

Abstract
Two collagen receptors, integrins α1β1 and α2β1, can regulate distinct functions in cells. Ligation of α1β1, unlike α2β1, has been shown to result in recruitment of Shc and activation of the Ras/ERK pathway. To identify the downstream signaling molecules activated by α2β1 integrin, we have overexpressed wild-type α2, or chimeric α2 subunit with α1 integrin cytoplasmic domain in human osteosarcoma cells (Saos-2) lacking endogenous α2β1. The chimeric α2/α1 chain formed a functional heterodimer with β1. In contrast to α2/α1 chimera, forced expression of α2 integrin resulted in upregulation of α1 (I) collagen gene transcription in response to three-dimensional collagen, indicating that the cytoplasmic domain of α2 integrin was required for signaling. Furthermore, signals mediated by α2β1 integrin specifically activated the p38α isoform, and selective p38 inhibitors blocked upregulation of collagen gene transcription. Dominant negative mutants of Cdc42, MKK3, and MKK4 prevented α2β1 integrin–mediated activation of p38α. RhoA had also some inhibitory effect, whereas dominant negative Rac was not effective. Our findings show the isoform-specific activation of p38 by α2β1 integrin ligation and identify Cdc42, MKK3, and MKK4 as possible downstream effectors. These observations reveal a novel signaling mechanism of α2β1 integrin that is distinct from ones previously described for other integrins.
Keywords: collagen, integrin, cytoplasmic domain, p38 MAPK
Integrin type receptors bind extracellular matrix (ECM)1 molecules and mediate cell adhesion, migration, and invasion during development, tissue repair, tumor invasion, and metastasis. They also act in concert with growth factor or cytokine receptors to regulate cell proliferation, differentiation, and survival (Hynes 1992; Clark and Brugge 1995). The relatively short cytoplasmic domains of the integrin α and β subunits do not have any intrinsic enzymatic activity, but integrin signaling is achieved by coupling signaling molecules, such as tyrosine kinases, to the cytoplasmic and transmembrane domains of the integrin subunits (Yamada and Miyamoto 1995). The β cytoplasmic domains share conserved regions, whereas the α cytoplasmic domains have highly divergent amino acid sequences (Laflamme et al. 1997). Integrins activate signaling pathways that are either common to all integrins or heterodimer specific (Giancotti 1997). Early integrin-triggered signaling events seem to be mediated by the cytoplasmic domains of the β subunits. These include the tyrosine phosphorylation of the focal adhesion kinase (FAK) (Schaller et al. 1994) and recruitment of components of the actin-based cytoskeleton, such as α-actinin and talin (Horwitz et al. 1986; Otey et al. 1990). In the next step, a large number of proteins with enzymatic activity, like kinases and GTPases, are recruited to the sites of adhesion. Activation of these signaling proteins is mediated both by the β subunits and specifically by individual α subunits (Howe et al. 1998).
The functions of α cytoplasmic domains of various integrins are distinct. Cytoplasmic domains of α5 and αL are not required for cell adhesion unlike those of α2 and α4. In the context of cell adhesion and migration, some of the α cytoplasmic domains seem to be interchangeable (Kassner and Hemler 1993), whereas signaling events specific for each individual integrin heterodimer may be triggered by the α cytoplasmic domains (Z. Zhang et al. 1995; Sastry et al. 1996).
Four members of the integrin family are known to bind collagens, namely α1β1, α2β1, α3β1, and α10β1 (Camper et al. 1998). α1β1 and α2β1 are considered to be the two major collagen binding integrins, whereas α3β1 seems to function as an assisting receptor (DiPersio et al. 1995). The recently identified integrin α10β1 is expressed on chondrocytes, but little is known about its biology (Camper et al. 1998). α1β1 and α2β1 are concomitantly expressed by many cell types but they may regulate different functions. Three-dimensional ECM culture systems provide means to study cell–matrix interactions in a more natural environment than the traditional monolayer culture (Grinnell 1994). The studies with three-dimensional collagenous matrices have shown that ligation of α2β1 triggers MMP-1 expression (Langholz et al. 1995; Riikonen et al. 1995a) and induces collagen gel contraction (Chan et al. 1992; Riikonen et al. 1995b). In fibroblasts, contact to collagen also leads to the activation of protein kinase (PK) C-ζ and NFκB and that correlates with the upregulation of α2β1 integrin and MMP-1 gene expression (Xu and Clark 1997), suggesting that these phenomena are mediated via α2β1. On the other hand, α1β1 integrin mediates downregulation of collagen α1(I) mRNA levels both in vitro and in vivo (Langholz et al. 1995; Riikonen et al., 1995; Gardner et al. 1999). A subset of integrins, including α1β1, have been shown to activate the mitogen-activated protein kinase (MAPK) extracellular signal-related kinase (ERK) via recruitment of Shc and activation of Ras (Wary et al. 1996; Mainiero et al. 1997; Pozzi et al. 1998), suggesting that α1β1 integrin is the collagen receptor that regulates cell proliferation in collagenous matrices. In contrast, α2β1 appears to be unable to recruit Shc and activate this specific signaling pathway.
We have previously observed that overexpression of α2β1 integrin prevents α1β1-mediated downregulation of collagen α1(I) mRNA levels when cells are brought into the contact with collagen (Riikonen et al. 1995a), but the molecular mechanism of this phenomenon has been unclear. To study the structural requirements of signaling via the distinct collagen receptors, we constructed a chimeric α chain composed of the extracellular and transmembrane domains of α2 and the cytoplasmic domain of α1. Using this construct, we show here that the upregulation of collagen α1(I) mRNA levels is due to active signaling requiring the cytoplasmic domain of α2 subunit.
We have recently shown that in normal dermal fibroblasts three-dimensional collagen activates three distinct classes of MAPKs, i.e., ERK, c-jun NH2-terminal kinase (JNK), and p38 (Ravanti et al. 1999). Ligation of α1β1 has been shown to activate the ERK kinase pathway, but α2β1 has not been previously linked to a specific MAP kinase pathway. Here, we report that the α2 cytoplasmic domain–dependent regulation of type I collagen mRNA levels requires p38 activity. We also show that the ligation of α2β1 leads to the activation of p38 MAPK, especially its p38α isoform. The activation is dependent on the α2 chain cytoplasmic domain and the function of the downstream effectors Cdc42, MKK3, and MKK4 is required. These results indicate a crucial role for the p38 pathway in integrin α2β1 signaling and provide novel insight on molecular mechanisms of integrin-specific signal transduction.
Materials and Methods
Reagents
Herbimycin A, bisindoylmaleimide, KT5720, KT5723, SB 203580, PD 98059, and SKF 86002 were obtained from Calbiochem-Novabiochem Corp. Cycloheximide was obtained from Sigma Chemical Co. (R)-(+)-perillyl alcohol (POH) was obtained from Aldrich and anisomycin was obtained from Boehringer Mannheim. α2 integrin cDNA corresponding to nucleotides 1–4559 in the published sequence (Takada and Hemler 1989) and the X2C5PFNeo plasmid (Kawaguchi and Hemler 1993) were provided by Dr. M. Hemler (Dana Farber, Boston, MA). The pAWneo2 expression vector was provided by Dr. A. Weiss (University of California San Francisco, San Francisco, CA) (Ohashi et al. 1985). The oligonucleotides used were purchased from Kebo Lab. The glutathione-S-transferase (GST)–tagged ATF2 (residues 1–109) (Gupta et al. 1995), the pRSV-MKK3(ala) and pcDNA-MKK4(ala) expression plasmids (Raingeaud et al. 1996; Whitmarsh et al. 1997) were provided by Dr. R. Davis (University of Massachusetts, Worcester, MA), and flag-tagged p38 isoform expression vectors (New et al. 1998) were provided by Dr. J. Han (Scripps Research Institute, La Jolla, CA). The pCEV29RhoAAsn19, pCEV29Rac1Asn17, and pCEVCdc42Asn17 were provided by Dr. J.C. Lacal (CSIC, Madrid, Spain). Bacterial expression of GST-ATF2 was done as described (Smith and Johnson 1988). The c-Jun protein was provided by Dr. E. Coffey (Åbo Akademi University, Turku, Finland). Bacterial expression of GST-ATF2 was done as described (Smith and Johnson 1988). Antibody against α2 integrin, 12F1 (Pischel et al. 1987) was provided by Dr. V. Woods (University of California, Medical Center, San Diego, CA). Polyclonal antisera against α1 integrin cytoplasmic tail (AB1934) was purchased from Chemicon International, Inc., and antibody against α1 integrin used in flow cytometry, SR-84, was a gift from Dr. W. Rettig (Boehringer Ingelheim, Germany).
Cell Culture
Human osteosarcoma cell line Saos-2 was obtained from the American Type Culture Collection. The cell cultures were maintained in DME supplemented with heat inactivated 10% FCS (GIBCO-BRL), 2 mM glutamine, 100 IU/ml penicillin-G, and 100 μg/ml streptomycin.
Plasmid Constructs and Stable Transfections
The α2 integrin expression construct was prepared as described previously (Riikonen et al. 1995a). The mutant α2 subunit, in which the cytoplasmic tail has been replaced with the corresponding α1 integrin sequence, was prepared in the following way. A silent point mutation (nucleotide 3488 in the published sequence; Takada and Hemler 1989) that introduces a new NheI recognition site was made with the Altered sites II in vitro mutagenesis system (Promega) according to the manufacturer's instructions. The mutant oligo used was ACCAGAGCTAGCAGCAAAAGG. The α2 cDNA was digested with NheI and SacI cleaving the region corresponding to the α2 cytoplasmic tail. The α1 DNA was obtained by annealing sense and antisense synthetic nucleotides corresponding to nucleotides 3506–3543 in the published α1 sequence (Briesewitz et al. 1993) flanked with corresponding α2 sequences; antisense oligo (TTTTGCTGCTAGCTCTGGTTGCAATTTTATGGAAGCTCGGATTCTTCAAAAGACCA-CTGAAAAAGAAAATGGAGAAATGAGAGCTCAGTAGCTG), sense oligo (TCAGCTACTGAGCTCTCATTTCTCCATTTTCTTTTTCAGTGGTCTTTTGAAGAATCCGAGCTTCCATAAAATTGCAACCAGAGCTAGCAGCAAAA). This synthetic DNA fragment was digested with NheI and SacI restriction enzymes and ligated with the cleaved α2 cDNA. The correctness of the construct was checked by sequencing. Stable transfections were performed with the calcium polybren/DNA method on confluent 60-mm dishes. Incubation with 5 μg DNA and 5 μg polybren in 1 ml 10% FCS/DME per dish was carried out for 6 h, agitating the dishes once an hour. DMSO (30% in FCS) shock was done for 3 min, cells were washed twice with PBS, and culture medium was added. Neomycin analogue G418 (Life Technologies, Inc.) was added to the culture medium in a concentration of 400 mg/ml. G418-resistant cell clones were selected for 2–3 wk, isolated, and analyzed for their expression of α2 integrin. Control cells were transfected with the pAWneo2 plasmid only. Transfected cells were cultured in 10% FCS/DME containing 2 mM glutamine, 100 IU/ml penicillin-G, 100 μg/ml of streptomycin and 200 mg/ml G418.
Collagen Gels and Gel Contractions
Collagen gels were prepared from bovine dermal collagen, which contains 95% type I collagen and 5% type III collagen (Cellon). 8 vol of Cellon were mixed with 1 vol of 10× concentrated DME and 1 vol of 10× concentrated NaOH (0.05 M) in Hepes buffer (0.2 M) and kept on ice. Cells were trypsinized, resuspended in 1/10 gel volume culture media DME supplemented with 10% FCS, mixed into neutralized Cellon solution, and transferred into 6-well plates. The collagen was allowed to polymerize for 2 h at 37°C, after which the culture media containing 10% FCS was added, the gels were detached from the sides of the wells, and incubation was continued for the times indicated. Cells were also cultured on plastic as a monolayer in culture media containing 10% FCS. In experiments involving inhibitors, the cells were pretreated with the inhibitor for 30 min at room temperature before mixing the cells with the neutralized Cellon solution, also supplemented with the inhibitor at the concentrations indicated. After polymerization, culture media containing 10% FCS and the inhibitor was added and the gels were detached from the sides of the dishes. Incubation was continued for 48 h. When studying collagen gel contraction, cell culture wells were photographed after 48 h and the surface areas of the gels were measured from the prints.
Immunofluorescence
The cells grown on immunofluorescence glass slides (CML) covered or uncovered with collagen film (Cellon) were rinsed with PBS and fixed with methanol at −20°C for 5 min. The cells grown inside collagen gels were excised from culture wells, embedded in OCT compound (Tissue-Tek; Miles Scientific), and frozen in isopentane chilled with liquid nitrogen. Sections of 10-μM thickness were cut and picked up onto microscope slides and treated as described above. The slides containing the fixed samples were incubated in 2% BSA in PBS and monoclonal anti–CD49b antibody (Chemicon International Inc.) was added to the same solution and incubated 30 min at room temperature. After rinsing, the cells were incubated with anti–mouse–FITC conjugate (Dako A/S) for 30 min and mounted before observation and photography. For staining of the actin filaments, plastic coverslips (Nunc) were coated with PBS containing 16 μg/ml type I collagen overnight at 4°C and blocked with 1% BSA in PBS for 1 h at 37°C. Cells were allowed to adhere and spread on collagen coated coverslips for 24 h in DME. Coverslips were washed once with PBS and fixed with 2% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS for 10 min, and incubated with rhodamine-conjugated phalloidin (Sigma Chemical Co.) 1:1,000 in PBS for 30 min. Cells were washed with PBS and mounted before observation and photography.
Northern Blot Hybridizations
Total cellular RNA was isolated using the Qiagen RNeasy kit. Total RNA was separated in formaldehyde-containing agarose gels, transferred to nylon membranes (Zeta-probe; Bio-Rad Laboratories), and hybridized with 32P-labeled (Amersham) cDNA probes. The following cDNAs were used: human α2 integrin (Takada and Hemler 1989), human collagen α1(I) (Mäkelä et al. 1988), human collagen α2(I) (Mäkelä et al. 1990) and 28S (a gift from R. Penttinen, University of Turku), and rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Fort et al. 1985).
Flow Cytometry
Cells were grown to early confluence, detached with trypsin-EDTA, and trypsin activity was inhibited by medium supplemented with serum. Cells were washed with PBS, pH 7.4, and then incubated with PBS containing 10 mg/ml BSA, 1 mg/ml glycine, and 0.02% NaN3 for 20 min at 4°C. Cells were collected by centrifugation, exposed to saturating concentration of mAb against α2 integrin (12F1) or α1 integrin (SR-84) in BSA/PBS (BSA concentration 1 mg/ml) containing NaN3 for 30 min at +4°C, and stained with rabbit anti–mouse IgG coupled to fluorescein (1:20 dilution; Dako A/S) for 30 min at 4°C. Cells were washed twice with PBS containing NaN3 and suspended in the same buffer. To measure the amount of α2 integrin on the cell surfaces, the fluorescent excitation spectra were analyzed by using a FACScan apparatus (Becton Dickinson). Control samples were prepared by treating the cells without primary antibodies.
Immunoprecipitation of Integrin from Metabolically Labeled Cells
Cells were metabolically labeled with 100 μCi/ml of [35S]methionine (Tran[35S]-label, ICN Biomedicals Inc.) for 16 h in methionine-free minimum essential medium. Cell monolayers were rinsed on ice with a solution containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 25 mM Tris-HCl, pH 7.4, and then detached by scraping. Cell pellets were obtained by centrifugation at 500 g for 5 min. Cells were solubilized in 200 μl of the same buffer containing 100 mM N-octyl-β-d-glucopyranoside (Sigma Chemical Co.) on ice with occasional vortexing. Insoluble material was removed by centrifugation at 10,000 g for 5 min at 4°C. Radioactivity in cell lysates was counted and an equal amount of radioactivity was used in immunoprecipitation assays. Triton X-100 (0.5% vol/vol) and BSA (0.5 mg/ml) were added to the supernatants, which were precleared by incubation with 50 μl of packed protein A–Sepharose (Pharmacia LKB Biotechnology Inc.). Supernatants were immunoprecipitated with antiintegrin antibody (12F1 or AB1934) for 12 h at 4°C followed by incubation with secondary antibody (rabbit anti–mouse, DAKO), when 12F1 was used. Immune complexes were recovered by binding to protein A–Sepharose and washing the beads four times with 25 mM Tris-buffered isotonic saline, pH 7.4, containing 0.5% Triton X-100 and 1 mg/ml BSA and twice with 0.5 M NaCl and 25 mM Tris-HCl, pH 7.4. The immunoprecipitates were analyzed by electrophoresis on SDS-containing 6% polyacrylamide gels under nonreducing conditions followed by fluorography.
Transcriptional Nuclear Run-on Analyses
The cells were lysed with NP-40 (ICN) and the nuclei were isolated by centrifugation (12,000 g) for 3 s at 4°C. The nuclei were incubated in the presence of 100 μCi of [α-32P]UTP (3,000 Ci/mmol, NEN) for 30 min at room temperature as described previously (Banerji et al. 1984). Radiolabeled RNA was hybridized with 2 μg of nitrocellulose-fixed plasmids: cDNAs for human α1(I) collagen, human α2(I) collagen, GAPDH, and pBluescript (Stratagene). The hybridization and washing conditions used were as described previously (Sistonen et al. 1992). Quantitation was performed with GS-250 Molecular Image System (Bio-Rad Laboratories) and the results were corrected for the levels of GAPDH transcripts in the same samples.
Cell Spreading Assays
The coating of a 96-well immunoplate (Maxi Sorp; Nunc) was done by exposure to 0.2 ml of PBS, pH 7.4, containing 0.1 μg/cm2 (1.64 μg/ml) type I collagen (from lathyric rat skin, Boehringer Mannheim) for 12 h at 37°C. Residual protein absorption sites in all wells were blocked with 1% BSA in PBS for 1 h at 37°C. BSA was also used to measure the nonspecific binding. Cells were detached by using 0.01% trypsin and 0.02% EDTA. Trypsin activity was inhibited by washing the cells with 1 mg/ml of soybean trypsin inhibitor (Sigma Chemical Co.). In cell spreading assays, cells were suspended in DME with 50 mM cycloheximide (Sigma Chemical Co.), transferred into each well, and incubated for 35 min at 37°C. The wells were washed with PBS and fixed with 8% formaldehyde and 10% sucrose in PBS for 30 min. The total number of cells attached per one microscopic field and the percentage of spread cells were counted. A spread cell was characterized as one having a clearly visible ring of cytoplasm around the nucleus.
Assay of MAPK Activation
The activation of ERK1 and 2, JNK/SAPK, and p38 MAPK was determined by Western blotting using antibodies specific for the phosphorylated, activated forms of the corresponding MAPKs (New England Biolabs). The control blots for the total (phosphorylated and nonphosphorylated forms) protein levels were done by using antibodies recognizing the corresponding MAPKs (p38, ERK2, New England Biolabs; JNK1, Santa Cruz Biotechnology). Saos cell clones were either grown in monolayer for 24 h or seeded in collagen gels as described earlier. Once polymerized, the gels were detached from the dish and incubated for the time indicated. The cells were released from the gels as described above and lysed in 100 μl of Laemmli sample buffer. Cells grown in monolayer were washed once with warm PBS and lysed in 100 μl of Laemmli sample buffer. The positive control treatment for the JNK Western blot was done by treating cells in suspension with 10 μg/ml anisomycin (Boehringer Mannheim). The samples were sonicated, fractionated by 10% SDS-PAGE, and transferred to a Hybond ECL membrane (Amersham Corp.). Western blotting was performed as described previously (Reunanen et al. 1998), with phosphospecific antibodies in dilution 1:1,000. Specific binding of antibodies was detected with peroxidase-conjugated secondary antibodies and visualized by enhanced chemiluminescence (ECL) detection system (Amersham).
In Vitro p38 and JNK Kinase Assay and Western Blot Analysis of Flag-tagged Protein
Subconfluent Saos cell clones plated on 60-mm dishes were transfected using 4 μl of Fugene 6 transfection reagent (Boehringer Mannheim) and 2 μg of either eukaryotic expression vector alone (pcDNA3; Invitrogen Corp.) or the same vector containing the flag-tagged p38 isoform (New et al. 1998). Cotransfection of dominant negative signaling proteins was done by using 12–16 μl of Fugene 6, 3–8 μg of empty expression vector or the same vector containing the effector mutant and 2 μg of flag-tagged p38α. 36 h after transfections, the cells were treated with collagen gel for 3 h as described earlier or left untreated. Cells were solubilized with RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 20 mM sodium fluoride, 0.5 mM DTT, 1 mM PMSF in PBS, pH 7.4) supplemented with leupeptin, antipain, and pepstatin, 2 μg/ml each. The extracts were centrifuged 3,000 rpm for 15 min at 4°C. 1 μg of M2 antibody (Eastman Kodak Co.) conjugated to protein G–Sepharose (Pharmacia-LKB Biotechnology) was used for immunoprecipitation. The immunoprecipitates were washed twice with RIPA buffer, once with LiCl wash buffer (500 mM LiCl, 100 mM Tris, pH 7.6, 0.1% Triton X-100, 1 mM DTT) and once with kinase buffer (20 mM MOPS, pH 7.2, 2 mM EGTA, 10 mM MgCl2, 0.1% Triton X-100, 1 mM DTT). The kinase assay was initiated with 2 μg GST-ATF2 as substrate and 50 mM MgCl2, 25 μM ATP, 3 μCi γ-[32P]ATP in a final volume of 50 μl. The reactions were terminated after 30 min at 37°C by the addition of Laemmli sample buffer. The phosphorylation of substrate protein was examined after SDS-PAGE by autoradiography. For Western blotting, samples of lysed cells were fractionated by 10% SDS-PAGE and transferred to Hybond ECL membrane (Amersham Corp.). Western blotting was performed as described previously (Reunanen et al. 1998), with M2 antibody (Eastman Kodak Company) in dilution 1:750. Specific binding of antibodies was detected with peroxidase conjugated secondary antimouse antibody and visualized by the ECL detection system (Amersham).
The JNK kinase assay was done as described in Westermarck et al. 1998 with some modifications, namely the antibody used was anti-JNK1 (Santa Cruz Biotechnology) and the immunoprecipitation was done with protein G–Sepharose. The cells were brought into suspension, counted, and diluted so that equal number of cells from both clones were used. The cells were then either lysed immediately, treated with anisomycin for 10 min or seeded inside collagen for 3 h.
Immunoprecipitation of FAK and Western Blot Analysis
The cells were brought into suspension, counted, and diluted so that equal number of cells from both clones were used. The cells were either lysed immediately or allowed to adhere to fibronectin (1 μg/cm2 coated on a 60-mm dish overnight at +4°C and blocked with 0.1% heat-inactivated BSA for 1 h at +37°C) or seeded inside collagen for 1 h. The assay was done as described previously (Elenius et al. 1997). The antibody used for immunoprecipitation was polyclonal anti-FAK (Upstate Biotechnology Inc.). The filter was first blotted with antiphosphotyrosine (4G10; Upstate Biotechnology Inc.), stripped according to manufacturer's instructions, and reprobed with anti–FAK antibody. Alternatively, total cell lysates were fractionated by SDS-PAGE and the filter was first blotted with an antibody mixture detecting all the phosphorylated forms of FAK (Biosource, Hopkinton, MA), following stripping and re-probing with anti-FAK antibody.
Results
Chimeric α2/α1β1 Integrin Functions like the Wild-type α2β1 Receptor in the Formation of Focal Contacts
Initially, we made stable transfectants expressing wild-type α2 or chimeric α2/α1 subunit using Saos-2 cells, which have endogenous α1β1 but no α2β1 (Vihinen et al. 1996). Flow cytometric analysis using anti–α2 mAb (Fig. 1 B) confirmed that both the wild-type and the chimeric receptor were expressed on the surface of the transfected cells. Based on the flow cytometric measurements, we selected cell clones with similar expression levels of wild-type α2 or chimeric α2/α1 integrin to be used in the experiments (Fig. 1 B). Also, the cell clones used in the experiments and not shown in the Fig. 1 B were tested. In addition, we measured the expression levels of α1β1 from both mock-transfected and α2-transfected cells to ensure that the α2 cDNA transfections did not alter the expression of the endogenous α1β1. The average levels of α1 integrin on four independent single cell clones tested were 60.8 ± 3.8 (arbitrary units) in mock-transfected cells and 68.3 ± 8.6 in α2-transfected cells. The corresponding values for α2 integrin were 13.5 ± 0.7 (represents negative background level fluorescence) and 362 ± 54. Immunoprecipitation experiments were performed to confirm that the chimeric α2/α1 subunit was associated with the endogenous β1 subunit to form a typical heterodimeric αβ complex. The chimeric structure of the heterodimer was verified using an antibody recognizing the α1 cytoplasmic tail (not shown).
Figure 1.

(A) The cDNA constructs used in the transfections. (B) Flow cytometry analysis showing surface expression of α2 integrin subunit. The stable transfected cell clones were exposed to saturating concentration of monoclonal antibody against α2 integrin (12F1). Bound antibody was detected with FITC-conjugated rabbit anti–mouse IgG.
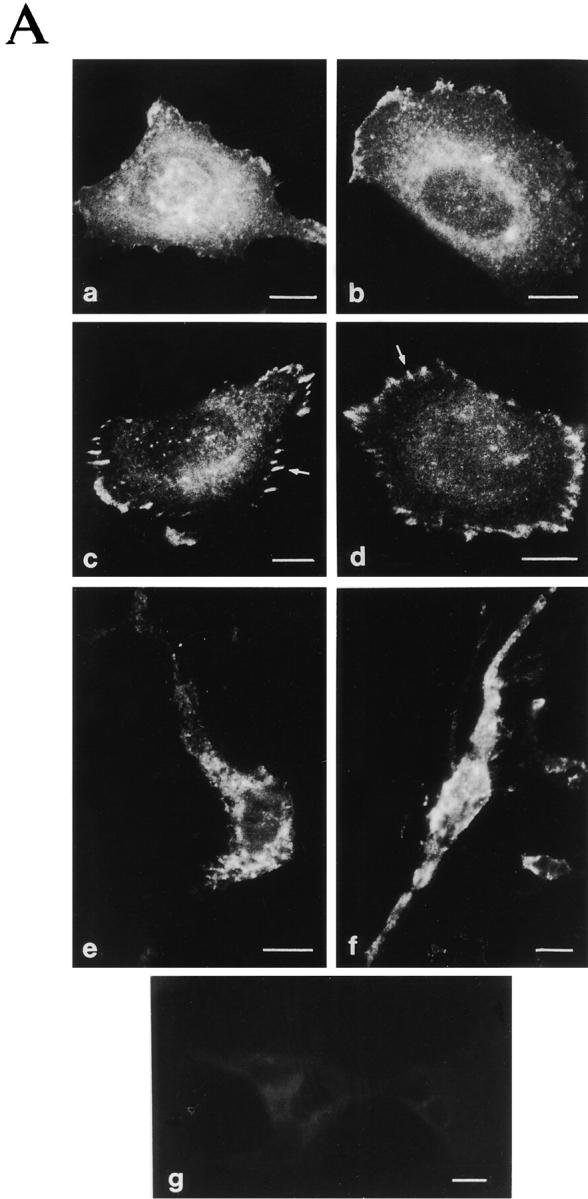
Deletion of the α2 cytoplasmic domain has been shown to result in ligand-independent recruitment of the integrin to preformed focal contacts (Kawaguchi et al. 1994). To study if swapping of the α2 cytoplasmic domain to the significantly shorter α1 tail results in indiscriminate integrin recruitment into focal adhesion sites formed by other integrins, we studied both wild-type α2 and chimeric α2/α1 in Saos-2 cells spread on serum proteins. Both clones showed a diffuse staining pattern indicating that the receptors do not localize to focal contacts ligand independently (Fig. 2 A, a and b). When these clones were allowed to adhere to and spread on collagen in serum-free conditions both wild-type and chimeric receptors were able to form focal contacts (Fig. 2 A, c and d) indicating that the α2 chain cytoplasmic tail can be replaced with the corresponding α1 sequence without affecting cellular localization of the receptor. Also, the formation of actin stress fibers on collagen was similar in both clones (Fig. 2 B). In cells cultured inside three-dimensional collagen, no aberrant clusters of integrins were seen and both receptors showed a diffuse staining pattern (Fig. 2 A, e and f).
Figure 2.
Characterization of the cellular location of α2 and α2/α1 integrin and formation of actin stress fibers. Stable Saos-2 transfectants were either grown in DME 10% FCS on immunofluorescence glass slides, plated on collagen coated glass slides, and allowed to adhere for 4 h in DME (serum-free) or grown inside a three-dimensional collagen gel for 48 h. (A) After incubation, the cells were fixed and stained for α2 integrin by using mAb followed by FITC-labeled rabbit anti–mouse antibody. Cells were washed, mounted, and examined on a microscope: (a) α2 clone 47 and (b) α2/α1 clone 18 on serum-derived fibronectin and vitronectin; (c) α2 clone 47 and (d) α2/α1 clone 18 on collagen; (e) α2 clone 47 and (f) α2/α1 clone 18 inside collagen gel; and (g) vector clone as a negative control. (B) Cells were stained for actin using fluorescein-conjugated phalloidin (a) α2 clone 47 and (b) α2/α1 clone 18 on collagen.

α2 Cytoplasmic Tail Is Required for Fast Spreading on Collagen and Collagen Gel Contraction
Cell adhesion to type I collagen was studied to determine the effect of swapping the tails on the adhesive properties of the integrin. Adhesion mediated by the chimeric α2/α1β1 receptor was found to be equivalent to that of the wild-type receptor (Fig. 3 A). Both the wild-type α2-transfected cells and the α2/α1 chimera–transfected cells adhered to collagen at the early time point studied (1 h) more efficiently than the mock-transfected cells. When studying focal contact formation by α2 wild-type and chimeric α2/α1–transfected clones, we noticed that even though cell adhesion to collagen was not influenced by the replacement of the α2 cytoplasmic tail with the α1 tail, the efficiency of the clones to spread on collagen was somewhat different. The cells expressing α2 wild-type receptor spread faster than cells expressing α2/α1 chimeric receptor, so that after 30 min on collagen 61.5 ± 6% of adherent α2-transfected cells had spread, whereas only 43 ± 8.3% of adherent α2/α1–transfected cells had spread (Fig. 3 B). At this early time point, mock-transfected cells expressing only α1β1 had just begun to adhere and virtually no spreading was seen. At later time points (i.e., 2 h and later), also the mock-transfected cells spread efficiently on collagen. Since both α2 wild-type and α2/α1 chimeric receptor were expressed at similar levels on the cell surface (Fig. 1 B), the small difference seen in cell spreading may indicate that the cytoplasmic tail of α2 functions in linking the integrin to the cytoskeleton in a manner promoting spreading on collagen. Therefore, we wanted to study the ability of α2 cytoplasmic tail to mediate cytoskeleton-dependent events and assayed the ability of α2- and α2/α1–transfected cells to contract collagen gels. As expected, vector control cells, having endogenous α1β1 but no α2β1, showed very weak contraction during the 72-h experiment (area of the gel 1.8 ± 0.35-fold reduced), whereas wild-type α2β1-expressing cells contract the gel efficiently (area of the gel 4.0 ± 1.0-fold reduced) (Fig. 3 C). The cytoplasmic tail of α2 seemed to be essential for linkage of the integrin to the cytoskeletal machinery since the cells expressing chimeric α2/α1 failed to contract the collagen gel (Fig. 3 C).
Figure 3.

Cell–collagen interaction of stable transfected human osteosarcoma Saos-2 expressing wild-type α2 integrin, chimeric α2/α1 integrin or empty vector. The data shown are the mean values ± SD of a representative experiment done in triplicate. (A) Cell adhesion to type I collagen was studied. 10,000 cells were suspended in DME (serum-free), then transferred into wells coated with indicated concentrations of type I collagen. After 1 h the nonadherent cells were washed out and the adherent ones were stained with crystal violet. Cell-bound stain was dissolved and measured spectrophotometrically. (B) Cell spreading on type I collagen and fibronectin was studied. 10,000 cells were suspended in DME with 50 μM cycloheximide, and then transferred into wells coated with different matrixes. After 30 min the nonadherent cells were washed out and the adherent ones were fixed. The percentage of spread cells was counted. (C) The ability of various collagen receptor integrins to mediate collagen gel contraction was tested using stable transfected clones of human osteosarcoma Saos-2 cells (mock-, α2-, or α2/α1–expressing clones). 5 × 105 cells were seeded inside a collagen gel, the polymerized gel was detached from the sides of the dish and the cells were cultured for 3 d in DME 10% FCS. The gels were photographed and surface areas of the gels were measured.
Integrin α2 Cytoplasmic Tail Is Required for the Upregulation of Collagen α1(I) and α2(I) Gene Expression at the Transcriptional Level
Overexpression of the wild-type α2 in Saos-2 cells resulted in upregulation of the mRNA levels of both collagen α1(I) (1.4–4.3-fold) and α2(I) (3.3-fold) in response to three-dimensional collagen matrix. To test the generality of this observation, all together three independent single cell clones overexpressing α2β1 were tested (not shown). In contrast, vector control cells showed a downregulation ranging from 0.8 to 2.4-fold for collagen α1(I) mRNA (Fig. 4a and Fig. b). Interestingly, the chimeric receptor failed to mediate upregulation of the collagen mRNAs in response to three-dimensional collagen matrix as seen with the Northern blot hybridization of total RNA isolated from two single cell clones pooled together. The mRNA levels of collagen α1(I) in cells expressing the chimeric receptor were downregulated by 1.5–1.8-fold in response to collagen compared with the average 3.6-fold upregulation seen in cells expressing the α2 wild-type receptor (Fig. 4a and Fig. b). Also, expression of collagen α2(I) was downregulated in the α2/α1–transfected cells. These results were again confirmed to be reproducible with RNA from a third independent single cell clone (not shown). Altogether, cells transfected with the chimera responded to collagen identically to the mock-transfected cells. Integrin α1β1 is known to function as a negative regulator of collagen (Langholz et al. 1995; Riikonen et al. 1995a). The transmembrane region of this receptor has been suggested to signal through interaction with caveolin (Wary et al. 1996), whereas the possible signaling function of α1 cytoplasmic domain cannot be excluded. In the α2/α1–transfected cells, no further reduction of collagen levels was seen when compared with the mock-transfected cells having endogenous α1β1 integrin, suggesting that the short α1 cytoplasmic tail is not alone sufficient to regulate collagen gene expression. To test this further, we transfected Saos-2 cells with the X2C5PFNeo plasmid coding for α2 subunit with α5 cytoplasmic tail (Kawaguchi and Hemler 1993). Again, similarly as both the mock-transfected and the α2/α1–transfected cells, collagen mRNA levels were decreased in response to collagen (not shown).
Figure 4.
Effect of matrix on the expression level of collagen α1(I) and collagen α2(I) mRNAs, transcriptional regulation and mRNA stability of collagen α1(I) in Saos-2 cells expressing either wild-type α2 or chimeric α2/α1 integrin. Mock-, α2-, and α2/α1–transfected cells were cultured in a monolayer (M) or inside collagen gel (C) for 48 h. Total RNA was isolated, separated by electrophoresis, and transferred to filters. Specific mRNA levels were analyzed with corresponding cDNA probes. 28S ribosomal RNA was used as a control. (A) Autoradiograms of a representative experiment and (B) quantitative analysis of an identical separately performed experiment was done by an image analyzer. (C) Nuclear run-on assay was performed with nuclei from Saos-2 cells grown either in monolayer or inside three-dimensional collagen gels 48 h before harvest. Nascent 32P-labeled RNA was hybridized to nitrocellulose filter-immobilized cDNA probe. Quantitative analysis was performed with an image analyzer system. The values for the collagen α1(I) gene transcription were corrected to GAPDH transcription in the same sample. The data shown are mean ± range of a representative experiment done in duplicate. (D) mRNA stability assay using actinomycin. Cells were cultured for 24 h before adding actinomycin D (6.4 μg/ml) either inside collagen gel or in monolayer. Total RNA was isolated from cells after 8 h. Autoradiograms from Northern blot analysis using collagen α1(I) cDNA and 28S cDNA as control are shown. Quantitative analysis of the experiment was done with an image analyzer system.



To assess the contribution of increased transcription of collagen α1(I) on this elevation seen in mRNA levels, we performed nuclear run-on experiments. Nuclei were isolated from α2- or α2/α1–transfected cells grown either in monolayer or in three-dimensional collagen gel for 48 h. The rate of the collagen gene transcription was compared with that of GAPDH. A 1.8-fold increase of transcription was seen in α2-transfected clones cultured in three-dimensional collagen when compared with the rate of transcription in cells grown in monolayer (Fig. 4 C). In contrast, cells transfected with the chimeric α2/α1 chain showed a 1.5-fold decrease in transcription rate (Fig. 4 C). Even though the increased transcription rate of collagen α1(I) gene accounted for most of the upregulation seen in α2β1-overexpressing clones, we also wanted to investigate the stability of the mRNAs. We used actinomycin D to block transcription in cell clones grown in monolayer and in collagen gels. Type I collagen mRNAs seem to have relatively long half-lives (>8 h) in Saos-2 cells transfected with wild-type α2, but no obvious difference was seen between the various culture conditions. Both in monolayer and in three-dimensional matrix collagen, mRNA levels were reduced by ∼50% after 8 h treatment with actinomycin D (Fig. 4 D).
α2β1 Integrin-mediated Upregulation of Collagen α1(I) Expression in Three-Dimensional Collagen Requires p38 MAP Kinase Pathway
To identify the downstream components of α2β1-mediated upregulation of collagen gene expression, we tested specific inhibitors at concentrations sufficient to inhibit various signaling kinases (Alessi et al. 1995; Dudley et al. 1995; Broberg and Heino 1996). The cells were exposed to inhibitors before seeding them inside collagen. Of the various compounds tested, the p38 inhibitor, SB203580, showed the most potent inhibition of collagen α1(I) gene expression (5.5-fold) in α2β1-overexpressing cells grown in three-dimensional collagen gel (Fig. 5 A). The compounds that showed some inhibition (about twofold) included tyrosine kinase inhibitor herbimycin A, PKG inhibitor KT5823, PKA inhibitor KT5720, and Ras farnesylation inhibitor (R)-(+)-Perillyl alcohol (POH), whereas MEK inhibitor PD98059 had no effect. High concentrations of the PKC inhibitor bisindolylmaleimide resulted in downregulation of collagen mRNA levels (twofold at 5 μM and 4.1-fold at 20 μM concentrations), but this effect was seen in both α2- and mock-transfected cells and was therefore considered to be nonspecific. In addition, phosphatidyl-inositol-3-kinase (PI-3K) inhibitor, wortmannin treatment resulted in a slight reduction of collagen α1(I) mRNA in α2-transfected cells (not shown) and this effect was smaller in vector-transfected cells (Fig. 5 A). The small effects of various inhibitors suggest that corresponding signaling proteins might participate in integrin signaling, but this hypothesis was not studied further. Another specific inhibitor targeting the p38 MAP kinase pathway, SKF86002, was tested to confirm the result obtained with SB203580. Treatment with this compound also resulted in concentration-dependent inhibition of α2β1-mediated upregulation of collagen α1(I) mRNA levels (6.1-fold at 10 μM and 9.5-fold at 20 μM). 20 μM SB203580 was a potent inhibitor of collagen mRNA levels in α2-transfected cells inside collagen (Fig. 5 B), but it had no effect on mRNA levels in mock-transfected cells inside collagen (Fig. 5 A) or α2-transfected cells in monolayer (Fig. 5 C), excluding the possibility that the compound could function as a general downregulator of collagen gene expression.
Figure 5.
Effects of different signaling pathway inhibitors on the expression level of collagen α1(I) and collagen α2(I) mRNAs. Cells were cultured in a monolayer or inside collagen gel for 2 d in the presence of inhibitors indicated (20 μM SB203580, 40 μM PD98059, 35 nM herbimycin A, 150 nM KT5723, 100 nM KT5720, 100 μM and 500 μM (R)-(+)-Perillyl alcohol (POH), 10 and 20 μM SKF86002). Total RNA was isolated, separated by electrophoresis, and transferred to filters. Specific mRNA levels were analyzed with corresponding cDNA probes. 28S ribosomal RNA was used as a control. (A) Cells from two separate single cell α2 clones 45 and 47 were pooled together for the study, autoradiograms are shown. (B) The effect of SB203580 on the expression level of collagen α1(I) mRNA was studied with mock-transfected clones 3 and 15, and α2 clones 45 and 47 cultured inside collagen gel for 48 h, quantitative analysis of the expression levels relative to collagen gel control levels are shown. The data are mean of two separate experiments each with a different single cell clone ± range. (C) Autoradiogram showing the effect of SB203580 on the expression level of collagen α1(I) mRNA in α2 clone 47 grown in a monolayer for 48 h.



Three-dimensional Collagen Gel Induce Isoform Specific Activation of p38α in Saos-2 Cells and the Efficient Activation Requires α2 Cytoplasmic Tail
The ability of the selective p38 inhibitors, SB203580 and SKF86002, to inhibit α2 cytoplasmic tail–dependent upregulation of collagen mRNA levels lead us to study how p38 activity is regulated in response to collagenous matrix in these cells. We examined the activation of p38 by using a phosphospecific antibody that recognizes phosphorylated p38α and p38β isoforms. The cells expressing wild-type α2 showed a marked activation (fivefold) already at 2 h after seeding the cells inside collagen. The activation gradually decreased during the next 12 h, but remained at levels threefold higher than the 0 h time point (Fig. 6 A). To study whether this activation was due to signaling via the α2β1, we analyzed the levels of phosphorylated p38 in cells expressing chimeric α2/α1 chain. In three separate experiments using different single cell clones, the wild-type α2-expressing cells showed in average 2.3-fold higher levels of active p38 2 h after seeding the cells inside collagen gel than cells expressing chimeric α2/α1 chain. Protein levels of p38 remained constant at all time points, as shown with the control antibody recognizing both activated and inactivated forms of p38β (Fig. 6 B). At a later (24 h) time point, the p38 activation persisted and α2-transfected cells showed a higher level of activation than the cells expressing chimeric α2/α1 chain (Fig. 6 C). To confirm this difference in the levels of active p38 at the 24-h time point, the experiment was repeated five times using four or five parallel samples and two individual single cell clones of both transfections. In all experiments, p38 was activated in response to three-dimensional collagen and, in cells expressing the wild-type α2 chain, the levels were on average 1.9-fold higher (range, 1.3–3.5-fold). The difference in levels of active p38 between the α2 and α2/α1 cells were found to be statistically significant when the results of all five experiments were combined together (two-way analysis of variance; P < 0.0001). Finally, to confirm that this difference is the result of signals dependent on the α2 cytoplasmic tail in response to collagen, we tested the levels of active p38 in the same cell clones when grown in monolayer. The overall levels of active p38 were relatively low and no significant difference between the clones was detected (Fig. 7 D).
Figure 6.
The regulation and isoform-specific activation of p38 by α2β1 integrin in response to matrix. (A) Cells from two separate α2 single cell clones (45 and 47) were pooled together and seeded in collagen gel and incubated for different periods of time, as indicated. The levels of activated p38α and p38β (p38-P) were determined by Western blot analysis using a phosphospecific antibody for p38α and β, antibody recognizing all forms of p38 was used as a control. The levels of activated p38 were quantitated by using an image analyzer system and are shown relative to the levels of total p38 present in each sample. (B) To test the role of the α2 cytoplasmic tail, the experiment was done with 0 h in the 2-h time points using both α2-transfected clones (45 and 47) and α2/α1–transfected clones (12 and 2). The levels of activated p38 were quantitated by using an image analyzer system and are shown relative to the levels of total p38 present in each sample. The data are mean of three separate experiments with different single cell clones ± SD. Autoradiogram of a representative experiment done in duplicate showing p38-P levels at 2-h time point is shown. (C) Levels of active p38 after 24 h culture of cells inside collagen gel. The data shown are mean ± SD of a representative experiment done in quadruplicate. **, P < 0.001. (D) Activation of p38 in α2 and α2/α1–transfected Saos-2 cells grown on a monolayer. Cells from two separate α2 cell clones (45 and 47) or α2/α1 clones (2 and 12) were pooled together and grown in a monolayer for 24 h. (E) Equal numbers of stable transfected Saos-2 cells were transiently transfected with flag-tagged p38 isoforms (α, β2, γ, or δ) and treated and assayed for p38 kinase activity. The levels of phosphorylated ATF-2 were quantitated using an image analyzer system and are shown as levels of ATF-2 phosphorylation with background correction (signal from control transfected cells subtracted). α2 clone 45 and α2/α1 clone 12 were transfected with either the control vector or the p38 isoforms indicated (F) α2 clones (45 and 47) and α2/α1 clones (2 and 12) were transfected with control vector, p38α, or p38β2. Expression of the two flag-tagged isoforms was checked with Western blot analysis using flag-tag recognizing antibody M2.



Figure 7.

The effect of p38 inhibitors SB203580 and SKF86002 on the various p38 isoforms. Equal numbers of stable transfected Saos-2 cells were transiently transfected with flag-tagged p38 isoforms (α, β2, γ, or δ). 36 h after transfection, the cells were detached from the plate and seeded inside collagen gel. After 3 h of incubation, the cells were collected and lysed, and flag-tagged p38 was immunoprecipitated. Before adding the ATF-2 protein as a substrate to the kinase reaction the inhibitors or DMSO (control) were added. Levels of phosphorylated ATF-2 are shown.
To date, the p38 MAP kinase group is known to include five isoforms: p38α (Lee et al. 1994), p38β (Jiang et al. 1996), p38β2 (Enslen et al. 1998), p38γ (Cuenda et al. 1997), and p38δ (Jiang et al. 1997; X.S. Wang et al. 1997). To study whether α2β1 cytoplasmic tail could specifically activate some isoform of p38, we overexpressed various forms of flag-tagged p38 kinases (New et al. 1998) in Saos cell stable clones expressing either wild-type α2 or chimeric α2/α1. The transfected cells were seeded inside a collagen gel, after 3 h the cells were collected, flag-tagged p38 was immunoprecipitated, and an in vitro kinase assay was performed. As seen in Fig. 6 E, p38α isoform was activated efficiently in α2-transfected cells (activity 0.47 units; arbitrary units = densitometric units − background), whereas in α2/α1–transfected cells the activity was 0.03. No activation of p38β2 was detected (α2 clone 45 = 0.04 and α2α1 clone 12 = 0.02). p38γ showed high activity in both cell clones (α2 clone 45 = 0.24 and α2/α1 clone 12 = 0.32) and p38δ activity was high in both clones (α2 clone 45 = 0.64 and α2/α1 clone 12 = 0.23). The results were confirmed with two individual α2- or α2/α1–expressing clones (Fig. 6 F). The expression levels of the transiently transfected kinases in various clones were equal as shown by Western blot analysis done by using antibody against the flag-tag.
The Effect of p38 Inhibitors SB203580 and SKF86002 on the Various p38 Isoforms Expressed in α2-Transfected Saos-2 Cells
As seen in Fig. 6E and Fig. F, p38α was activated only in α2-transfected cells and no activation of p38β2 was seen. However, equally high activity of p38γ was seen in both α2- and α2/α1–transfected cells and the p38δ activity was higher in α2 cells than in α2/α1 cells. To study which of these activated kinases are relevant to the α2-mediated upregulation of collagen, shown to be inhibited by chemical inhibitors (SB203580 and SKF86002), we tested the effect of these inhibitors in an in vitro kinase assay. α2-transfected Saos-2 cells were transiently transfected with flag-tagged p38 isoforms (α, β2, γ, or δ), the cells were treated with collagen and the kinase activity of each isoform was measured in the presence or absence of the inhibitory compound. In accordance with previously published experimental data and recent structural evidence (Kumar et al. 1997; Whitmarsh et al. 1997; Lisnock et al. 1998) the inhibitor SB203580 had no effect on the γ and δ isoforms of p38 (Fig. 7). Previously, SKF86002 has been shown to inhibit p38α (Lee et al. 1994) and we show here that it has no effect on the γ and δ isoforms. From these results we can conclude that it is the p38α isoform that is essential in the α2β1 integrin–dependent upregulation of collagen.
Collagen Gel Induces Transient Activation of ERK1 and 2 but not JNK/SAPK in Both the α2- and α2/α1–Transfected Cells
We have recently shown that seeding dermal fibroblasts inside three-dimensional collagen gels results in activation of ERK1 and 2, JNK/SAPK, and p38 MAPKs (Ravanti et al. 1999). Therefore, we examined also the activation of ERK1 and 2 and JNK/SAPK in both α2- and α2/α1–transfected Saos-2 cells by Western blot analysis of cellular proteins, using phosphospecific antibodies to detect activated forms of these MAP kinases. The levels of activated ERK2 were increased 2 h after seeding the cells inside collagen (3-fold in α2 and 1.5-fold in α2/α1 cells) and they increased further (up to 4-fold in α2 and 7-fold in α2/α1 cells) at 6 h time point. This activation in response to the collagen gel was transient in both clones since no phosphorylated ERK1 or 2 was detected at 12-h time point. Protein levels of ERK2 remained constant at all time points, as shown with the antibody recognizing all forms of ERK2 (Fig. 8 A). Low levels of ERK1 were also detected at 2-, 4-, and 6-h time points. No induction in the levels of phosphorylated JNK/SAPK was seen in these cells in response to three-dimensional collagen. Some activated JNK/SAPK was seen at 0-h time point, immediately after trypsinization but no active protein was detected inside collagen gel. Treatment with anisomycin was used as a positive control for JNK activation. Protein levels of JNK1 remained constant at all time points, as shown with the antibody recognizing all forms of JNK1 (Fig. 8 B). The results with the phosphospecific antibodies were confirmed with a JNK in vitro kinase assay. Endogenous kinase was immunoprecipitated with anti–JNK1 antibody recognizing also JNK2 and 3 and recombinant c-Jun protein was used as a substrate (Fig. 8 C).
Figure 8.

Regulation of ERK and JNK MAPKs in response to collagen gel. Cells from two separate α2 or α2/α1 single cell clones (45, 47 and 2, 12) were pooled together and either lysed immediately (0 h sample) or seeded in collagen gel and incubated for different periods of time, as indicated. (A) The levels of activated ERK1 and 2 (ERK1-P and ERK2-P) were determined by Western blot analysis using a phosphospecific antibody for ERK1 and 2 and an antibody recognizing all forms of ERK2 was used as a control. The levels of activated ERK1 and 2 were quantitated using an image analyzer system and are shown relative to the levels of total ERK2 present in each sample. (B) The levels of activated JNK1 and 2 were determined by Western blot analysis using a phosphospecific antibody for JNK1 and 2, and an antibody recognizing all forms of JNK1 was used as a control. The positive control treatment (+) was 10 min anisomycin (10 μg/ml). For the kinase assay, equal numbers of stable transfected Saos-2 cells (α2 or α2/α1) were either lysed immediately after trypsinization, treated with collagen for 3 h or anisomycin (10 μg/ml) for 10 min, and the JNK kinase activity was assayed.
Three-dimensional Collagenous Matrix Fails to Activate FAK
Ligation of integrins leads to activation of FAK. To check whether FAK would play a role in the activation of p38 in response to three-dimensional collagen, we allowed both α2- and α2/α1–transfected cells to interact with collagen gels for 1, 2, or 3 h. (Fig. 9 or not shown). The polymerization of the collagen gel takes place in 1 h, so shorter time points could not be studied. No phosphorylated FAK was detected in cells treated with collagen, in contrast to cells from both clones adhering to fibronectin (Fig. 9). Some phosphorylation of FAK was seen in cells lyzed immediately after trypsinization. The experiment was repeated with similar results by using antibodies recognizing all phosphorylated forms of FAK (Fig. 9). In the lower panel of Fig. 9 phosphorylated FAK is the upper band. The lower band seen in all samples could not be recognized by anti–FAK antibody and its identity is unknown.
Figure 9.

Three-dimensional collagenous matrix fails to activate FAK in Saos-2 cells. Cells from two separate α2 or α2/α1 single cell clones (45, 47 and 2, 12) were pooled together and either lysed immediately (0 h sample), seeded in collagen gel, or allowed to adhere to fibronectin for 1 h. FAK was immunoprecipitated and immunoplotted with an antiphosphotyrosine antibody (4G10). Alternatively, cell lysates were fractionated on SDS-PAGE and immunoblotted with phosphospecific FAK antibody. The blots were reprobed with anti–FAK antibody to show the levels of FAK protein. Note that in the lower panel the anti-phospho FAK antibody recognizes two bands, but it is the upper one that comigrates with the band recognized with the anti–FAK antibody. The origin of the lower band is unknown.
Effect of Inhibitors of GTPases and MAPKKs on α2β1-mediated Activation of p38α
To investigate possible downstream effectors of α2β1 integrin in the activation of p38α, we used dominant negative mutants of the Rho family GTPases and the p38 upstream kinases, MKK3 and MKK4. The effector mutants or an empty vector in control cells were cotransfected with the flag-tagged p38α into the α2-expressing cells and, 36 h after transfection, the cells were exposed to three-dimensional collagen and an in vitro p38 kinase assay was performed. Of the Rho family GTPases Cdc42 seemed essential for α2β1-mediated signaling since dominant negative Cdc42 constantly resulted in an inhibition of p38α activity (Fig. 10 B). Similar inhibition was not seen when the cells were transfected with wild-type Cdc42, used as a control. In three separate experiments, 4 μg/plate dominant negative Rac slightly decreased p38α activity (75 ± 12% of control) and mutant RhoA was only somewhat more effective (70 ± 27% of control). The experiment was also repeated with higher plasmid concentration (8 μg/plate) and p38α activity was unaltered in dominant negative Rac transfected cells (116% of control). Mutant RhoA showed some inhibition (66% of control). Again, dominant negative Cdc42 was the most efficient (17% of control). The dominant negative forms of the MAPK kinases known to function upstream of p38, namely MKK3 and MKK4, both had an inhibitory effect. Dominant negative MKK3 inhibited p38α activity by 90–91% and dominant negative MKK4 by 76–90% (Fig. 10). These results indicate that the activity of Cdc42 and the MAPK kinases MKK3 and MKK4 are necessary for the α2β1 integrin-mediated p38α activation.
Figure 10.
A possible role for RhoA, Cdc42, MKK3, and MKK4 in integrin α2β1-mediated activation of p38α. Equal numbers of stable α2-transfected Saos-2 cells were transiently cotransfected with flag-tagged p38α (2 μg) and the dominant negative form of the protein indicated (4 μg). The cells were treated and assayed for p38 kinase activity. The possible effect of the cotransfected plasmid on p38α-flag expression was checked with Western blot analysis of the cell lysate using flag-tag recognizing antibody M2. An autoradiogram of a representative experiment is shown. (B) Various concentrations of dominant negative (DN) Cdc42 were tested in cotransfections with 2 μg of p38α-flag. Each experiment was repeated 2 to 4 times and mean values ± SEM are shown. Transfection of wild-type (wt) Cdc42 was used as a control.


Discussion
The integrins provide a physical linkage between the cytoskeleton and ECM and they transduce signals initiated by extracellular interactions. Since integrins have no intrinsic kinase activity they need to recruit other proteins, i.e., kinases to trigger signaling (for reviews see Clark and Brugge 1995; Yamada and Miyamoto 1995). A rapidly increasing number of molecules has been shown to interact with integrins (for review see Hemler 1998). One obvious site for interaction is the cytoplasmic domain of an integrin subunit and several proteins binding specifically to either the α or the β cytoplasmic tail have been identified. Interactions mediated by the membrane spanning region of integrins have also been shown (Lindberg et al. 1993; Berditchevski et al. 1995, Berditchevski et al. 1996; Wary et al. 1996). These interactions may be used differentially by different integrins establishing the bases for receptor-specific signal transduction.
Integrin α subunit–specific interactions with other cellular proteins are of special interest because they may explain the distinct signaling functions of integrin heterodimers sharing a common β1 subunit (Werb et al. 1989). The two collagen receptors, α1β1 and α2β1 integrins, have distinct effects on cellular signaling and gene expression (Langholz et al. 1995; Riikonen et al. 1995a; Ravanti et al. 1999). Here, the function of their α cytoplasmic domains was studied by swapping the α1 tail into α2 and expressing the chimeric integrin in cells negative for α2 integrin.
The swapping of the α1 and α2 subunit cytoplasmic domains did not affect the localization to focal adhesions or the ability to mediate cell adhesion to collagen. This is in agreement with previously published data where the specific sequence of the α tail seemed less important than the number of residues present. Four to seven residues after the conserved GFFKR sequence were needed to be included for optimal adhesive activity (Kassner et al. 1994). Here, fast cell spreading and the ability to contract three-dimensional collagen gels were impaired if the cells expressed chimeric α2/α1 instead of the α2 integrin. Previously, α2, α4, and α5 cytoplasmic tails have been shown to be interchangeable with respect to their positive contributions towards cell adhesion (Kawaguchi and Hemler 1993), while the ability to mediate collagen gel contraction and cell migration is more restricted to a specific set of α cytoplasmic tails (Chan et al. 1992). Collagen gel contraction assays are used to study cell–type I collagen interaction. It is generally thought that the reorganization of collagen fibrils, seen as contraction of floating collagen gels, is dependent on the expression of α2β1 on the cell surface (Klein et al. 1991; Schiro et al. 1991; Riikonen et al. 1995b). However, there are more recent reports indicating that α1β1 can have an essential role in the contraction process, especially in smooth muscle cells and liver myofibroblasts (Gotwals et al. 1996; Racine-Samson et al. 1997), suggesting that α1 cytoplasmic tail, unlike the ones of α4 and α5, could mediate the same interactions as the α2 tail. In contrast to these reports, our results do not support the idea that α1β1 or chimeric α2/α1β1 could mediate collagen gel contraction. This is also the case with α1- and α2/α1–transfected CHO cells (Ivaska, J., and J. Heino, unpublished results). One explanation could be that α1β1 needs an interaction with a certain cytoplasmic structure to be able to mediate contraction, and that this component is only present in a subset of cell types.
Synergy between integrin-mediated signaling and signals initiated by growth factors has been well established (Schlaepfer et al. 1994; Miyamoto et al. 1995, Miyamoto et al. 1996). Pathways first identified to be activated by mitogens, the MAP kinase cascades, have now been shown to be activated by integrin ligation (Chen et al. 1994; Morino et al. 1995; MacKenna et al. 1998). Furthermore, integrin specificity in MAP kinase activation has been established. A subset of integrins, namely α1β1, α5β1, α6β4, and αVβ3, has been shown to activate the Ras-ERK pathway via recruitment of the adaptor protein Shc (Wary et al. 1996; Giancotti 1997). Other integrins (α2β1, α3β1, and α6β1) were unable to induce ERK activation in these studies (Wary et al. 1996).
Given the difference between α1β1 and α2β1 integrins in activation of Ras-ERK pathway it is not surprising that ligation of these two receptors have different effects on cellular gene expression. Both α1β1 and α2β1 have been shown to regulate collagen mRNA levels in response to contact with three-dimensional collagen. Integrin α1β1 can function as a negative regulator of collagen synthesis (Langholz et al. 1995; Riikonen et al. 1995a; Gardner et al. 1999; Ravanti et al. 1999), whereas overexpression of α2β1 in osteosarcoma cells results in an upregulation of collagen mRNA levels (Riikonen et al. 1995a). Integrin α2β1 seems to be a positive regulator of MMP-1 and MMP-13 (collagenase-3) expression as well. Further, supporting the differential signaling by these two receptors, α1β1 is not involved in MMP-1 upregulation and it seems to be a less potent upregulator of MMP-13 than α2β1 (Ravanti et al. 1999). Activation of Ras-Raf-ERK pathway by α1β1 integrin may lead to reduced collagen synthesis since Ras-Raf activation has been shown to downregulate type I collagen gene expression (Davis et al. 1996). Signaling through α2β1 is less well characterized than α1β1 signaling. MMP-1 upregulation in fibroblasts cultured inside three-dimensional collagen is mediated by PKC-ζ and NFκB, but these pathways have not been directly connected to α2β1 (Xu and Clark 1997). Three-dimensional collagen mediates PKC-ζ activation in 4 h in CHO cells even though they lack collagen receptor integrins (Ivaska, J., and J. Heino, unpublished results) proposing that the PKC-ζ activation might at least partly be due to a change in cell shape rather than active signaling through collagen binding integrins.
Previously, it has been shown that cell contact with either two- or three-dimensional collagen induces activation of ERK1 and ERK2 (Roeckel and Krieg 1994; Langholz et al. 1997). We have recently shown the participation of p38 MAPK pathway in induction of MMP-13 expression in response to cell–collagen interaction (Ravanti et al. 1999). Here, we provide several lines of evidence that integrin α2β1 regulates collagen gene transcription by activating p38α in response to collagen, and that this signaling requires the α2 cytoplasmic domain. First, expression of wild-type α2 chain in cells exposed to collagen leads to upregulation of collagen mRNA levels. Chimeric receptor in which the cytoplasmic domain of α2 is replaced with the corresponding sequence in α1, is not able to upregulate collagen synthesis in response to three-dimensional matrix. Second, regulation of collagen gene transcription was shown to require p38 MAPK activity based on the use of two p38 kinase inhibitors, SB203580 and SKF86002. Third, contact with three-dimensional collagen results in only a transient activation of ERK1 and 2, no evident activation of SAPK/JNK, but persistent activation of p38 kinase. Activation of p38 MAPK requires intact α2 subunit since levels of activated p38 were significantly lower in cells expressing chimeric α2/α1 receptor. Finally, using transient transfections of flag-tagged isoforms of p38 and dominant negative signaling proteins, we were able to show that α2 cytoplasmic domain specifically activates the α isoform and has no effect on the p38β2 isoform. We also show that the activity of Cdc42 and the MAPK kinases MKK3 and MKK4 is necessary for the α2β1 integrin-mediated p38α activation.
The data presented clearly show that the p38α isoform is essential in the α2β1 integrin–dependent upregulation of collagen expression. The facts to support this areas follows. First, the p38α isoform is activated in response to collagen in the α2-transfected but not the α2/α1–transfected cells. Second, the p38β2 isoform is not activated in these cells. Third, even though p38γ showed high activity in both clones and p38δ was more efficiently activated in the α2-transfected cells, these isoforms cannot be responsible for the upregulation of collagen gene expression since the p38 inhibitors used have no effect on these kinases (Fig. 7). Together, these findings demonstrate that the cytoplasmic sequence of α2 integrin subunit regulates the ability of α2β1 integrin to activate p38 kinase in an isoform-specific manner and suggest a novel signaling mechanism for α2β1.
An issue that arises from the data is the mechanism by which α2β1 integrin activates the p38 pathway. Integrins are known to activate the Rho family of GTPases. Integrin ligation to the ECM leads to the activation of Cdc42 that subsequently activates Rac; Rho, on the other hand, has been shown to be activated independently of Cdc42 by integrin ligation (Price et al. 1998). Of the GTPases Cdc42 has been shown to activate both the p38 and JNK pathways and Rac1 has been shown to activate p38 (Bagrodia et al. 1995; Coso et al. 1995; Minden et al. 1995; S. Zhang et al. 1995). Cdc42 and Rac1 have been shown to induce integrin-mediated cell motility on collagen and invasiveness through collagen gels (Keely et al. 1997), suggesting a role for these small GTPases in the inside-out signaling regulating the function of the collagen binding integrins. Here we identify Cdc42 to be important for α2β1-mediated signaling and also show that the MAPK kinases MKK3 and MKK4 may be involved.
The signaling molecules downstream of Cdc42 and upstream of the MAPK kinases remain to be clarified in further studies. The various p38 isoforms seem to be differentially activated by upstream MAPKKs: MKK3, MKK4, and MKK6 (Cohen et al. 1997; Jiang et al. 1997), which in turn are activated by MAPKKKs (Denhardt 1996). MAPKKKs shown to be effective in the p38 pathway involve ASK and TAK1 (transforming growth factor–activated kinase) (Derijard et al. 1995; Yamaguchi et al. 1995; W. Wang et al. 1997) but little is known about their upstream effectors. PI-3K has been shown to function in integrin-mediated cell migration and invasion and it may function upstream of Rac1 and Cdc42 (Keely et al. 1997). In fact, a lipid product of PI-3K has been shown to interact with Rac1 (Missy et al. 1998). A recent study shows that Cdc42 controls integrin-dependent activation of Akt (Clark et al. 1998), a kinase whose activity is regulated by PI-3K (Bondeva et al. 1998). Very recently a mechanism was suggested in which PI-3K activates NFκB (Beraud et al. 1999), a transcription factor that has been shown to be activated in fibroblasts in response to collagen gel (Xu and Clark 1997). In our experiments, the PI-3K inhibitor, wortmannin, slightly reduced α2β1-mediated upregulation of collagen α1(I) thereby leaving open the possibility that PI-3K may be one of the upstream effectors of the signaling pathway described here.
Other candidates for upstream effectors that could mediate α2β1-related activation of p38 include p21 activated kinases (PAKs), the best characterized effectors of Cdc42 and ACKs (activated Cdc42-binding kinases) recently shown to be activated by cell adhesion via integrin β1 (Yang et al. 1999). In a study published by Bourdoulous et al. 1998, alteration in integrin occupancy by fibronectin led to upregulation of ACK and p38 MAPK while a concomitant inhibition of PAK and JNK/SAPK was seen. This is interesting from the point of view of this discussion since similar effects on the MAPKs were seen in the α2-transfected Saos cells in response to collagen. In addition, TAK1 might be an interesting candidate, since it is activated by TGF-β and has been shown to activate both MKK3 and MKK6 (Yamaguchi et al. 1995; Enslen et al. 1998). On the other hand, TGF-β has many effects on the cell, one of them being the upregulation of collagen levels (Massagué 1990). How is integrin signaling then transduced to these molecules, especially to Cdc42? One attractive model for α2β1 signaling would be the phosphorylation of the α2 cytoplasmic domain. Phosphorylation in response to integrin ligation could generate a binding site for an effector kinase inside the cell. However, our preliminary data have failed to convincingly show α2 phosphorylation in response to binding to collagen (Ivaska, J., and J. Heino, unpublished results).
It is evident that the two collagen receptors studied here function in close collaboration to regulate cell behavior in response to collagenous matrix. Impaired regulation of collagen turnover may lead to pathological conditions. For example, skin fibroblasts from scleroderma patients show upregulated collagen synthesis and concomitantly reduced expression of α1β1 (Ivarsson et al. 1993). A striking example of correlated functions of integrins α1β1 and α2β1 is seen in α1 null mice, in which the absence of α1β1 leads to enhanced collagen synthesis in skin. However, simultaneously collagenase-3 expression is increased, possibly via increased α2β1 ligation, leading to a situation in which the collagen accumulation is seen only if the degradation of collagen is prevented (Gardner et al. 1999). Previous studies have shown the regulation of specific MAPK pathways by α1β1 integrin (Wary et al. 1996) and also introduced the dual role of MAPK pathways in the regulation of collagen production (Davis et al. 1996). Here, we provide new information which connects α2β1 integrin to the regulation of p38α via Cdc42 and MAPKKs, MKK3 and MKK4, and show how α2β1 functions in the regulation of the delicate balance of collagen accumulation in tissues.
Acknowledgments
We wish to thank Drs. E. Vuorio (University of Turku, Finland), R. Penttinen, M. Hemler, J.C. Lacal, A. Weiss, E. Marcantonio, R. Davis, P. Fort, and J. Han for cDNAs and vectors used in this study, and Drs. W. Rettig, W. Woods, and E. Coffey for the antibodies and c-Jun protein used in this study. The technical assistance by M. Potila, T. Heikkilä, and U. Paasio is gratefully acknowledged. We also wish to thank Dr. J. Eriksson and I. Elo for their valuable help with the p38 kinase assays and J. Hakalax for statistical evaluation of the results.
J. Ivaska has a fellowship from the Turku Graduate School in Biomedical Science. This study was financially supported by grants from the Pharmacal Research Foundation, the Academy of Finland, the Sigrid Jusélius Foundation, the Finnish Cancer Association, the Turku University Central Hospital, and the Technology Development Centre in Finland.
Footnotes
1.used in this paper: ECM, extracellular matrix; ERK, extracellular signal–related kinase; FAK, focal adhesion kinase; JNK, c-jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MAPKK, MAPK kinase; PI-3K, phosphatidylinositol-3-kinase; PK, protein kinase
References
- Alessi D.R., Cuenda A., Cohen P., Dudley D.T., Saltiel A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Bagrodia S., Derijard B., Davis R.J., Cerione R.A. Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 1995;270:27995–27998. doi: 10.1074/jbc.270.47.27995. [DOI] [PubMed] [Google Scholar]
- Banerji S.S., Theodorakis N.G., Morimoto R.I. Heat shock-induced translational control of HSP70 and globin synthesis in chicken reticulocytes. Mol. Cell Biol. 1984;4:2437–2448. doi: 10.1128/mcb.4.11.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud C., Henzel W.J., Baeuerle P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl. Soc. Sci. USA. 1999;96:429–434. doi: 10.1073/pnas.96.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F., Bazzoni G., Hemler M.E. Specific association of CD63 with the VLA-3 and VLA-6 integrins. J. Biol. Chem. 1995;270:17784–17790. doi: 10.1074/jbc.270.30.17784. [DOI] [PubMed] [Google Scholar]
- Berditchevski F., Zutter M.M., Hemler M.E. Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins) Mol. Biol. Cell. 1996;7:193–207. doi: 10.1091/mbc.7.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeva T., Pirola L., Bulgarelli-Leva G., Rubio I., Wetzker R., Wymann M.P. Bifurcation of lipid and protein kinase signals of PI3Kγ to the protein kinases PKB and MAPK. Science. 1998;282:293–296. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- Bourdoulous S., Orend G., MacKenna D.A., Pasqualini R., Ruoslahti E. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J. Cell Biol. 1998;143:267–276. doi: 10.1083/jcb.143.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briesewitz R., Epstein M.R., Marcantonio E.E. Expression of native and truncated forms of the human integrin α1 subunit. J. Biol. Chem. 1993;268:2989–2996. [PubMed] [Google Scholar]
- Broberg A., Heino J. Integrin α2β1-dependent contraction of floating collagen gels and induction of collagenase are inhibited by tyrosine kinase inhibitors. Exp. Cell Res. 1996;228:29–35. doi: 10.1006/excr.1996.0295. [DOI] [PubMed] [Google Scholar]
- Camper L., Hellman U., Lundgren-Åkerlund E. Isolation, cloning, and sequence analysis of the integrin subunit α10, a β1-associated collagen binding integrin expressed on chondrocytes. J. Biol. Chem. 1998;273:20383–20389. doi: 10.1074/jbc.273.32.20383. [DOI] [PubMed] [Google Scholar]
- Chan B.M., Kassner P.D., Schiro J.A., Byers H.R., Kupper T.S., Hemler M.E. Distinct cellular functions mediated by different VLA integrin alpha subunit cytoplasmic domains. Cell. 1992;68:1051–1060. doi: 10.1016/0092-8674(92)90077-p. [DOI] [PubMed] [Google Scholar]
- Chen Q., Kinch M.S., Lin T.H., Burridge K., Juliano R.L. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J. Biol. Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Clark E.A., Brugge J.S. Integrins and signal transduction pathwaysthe road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Clark E.A., King W.G., Brugge J.S., Symons M., Hynes R.O. Integrin-mediated signals regulated by members of the Rho family of GTPases. J. Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.S., Schmidtmayerova H., Dennis J., Dubrovsky L., Sherry B., Wang H., Bukrinsky M., Tracey K.J. The critical role of p38 MAP kinase in T cell HIV-1 replication. Mol. Med. 1997;3:339–346. [PMC free article] [PubMed] [Google Scholar]
- Coso O.A., Chiariello M., Yu J.C., Teramoto H., Crespo P., Xu N., Miki T., Gutkind J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Cuenda A., Goedert M., Craxton M., Jakes R., Cohen P. Activation of the novel MAP kinase homologue SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6) Biochem. Soc. Trans. 1997;25:S569. doi: 10.1042/bst025s569. [DOI] [PubMed] [Google Scholar]
- Davis B.H., Chen A., Beno D.W. Raf and mitogen-activated protein kinase regulate stellate cell collagen gene expression. J. Biol. Chem. 1996;271:11039–11042. doi: 10.1074/jbc.271.19.11039. [DOI] [PubMed] [Google Scholar]
- Denhardt D.T. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cellthe potential for multiplex signalling. Biochem. J. 1996;318:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B., Raingeaud J., Barrett T., Wu I.H., Han J., Ulevitch R.J., Davis R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- DiPersio C.M., Shah S., Hynes R.O. α3Aβ1 integrin localizes to focal contacts in response to diverse extracellular matrix proteins. J. Cell Sci. 1995;108:2321–2336. doi: 10.1242/jcs.108.6.2321. [DOI] [PubMed] [Google Scholar]
- Dudley D.T., Pang L., Decker S.J., Bridges A.J., Saltiel A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Soc. Sci. USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenius K., Paul S., Allison G., Sun J., Klagsbrun M. Activation of HER4 by heparin-binding EGF-like growth factor stimulates chemotaxis but not proliferation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1268–1278. doi: 10.1093/emboj/16.6.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H., Raingeaud J., Davis R.J. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Fort P., Marty L., Piechaczyc M., el Sabrouty S., Dani C., Jeanteur P., Blanchard J.M. Various rat adult tissues express only one major mRNA species from glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner H., Broberg A., Pozzi A., Laato M., Heino J. Absence of integrin α1β1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J. Cell Sci. 1999;112:263–272. doi: 10.1242/jcs.112.3.263. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G. Integrin signalingspecificity and control of cell survival and cell cycle progression. Curr. Opin. Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- Gotwals P.J., Chi Rosso G., Lindner V., Yang J., Ling L., Fawell S.E., Koteliansky V.E. The α1β1 integrin is expressed during neointima formation in rat arteries and mediates collagen matrix reorganization. J. Clin. Invest. 1996;97:2469–2477. doi: 10.1172/JCI118693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 1994;124:401–404. doi: 10.1083/jcb.124.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Campbell D., Derijard B., Davis R.J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Hemler M.E. Integrin associated proteins. Curr. Opin. Cell Biol. 1998;10:578–585. doi: 10.1016/s0955-0674(98)80032-x. [DOI] [PubMed] [Google Scholar]
- Horwitz A., Duggan K., Buck C., Beckerle M., Burridge K. Interaction of plasma membrane fibronectin receptor with talin, a transmembrane linkage. Nature. 1986;320:531–533. doi: 10.1038/320531a0. [DOI] [PubMed] [Google Scholar]
- Howe A., Aplin A.E., Alahari S.K., Juliano R.L. Integrin signaling and cell growth control. Curr. Opin. Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversality, modulation, and signalling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ivarsson M., McWhirter A., Black C.M., Rubin K. Impaired regulation of collagen pro α1(I) mRNA and change in pattern of collagen-binding integrins on scleroderma fibroblasts. J. Invest. Dermatol. 1993;101:216–221. doi: 10.1111/1523-1747.ep12364810. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Chen C., Li Z., Guo W., Gegner J.A., Lin S., Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J. Biol. Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Gram H., Zhao M., New L., Gu J., Feng L., Di Padova F., Ulevitch R.J., Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J. Biol. Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- Kassner P.D., Hemler M.E. Interchangeable α chain cytoplasmic domains play a positive role in control of cell adhesion mediated by VLA-4, a β1 integrin. J. Exp. Med. 1993;178:649–660. doi: 10.1084/jem.178.2.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner P.D., Kawaguchi S., Hemler M.E. Minimum α chain cytoplasmic tail sequence needed to support integrin-mediated adhesion. J. Biol. Chem. 1994;269:19859–19867. [PubMed] [Google Scholar]
- Kawaguchi S., Hemler M.E. Role of the α subunit cytoplasmic domain in regulation of adhesive activity mediated by the integrin VLA-2. J. Biol. Chem. 1993;268:16279–16285. [PubMed] [Google Scholar]
- Kawaguchi S., Bergelson J.M., Finberg R.W., Hemler M.E. Integrin alpha 2 cytoplasmic domain deletion effectsloss of adhesive activity parallels ligand-independent recruitment into focal adhesions. Mol. Biol. Cell. 1994;5:977–988. doi: 10.1091/mbc.5.9.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Klein C.E., Dressel D., Steinmayer T., Mauch C., Eckes B., Krieg T., Bankert R.B., Weber L. Integrin α2β1 is upregulated in fibroblasts and highly aggressive melanoma cells in three-dimensional collagen lattices and mediates the reorganization of collagen I fibrils. J. Cell Biol. 1991;115:1427–1436. doi: 10.1083/jcb.115.5.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., McDonnell P.C., Gum R.J., Hand A.T., Lee J.C., Young P.R. Novel homologues of CSBP/p38 MAP kinaseactivation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- Laflamme S.E., Homan S.M., Bodeau A.L., Mastrangelo A.M. Integrin cytoplasmic domains as connectors to the cell's signal transduction apparatus. Matrix Biol. 1997;4:153–163. doi: 10.1016/s0945-053x(97)90003-2. [DOI] [PubMed] [Google Scholar]
- Langholz O., Rockel D., Mauch C., Kozlowska E., Bank I., Krieg T., Eckes B. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by α1β1 and α2β1 integrins. J. Cell Biol. 1995;131:1903–1915. doi: 10.1083/jcb.131.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langholz O., Roeckel D., Petersohn D., Broermann E., Eckes B., Krieg T. Cell-matrix interactions induce tyrosine phosphorylation of MAP kinases ERK1 and ERK2 and PLCγ in two-dimensional and three-dimensional cultures of human fibroblasts. Exp. Cell Res. 1997;235:22–27. doi: 10.1006/excr.1997.3640. [DOI] [PubMed] [Google Scholar]
- Lee J.C., Laydon J.T., McDonnell P.C., Gallagher T.F., Kumar S., Green D., McNulty D., Blumenthal M.J., Heys J.R., Landvatter S.W. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Lindberg F.P., Gresham H.D., Schwarz E., Brown E.J. Molecular cloning of integrin-associated proteinan immunoglobulin family member with multiple membrane-spanning domains implicated in α5β3-dependent ligand binding. J. Cell Biol. 1993;123:485–496. doi: 10.1083/jcb.123.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisnock J., Tebben A., Frantz B., O'Neill E.A., Croft G., O'Keefe S.J., Li B., Hacker C., de Laszlo S., Smith A. Molecular basis for p38 protein kinase inhibitor specificity. Biochemistry. 1998;37:16573–16581. doi: 10.1021/bi981591x. [DOI] [PubMed] [Google Scholar]
- MacKenna D.A., Dolfi F., Vuori K., Ruoslahti E. Extracellular signal-regulated kinase and c-jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 1998;101:301–310. doi: 10.1172/JCI1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainiero F., Murgia C., Wary K.K., Curatola A.M., Pepe A., Blumemberg M., Westwick J.K., Der C.J., Giancotti F.G. The coupling of α6β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä J.K., Raassina M., Virta A., Vuorio E. Human pro α1(I) collagencDNA sequence for the C-propeptide domain. Nucleic Acids Res. 1988;16:349. doi: 10.1093/nar/16.1.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä J.K., Vuorio T., Vuorio E. Growth-dependent modulation of type I collagen production and mRNA levels in cultured human skin fibroblasts. Biochim. Biophys. Acta. 1990;1049:171–176. doi: 10.1016/0167-4781(90)90037-3. [DOI] [PubMed] [Google Scholar]
- Massagué J. The transforming growth factor-β family. Annu. Rev. Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- Minden A., Lin A., Claret F.X., Abo A., Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Missy K., Van Poucke V., Raynal P., Viala C., Mauco G., Plantavid M., Chap H., Payrastre B. Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J. Biol. Chem. 1998;273:30279–30286. doi: 10.1074/jbc.273.46.30279. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Akiyama S.K., Yamada K.M. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Gutkind J.S., Yamada K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activationroles of integrin aggregation and occupancy of receptors. J. Cell. Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino N., Mimura T., Hamasaki K., Tobe K., Ueki K., Kikuchi K., Takehara K., Kadowaki T., Yazaki Y., Nojima Y. Matrix/integrin interaction activates the mitogen-activated protein kinases, p44erk-1 and p42erk-2. J. Biol. Chem. 1995;270:269–273. doi: 10.1074/jbc.270.1.269. [DOI] [PubMed] [Google Scholar]
- New L., Jiang Y., Zhao M., Liu K., Zhu W., Flood L.J., Kato Y., Parry G.C., Han J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3372–3384. doi: 10.1093/emboj/17.12.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi P.S., Mak T.W., Van den Elsen P., Yanagi Y., Yoshikai Y., Calman A.F., Terhorst C., Stobo J.D., Weiss A. Reconstitution of an active surface T3/T-cell antigen receptor by DNA transfer. Nature. 1985;316:606–609. doi: 10.1038/316606a0. [DOI] [PubMed] [Google Scholar]
- Otey C.A., Pavalko F.M., Burridge K. An interaction between α-actinin and the β1 integrin subunit in vitro. J. Cell Biol. 1990;111:721–729. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pischel K.D., Hemler M.E., Huang C., Bluestein H.G., Woods V.L., Jr. Use of the monoclonal antibody 12F1 to characterize the differentiation antigen VLA-2. J. Immunol. 1987;138:226–233. [PubMed] [Google Scholar]
- Pozzi A., Wary K.K., Giancotti F.G., Gardner H.A. Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J. Cell Biol. 1998;142:587–594. doi: 10.1083/jcb.142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price L.S., Leng J., Schwartz M.A., Bokoch G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine-Samson L., Rockey D.C., Bissell D.M. The role of α1β1 integrin in wound contraction. A quantitative analysis of liver myofibroblasts in vivo and in primary culture. J. Biol. Chem. 1997;272:30911–30917. doi: 10.1074/jbc.272.49.30911. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Whitmarsh A.J., Barrett T., Derijard B., Davis R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanti L., Heino J., López-Otín C., Kähäri V-M. Induction of collagenase-3 (MMP-13) expression in human skin fibroblasts by three-dimensional collagen is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 1999;274:2446–2455. doi: 10.1074/jbc.274.4.2446. [DOI] [PubMed] [Google Scholar]
- Reunanen N., Westermarck J., Häkkinen L., Holmstrom T.H., Elo I., Eriksson J.E., Kähäri V.-M. Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J. Biol. Chem. 1998;273:5137–5145. doi: 10.1074/jbc.273.9.5137. [DOI] [PubMed] [Google Scholar]
- Riikonen T., Westermarck J., Koivisto L., Broberg A., Kähäri V.M., Heino J. Integrin α2β1 is a positive regulator of collagenase (MMP-1) and collagen α1(I) gene expression J. Biol. Chem. 270 1995. 13548 13552a [DOI] [PubMed] [Google Scholar]
- Riikonen T., Koivisto L., Vihinen P., Heino J. Transforming growth factor-β regulates collagen gel contraction by increasing α2β1 integrin expression in osteogenic cells J. Biol. Chem. 270 1995. 376 382b [DOI] [PubMed] [Google Scholar]
- Roeckel D., Krieg T. Three-dimensional contact with type I collagen mediates tyrosine phosphorylation in primary human fibroblasts. Exp. Cell Res. 1994;211:42–48. doi: 10.1006/excr.1994.1056. [DOI] [PubMed] [Google Scholar]
- Sastry S.K., Lakonishok M., Thomas D.A., Muschler J., Horwitz A.F. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J. Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M.D., Hildebrand J.D., Shannon J.D., Fox J.W., Vines R.R., Parsons J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiro J.A., Chan B.M., Roswit W.T., Kassner P.D., Pentland A.P., Hemler M.E., Eisen A.Z., Kupper T.S. Integrin α2β1 (VLA-2) mediates reorganization and contraction of collagen matrices by human cells. Cell. 1991;67:403–410. doi: 10.1016/0092-8674(91)90191-z. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks S.K., Hunter T., van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Sistonen L., Sarge K.D., Phillips B., Abravaya K., Morimoto R.I. Activation of heat shock factor 2 during hemin-induced differentiation of human erythroleukemia cells. Mol. Cell Biol. 1992;12:4104–4111. doi: 10.1128/mcb.12.9.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B., Johnson K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione-S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Takada Y., Hemler M.E. The primary structure of the VLA-2/collagen receptor α2 subunit (platelet GPIa)homology to other integrins and the presence of a possible collagen-binding domain. J. Cell Biol. 1989;109:397–407. doi: 10.1083/jcb.109.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen P., Riikonen T., Laine A., Heino J. Integrin α2β1 in tumorigenic human osteosarcoma cell lines regulates cell adhesion, migration, and invasion by interaction with type I collagen. Cell Growth Differ. 1996;7:439–447. [PubMed] [Google Scholar]
- Wang W., Zhou G., Hu M.C.T., Yao Z., Tan T.H. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 1997;272:22771–22775. doi: 10.1074/jbc.272.36.22771. [DOI] [PubMed] [Google Scholar]
- Wang X.S., Diener K., Manthey C.L., Wang S., Rosenzweig B., Bray J., Delaney J., Cole C.N., Chan-Hui P.Y., Mantlo N., Lichenstein H.S., Zukowski M., Yao Z. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J. Biol. Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- Wary K.K., Mainiero F., Isakoff S.J., Marcantonio E.E., Giancotti F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Werb Z., Tremble P.M., Behrendtsen O., Crowley E., Damsky C.H. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J. Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J., Holmstrom T., Ahonen M., Eriksson J.E., Kähäri V.M. Enhancement of fibroblast collagenase-1 (MMP-1) gene expression by tumor promoter okadaic acid is mediated by stress-activated protein kinases Jun N-terminal kinase and p38. Matrix Biol. 1998;17:547–557. doi: 10.1016/s0945-053x(98)90107-x. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Yang S.H., Su M.S., Sharrocks A.D., Davis R.J. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol. Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Clark R.A. A three-dimensional collagen lattice induces protein kinase C-ζ activityrole in α2 integrin and collagenase mRNA expression. J. Cell Biol. 1997;136:473–483. doi: 10.1083/jcb.136.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.M., Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr. Opin. Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Yang W., Lin Q., Guan J.L., Cerione R.A. Activation of the Cdc42-associated tyrosine kinase-2 (ACK-2) by cell adhesion via integrin β1. J. Biol. Chem. 1999;274:8524–8530. doi: 10.1074/jbc.274.13.8524. [DOI] [PubMed] [Google Scholar]
- Zhang S., Han J., Sells M.A., Chernoff J., Knaus U.G., Ulevitch R.J., Bokoch G.M. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J. Biol. Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Vuori K., Reed J.C., Ruoslahti E. The α5β1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Soc. Sci. USA. 1995;92:6161–6165. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]