

SUMMARY
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel localized at apical cell membranes and exists in macromolecular complexes with a variety of signaling and transporter molecules. Here we report that the multidrug resistance protein 4 (MRP4), a cAMP transporter, is functionally and physically associates with CFTR. Adenosine-stimulated CFTR-mediated chloride currents are potentiated by MRP4 inhibition, and this potentiation is directly coupled to attenuated cAMP efflux through the apical cAMP transporter. CFTR single-channel recordings and FRET-based intracellular cAMP dynamics suggest that a compartmentalized coupling of cAMP transporter and CFTR occurs via the PDZ scaffolding protein, PDZK1, forming a macromolecular complex at apical surfaces of gut epithelia. Disrupting this complex abrogates the functional coupling of cAMP transporter activity to CFTR function. MRP4 knockout mice are more prone to CFTR-mediated secretory diarrhea. Our findings have important implications for disorders such as inflammatory bowel disease and secretory diarrhea.
INTRODUCTION
The cyclic nucleotides (e.g., cAMP) are important second messengers involved in the cellular responses to a wide variety of signals in every living cell. In epithelial cells lining the gut, kidney, and lung, cAMP also plays key roles in extracellular regulation of fluid homeostasis (Jackson and Raghvendra, 2004). Tight regulation of intracellular cAMP levels is critical, as excessive cAMP production within cells leads to over-stimulation of certain secretory events, dysregulation of cell function, or even cell toxicity. Increased intracellular cAMP is restored to basal levels via hydrolysis into 5’-AMP by phosphodiesterases (PDEs) (Jackson and Raghvendra, 2004). Recently, the molecular identity of a cAMP efflux transporter has been discovered. MRP4 (ABCC4) is a member of the ATP-binding cassette (ABC) transporter superfamily, whose gene products are capable of transporting substrates from the inner membrane leaflet to the outer membrane leaflet (Dean et al., 2001). MRP4 has been shown to function as a high affinity efflux pump for cAMP (Chen et al., 2001; van Aubel et al., 2002; Wielinga et al., 2003), and is expressed in epithelial cells lining lung, kidney, intestine, etc., and localized on both the apical membranes (van Aubel et al., 2002) and basolateral membranes (Lai and Tan. 2002) of polarized cells.
The concept of spatially restricted and tightly modulated compartmentalization of cAMP signaling events was formulated over 20 years ago (reviewed in Steinberg and Brunton, 2001; Cooper, 2005). It is well documented that cAMP signalling specificity relies substantially on the organization of macromolecular signaling complexes that effectively assemble multiple proteins (from receptors to targets) into three-dimensional arrays at subcellular locations and that proximity of receptors to their ultimate targets guarantees response velocity and signaling specificity (Davare et al., 2001; Huang et al., 2001; Naren et al., 2003; Li et al., 2005). The assembly of this signaling complex ensures a specific and rapid signaling from the receptor to the channel. Stimulation of β2 adrenergic receptors (β2AR) in airway epithelial cells also activates chloride transport mediated by cystic fibrosis transmembrane conductance regulator (CFTR), the product of the gene mutated in patients with cystic fibrosis (Li and Naren, 2005). Regulation of CFTR Cl− channel is accomplished through activation of this apical surface receptor that couples to adenylate cyclase (AC) and raises cellular cAMP. Huang and colleagues reported that signaling elements compartmentalized in apical compartments that activate CFTR in polarized lung epithelial cells (Huang et al., 2001). They observed that maximal stimulation of CFTR-mediated Cl− secretion by adenosine, a ligand for A2b adenosine receptors (A2bAR) that couple to membrane-bound AC, was accompanied by no measurable change in total cellular cAMP, signifying highly localized regulation of CFTR by A2bAR in the apical cell membrane (Huang et al., 2001).
The functional significance of the macromolecular complex containing CFTR and its interacting partners in subcellular compartments was reconfirmed (Li et al., 2005). Most recently, we demonstrated that a type 2 lysophosphatidic acid (LPA) receptor forms a macromolecular complex with CFTR mediated through a PDZ scaffolding protein (NHERF2) at the apical surfaces of gut epithelial cells (Li et al., 2005). This assembly of multiprotein complex forms the basis for the functional coupling between LPA signaling and CFTR-mediated chloride transport. In the present study, we identified a novel functional coupling of a cAMP efflux transporter (MRP4) to CFTR activity that is physically mediated through PDZK1/CAP70 scaffolding protein (Wang et al., 2000) in a spatially segregated microdomain, and this interaction has important implications in diseases such as inflammatory bowel disease and secretory diarrhea.
RESULTS
Inhibition of cAMP Transporter (MRP4) Potentiates the Function of Colocalized CFTR Cl− Channel at the Apical Surface of Gut Epithelial Cells
CFTR is localized at the apical membrane of epithelial cells lining the gut (Li and Naren, 2005). The functional activity of this Cl− channel is regulated by PKA following a rise in the local concentration of cAMP. We hypothesized that inhibition of the plasma membrane cAMP efflux transporter MRP4 would enhance the cAMP signal, thus magnify CFTR function. In this study, gut epithelial cells were permeabilized at the basolateral surface with alpha-toxin derived from Staphylococcus aureus; the cells were then pretreated with MK571, a potent MRP4 inhibitor (Reid et al., 2003). Alpha-toxin forms pores that are permeable to molecules <5,000 Da, therefore effectively removing the basolateral membrane as a barrier to cAMP without affecting the functional integrity of the apical membrane (Reddy and Quinton, 1996). The cell membrane impermeable agonist (cAMP) was added to the permeabilized surface of polarized gut epithelial cell monolayers to elicit CFTR-mediated short-circuit currents (Isc). In basolateral-permeabilized cells pretreated with MK571, a low dose of cAMP (10 μM) elicited a near maximal CFTR-mediated Isc, which was not boosted further by higher dose of cAMP (50 μM) (Figure 1A, top). Whereas in untreated (or vehicle-treated) cells, 10 μM of cAMP induced only a very small Isc response (only 30–40% magnitude of the MK571-treated cells), the currents can be further increased to maximal level by higher dose of cAMP (50 μM) (Figure 1A, bottom). However, at a dose of 20 μM cAMP, maximal CFTR-dependent Isc responses were observed both in the presence and absence of MK571 (Figure 1B). The short-circuit currents potentiated by MK571 in basolateral-permeabilized gut epithelial cells were inhibited by various CFTR inhibitors (glybenclamide as in Figure 1A; a CFTR specific inhibitor, CFTRinh-172 as in Figure S1A in Supplemental Data). Experiments were also performed to demonstrate that cAMP did not activate CFTR in nonpermeabilized cells when added at either apical or basolateral surfaces (Figure S2A, bottom). Also, in cells permeabilized at the basolateral side, cAMP did not activate CFTR from the apical surface (Figure S2A, top).
Figure 1. The MRP4 Inhibitor MK571 Potentiates CFTR-mediated Short-Circuit Currents.
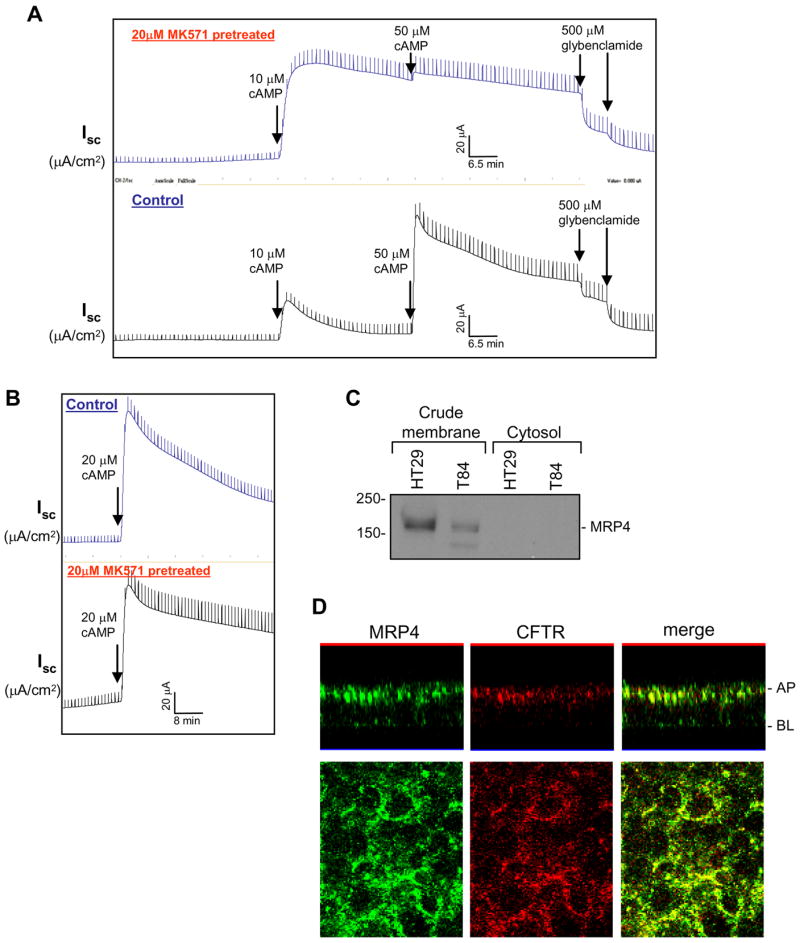
(A and B) Representative traces of CFTR-mediated Isc in response to cAMP in basolateral-permeabilized HT29-CL19A cells. Basolateral membranes of the polarized cells were permeabilized with 100 μg/ml alpha-toxin for 30 min. Cells were pretreated with 20 μM MK571 at both sides before cAMP was added to the basolateral side. A CFTR inhibitor, glybenclamide, was added to the apical side at the end of the experiment.
(C) MRP4 expression in crude plasma membrane prepared from colonic epithelial cells HT29-CL19A and T84 cells.
(D) Localization of MRP4 and CFTR in polarized HT29-CL19A cells by confocal fluorescence microscopy. Top, x-z axis images; bottom, x-y axis images. AP, apical membrane; BL, basolateral membrane.
We consistently observed that, in MK571-pretreated cells, the cAMP-activated state of the CFTR Cl− channel is sustained for a relatively longer period of time compared to untreated cells, i.e., the deactivation of the channel was relatively slower in the presence of MK571 (Figures 1A and 1B; and Figure S1A). This potentiation (enhancement) of CFTR function in response to exogenous cAMP by the MRP4 inhibitor (MK571) raises the possibility that attenuated or blocked apical MRP4-mediated cAMP efflux by MK571 may help accumulate higher levels of the second messenger in the close proximity to CFTR Cl− channel, thus resulting in increased CFTR Cl− currents as opposed to the untreated control.
Using an MRP4-specific antibody, we detected MRP4 expression by immunoblotting in the crude membranes prepared from gut epithelial cells (HT29-CL19A and T84 cells) (Figure 1C). Immunoreactivity for MRP4 was not observed in the cytosol (Figure 1C). Immunofluorescent confocal microscopy demonstrated that MRP4 is localized to both the apical and basolateral membranes of HT29-CL19A cells (Figure 1D, left), with a higher abundance on the apical plasma membrane where it colocalizes partially with CFTR (Figure 1D, right).
cAMP Transporter Function at the Apical Plasma Membrane of Gut Epithelial Cells is Inhibited by MK571
Given that MRP4 is localized in part at the apical membranes of epithelial cells, the unidirectional transport of etheno-cAMP (a fluorescent analog of cAMP) and [3H]cAMP across the apical membranes of basolateral-permeabilized polarized gut epithelial cells was monitored. Etheno-cAMP, like cAMP, is cell impermeant and induced CFTR-mediated Cl− currents in basolaterally permeabilized epithelial cells only (Figure S3A, bottom). As MK571 itself has significant autofluorescence when excited at the same wavelength as etheno-cAMP (ex315-nm/em420-nm), we used an MRP4 substrate, 9-(2-phosphonyl methoxyethyl) adenine (PMEA), which is a stable monophosphorylated nucleotide analog (Schuetz et al., 1999), to compete with etheno-cAMP for MRP4-mediated efflux across the apical membrane of polarized epithelial cells. As illustrated in Figure S3B, PMEA significantly decreased the etheno-cAMP transport across the apical membrane of HT29-CL19A cells. The inhibitory effect of PMEA on etheno-cAMP efflux demonstrated a dose-dependence (Figure S3B). Likewise, both MK571 and PMEA dose-dependently decreased the unidirectional transport of [3H]cAMP from the basolateral chamber into the apical chamber through the apical membrane MRP4 transporter in HT29-CL19A cells (Figures 2A and 2B). These data clearly show that competitive interactions with an MRP4 inhibitor (MK571) or substrate (PMEA) reduces cAMP efflux across the intact apical MRP4 transporter when the basolateral membrane barrier to cAMP is removed in polarized epithelial cells.
Figure 2. cAMP Transport across the Apical Plasma Membrane is Inhibited by MRP4 Inhibitor and Substrate.

(A and B) Unidirectional transport of [3H]cAMP by the apical MRP4 in basolateral-permeabilized HT29-CL19A cells pretreated with MRP4 inhibitor MK571 (A) or MRP4 substrate PMEA (B). A.U. = arbitrary units. Data represent the mean ± SEM (n=4).
Inhibition of cAMP Transporter Activity Potentiates CFTR Function in a Compartmentalized Fashion
cAMP-elevating ligand adenosine (ADO) has been reported to stimulate CFTR-mediated Cl− currents in a compartmentalized manner at the apical cell membrane of polarized pithelial cells (Huang et al., 2001). ADO is produced at almost 1 μM in unstressed tissue, whereas in inflamed or ischemic tissues it can be as high as 100 μM (Hasko and Cronstein, 2004). Under conditions of inflammation (e.g., inflammatory bowel disease), ADO is generated at sites of tissue stress and injury and secreted into the gut, which would eventually activate CFTR (by acting on certain adenosine receptors and causing increased cAMP production, see Li et al., 2005) and lead to increased chloride and fluid secretion into the gut lumen (Barrett and Keely, 2000). Because inhibition of MRP4 by MK571 reduced the cAMP efflux via apical MRP4 transporter (Figure 2A), and also potentiated CFTR Cl− channel function in response to exogenous cAMP stimulation in basolateral-permeabilized epithelial cells (Figure 1A and Figure S1A), we sought to monitor the effect of MK571 on CFTR-mediated Isc across intact (nonpermeabilized) polarized gut epithelial cell monolayers in response to ADO. MK571 potentiated CFTR channel function in response to ADO stimulation, and the potentiating effect of MK571 on CFTR-mediated Cl− currents was more prominent when CFTR was activated with lower concentration of ADO (at 3 μM ADO, ∼ 2-fold Isc for MK571-treated cells compared to the control; at 50 μM ADO, ∼ 1.4-fold Isc for MK571-treated cells compared to the control) (Figure 3A; and Figure S1B). However, MK571 failed to significantly potentiate the channel function when CFTR was maximally stimulated with a relatively higher dose of ADO (100 μM) (Figure S2B) or 20 μM forskolin (FSK), the adenylyl cyclase stimulator (Figure 3A), which causes a global increase in intracellular cAMP level (Li et al., 2005). It is worthy to note that MK571 itself can induce a very small Isc (∼ 3 5 μA/cm2) even in the absence of ADO stimulation (Figure 3A, bottom).
Figure 3. MRP4 Inhibition Potentiates CFTR-mediated Cl− Currents in a Compartmentalized Manner.

(A) CFTR-mediated Isc in response to adenosine (ADO) and forskolin (FSK) in MK571-pretreated HT29-CL19A cells (bottom) or vehicle (DMSO)-treated cells (top). ADO, FSK, and a CFTR inhibitor, DPC, were added to the apical side.
(B) CFTR-mediated Isc in response to ADO in MRP4 siRNA transfected HT29-CL19A cells (bottom) or control siRNA transfected cells (top). A CFTR-specific inhibitor, CFTRinh-172, was added to the apical side.
(C) The protein levels of MRP4, CFTR, and PDZK1 in the cells from (B) were assessed by Western blotting using corresponding antibodies as described in the Supplemental Experimental Procedures.
(D) Cell-attached single-channel recordings of CFTR currents in vehicle-treated (circle) or MK571-pretreated (square) HT29-CL19A cells in response to ADO. NPo: open-state probability of the channel. Numbers in the parenthesis indicate the number of patches for each condition. Error bars show SEM.
(E) Dose-dependence curve of CFTR NPo at different concentrations of MK571 when cells were activated by 1.5 μM of ADO in HT29-CL19A cells.
To test the hypothesis that the potentiating effect of MK571 on CFTR-mediated Cl− currents derives from the inhibitory effect of MK571 on MRP4 function, we used MRP4 siRNA oligonucleotides to specifically knock down the endogenous MRP4 protein amount, then monitored the CFTR-mediated Cl− currents in response to ADO stimulation. MRP4 siRNA significantly enhanced the ADO-stimulated Isc in HT29-CL19A epithelial cells compared to control siRNA, with the enhancing effect occurring at the lower dose (3 μM) of ADO stimulation (Figure 3B; and Figure S1C). When CFTR was stimulated with a relatively higher dose of ADO (20–100 μM), there is no difference in CFTR-mediated Isc between control cells and MRP4 knock-down cells (Figure 3B). The specificity of MRP4 siRNA was also checked to show that MRP4 siRNA did not affect the endogenous CFTR or PDZK1 protein levels (Figure 3C). The Cl− currents potentiated by MK571 or MRP4 siRNA in nonpermeabilized gut epithelial cells were inhibited by CFTR blockers, DPC (Figure 3A), and CFTR specific inhibitor, CFTRinh-172 (Figure 3B; and Figures S1B and S1C), suggesting that the enhanced short-circuit currents by MK571 or MRP4 siRNA were mediated through CFTR Cl− channel.
Cell-attached, CFTR single-channel recordings confirmed that MK571 leads to compartmentalized accumulation of cAMP at or near the plasma membrane. Varying concentrations (0–40 μM) of ADO were applied in the patch pipette either to evoke a localized generation of intracellular cAMP (< 20 μM ADO) or to elicit a significantly higher elevation (> 20 μM ADO) of the second messenger which might activate CFTR function sub-maximally or maximally (Huang et al., 2001; Li et al, 2005). In gut epithelial cells pretreated with MK571, the stimulatory effect of ADO on the CFTR single-channel activity (channel open-state probability, NPo) was compared with the untreated cells (Figure 3D; and Figure S4). The single channel activity was significantly increased at concentrations of 1.5–10 μM ADO in the presence of MK571 compared to the control (Figure 3D; Figure S4). In contrast, at doses of ADO >20 μM in the pipette (leading to a significantly higher increase in intracellular cAMP, which might activate CFTR function sub-maximally or maximally), MK571 no longer potentiated channel function. It is to be noted that the NPo of CFTR Cl− channels was significantly higher in MK571 pretreated cells (NPo = 0.2) compared to untreated cells (NPo = 0) even in the absence of ADO stimulation (Figure 3D), which is consistent with the above-described observation that MK571 itself can induce a very small Isc (∼ 3 5 μA/cm2) even in the absence of ADO stimulation (Figure 3A, bottom). This suggests that MK571 itself, by inhibiting MRP4 cAMP transporter, might restrain a certain amount of localized cAMP within a microdomain, which is enough to elicit certain level of CFTR stimulation. The potentiation of CFTR function by MK571 also demonstrates a dose-dependence, with a linear increase range falling within 0–30 μM of MK571 used, when the CFTR channel was stimulated in a localized pattern by only 1.5 μM ADO (Figure 3E). MK571 potentiates CFTR activity with concentrations as low as 10 μM, and is maximized by 30 μM (Figure 3E). All of the above data suggest that MK571 potentiates CFTR-mediated Cl− currents in a locally-restricted manner when CFTR is not maximally activated.
To further characterize the cAMP location after inhibition of the cAMP transporter, we used a FRET-based cAMP indicator, CFP-EPAC-YFP (Ponsioen et al., 2004), to monitor the effect of MK571 on intracellular cAMP signaling and dynamics in response to cAMP-elevating agents (such as ADO or forskolin) in live cells. This highly sensitive, unimolecular fluorescent indicator for cAMP can display significant FRET change, which rapidly diminishes following a rise in intracellular cAMP and increases again in response to a fall in cAMP, making it an ideal fluorescent probe for monitoring cAMP dynamics in living cells (Ponsioen et al., 2004). In gut epithelial cells (T84) stably expressing CFP-EPAC-YFP indicator for intracellular cAMP, forskolin, the potent cAMP-raising agent, evoked an average of 29% increase of CFP/FRET emission ratio (CFP/FRET emission ratio corresponding to intracellular cAMP signals; also see Supplemental Experimental Procedures for detail) (Figure S5), which indicates a global increase of intracellular cAMP. This is consistent with the observations of Ponsioen et al. (2004). As shown in Figure 4A (top), MK571 itself also induced a small but substantial increase of CFP/FRET emission ratio (∼ 8%; indicating increased cAMP level), as observed from short-circuit current measurements (Figure 3A, bottom) and CFTR single-channel recordings (Figure 3D). Interestingly, the most prominent increase of CFP/FRET emission ratio, after blocking the cAMP efflux transporter MRP4, occurred at the edge area of the cells which indicates a localized (spatially restricted) cAMP accumulation near the plasma membrane (i.e., subcellular cAMP heterogeneity) (Figure 4A, top).
Figure 4. MRP4 Inhibition Enhances Intracellular cAMP Signals.

(A) The monochrome CFP image showing cytosolic distribution of the fluorescent EPAC probe in T84 cells transfected with CFP-EPAC-YFP; and representative pseudocolor images of CFP/FRET emission ratio before (time = 0 min) and after the addition of 20 μM MK571 and/or 2 μM ADO (time = 5 min). The images in each panel were captured from the same field of view. Color bar shows magnitude of the emission ratio. Scale bar: 10 μm.
(B) Kinetics of cAMP changes (represented by the normalized CFP/FRET emission ratio) recorded in the cells shown in (A). Arrow indicates addition of the reagents.
(C) CFP image and representative pseudocolored CFP/FRET ratio images before (time = 0 min) and after the addition of 20 μM MK571 and/or 100 μM ADO (time = 5 min). The probe was excluded from the nuclear compartments, although in these nonconfocal images, a signal emanating from above and below the nucleus gives the appearance of a nuclear ratio change. Scale bar: 10 μm.
(D) Kinetics of cAMP changes (normalized CFP/FRET emission ratio) recorded in the cells shown in (C). Arrow indicates addition of the reagents.
(E) The summary of all the experiments performed in the same conditions as in (A)-(D) and in Figure S5. Data represent the mean ± SEM (n=4–6).
As expected, ADO elicited dose-dependent increase in CFP/FRET emission ratio. A lower dose of ADO (2 μM) elicited a smaller increase of the ratio (∼7%; Figure 4A, middle, and Figure 4B and 4E) compared to a higher dose of ADO (100μM) in which a much higher ratio was observed (∼24%; Figure 4C, top, and Figure 4D and 4E). Interestingly, MK571 led to a significant further increase in cAMP level in addition to the cAMP increase caused by a lower dose of ADO (2 μM) (from ∼7% to ∼17%; Figure 4A, middle and bottom, Figures 4B and 4E). However, MK571 failed to execute any significant additional increase of cAMP on top of the cAMP increase caused by a higher dose of ADO (100μM) (from ∼24% to ∼25%; Figures 4C, 4D, and 4E). These results reveal a transition from compartmentalized cAMP elevation (Figure 4A) to global increase in intracellular cAMP (Figure 4C; indicated by the uniform increase of the emission ratio in the entire cytoplasm) which is similar to the effect seen with 20 μM forskolin (from ∼28.8% to ∼29%; Figure S5, Figure 4E). This finding is consistent with the data from the above functional studies, the short-circuit current measurements (Figures 1A, 1B, and 3A) and cell-attached single-channel recordings (Figure 3D). These results imply that the increased cAMP accumulation through inhibiting cAMP efflux (via MRP4 transporter) demonstrates the biggest magnitude only when it is happening in a compartmentalized, spatially-restricted microdomain near the plasma membrane, suggesting a spatial cAMP heterogeneity within a single cell.
Inhibition of cAMP Transporter in vivo Induces Secretory Diarrhea in Mice
CFTR plays a critical role in cholera toxin (CTX)-induced intestinal fluid secretion (secretory diarrhea) (Clarke et al., 1992; Li et al., 2005). Given that MK571 inhibits MRP4 activity and thus cAMP transport, leading to CFTR Cl− channel activation as demonstrated in above studies, we therefore hypothesized that inhibition of MRP4 can induce secretory diarrhea in mice. To test this hypothesis, the effect of MK571 on CTX-induced CFTR-mediated intestinal fluid secretion was monitored in a closed-loop model of secretory diarrhea in wild-type and MRP4 knockout mice. CTX (0.5 μg) elicited a fluid accumulation in ligated ileal loops of both wild-type and MRP4 knockout mice (Figure 5A and 5B). MK571 treatment significantly increased the fluid accumulation in the toxin-treated intestinal loops in wild-type mice (Figure 5A) but failed to do so in MRP4 knockout mice (Figure 5B). Interestingly, we observed that, compared to wild-type mice, PBS control loop in MRP4 knockout mice demonstrated a slightly, also significantly, higher level of fluid secretion (Figure 5C; ∼24%, P<0.05, n=10–11). In addition, CTX injected loop in MRP4 knockout mice also demonstrated a significantly higher level of fluid accumulation compared to wild-type mice (Figure 5C; ∼27%, P<0.01, n=12–13). The MK571-potentiated, toxin-induced intestinal fluid secretion is mediated through CFTR Cl− channel because the CFTR specific inhibitor, CFTRinh-172, significantly inhibited the fluid secretion (Figure 5D).
Figure 5. MRP4-deficient Mice are more Prone to CFTR-mediated Secretory Diarrhea.

(A and B) Representative mouse ileal loops 6 h after luminal injection with CTX (0.5 μg) along with or without MK571 (20 μM) (picture) in wild-type mice (A) or MRP4 knockout mice (B). The bar graphs (bottom) show the averaged loop weight from wild-type mice (A) or MRP4 knockout mice (B). Scale bar: 10 mm. Numbers in the parenthesis indicate the repeated times for each experimental condition. Error bars show SEM (n=10–14). NS, no significance.
(C) The bar graphs showing the averaged loop weight injected with PBS together with or without CTX (0.5 μg) from wild-type or MRP4 knockout mice. Data represent the mean ± SEM (n=10–13).
(D) Representative mouse ileal loops 6 h after luminal injection with CTX (0.5 μg) + MK571 (20μM) along with or without CFTR specific inhibitor, CFTRinh-172 (20 μM), in wild-type mice.
Cumulatively, our findings provide clear evidence that, i) the potentiating effect on CTX-induced fluid secretion by MK571 is attributed to the inhibitory effect of MK571 on MRP4 transporter activity; and ii) MRP4-deficient mice are more prone to CFTR-mediated secretory diarrhea. The latter argument is also supported by the MRP4 siRNA approach as described above to specifically knock down endogenous MRP4 protein amount followed by CFTR-mediated short-circuit current measurement (Figure 3B; and Figure S1C). The data from the intestinal fluid secretion study are consistent with the findings from short-circuit current study, single-channel recordings, as well as the FRET-based intracellular cAMP dynamics described above.
A Macromolecular Complex of MRP4 C-tail and CFTR is Mediated by PDZK1
Given that we observed a functional coupling between inhibition of MRP4 transporter activity and potentiation of CFTR Cl− channel function, we next asked if a physical association between CFTR and MRP4 could be detected as well. Like CFTR, MRP4 also possesses a consensus PDZ motif at its C-terminus (-TALCOOH). Therefore it is possible that MRP4 can interact with CFTR via PDZ motif-based interaction. We therefore explored the binding affinity between MRP4 and several PDZ proteins that are reported to localize to the apical membrane of epithelial cells (Hung and Sheng, 2002; Li and Naren, 2005). Pull-down assays demonstrated that MRP4 binds to several PDZ proteins, while with the highest affinity to PDZK1 (Figure 6A). In order to quantify the binding affinity (EC50) between MRP4 and PDZK1, two purified proteins were used in a binding kinetics study which demonstrated that MRP4 interacts with PDZK1 directly with a binding constant of EC50=3.75 nM (Figure 6B).
Figure 6. A Macromolecular Complex Is Formed between MRP4 C-tail, PDZK1, and CFTR.

(A) MRP4 was pulled down by various GST-PDZ scaffolding proteins from lysates of HEK293 cells overexpressing MRP4.
(B) Binding of PDZK1 and GST-MRP4 C-tail 50 amino acids (EC50=3.75 nM). The inset shows a representative immunoblot of the dose-dependent interaction between His-PDZK1 and GST-MRP4-C50.
(C) PDZK1 expression in HT29-CL19A and T84 colonic epithelial cells by immunoblotting.
(D) A pictorial representation of the macromolecular complex assay (top; see Supplemental Experimental Procedures for details). A macromolecular complex was detected in vitro with three purified proteins (GST-MRP4-C50, His-PDZK1, and Flag-CFTR) in a dose-dependent manner.
(E) Cell lysates from HT29-CL19A were co-immunoprecipitated using anti-CFTR antibody (R1104) and probed for PDZK1 and MRP4. MRP4 blot was visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) because of the weak signal.
We generated polyclonal antibody against PDZK1 protein and detected expression of PDZK1 in both HT29-CL19A and T84 gut epithelial cells (Figure 6C). PDZK1 has been reported to interact with CFTR through PDZ-based interaction (Wang et al., 2000). Given that both MRP4 and CFTR interact with PDZK1 and that these two transporters are colocalized to the apical membrane of gut epithelial cells (Figure 1D), we hypothesized that the interaction between MRP4 and CFTR was likely mediated by PDZK1. To address this, we assembled a macromolecular complex of MRP4 C-tail, PDZK1, and CFTR in vitro, which is represented schematically in Figure 6D (top). A macromolecular complex was formed between MRP4 C-terminal 50 amino acids, PDZK1, and CFTR (Figure 6D, bottom). The complex formation increased dose-dependently with increasing amounts of the intermediary protein, PDZK1 (Figure 6D, bottom). We next immunoprecipitated CFTR from cultured gut epithelial cells that endogenously express all three proteins, and demonstrated that both MRP4 and PDZK1 can be co-immunoprecipitated with CFTR (Figure 6E). These studies suggest that a macromolecular complex consisting of endogenous MRP4-PDZK1-CFTR is likely present in the apical surface of gut epithelial cells, which forms the bases for the functional coupling of MRP4 transporter activity to CFTR channel function we observed in this study.
Disrupting the Macromolecular Complex Inhibits the Functional Coupling between MRP4 and CFTR
To demonstrate the importance of a macromolecular complex containing MRP4, PDZK1, and CFTR in the MK571 potentiation of CFTR function, we disrupted the complex by using competitive MRP4 PDZ-motif peptides (containing the last 10 amino acids including the tripeptide PDZ-motif at the extreme C-terminus). We synthesized various peptides, with and without a biotin-conjugate at the N-terminus (MRP4-C10 wt), and mutant peptides with the alanine substitution point mutation of the last amino acid within the tripeptide PDZ-motif (MRP4-C10-L1325A) (Figures 7A) and the alanine substitution mutations of the whole tripeptide PDZ-motif (MRP4-C10-AAA) (Figure S6A). The biotin-conjugate facilitated detecting the peptides by using FITC-streptavidin. The competing efficacy of the peptides was confirmed by the pull-down in vitro binding studies, which showed that the synthetic wild-type peptides substantially competed with GST fusion protein of MRP4 C-terminal 50 amino acids for binding to PDZK1 (Figure 7A and Figure S6A) compared to the controls. The mutant peptides demonstrated significantly less efficacy of competition (Figure 7A and Figure S6A). This clearly shows that the tripeptide PDZ-motif (-TALCOOH) at the extreme C-terminus of MRP4 transporter is critical for the physical interaction between MRP4 and PDZK1. Using a high-efficient peptide delivery system (Chariot system; see Li et al., 2005), we delivered these peptides into polarized gut epithelial cells before short-circuit current measurements stimulated by ADO. Our data clearly demonstrated that the MRP4-specific peptides significantly attenuated the MK571-elicited potentiation of CFTR-mediated currents in gut epithelial cells (Figure 7B). At the end of the experiment, the epithelial cells were fixed and immunostained using FITC-streptavidin to confirm the delivered peptide in the cells (Figure 7C, right).
Figure 7. MRP4 Peptides That Disrupt MRP4 interaction with PDZK1 Attenuate Elevation of CFTR Cl− Currents Mediated by MRP4 inhibition.

(A) PDZK1 binding to MRP4 in the presence of biotin-conjugated MRP4 C-tail peptides (last 10 amino acids; wild-type and mutant L1325A). The bar graphs (bottom) show the averaged band density from respective groups (n=3).
(B) Representative Isc traces in response to ADO in HT29-CL19A polarized cells. MRP4 peptides (2 μM) were delivered to the cells before Isc measurement. Cells in all groups were pretreated with 20 μM MK571 for 30 min before activating with ADO. Bar graphs summarize the averaged maximal Isc after each dosage of ADO as percentage of the control. Results are presented as mean ± SEM (n = 3–4 for each group). NS, no significance; **P < 0.01; ***P < 0.001 compared with the control for each ADO dosage.
(C) Immunofluorescence micrographs of HT29-CL19A cells from (B) showing the biotin-conjugated MRP4 peptides (green; right) delivered into the polarized cells. The nucleus was stained with propidium iodide (red). FITC-streptavidin was used to detect the biotin conjugated with the peptides.
(D) Schematic representation of the compartmentalized signaling involved in the spatiotemporal coupling of cAMP transporter (MRP4) to CFTR Cl− channel in the gut epithelia. Refer to the Discussion for details.
To rule out the possibility that the MRP4 peptides might interfere with other PDZ domain interaction (or these peptides might inhibit PDZK1 interactions with other target proteins), and to confirm that MRP4 peptide does disrupt local macromolecular complex in these peptide-delivered-epithelial cells, we also performed the co-immunoprecipitation using PDZK1 antibody. The co-immunoprecipitated CFTR and MRP4 amounts were checked by immunoblotting. As the data showed, MRP4 wild-type peptide significantly reduced the endogenous MRP4 protein amount that was co-immunoprecipitated with PDZK1 in HT29-CL19A epithelial cells (Figure S6B, right). However, MRP4 wild-type peptide did not affect CFTR amount co-immunoprecipitated with PDZK1 (Figure S6B, left), which suggests the specificity of the MRP4 peptide. MRP4-L1325A mutant peptide behaved in a similar fashion to biotin control (Figure S6B).
DISCUSSION
Secretory epithelia perform vectorial transport of salt and water molecules by coordinated actions of the transporters expressed in polarized epithelial membranes, and CFTR is one of the key membrane proteins regulating overall fluid movements. Hypo- or hyperfunctioning of CFTR can cause aberrant membrane transport and may result in life-threatening diseases, such as cystic fibrosis or secretory diarrhea (Kunzelmann and Mall, 2002). Therefore, the fine-tuned regulation of salt and water transport is essential in epithelial and body homeostasis.
Spatiotemporal Coupling of cAMP Transporter Function and CFTR Cl− Channel Activity
In the present study, we found that inhibition of a cAMP transporter MRP4 by MK571 potentiated CFTR-mediated Cl− currents. We also observed that potentiation of CFTR function by MK571 was most prominent when CFTR was activated by lower concentrations of ADO (< 20 μM) but not with higher doses of agonists (> 20 μM). This suggests that lower doses of ADO may lead to compartmentalized cAMP accumulation, which is further enhanced by the reduced or inhibited cAMP efflux through MRP4 by MK571. This finding is consistent with the previous observation that cAMP is generated in a compartmentalized pocket upon stimulation by 2 μM ADO, which is enough to activate CFTR function in proximity, and that LPA, a cAMP-lowering agent, significantly inhibits CFTR function (Li et al., 2005). Here the concept of cAMP compartmentalization was recapitulated with the treatment of MK571, which significantly potentiated CFTR function when it was stimulated by lower doses of ADO. It has been reported that inhibiting the transport of MRP4 does not lead to substantial increases in intracellular cyclic nucleotide levels (globally); neither does overexpression of MRP4 lead to a substantial decrease in intracellular cAMP levels, even when these levels are high (Wielinga et al., 2003). This is probably because the inhibition of MRP4 resulted only in localized fluctuations of cyclic nucleotide levels but not global levels.
Based on previous findings and the data obtained from this study, we propose a model depicting the spatiotemporal coupling of cAMP transporter to CFTR Cl− channel function in the gut epithelia (Figure 7D). Underneath the apical plasma membrane, highly localized compartments exist and contain a series of signaling molecules such as adenosine receptor (AR); G protein (Gs); AC; PKA and its anchoring proteins AKAPs; CFTR; cAMP transporter (MRP4); and PDZ scaffolding proteins (in this case, PDZK1) which functions to physically connect CFTR to MRP4. This macromolecular signaling complex provides a potential anatomical basis for the generation and modulation of local cAMP compartments. When an agonist (in this case, ADO) binds AR, a series of G-protein-mediated reactions leads to activation of AC present in the apical membrane. Sufficient cAMP (red spheres; Figure 7D) is locally generated in a diffusionally restricted apical microdomain (but not in other cellular compartments). cAMP activates PKA, which is anchored also to the apical membrane by AKAP (i.e., ezrin), and phosphorylates CFTR Cl− channel in close vicinity, resulting in an increase of Cl− currents. The CFTR-mediated Cl− currents can be further increased (potentiated) through the additional increase of local cAMP resulting from the reduced or blocked efflux via a neighboring apical membrane cAMP transporter (MRP4) in the same subcellular compartment.
In general, it is believed that PDEs provide the sole means for degrading cAMP in cells and play a vital role in shaping intracellular gradients of the second messenger (Cooper, 2005; Jackson and Raghvendra 2004). Here, we identified additional means of regulating cAMP levels in a microdomain underneath the surface membrane, an efflux path for cAMP via MRP4 transporter in the close vicinity of CFTR-containing signaling complex. The interaction between CFTR and MRP4 provides an additional layer of mechanism to regulate CFTR function, which is important in maintaining epithelial and body homeostasis.
Macromolecular Signaling Complex in the Gut
PDZ domains are conserved protein-interaction modules of ∼80–90 amino acids in length that fold to form peptide-binding clefts and typically mediate interactions with the carboxyl termini of target proteins that terminate in consensus PDZ-binding sequences (i.e., PDZ-motif) (Hung & Sheng, 2002; Li and Naren, 2005). PDZK1 has been reported to interact with CFTR, SLC26A3 [downregulated in adenoma (DRA)], SLC26A6 [putative anion transporter 1 (PAT1)], and Na+/H− exchanger NHE3, the major transport proteins for intestinal anion secretion and salt absorption (Lamprecht and Seidler, 2006). Many studies have reported the association of these PDZ proteins with a wide variety of ion channels, receptors, transporters, and signaling proteins in the apical surfaces of cells, suggesting that apical membrane PDZ proteins could facilitate the formation of multiprotein complexes clustered within microdomains that modulate trafficking, transport, and signaling in polarized epithelial cells (Wang et al., 2000; Lamprecht and Seidler, 2006).
Compartmentalization of signaling complexes containing CFTR, PDZK1, and MRP4 (Figure 7D) has important implications. Our present study, for the first time, demonstrated a physically and functionally interacting macromolecular complex containing two ABC transporters mediated by a PDZ scaffolding protein, which may have significant in vivo physiological or pathophysiological relevance. Under normal conditions, MRP4 pumps out cAMP, which is in proximity to CFTR, thus modulating CFTR function stimulated by cAMP-producing pathways (such as ADO). Under aberrant conditions (such as inflammatory bowel disease with diarrhea or irritable bowel syndrome with constipation), the expression levels and/or subcellular localization of CFTR and/or MRP4 may be dysregulated. The ratio of CFTR versus MRP4 may be altered, thereby resulting in elevated or reduced cAMP accumulation in the compartment and leading to enhanced or attenuated CFTR-mediated Cl− transport, which consequently results in secretory diarrhea in inflammatory bowel disease or constipation occurring in irritable bowel syndrome. In inflamed colonic mucosa of mice with inflammatory bowel disease, CFTR expression was increased 2.5-fold compared with noninflamed samples (Lohi et al., 2002). Elevated CFTR expression and enhanced cAMP-dependent Cl− secretion have been demonstrated in hyperproliferated mouse intestine (Umar et al., 2000). In CFTR knockout mice, ileal mucosal bile acid absorption is increased significantly compared to wild type mice (Stelzner et al., 2001). Interestingly, MRP4 is reported to be an alternative bile acid export pump (Rius et al., 2003). This suggests a possible up-regulation of MRP4 function in the absence of CFTR. Our present studies have significant in vivo implications for diseases such as secretory diarrhea, inflammatory bowel disease, and irritable bowel syndrome.
EXPERIMENTAL PROCEDURES
Short-circuit Current (Isc) Measurements
Polarized colonic epithelial cell (HT29-CL19A and T84) were grown to confluency on permeable supports before mounted in a Ussing chamber, and short-circuit currents (Isc) were measured as reported previously (Li et al., 2005). MK571 (5–50 μM) was added to both the apical and basolateral sides for 30 min before ADO was added to apical side. In some experiments, cells were permeabilized at the basolateral side with 100 μg/ml alpha-toxin for 30 min at 37°C before Isc measurement. CFTR inhibitor DPC (500 μM), glybenclamide (500 μM), or CFTRinh-172 (1–10 μM) were added into the apical side.
[3H]-cAMP and etheno-cAMP Transport Assay across Apical Cell Membranes
HT29-CL19A and T84 cells were permeabilized at the basolateral side with alpha-toxin as above, and incubated with MK571 or PMEA at 37°C for 30 min. [3H]-cAMP or etheno-cAMP was added into the basolateral chamber containing 4 mM ATP (Mg salt). For [3H]-cAMP transport assay, an aliquot of 40 μl apical solution was collected at various time points, and the radioactivity was measured using a liquid scintillation counter (LS5000TA; Beckman Coulter, Fullerton, CA). For etheno-cAMP transport assay, an aliquot of 40 μl apical solution was collected, and the fluorescence was measured at wavelength ex315-nm/em420-nm using a fluorescence spectrophotometer (F-2500; Hitachi, Tokyo, Japan).
Co-immunoprecipitation and Immunoblotting
Cells were harvested and processed for co-IP and immunoblot as described previously (Li et al., 2005). Cells were solubilized in RIPA buffer for immunoblot alone or PBS-0.2% Triton X-100 for co-immunoprecipitation, and lysates were spun at 15,000 g for 15 min at 4°C to pellet insoluble material and processed for co-IP and immunoblot as described in detail in Supplemental Experimental Procedures.
Cell-attached Single-channel Recordings
Single-channel recordings were obtained from HT29-CL19A cells as reported previously (Li et al., 2005). CFTR channels were activated with adenosine (0–40 μM) included in the pipette as indicated. Single-channel currents were continuously recorded for 5 min at a test potential of +100 mV (referenced to the cell interior) delivered from the recording electrode and were filtered at 1 kHz and sampled at 2 kHz (Li et al., 2005; details in Supplemental Experimental Procedures).
FRET Imaging Microscopy and Image Analysis
Cells (expressing CFP-EPAC-YFP) for FRET imaging were seeded in 35-mm glass bottom dishes (MatTek) and grown for 24–48 h, washed with HBSS before mounted on an Olympus microscopy system for FRET imaging. Images were recorded with a cooled CCD camera Hamamatsu ORCA285 (Hamamatsu, Japan) mounted on the Olympus microscope IX51 (U-Plan Fluorite 60 × 1.25 NA oil-immersion objective), and the system was controlled by SlideBook 4.1 software (Intelligent Imaging Innovations, Denver, CO) with ratio and FRET modules used to obtain and analyze the images. Excitation light was provided by a 300W Xenon lamp and attenuated with a ND filter with 50% light transmission. Images were captured using a JP4 CFP/YFP filter set (Chroma, Brattleboro, VT) including a 430/25-nm excitation filter, a double dichroic beam splitter, and two emission filters (470/30-nm for CFP and 535/30-nm for FRET) alternated by a filter-changer Lambda 10–3 (Sutter Instruments, Novato, CA). Time-lapse images were acquired with 4 × 4 binning mode, 200–400 ms exposure time, and 1-min intervals to reduce photobleaching of the fluorophores. Acquired fluorescent images were background-subtracted, and multiple regions of interest (ROIs) on the cell periphery were selected for quantitative data analysis (∼20–30 ROIs per cell, and 4–6 cells per condition were averaged). The emission ratio images (CFP/FRET) were generated at different time points on a pixel-by-pixel basis by the SlideBook ratio module as formulated below,
The ratio (R) was normalized as Rt/R0, where Rt is the ratio at time point t and R0 is the ratio at time point = 0 (right before the addition of first test compound). The cell images are presented in pseudocolor to highlight the changes in the ratio of CFP/FRET fluorescence intensity.
Macromolecular Complex Assembly
The in vitro complex formation (Li et al., 2005) was performed by mixing GST-His-S-MRP4-C terminal 50 a.a. fusion protein (20 μg) with various amounts of His-S-PDZK1 (0-40 μg) and 0.5 μg purified Flag-tagged wt-CFTR at 4 oC for 3 h (Li et al., 2005; details in Supplemental Experimental Procedures).
MRP4 Peptide in vitro Competitive Binding
MRP4 peptides (biotin-conjugated wild-type or L1325A mutant peptide, or non-conjugated wild-type or C10-AAA mutant peptide; 2 μM each) were mixed with His-PDZK1 (1.3 nM and 6.6 nM) at 22–24°C for 1 hr before GST-MRP4-C50 (immobilized on glutathione beads) was added and continued to mix for another 1 hr. The beads were washed, and protein complex was eluted from the beads, separated by SDS-PAGE and immunoblotted with rabbit anti-PDZK1 polyclonal antibody as described above.
Delivery of MRP4 Peptide into Polarized Epithelial Cells followed by Immunocytochemistry and Co-IP
Delivery of MRP4-specific peptide was performed as reported before (Li et al., 2005) and described in detail in Supplemental Experimental Procedures. After Isc measurement, the cells were fixed and immunostained for the peptides using FITC-conjugated streptavidin. In some experiments, after peptide delivery and Isc measurement, cells in the filters were scraped into RIPA buffer, and co-IP using rabbit PDZK1 antibody was performed. Endogenous CFTR and MRP4 protein amounts in the co-immunoprecipitated complex were checked by immunoblotting using anti-CFTR (R1104 monoclonal) and anti-MRP4 (M4I-10, rat monoclonal) antibodies as described above.
MRP4 siRNA to Knock Down MRP4 in Polarized Epithelial Cells
MRP4 siRNA and Lipofectamine 2000 transfection reagent were prepared and mixed according to the manufacturer’s instruction. siRNA-Lipofectamine 2000 complexes were added to HT29-CL19A cells grown to confluency on transwells, and incubated at 37°C in a CO2 incubator. After 60–72 hrs, the transwells were mounted in a Ussing chamber for Isc measurement in response to ADO stimulation. After the Ussing chamber study, the transwells were washed with PBS, and the cells were scraped into RIPA lysis buffer. The protein levels of MRP4, CFTR, and PDZK1 were assessed by Western blotting using corresponding antibodies.
Statistical Analysis
Results are presented as mean ± SEM for the indicated number of experiments. Statistical analysis was performed using Student’s t-test and one-way ANOVA. A value of P < 0.05, P < 0.01, or P < 0.001 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. David Armbruster for critically reading the manuscript and Ms. Lillian Zalduondo for kind assistance with mice ileal loop experiments. We also thank Dr. Gabor Tigyi for help in obtaining constructs, and Danny Morse for graphics. This work was supported by grants from NIH (to A.P.N., J.D.S., and D.J.N.), American Lebanese Syrian Associated Charities (to J.D.S.), American Lung Association (to A.P.N.), and American Heart Association - Greater Southeast Affiliate (#0765185B to C.L.).
References
- Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- Chen ZS, Lee K, Kruh GD. Transport of cyclic nucleotides and estradiol 17-beta-D-glucuronide by multidrug resistance protein 4. Resistance to 6-mercaptopurine and 6-thioguanine. J Biol Chem. 2001;276:33747–33754. doi: 10.1074/jbc.M104833200. [DOI] [PubMed] [Google Scholar]
- Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene targeted mouse model of cystic fibrosis. Science. 1992;257:1125–1128. doi: 10.1126/science.257.5073.1125. [DOI] [PubMed] [Google Scholar]
- Cooper DM. Compartmentalization of adenylate cyclase and cAMP signalling. Biochem Soc Trans. 2005;33:1319–1322. doi: 10.1042/BST0331319. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci U S A. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- Jackson EK, Raghvendra DK. The extracellular cyclic AMP-adenosine pathway in renal physiology. Annu Rev Physiol. 2004;66:571–599. doi: 10.1146/annurev.physiol.66.032102.111604. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82:245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- Lai L, Tan TM. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem J. 2002;361:497–503. doi: 10.1042/0264-6021:3610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht G, Seidler U. The emerging role of PDZ adapter proteins for regulation of intestinal ion transport. Am J Physiol Gastrointest Liver Physiol. 2006;291:G766–G777. doi: 10.1152/ajpgi.00135.2006. [DOI] [PubMed] [Google Scholar]
- Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther. 2005;108:208–223. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Lohi H, Makela S, Pulkkinen K, Hoglund P, Karjalainen-Lindsberg ML, Puolakkainen P, Kere J. Upregulation of CFTR expression but not SLC26A3 and SLC9A3 in ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2002;283:G567–G575. doi: 10.1152/ajpgi.00356.2001. [DOI] [PubMed] [Google Scholar]
- Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan, et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci U S A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MM, Quinton PM. Deactivation of CFTR GCl by endogenous phosphatases in the native sweat duct. Am J Physiol. 1996;270:C474–C480. doi: 10.1152/ajpcell.1996.270.2.C474. [DOI] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, De Haas M, Van Deemter L, Wijnholds J, Balzarini J, Borst P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63:1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology. 2003;38:374–384. doi: 10.1053/jhep.2003.50331. [DOI] [PubMed] [Google Scholar]
- Schuetz JD, Connelly MC, Sun D, Paibir SG, Flynn PM, Srinivas RV, Kumar A, Fridland A. MRP4: A previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat Med. 1999;5:1048–1051. doi: 10.1038/12487. [DOI] [PubMed] [Google Scholar]
- Steinberg SF, Brunton LL. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- Stelzner M, Somasundaram S, Lee SP, Kuver R. Ileal mucosal bile acid absorption is increased in Cftr knockout mice. BMC Gastroenterol. 2001;1:10–16. doi: 10.1186/1471-230X-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Scott J, Sellin JH, Dubinsky WP, Morris AP. Murine colonic mucosa hyperproliferation. I Elevated CFTR expression and enhanced cAMP-dependent Cl− secretion . Am J Physiol Gastrointest Liver Physiol. 2000;278:G753–G764. doi: 10.1152/ajpgi.2000.278.5.G753. [DOI] [PubMed] [Google Scholar]
- van Aubel RA, Smeets PH, Peters JG, Bindels RJ, Russel FG. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol. 2002;13:595–603. doi: 10.1681/ASN.V133595. [DOI] [PubMed] [Google Scholar]
- Wang S, Yue H, Derin RB, Guggino WB, Li M. Accessory protein facilitated CFTR-CFTR interaction, a molecular mechanism to potentiate the chloride channel activity. Cell. 2000;103:169–179. doi: 10.1016/s0092-8674(00)00096-9. [DOI] [PubMed] [Google Scholar]
- Wielinga PR, van der Heijden I, Reid G, Beijnen JH, Wijnholds J, Borst P. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J Biol Chem. 2003;278:17664–17671. doi: 10.1074/jbc.M212723200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.