 2.5 × 105 cells/ml, and 2 ml of the suspension was filled into the upper reservoir. Cells were allowed to migrate through the filter for 22 h at 37°C. Cells on the upper surface were scraped off, and those on the lower surface were washed once with PBS and subsequently fixed with 10% formaldehyde. Staining was performed with crystal violet for 30 min, the bound dye was extracted with 1 ml 2% SDS, and the absorption was determined at 630 nm. The absorption measured from filter inserts coated with BSA only served as a reference.
Western blotting
Preparation of cell extracts and Western blotting were as described previously (Weinhold et al., 2000).
Quantitative RT-PCR
Total RNA preparation (RNeasy kit; QIAGEN) and first strand cDNA synthesis (Superscript II, GIBCO BRL) were done according to the manufacturer's protocols. 1 μg of total RNA treated with DNase I was used for RT. One twentieth of the RT reaction was included in a 25 μl PCR reaction. For a quantitative analysis, SYBR green PCR technology was used (PerkinElmer). Real-time detection of the PCR product was monitored by measuring the increase in fluorescence caused by the binding of SYBR green to double-stranded DNA with ABI PRISM 7700 Sequence Detector. To calculate relative quantification values, a threshold cycle (Ct), the cycle at which a statistically significant increase in fluorescence occurs, was derived from the resulting PCR profiles of each sample. Ct is a measure for the amount of template present in the starting reaction. To correct for different amounts of total cDNA in the starting reaction, Ct values of an endogenous control (Hprt) were subtracted from those of the corresponding sample, resulting in ΔCt. The relative quantitation value is expressed as 2-ΔCt. All RT-PCR data sets shown (Figs. 2 B and 8 B) provide representative results of several independent mRNA preparations and amplifications, as indicated in the figure legends.
Primers used: Hprt, F(gcctaagatgagcgcaagttg); B(tactaggcagatggccacagg); Egr-1, F(gccgagcgaacaacccta); B(tccaccatcgccttctcatt); Zyxin, F(ggctgctacaccgacactttg); B(ctcagcatgcggtcagtgat); Vinculin, F(ccaaggtcagagaagccttcc); B(cgtagctgttcaaggtctggtg); β -actin, F(ggcgcttttgactcaggatt); B(gggat-gtttgctccaaccaa); Talin, F(gctcgggcgttagcagtc); B(atggaatctgagacagtccgaga); β 1-integrin, F(tggctttgatgcaatcatgc); B(tccgtggaaaacaccagca).
FACS analysis
ES cells were collected, fixed in 4% formaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 5 min, and blocked in 1% BSA for 1 h. After three washes in PBS, the cells were incubated for 30 min in a mixture of 1 U Oregon green phalloidin and 0.3 μM Texas red DNase I (both from Molecular Probes) in 1% BSA. For a reference, incubation was performed in the absence of any fluorescent dye. For antibody staining, cells were incubated for 1 h with primary antibody, washed once with PBS, and incubated for an additional 30 min with secondary antibody solution. Actin staining reagents were included in the secondary antibody solution. Rabbit IgG served as isotype control. After washing, cells were subjected to FACscan analysis (Becton Dickinson). Dead cells were gated out and 104 cells were counted for each sample. Data analysis was performed with CellQuest software (Becton Dickinson).
F-actin sedimentation assay
Actin sedimentation assays were performed in principle as described (Jahraus et al., 2001). ES cell pellets were resuspended in 2 ml of lysis buffer (10 mM Hepes, pH 7.6, 100 mM KCl, 1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF) and the cells broken using a probe sonicator. Nuclei were removed by centrifugation (3,000 g, 30 min) and the supernatant (low-speed extract) subjected to a high-speed centrifugation step (400,000 g, 1 h). The clear supernatant (high-speed extract) was collected and concentrated by using Centriprep-10 spin columns (Amicon Corp.). Pellets were subsequently solved in 1% Triton X-100. Equal amounts of low-speed extracts, high-speed extracts, and high-speed pellets were resolved on a 10% SDS-PAGE, blotted, and probed with a polyclonal rabbit anti-pan actin antiserum (gift of H. Knauth, MPI Tübingen). Relative actin levels in each sample were quantified by normalizing Western blot signals to Coomassie staining.
Indirect immunofluorescence
Cells were grown on gelatine-coated coverslips in complete growth medium for 48 h. The samples were then fixed in 4% formaldehyde and permeabilized in 0.2% Triton X-100. Nonspecific binding was blocked by incubating the cells for 1 h in 1% BSA in PBS at 37°C, before staining with primary antibody for 1 h at 37°C. Incubation with fluorescence-conjugated secondary antibodies (Molecular Probes; 1:200 in 0.2% BSA) was performed for 30 min at 37°C. To visualize filamentous actin, 1 U Oregon green phalloidin (Molecular Probes) was included into the mixture. Coverslips were washed four times with PBS, once in water, air-dried, and mounted in Moviol. Image acquisition was done with LSM 510 (ZEISS).
Electron microscopy
Embryonic stem cells were grown on gelatine-coated coverslips for 2 h. Samples were fixed with 1.6% glutaraldehyde in 20 mM Hepes, pH 7.6, 120 mM NaCl for 5 min at room temperature and for 1 h at 4°C, postfixed with 1% osmium tetroxide in PBS for 1 h on ice, washed with H2O, and treated with 1% aqueous uranyl acetate for 1 h at 4°C. For scanning electron microscopy, cells were dehydrated in ethanol and critical point dried from CO2. The samples were sputter-coated with 8 nm gold palladium and examined at 20 kV accelerating voltage in a Hitachi S-800 field emission scanning electron microscope.
FAK activation
Cells were either transfected as described or left untreated and grown in complete medium for 48 h. The medium was then exchanged and cells were kept for 18 h in complete medium lacking FBS. Cells were then detached by trypsinization, and the reaction was stopped by adding 0.5 mg/ml soybean trypsin inhibitor (Sigma-Aldrich). After washing once with serum-free medium, cells were kept in suspension in serum-free medium for an additional hour at 37°C. Then, the cells were replated on fibronectin-coated tissue plates for 10–60 min at 37°C. Preparation of cell extracts for Western blotting was as described (Weinhold et al., 2000).
Acknowledgments
This work was supported by the DFG through grants NO 120/7-4 and SFB 446 (B7), and the Volkswagen-Stiftung.
Footnotes
Abbreviations used: ES, embryonic stem; FA, focal adhesion; FAK, FA kinase; IEG, immediate early gene; MLC, myosin light chain; MLCK, MLC kinase; RT, reverse transcription; SRE, serum response element; SRF, serum response factor; TCF, ternary complex factor.
References
- Affolter, M., J. Montagne, U. Walldorf, J. Groppe, U. Kloter, M. LaRosa, and W.J. Gehring. 1994. The Drosophila SRF homolog is expressed in a subset of tracheal cells and maps within a genomic region required for tracheal development. Development. 120:743–753. [DOI] [PubMed] [Google Scholar]
- Arsenian, S., B. Weinhold, M. Oelgeschläger, U. Rüther, and A. Nordheim. 1998. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 17:6289–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle, M.C. 1998. Spatial control of actin filament assembly: lessons from Listeria. Cell. 95:741–748. [DOI] [PubMed] [Google Scholar]
- Belkin, A.M., S.F. Retta, O.Y. Pletjushkina, F. Balzac, L. Silengo, R. Fassler, V.E. Koteliansky, K. Burridge, and G. Tarone. 1997. Muscle β1D integrin reinforces the cytoskeleton-matrix link: modulation of integrin adhesive function by alternative splicing. J. Cell Biol. 139:1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge, K., and M. Chrzanowska-Wodnicka. 1996. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12:463–518. [DOI] [PubMed] [Google Scholar]
- Carnac, G., M. Primig, M. Kitzmann, P. Chafey, D. Tuil, N. Lamb, and A. Fernandez. 1998. RhoA GTPase and serum response factor control selectively the expression of MyoD without affecting Myf5 in mouse myoblasts. Mol. Biol. Cell. 9:1891–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catala, F., R. Wanner, P. Barton, A. Cohen, W. Wright, and M. Buckingham. 1995. A skeletal muscle-specific enhancer regulated by factors binding to E and CArG boxes is present in the promoter of the mouse myosin light-chain 1A gene. Mol. Cell. Biol. 15:4585–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.Y., J. Croissant, M. Majesky, S. Topouzis, T. McQuinn, M.J. Frankovsky, and R.J. Schwartz. 1996. Activation of the cardiac α-actin promoter depends upon serum response factor, Tinman homologue, Nkx-2.5, and intact serum response elements. Dev. Genet. 19:119–130. [DOI] [PubMed] [Google Scholar]
- Chen, H., B.W. Bernstein, and J.R. Bamburg. 2000. Regulating actin-filament dynamics in vivo. Trends Biochem. Sci. 25:19–23. [DOI] [PubMed] [Google Scholar]
- Chihara, K., M. Amano, N. Nakamura, T. Yano, M. Shibata, T. Tokui, H. Ichikawa, R. Ikebe, M. Ikebe, and K. Kaibuchi. 1997. Cytoskeletal rearrangements and transcriptional activation of c-fos serum response element by Rho-kinase. J. Biol. Chem. 272:25121–25127. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka, M., and K. Burridge. 1996. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133:1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, M.L., E.A. Sahai, M. Yeo, M. Bosch, A. Dewar, and M.F. Olson. 2001. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3:339–345. [DOI] [PubMed] [Google Scholar]
- Critchley, D.R. 2000. Focal adhesions - the cytoskeletal connection. Curr. Opin. Cell Biol. 12:133–139. [DOI] [PubMed] [Google Scholar]
- Dalton, S., and R. Treisman. 1992. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 68:597–612. [DOI] [PubMed] [Google Scholar]
- Drees, B.E., K.M. Andrews, and M.C. Beckerle. 1999. Molecular dissection of zyxin function reveals its involvement in cell motility. J. Cell Biol. 147:1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees, B., E. Friederich, J. Fradelizi, D. Louvard, M.C. Beckerle, and R.M. Golsteyn. 2000. Characterization of the interaction between zyxin and Ena/VASP family of proteins: Implications for actin cytoskeleton organization. J. Biol. Chem. 275:22503–22511. [DOI] [PubMed] [Google Scholar]
- Fassler, R., M. Pfaff, J. Murphy, A.A. Noegel, S. Johansson, R. Timpl, and R. Albrecht. 1995. Lack of β 1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson, R.M., M.R. Micheau, A. Iwamoto, and N.G. Miyamoto. 1989. 5′ flanking and first intron sequences of the human β-actin gene required for efficient promoter activity. Nucleic Acids Res. 17:253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvagni, F., M. Lestingi, E. Cartocci, and S. Oliviero. 1997. Serum response factor and protein-mediated DNA bending contribute to transcription of the dystrophin muscle-specific promoter. Mol. Cell. Biol. 17:1731–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore, A.P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature. 381:531–535. [DOI] [PubMed] [Google Scholar]
- Gineitis, D., and R. Treisman. 2001. Differential usage of signal transduction pathways defines two types of SRF target gene. J. Biol. Chem. 7:7. [DOI] [PubMed] [Google Scholar]
- Graham, R., and M. Gilman. 1991. Distinct protein targets for signals acting at the c-fos serum response element. Science. 251:189–192. [DOI] [PubMed] [Google Scholar]
- Guillemin, K., J. Groppe, K. Ducker, R. Treisman, E. Hafen, M. Affolter, and M.A. Krasnow. 1996. The pruned gene encodes the Drosophila serum response factor and regulates cytoplasmic outgrowth during terminal branching of the tracheal system. Development. 122:1353–1362. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B.M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 84:345–357. [DOI] [PubMed] [Google Scholar]
- Herschman, H.R. 1991. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60:281–319. [DOI] [PubMed] [Google Scholar]
- Hill, C.S., J. Wynne, and R. Treisman. 1994. Serum-regulated transcription by serum response factor (SRF): a novel role for the DNA binding domain. EMBO J. 13:5421–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, C.S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 81:1159–1170. [DOI] [PubMed] [Google Scholar]
- Hines, W.A., J. Thorburn, and A. Thorburn. 1999. A low-affinity serum response element allows other transcription factors to activate inducible gene expression in cardiac myocytes. Mol. Cell. Biol. 19:1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttelmaier, S., O. Mayboroda, B. Harbeck, T. Jarchau, B.M. Jockusch, and M. Rudiger. 1998. The interaction of the cell-contact proteins VASP and vinculin is regulated by phosphatidylinositol-4,5-bisphosphate. Curr. Biol. 8:479–488. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 1999. Cell adhesion: old and new questions. Trends Cell Biol. 9:M33–M37. [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. a. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, T. Suda, T. Atsumi, J. Fujimoto, Y. Ikawa, T. Yamamoto, and S. Aizawa. 1995. b. Focal adhesion kinase is not essential for in vitro and in vivo differentiation of ES cells. Biochem. Biophys. Res. Commun. 209:300–309. [DOI] [PubMed] [Google Scholar]
- Jahraus, A., M. Egeberg, B. Hinner, A. Habermann, E. Sackman, A. Pralle, H. Faulstich, V. Rybin, H. Defacque, and G. Griffiths. 2001. ATP-dependent membrane assembly of F-actin facilitates membrane fusion. Mol. Biol. Cell. 12:155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen, F.E., and R. Prywes. 1994. Two pathways for serum regulation of the c-fos serum response element require specific sequence elements and a minimal domain of serum response factor. Mol. Cell. Biol. 14:5920–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, S., T. Piekenbrock, W.H. Goldmann, M. Barmann, and G. Isenberg. 1991. Talin binds to actin and promotes filament nucleation. FEBS Lett. 284:187–191. [DOI] [PubMed] [Google Scholar]
- Kovacs, A.M., and W.E. Zimmer. 1998. Cell-specific transcription of the smooth muscle gamma-actin gene requires both positive- and negative-acting cis elements. Gene Expr. 7:115–129. [PMC free article] [PubMed] [Google Scholar]
- Kuhn, R., K. Rajewsky, and W. Muller. 1991. Generation and analysis of interleukin-4 deficient mice. Science. 254:707–710. [DOI] [PubMed] [Google Scholar]
- Laurent, V., T.P. Loisel, B. Harbeck, A. Wehman, L. Grobe, B.M. Jockusch, J. Wehland, F.B. Gertler, and M.F. Carlier. 1999. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Cell Biol. 144:1245–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T.C., K.L. Chow, P. Fang, and R.J. Schwartz. 1991. Activation of skeletal α-actin gene transcription: the cooperative formation of serum response factor-binding complexes over positive cis-acting promoter serum response elements displaces a negative-acting nuclear factor enriched in replicating myoblasts and nonmyogenic cells. Mol. Cell. Biol. 11:5090–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, C.P., and G.K. Owens. 1999. Regulation of smooth muscle α-actin expression in vivo is dependent on CArG elements within the 5′ and first intron promoter regions. Circ. Res. 84:852–861. [DOI] [PubMed] [Google Scholar]
- Maekawa, M., T. Ishizaki, S. Boku, N. Watanabe, A. Fujita, A. Iwamatsu, T. Obinata, K. Ohashi, K. Mizuno, and S. Narumiya. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 285:895–898. [DOI] [PubMed] [Google Scholar]
- Mohun, T.J., M.V. Taylor, N. Garrett, and J.B. Gurdon. 1989. The CArG promoter sequence is necessary for muscle-specific transcription of the cardiac actin gene in Xenopus embryos. EMBO J. 8:1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseyeva, E.P., P.A. Weller, N.I. Zhidkova, E.B. Corben, B. Patel, I. Jasinska, V.E. Koteliansky, and D.R. Critchley. 1993. Organization of the human gene encoding the cytoskeletal protein vinculin and the sequence of the vinculin promoter. J. Biol. Chem. 268:4318–4325. [PubMed] [Google Scholar]
- Montaner, S., R. Perona, L. Saniger, and J.C. Lacal. 1999. Activation of serum response factor by RhoA is mediated by the nuclear factor-κB and C/EBP transcription factors. J. Biol. Chem. 274:8506–8515. [DOI] [PubMed] [Google Scholar]
- Murthy, K.K., K. Clark, Y. Fortin, S.H. Shen, and D. Banville. 1999. ZRP-1, a zyxin-related protein, interacts with the second PDZ domain of the cytosolic protein tyrosine phosphatase hPTP1E. J. Biol. Chem. 274:20679–20687. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Norman, C., M. Runswick, R. Pollock, and R. Treisman. 1988. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 55:989–1003. [DOI] [PubMed] [Google Scholar]
- Papadopoulos, N., and M.T. Crow. 1993. Transcriptional control of the chicken cardiac myosin light-chain gene is mediated by two AT-rich cis-acting DNA elements and binding of serum response factor. Mol. Cell. Biol. 13:6907–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff, M., X. Du, and M.H. Ginsberg. 1999. Calpain cleavage of integrin γ cytoplasmic domains. FEBS Lett. 460:17–22. [DOI] [PubMed] [Google Scholar]
- Priddle, H., L. Hemmings, S. Monkley, A. Woods, B. Patel, D. Sutton, G.A. Dunn, D. Zicha, and D.R. Critchley. 1998. Disruption of the talin gene compromises focal adhesion assembly in undifferentiated but not differentiated embryonic stem cells. J. Cell Biol. 142:1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A.J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70:389–399. [DOI] [PubMed] [Google Scholar]
- Salmon, E.D. 1989. Cytokinesis in animal cells. Curr. Opin. Cell Biol. 1:541–547. [DOI] [PubMed] [Google Scholar]
- Sartorelli, V., K.A. Webster, and L. Kedes. 1990. Muscle-specific expression of the cardiac α-actin gene requires MyoD1, CArG-box binding factor, and Sp1. Genes Dev. 4:1811–1822. [DOI] [PubMed] [Google Scholar]
- Schlaepfer, D.D., C.R. Hauck, and D.J. Sieg. 1999. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71:435–478. [DOI] [PubMed] [Google Scholar]
- Schratt, G., B. Weinhold, A.S. Lundberg, S. Schuck, J. Berger, H. Schwarz, R.A. Weinberg, U. Rüther, and A. Nordheim. 2001. Serum response factor is required for immediate-early gene activation yet is dispensable for proliferation of embryonic stem cells. Mol. Cell. Biol. 21:2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebbagh, M., C. Renvoize, J. Hamelin, N. Riche, J. Bertoglio, and J. Breard. 2001. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3:346–352. [DOI] [PubMed] [Google Scholar]
- Shaw, P.E., H. Schröter, and A. Nordheim. 1989. The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell. 56:563–572. [DOI] [PubMed] [Google Scholar]
- Shore, P., and A.D. Sharrocks. 1995. The MADS-box family of transcription factors. Eur. J. Biochem. 229:1–13. [DOI] [PubMed] [Google Scholar]
- Sieg, D.J., C.R. Hauck, D. Ilic, C.K. Klingbeil, E. Schaefer, C.H. Damsky, and D.D. Schlaepfer. 2000. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2:249–256. [DOI] [PubMed] [Google Scholar]
- Sieg, D.J., C.R. Hauck, and D.D. Schlaepfer. 1999. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 112:2677–2691. [DOI] [PubMed] [Google Scholar]
- Sobue, K., K. Hayashi, and W. Nishida. 1999. Expressional regulation of smooth muscle cell-specific genes in association with phenotypic modulation. Mol. Cell. Biochem. 190:105–118. [PubMed] [Google Scholar]
- Sotiropoulos, A., D. Gineitis, J. Copeland, and R. Treisman. 1999. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 98:159–169. [DOI] [PubMed] [Google Scholar]
- Tam, P.P., and R.R. Behringer. 1997. Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 68:3–25. [DOI] [PubMed] [Google Scholar]
- Toutant, M., C. Gauthier-Rouviere, M.Y. Fiszman, and M. Lemonnier. 1994. Promoter elements and transcriptional control of the chicken tropomyosin gene. Nucleic Acids Res. 22:1838–1845 (erratum published 23:540). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman, R., and G. Ammerer. 1992. The SRF and MCM1 transcription factors. Curr. Opin. Genet. Dev. 2:221–226. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L., and C. D'Souza-Schorey. 1997. Rho GTPases and signaling networks. Genes Dev. 11:2295–2322. [DOI] [PubMed] [Google Scholar]
- Volberg, T., B. Geiger, Z. Kam, R. Pankov, I. Simcha, H. Sabanay, J.L. Coll, E. Adamson, and A. Ben-Ze'ev. 1995. Focal adhesion formation by F9 embryonal carcinoma cells after vinculin gene disruption. J. Cell Sci. 108:2253–2260. [DOI] [PubMed] [Google Scholar]
- Watanabe, N., P. Madaule, T. Reid, T. Ishizaki, G. Watanabe, A. Kakizuka, Y. Saito, K. Nakao, B.M. Jockusch, and S. Narumiya. 1997. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16:3044–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, L., L. Wang, J.A. Carson, J.E. Agan, K. Imanaka-Yoshida, and R.J. Schwartz. 2001. β1 integrin and organized actin filaments facilitate cardiomyocyte-specific RhoA-dependent activation of the skeletal α-actin promoter. FASEB J. 15:785–796. [DOI] [PubMed] [Google Scholar]
- Wei, L., W. Zhou, J.D. Croissant, F.E. Johansen, R. Prywes, A. Balasubramanyam, and R.J. Schwartz. 1998. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 273:30287–30294. [DOI] [PubMed] [Google Scholar]
- Weinhold, B., G. Schratt, S. Arsenian, J. Berger, K. Kamino, H. Schwarz, U. Rüther, and A. Nordheim. 2000. Srf(−/−) ES cells display non-cell autonomous impairment in mesodermal differentiation. EMBO J. 19:5835–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W., H. Baribault, and E.D. Adamson. 1998. Vinculin knockout results in heart and brain defects during embryonic development. Development. 125:327–337. [DOI] [PubMed] [Google Scholar]
- Zilberman, A., V. Dave, J. Miano, E.N. Olson, and M. Periasamy. 1998. Evolutionarily conserved promoter region containing CArG–like elements is crucial for smooth muscle myosin heavy chain gene expression. Circ. Res. 82:566–575. [DOI] [PubMed] [Google Scholar]