

Abstract
Physiological roles of the members of the synaptophysin family, carrying four transmembrane segments and being basically distributed on intracellular membranes including synaptic vesicles, have not been established yet. Recently, mitsugumin29 (MG29) was identified as a novel member of the synaptophysin family from skeletal muscle. MG29 is expressed in the junctional membrane complex between the cell surface transverse (T) tubule and the sarcoplasmic reticulum (SR), called the triad junction, where the depolarization signal is converted to Ca2+ release from the SR. In this study, we examined biological functions of MG29 by generating knockout mice. The MG29-deficient mice exhibited normal health and reproduction but were slightly reduced in body weight. Ultrastructural abnormalities of the membranes around the triad junction were detected in skeletal muscle from the mutant mice, i.e., swollen T tubules, irregular SR structures, and partial misformation of triad junctions. In the mutant muscle, apparently normal tetanus tension was observed, whereas twitch tension was significantly reduced. Moreover, the mutant muscle showed faster decrease of twitch tension under Ca2+-free conditions. The morphological and functional abnormalities of the mutant muscle seem to be related to each other and indicate that MG29 is essential for both refinement of the membrane structures and effective excitation-contraction coupling in the skeletal muscle triad junction. Our results further imply a role of MG29 as a synaptophysin family member in the accurate formation of junctional complexes between the cell surface and intracellular membranes.
Keywords: excitation-contraction coupling, sarcoplasmic reticulum, synaptophysin family, transverse tubule, triad junction
Synaptophysin is known as a major membrane protein on neurotransmitter-containing vesicles in the presynaptic regions of neurons (Jahn and Sudhof 1994; Matthews 1996). Recent DNA cloning studies have identified at least five synaptophysin-related proteins, namely synaptoporin (Knaus et al. 1990), synaptogyrin (Stenius et al. 1995), pantophysin (Haass et al. 1996), cellugyrin (Janz and Sudhof 1998), and mitsugumin29 (MG29) (Takeshima et al. 1998). Synaptophysin has been the best characterized among the family members. Synaptophysin forms an oligomeric structure and exhibits ionic channel activity in lipid bilayers (Thomas et al. 1988). Antibody and antisense oligonucleotides to synaptophysin inhibit neurotransmitter release (Alder et al. 1992a,Alder et al. 1992b). Therefore, it has been proposed that synaptophysin might form a protein component of the fusion pore during exocytosis in the presynaptic nerve ending (Betz 1990). However, neurons from mutant mice lacking synaptophysin show normal neurotransmitter release (McMahon et al. 1996), and no synaptophysin-related protein is found in the yeast genome, indicating that synaptophysin is unlikely to be essential for exocytosis. Thus, the physiological roles of synaptophysin and other family members have not been established yet.
In skeletal muscle, excitation-contraction (E-C) coupling requires the signal transduction system to convert depolarization of the cell surface transverse (T) tubule into Ca2+ release from the sarcoplasmic reticulum (SR) without the entry of extracellular Ca2+ (Schneider 1994). The two major molecules participating in this signal conversion are the dihydropyridine receptor (DHPR) as the T tubular voltage sensor (Tanabe et al. 1988), and the ryanodine receptor (RyR) as the SR Ca2+ release channel (Takeshima et al. 1994). Both receptor proteins are thought to be linked to each other functionally and mechanically at the junctional membrane region, called the triad junction, where the T tubule faces the SR membrane on both sides (Franzini-Armstrong and Jorgensen 1994). On the other hand, previous studies have suggested that an as-yet unidentified component(s) other than DHPR and RyR is required for the formation of the triad junction, which may provide a structural foundation for the direct interaction of the receptor molecules (Takekura et al. 1995; Ikemoto et al. 1997; Suda et al. 1997). In this context, we started to identify protein components unique to the triad junction using the mAb technique, and recently found several novel transmembrane proteins (Nishi et al. 1998; Takeshima et al. 1998). Of the identified proteins, MG29 shows high sequence homologies and shares structural features with the synaptophysin family members. Immunochemical and RNA blot experiments showed that MG29 is expressed abundantly in the skeletal muscle and at moderate levels in renal tubular cells (Shimuta et al. 1998). MG29 starts to appear on the developing SR before the onset of T tubule formation during skeletal muscle maturation, and then is localized to the triad junction in mature muscle cells. Furthermore, overexpression of MG29 in an amphibian expression system resulted in the generation of abnormal tubular ER (Komazaki et al. 1999). The molecular structure and subcellular distribution suggest that MG29 may be involved in the formation of specialized SR–ER network systems and in the communication between the SR–ER and cell surface membranes.
In recent years, knockout mice have been used to understand the molecular basis of physiological functions of specific gene products. We report here the generation and characterization of mutant mice lacking MG29. Although MG29-deficient muscle retained the triad junctions, the results obtained clearly indicate that MG29 is important for the proper organization of both the intracellular and cell surface membranes, and for the signal transduction of E-C coupling in skeletal muscle cells.
Materials and Methods
Generation of Mutant Mice
The targeting vector was constructed using the mouse genomic DNA fragments obtained in our previous experiments (Shimuta et al. 1998): the 1.1-kb XhoI/SalI fragment from pMC1 Neo polyA (Stratagene), the 0.7-kb XhoI/SalI fragment from pMC1-DT-A (Yagi et al. 1990), and pBluescript SK(−) (Stratagene). The short arm of the vector is the ∼2.2-kb genomic fragment containing the partial sequence of exon 1 and full sequence of exon 2, and the long arm is the ∼7.8-kb fragment containing the 5′-flanking sequence in the gene (see Fig. 1 A). The vector was linearized with XhoI and transfected into J1 embryonic stem (ES) cells (Li et al. 1992). Of ∼350 G418-resistant ES clones screened by Southern blot hybridization, two clones carried the expected homologous mutation. Chimeric mice produced with the ES clone numbered 171 were crossed with C57BL/6J mice and could transmit the mutant gene to their pups (F1). F2 mice obtained by crossing the heterozygous F1 mice were used for the biochemical, morphological, and physiological analyses in this study. To determine the mouse genotype, PCR was carried out using mouse genomic DNA as the template (see Fig. 1 A); the nucleotide sequences of the synthetic primers used are P29-1 (TACGCGCGGAAAAAGGGGAGAGCAAGG), neo-5′a (GCCACACGCGTCACCTTAATATGCG), and P29-2 (CTTACCTGCTGGCGCGGAGACTTGTCC).
Figure 1.

Generation of MG29-knockout mice. Homologous recombination at the MG29 locus (A). Restriction enzyme maps of the wild-type allele, targeting vector, and mutant allele (MG29m1) are illustrated. The exons in the gene (E1–E3) are indicated by filled boxes, and the neomycin resistance gene (neo) and diphtheria toxin gene (DTA) are indicated by open boxes. The directions of transcription are indicated by arrows. The genomic DNA probe and PCR primers for detection of the mutant gene are indicated by a hatched box and open arrows, respectively. The predicted sizes of the DNA fragments in Southern blot analysis and PCR are also shown. Southern blot analysis of DNAs from mice bearing the MG29m1 mutation (B). Genomic DNAs digested with BamHI were analyzed using the hybridization probe indicated in A. Size markers are indicated in kb pairs. Blot hybridization analysis of skeletal muscle RNAs from mice bearing the MG29m1 mutation (C). Mouse MG29 cDNA was used as the hybridization probe, and size markers are indicated in kb. Immunoblot analysis of skeletal muscle microsomal proteins from mice bearing the MG29m1 mutation (D). Microsomal proteins from hind limb muscles were analyzed with antibody against mouse MG29, and size markers are indicated in kD.
Blot Analysis
Genomic DNA and total RNA were prepared from mice tissues, and Southern and Northern blot hybridization analyses were performed as described previously (Takeshima et al. 1994). A peptide derived from the primary structure of mouse MG29, SSTESPGRTSDKSPR (in one-letter code), was synthesized and coupled to keyhole limpet hemocyanin (KLH) as a carrier protein using glutaraldehyde. An adult rabbit was repeatedly immunized with the peptide–KLH complex, and antibody against mouse MG29 was recovered from the antiserum using a protein G–Sepharose column (Amersham Pharmacia Biotech). Total microsomal proteins were prepared from hind limb muscle of mice (Saito et al. 1984) and were subjected to immunoblot analysis as described previously (Takeshima et al. 1994).
Morphological Analysis
Skeletal muscle preparations were treated with a prefixative solution (2.5% glutaraldehyde and 0.1 M sodium cacodylate, pH 7.4). For specific staining of the T tubule (Franzini-Armstrong 1991), skeletal muscle was treated with the prefixative solution supplemented with 50 mM CaCl2. The prefixed muscle was then postfixed with a buffer containing 1% OsO4 and 0.1 M sodium cacodylate, pH 7.4. The fixed muscle was washed, dehydrated with alcohol and acetone, and embedded in Epon. Thin sections were prepared, stained with uranyl acetate and lead citrate, and observed under an electron microscope (JEM-200CX, JEOL).
Muscle Contraction Measurement
Skeletal muscle preparations of whole extensor digitorum longus (EDL) muscle and diaphragm strips, including a part of the rib and central tendon, were dissected from mice. The muscle preparations were mounted between a force transducer (UL-100; Minebea Co.) and fixed hock in a chamber containing Krebs solution (120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM NaH2PO4, 25 mM NaHCO3, and 11 mM glucose) bubbled with 95% O2 and 5% CO2 at 25°C. The preparations were stretched with a resting tension of 0.7 g and field-stimulated with supramaximal voltage. For inducing tetanic contractions, train stimulations of total 50 times were delivered at various frequencies. The effects of Ca2+ removal from the Krebs solution were examined on twitches evoked at the rate of 0.5 Hz for 20 min.
Results and Discussion
Generation of Mutant Mice Lacking MG29
Genomic DNA segments containing the mouse MG29 gene isolated in our previous experiments were used for constructing a targeting vector (Fig. 1 A). In the vector, the genomic sequence containing parts of the putative promoter and first exon is replaced by the neomycin resistance gene for positive selection, and the diphtheria toxin gene is attached at the 3′ terminus of the genomic sequence for negative selection. ES cells were transfected with the targeting vector, and the resultant G418-resistant clones were screened by Southern blot hybridization. The ES clones harboring the introduced mutant gene (MG29m1) were shown to have the expected pattern of arrangement in genomic DNA by the blot analysis using several restriction enzymes and specific probes for the gene (Fig. 1 B). Chimeric male mice were generated by injecting the ES cells into blastocysts, and bred to yield mice carrying the mutant gene. Mice homozygous for the mutation, obtained by crossing between heterozygous mice, did not exhibit lethality. Blot analyses showed absence of both MG29 mRNA (Fig. 1 C) and its protein product (Fig. 1 D) in skeletal muscle from the homozygous mutants, demonstrating that the introduced mutation is a null mutation of the gene.
The MG29-deficient mice exhibited no abnormalities in health or reproduction under our conventional housing conditions. Because MG29 is expressed at high levels in skeletal muscle cells and at moderate levels in renal tubular cells (Shimuta et al. 1998; Takeshima et al. 1998), we expected physiological defects in skeletal muscle and kidney in the mutant mice. However, light microscopic observations did not reveal any morphological abnormalities in the kidneys from the mutant mice (data not shown). This observation, together with normal health of the mutant mice, indicates that the loss of MG29 causes no significant defects in the kidney. This report deals with abnormalities in skeletal muscle from the mutant mice.
Reduced Body Weight in MG29-deficient Mice
The MG29-deficient mice were slightly smaller in body size and lighter in body weight than their wild-type littermates (Fig. 2). The heterozygous mutants showed values intermediate between those of the wild-type and homozygous mice. The reduced body weight in the mutant mice may be mainly due to lightened skeletal muscle systems as shown below.
Figure 2.

Reduced body weight in the MG29-deficient mice. Male mice were weaned at 4 wk of age, and their body weight was measured every week. Each value (n = 16) represents the mean ± SEM. Significant differences between the groups were observed by t test (asterisk indicates P < 0.05; two asterisks indicate P < 0.01). The values for the heterozygous mutants were intermediate between those of the homozygous mutants and wild-type mice (data not shown). Results observed in the female mice were essentially similar (data not shown).
To survey gross defects of motor function in the MG29-deficient mice, four behavioral tasks that reflect skeletal muscle performance were used. In any of the tasks (the fixed-bar test, rotarod test, open-field locomotion test, and forced swimming test), no obvious differences were detected between the mutant and wild-type mice (Kuriyama, K., S. Shibata, and H. Takeshima, unpublished observations). The results indicate that the loss of MG29 produces no significant defects in motor coordination or motor ability on the whole-animal level.
Morphological Abnormalities in MG29-deficient Skeletal Muscle
Because MG29 is expressed predominantly in skeletal muscle, we examined the morphological abnormalities in hind limb muscle from the MG29-deficient mice (young adults, 8–9 wk old). The averaged wet weight of whole EDL muscle from the mutant mice was significantly reduced in comparison with that of controls (see Table ). Therefore, sections of EDL muscle were examined by light microscopy and EM (Fig. 3). The cross-sectional areas of muscle cells from MG29-deficient mice (mean ± SD, 705 ± 315 μm2, n = 399 cells from two typical EDL bundles) were significantly smaller (P < 0.001, t test) than those from wild-type mice (1154 ± 526 μm2, n = 382 cells from two bundles). However, there was no significant difference in the cell number in the muscle bundles between the mutant and wild-type mice; EDL bundle from either genotype was composed of ∼900 muscle cells. EM analysis of EDL muscle showed that the cytoplasmic region was full of contractile filaments in both MG29-deficient and wild-type muscle cells, and no significant difference was observed in the sectional areas of myofibrils between the genotypes. Therefore, the reduced weight of mutant EDL muscle is considered to be caused mainly by the smaller cell size.
Table 1.
Properties of EDL Muscles from Wild-type and MG29-deficient Mice
| Wet weight | Twitch/weight | Tetanus/weight | |
|---|---|---|---|
| mg | g/mg | g/mg | |
| Wild-type muscle | 9.6 ± 1.5 | 0.51 ± 0.059 | 2.6 ± 0.55 |
| MG29-deficient muscle | 7.1 ± 0.85** | 0.36 ± 0.064** | 2.9 ± 0.30 |
Data (means ± SD) were derived from at least eight muscle preparations from 8–9-wk-old mice. The twitch and tetanic responses were evoked by electrical stimuli at 0.5 and 200 Hz, respectively. Statistical differences are indicated by asterisks (P < 0.01 in t test).
Figure 3.

Histological analysis of MG29-deficient EDL muscle. Toluidine blue–stained cross sections of EDL muscles from hind limb of 8-wk-old wild-type (A) and MG29-deficient mice (B) are shown. The cross-sectional areas of mutant muscle cells are significantly reduced compared with those of the control muscle cells. However, no significant differences between the genotypes are observed in the number of muscle cells and myofibril density within the cells. Bar, 100 μm.
Abnormalities in the T tubules in the mutant muscle cells were examined by the well-established method of T tubule–specific staining (Fig. 4). In cross sections, the T tubules in wild-type muscle were flat and elliptical (short axis 0.01–0.03 μm and long axis 0.08–0.12 μm), whereas the T tubules in MG29-deficient muscle were almost round (diameter 0.1–0.2 μm). In wild-type muscle the T tubules ran straight in a right-angled direction to myofibrils at the interphases between the A- and I-bands (A-I junction). However, abnormal running route of the T tubules was conspicuous in MG29-deficient muscle. Such abnormal orientation of the T tubules was not detected in wild-type muscle.
Figure 4.
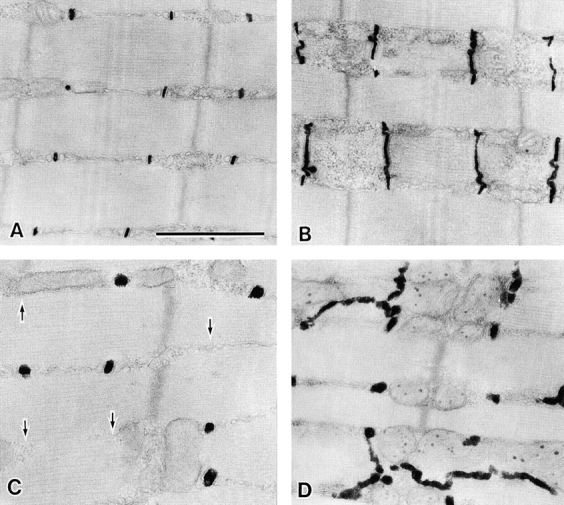
Abnormal T tubular system in MG29-deficient skeletal muscle. Electron micrographs of the longitudinal sections of T tubule–stained hind limb muscles from 8-wk-old wild-type (A and B) and MG29-deficient (C and D) mice. The stained T tubules are swollen and run in irregular directions in the mutant muscle cells. Arrows in C indicate the missing T tubules at the A-I junctions. Bar, 1 μm.
Next, we analyzed the triad junctions from the MG29-deficient muscle cells (Fig. 5). In wild-type muscle, the triad junction between the T tubule and SR was well-established, and the SR network was well-organized. However, the SR network was poorly formed in MG29-deficient muscle. For example, vacuolated, highly fragmented, or simple tubular SR structures were frequently observed in mutant muscle; such irregular structures were detected in 8% of the SR examined in wild-type muscle (n = 231) and 55% of those in mutant muscle (n = 442). As could be expected from the abnormal routes of T tubules (Fig. 4), it was further confirmed that the lack of triad junctions in the A-I junctions was frequent in MG29-deficient muscle. The ratios for the absence of triads were 1.4% in wild-type muscle (triad junctions observed, n = 1,665) and 24% in MG29-deficient muscle (n = 2,616).
Figure 5.

Abnormal triad junctions in MG29-deficient skeletal muscle. The triad junctions and SR in hind limb muscles from 8-wk-old wild-type (A) and MG29-deficient (B and C) mice are shown. The tubular network of the SR is well-developed in wild-type muscle cells, whereas the vacuolar SR shown in B and simple tubular SR shown in C are frequently observed in mutant muscle cells. Insets show a cross section of the triad junction. Arrows and arrowheads indicate the triad junctions and M bands, respectively. Bars: 0.5 μm (A); 0.1 μm (inset).
The morphological abnormalities detected in mutant hind limb muscle, such as swollen and irregularly running T tubules, partial misformation of triad junctions, and poorly developed SR networks were also observed in mutant diaphragm muscle (data not shown). The results demonstrate that the ultrastructural abnormalities around the triad junction were shared by both fast and slow muscle fibers in the MG29-deficient mice.
Irregular Contractile Properties of MG29-deficient Skeletal Muscle
To survey functional abnormalities of MG29-deficient muscle, we examined the contractile properties of EDL and diaphragm muscles, containing mainly fast and slow muscle fibers, respectively. Both mutant muscle preparations showed contractile responses evoked by electrical stimulation, even in a Ca2+-free solution, and no obvious difference in the basic mechanism of E-C coupling was observed between mutant and wild-type muscles. However, two major abnormalities were found in mutant muscle as described below.
Because of the different wet weight between mutant and control EDL muscles, the developed force was normalized to the weight, assuming that there was no difference in the length of the muscle preparations between the genotypes. MG29-deficient EDL muscle developed less normalized twitch tension than wild-type muscle, although no significant difference was observed in the maximum force evoked by tetanic stimulation between the genotypes (Table ). Therefore, the twitch/tetanus ratio of mutant muscle (mean ± SD, 0.12 ± 0.015) was significantly smaller than that of control muscle (0.21 ± 0.036). Furthermore, the force-frequency curve was shifted right in mutant muscle compared with that in wild-type muscle (Fig. 6). In experiments using diaphragm preparations, a similar right-shifted force-frequency curve was observed in mutant muscle, although the degree of differences between the genotypes was less impressive than that in EDL muscle (data not shown). The twitch/tetanus ratios of diaphragm muscles from the mutant and wild-type mice were 0.23 ± 0.016 and 0.26 ± 0.011, respectively (P < 0.01, t test). The right-shifted force-frequency relationship in the mutant muscle does not seem to reflect an increase in the number of immature or slow muscle fibers, which exhibit left-shifted curves compared with mature or fast muscle fibers (Close 1964). These observations show that the loss of MG29 partly impairs E-C coupling at low-frequency stimuli. Because the differences in the developed force between the genotypes diminished at higher-frequency stimuli, MG29 deficiency probably reduced the efficiency of signal conversion during E-C coupling.
Figure 6.

Irregular force-frequency relationship in MG29-deficient skeletal muscle. Isometric tension at several frequencies was determined in EDL muscles from 8–9-wk-old mice. Typical recording from wild-type and mutant muscles are shown in A. Bars, 1 g/mg wet weight. Force-frequency relationships in MG29-deficient and wild-type muscles are shown in B. Each value normalized to the maximum force represents the mean ± SD. (n = 8–9 from five mice). Significant differences between the groups were observed by t test (asterisk indicates P < 0.05; two asterisks indicate P < 0.01).
It has been reported that dantrolene, a muscle relaxant drug, preferentially inhibits twitch and shifts the force-frequency curve to the right in skeletal muscle tension measurements (Leslie and Part 1981). Therefore, MG29-deficient and dantrolene-treated muscles show similar contractile properties. The pharmacological action of dantrolene is still unclear, and the results here raise the possibility that MG29 may be a target site of dantrolene. However, dantrolene was still effective in MG29-deficient muscle (data not shown).
Next, we examined the effects of Ca2+ removal from the bath solution on twitches of diaphragm, which is composed of only several muscle layers. Under nominally Ca2+-free conditions, mutant diaphragm muscle showed a more rapid decrease of twitch tension than control muscle (Fig. 7). The apparent rate constants of the decay in wild-type and MG29-deficient muscles were roughly estimated to be 0.01 and 0.02 (min−1), respectively. The tension of EDL muscle was highly resistant under Ca2+-free conditions, probably because EDL muscle is composed of multiple layers of muscle fibers and diffusion of the bathing solution is highly restricted. Faster decrease of twitch tension under nominally Ca2+-free conditions was also observed in mutant EDL muscle (data not shown).
Figure 7.

Effects of Ca2+ withdrawal on twitches in MG29-deficient skeletal muscle. Twitch tension at 5, 10, and 20 min after Ca2+ removal from the extracellular solution was measured in diaphragm muscles from 8–9-wk-old mice. Each value normalized to tension under normal conditions represents the mean ± SD. (n = 7–9 from six mice). Significant differences between the groups were observed by t test (asterisks indicate P < 0.01).
Relationship between Morphological and Physiological Abnormalities
The partial misformation of the triad junction and the abnormalities of SR structures may correlate well with the reduced twitch tension in MG29-deficient muscle. The smaller twitch tension is probably caused by the low efficiency of signal conversion during E-C coupling in the mutant muscle, because no significant differences were detected in normalized maximum tension and density of cellular contractile filaments between the genotypes. It may be that the partial loss of the triad junctions directly reduces the efficiency of E-C coupling, and that the abnormal SR structures decrease the available Ca2+ for twitch responses in the mutant muscle. The apparently normal phenotype of the mutant mice in the behavioral tasks to test motor performance is likely due to compensatory mechanisms by motor neurons and/or upper motor centers in the brain.
The faster decrease of twitch tension under Ca2+-free conditions and the swollen T tubules in MG29-deficient muscle may be related. Although E-C coupling is retained under Ca2+-free conditions in skeletal muscle, contraction responses gradually decrease, likely due to the reduction of Ca2+ content in the SR or the inactivation of DHPR as the T tubular voltage sensor. Assuming that passive diffusion restricted by the area of the T tubule rather than that of the sarcolemma was mainly responsible for the Ca2+ leak in skeletal muscle, it might be reasonable to expect quick Ca2+ depletion in the mutant muscle bearing the expanded T tubules. Alternatively, the abnormal T tubular structures might affect the functions of DHPRs in the mutant muscle.
Proposed Role of MG29 as a Synaptophysin Family Member
In view of the abundant expression of MG29 in the triad junction (Takeshima et al. 1998), the lack of a failure in membrane organization or E-C coupling in mutant skeletal muscle is rather surprising. We need to consider the possibility that the functions of MG29 are partly compensated by related molecules. Indeed, of the synaptophysin family members, pantophysin and cellugyrin are ubiquitously expressed in various cell types (Haass et al. 1996; Janz and Sudhof 1998). It has been suggested that synaptophysin may play a role in the structural organization of the synaptic vesicle as a small and highly curved organelle (Jahn and Sudhof 1994). This possibility may not be supported by MG29-deficient muscle, in which curvilineal membrane structures were preserved in the triad junction. On the other hand, the abnormalities of the mutant muscle described above suggest another important biological function of MG29. After the random formation of junction membrane structures between the disorganized SR and primitive T tubules, the membrane systems gradually mature during muscle differentiation. The processes include the rearrangement of the transverse-oriented T tubules, flattening of the junctional T tubular regions, specific localization of the triad junctions at the A-I junctions and development of differentiated regions of the SR (Franzini-Armstrong 1991; Flucher 1992). Since the formation of junctional membrane complexes seems to be normal in MG29-deficient muscle, our results indicate that the loss of MG29 specifically impairs the maturation processes to generate the refined membrane systems. It is unlikely that the deficiency of MG29 simply delays the maturation or induces degeneration after the maturation, because skeletal muscle cells from the 4-, 8-, and 12-wk-old mutant mice essentially shared the same abnormalities around the triad junction (Komazaki, S., unpublished observations). These observations suggest that synaptophysin family members, including MG29, may be important for the structural refinement of junctional membranes commonly observed in the triad junction, subsurface cisternae, and presynaptic regions.
MG29 is recovered in both SR and T tubular fractions in biochemical membrane preparations (Takeshima et al. 1998; Brandt and Caswell 1999), and MG29 deficiency results in abnormalities of membrane structures in both the SR and T tubular systems as described above, indicating that MG29 is distributed on both the membranes in intact skeletal muscle cells. The molecular action of MG29 during the refinement of the membrane structures is still uncertain, and at the present several possibilities may be proposed in the explanation. For example, MG29 might be involved in the transport between the SR and T tubule to support the refined structures of junctional membranes, although the vesicular transport system in the triad junction is not known. Alternatively, it may be possible to presume that MG29-mediated reactions may stiffen the junctional T tubular region to generate the flattened structure and may facilitate the translocation of the stiffened T tubule to the A-I junctions. In either case, the presumed functions of MG29 may underlie intermolecular interaction. It would be important to identify the proposed binding partner(s) of MG29 in future studies.
Acknowledgments
We thank Izumi Dobashi, Akiko Sakamoto, and Miki Matsunaga for their help in some of the experiments.
This work was supported in part by grants from the Ministry of Education, Science, Sports and Culture, the Ministry of Health and Welfare, the TMFC, the Ichiro Kanehara Memorial Foundation and the Inamori Foundation.
Footnotes
Abbreviations used in this paper: DHPR, dihydropyridine receptor; E-C, excitation-contraction; EDL, extensor digitorum longus; ES, embryonic stem; MG29, mitsugumin29; SR, sarcoplasmic reticulum; T tubule, transverse tubule.
References
- Alder J., Lu B., Valtorta F., Greengard P., Poo M. Calcium-dependent transmitter secretion reconstituted in Xenopus oocytesrequirement for synaptophysin Science. 257 1992. 657 661a [DOI] [PubMed] [Google Scholar]
- Alder J., Xie Z.P., Valtorta F., Greengard P., Poo M. Antibodies to synaptophysin interfere with transmitter secretion at neuromuscular synapses Neuron. 9 1992. 759 768b [DOI] [PubMed] [Google Scholar]
- Betz H. Homology and analogy in transmembrane channel designlessons from synaptic membrane proteins. Biochemistry. 1990;29:3591–3599. doi: 10.1021/bi00467a001. [DOI] [PubMed] [Google Scholar]
- Brandt N.R., Caswell A.H. Localization of mitsugumin29 to transverse tubules in rabbit skeletal muscle. Arch. Biochem. Biophys. 1999;371:348–350. doi: 10.1006/abbi.1999.1444. [DOI] [PubMed] [Google Scholar]
- Close R. Dynamic properties of fast and slow skeletal muscle of the rat during development. J. Physiol. 1964;173:74–95. doi: 10.1113/jphysiol.1964.sp007444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher D.E. Structural analysis of muscle developmenttransverse tubules, sarcoplasmic reticulum, and the triad. Dev. Biol. 1992;154:245–260. doi: 10.1016/0012-1606(92)90065-o. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C. Simultaneous maturation of transverse tubules and sarcoplasmic reticulum during muscle differentiation in the mouse. Dev. Biol. 1991;146:353–363. doi: 10.1016/0012-1606(91)90237-w. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Jorgensen A.O. Structure and development of E-C coupling units in skeletal muscle. Annu. Rev. Physiol. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- Haass N.K., Kartenbeck J., Leube R.E. Pantophysin is a ubiquitously expressed synaptophysin homologue and defines constitutive transport vesicles. J. Cell Biol. 1996;134:731–746. doi: 10.1083/jcb.134.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto T., Komazaki S., Takeshima H., Nishi M., Noda T., Iino M., Endo M. Functional and morphological features of skeletal muscle from mutant mice lacking both type 1 and type 3 ryanodine receptors. J. Physiol. 1997;501:305–312. doi: 10.1111/j.1469-7793.1997.305bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R., Sudhof T.C. Synaptic vesicles and exocytosis. Annu. Rev. Neurosci. 1994;17:219–246. doi: 10.1146/annurev.ne.17.030194.001251. [DOI] [PubMed] [Google Scholar]
- Janz R., Sudhof T.C. Cellugyrin, a novel ubiquitous form of synaptogyrin that is phosphorylated by pp60c-src. J. Biol. Chem. 1998;273:2851–2857. doi: 10.1074/jbc.273.5.2851. [DOI] [PubMed] [Google Scholar]
- Knaus P., Marqueze-Pouey B., Scherer H., Betz H. Synaptoporin, a novel putative channel protein of synaptic vesicles. Neuron. 1990;5:453–462. doi: 10.1016/0896-6273(90)90084-s. [DOI] [PubMed] [Google Scholar]
- Komazaki S., Nishi M., Kangawa K., Takeshima H. Immunolocalization of mitsugumin29 in developing skeletal muscle and effects of the protein expressed in amphibian embryonic cells. Dev. Dyn. 1999;215:87–95. doi: 10.1002/(SICI)1097-0177(199906)215:2<87::AID-DVDY1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Leslie G.C., Part N.J. The action of dantrolene sodium on rat fast and slow muscle in vivo. Br. J. Pharmacol. 1981;72:665–672. doi: 10.1111/j.1476-5381.1981.tb09147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Matthews G. Neurotransmitter release. Annu. Rev. Neurosci. 1996;19:219–233. doi: 10.1146/annurev.ne.19.030196.001251. [DOI] [PubMed] [Google Scholar]
- McMahon H.T., Bolshakov V.Y., Janz R., Hammer R.E., Siegelbaum S.A., Sudhof T.C. Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc. Natl. Acad. Sci. USA. 1996;93:4760–4764. doi: 10.1073/pnas.93.10.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M., Komazaki S., Iino M., Kangawa K., Takeshima H. Mitsugumin23, a novel transmembrane protein on endoplasmic reticulum and nuclear membranes. FEBS Lett. 1998;432:191–196. doi: 10.1016/s0014-5793(98)00864-3. [DOI] [PubMed] [Google Scholar]
- Saito A., Seiler S., Cho A., Fleischer S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J. Cell Biol. 1984;99:875–885. doi: 10.1083/jcb.99.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M.F. Control of calcium release in functioning skeletal muscle fibers. Annu. Rev. Physiol. 1994;56:463–484. doi: 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Shimuta M., Komazaki S., Nishi M., Iino M., Nakagawara K., Takeshima H. Structure and expression of mitsugumin29 gene. FEBS Lett. 1998;431:263–267. doi: 10.1016/s0014-5793(98)00770-4. [DOI] [PubMed] [Google Scholar]
- Stenius K., Janz R., Sudhof T.C., Jahn R. Structure of synaptogyrin (p29) defines novel synaptic vesicle protein. J. Cell Biol. 1995;131:1801–1809. doi: 10.1083/jcb.131.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda N., Franzius D., Fleig A., Nishimura S., Bodding M., Hoth M., Takeshima H., Penner R. Ca2+-induced Ca2+ release in Chinese hamster ovary (CHO) cells co-expressing dihydropyridine and ryanodine receptors. J. Gen. Physiol. 1997;109:619–631. doi: 10.1085/jgp.109.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekura H., Takeshima H., Nishimura S., Takahashi M., Tanabe T., Flockerzi V., Hofmann F., Franzini-Armstrong C. Co-expression in CHO cells of two muscle proteins involved in excitation-contraction coupling. J. Muscle Res. Cell Motil. 1995;16:465–480. doi: 10.1007/BF00126431. [DOI] [PubMed] [Google Scholar]
- Takeshima H., Iino M., Takekura H., Nishi M., Kuno J., Minowa O., Takano H., Noda T. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369:556–559. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- Takeshima H., Shimuta M., Komazaki S., Ohmi K., Nishi M., Iino M., Miyata A., Kangawa K. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem. J. 1998;331:317–322. doi: 10.1042/bj3310317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Powell J.A., Numa S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- Thomas L., Hartung K., Langosch D., Rehm H., Bamberg E., Franke W.W., Betz H. Identification of synaptophysin as a hexameric channel protein of the synaptic vesicle membrane. Science. 1988;242:1050–1053. doi: 10.1126/science.2461586. [DOI] [PubMed] [Google Scholar]
- Yagi T., Ikawa Y., Yoshida K., Shigetani Y., Takeda N., Mabuchi I., Yamamoto T., Aizawa S. Homologous recombination at c-fyn locus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc. Natl. Acad. Sci. USA. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]