

Abstract
Here, we describe the isolation of adenine nucleotide translocase-1 (ANT-1) in a screen for dominant, apoptosis-inducing genes. ANT-1 is a component of the mitochondrial permeability transition complex, a protein aggregate connecting the inner with the outer mitochondrial membrane that has recently been implicated in apoptosis. ANT-1 expression led to all features of apoptosis, such as phenotypic alterations, collapse of the mitochondrial membrane potential, cytochrome c release, caspase activation, and DNA degradation. Both point mutations that impair ANT-1 in its known activity to transport ADP and ATP as well as the NH2-terminal half of the protein could still induce apoptosis. Interestingly, ANT-2, a highly homologous protein could not lead to cell death, demonstrating the specificity of the signal for apoptosis induction. In contrast to Bax, a proapoptotic Bcl-2 gene, ANT-1 was unable to elicit a form of cell death in yeast. This and the observed repression of apoptosis by the ANT-1–interacting protein cyclophilin D suggest that the suicidal effect of ANT-1 is mediated by specific protein–protein interactions within the permeability transition pore.
Keywords: cell death, ATP transport, transfection, apoptosis, membrane potential
Apoptosis is a form of cell death that plays a role in development, tissue homeostasis, and disease (White 1996). Its induction must be tightly regulated. Otherwise, serious consequences may follow. A hyperactive apoptosis induction might lead to degenerative diseases like Alzheimer's disease (Loo et al. 1993); a reduced activity can contribute to the multistep process of tumorigenesis since tumor cells are exposed to multiple proapoptotic stimuli (McGill 1997). Therefore, the induction of apoptosis is governed by an elaborate array of checks and balances in the cell. Eventually, a family of cysteine proteases, the so-called caspases, is activated (Salvesen and Dixit 1997). These enzymes can cleave specific substrates in the cell that leads to the typical apoptotic phenotype and the self-destruction of the cell.
The first indication that mitochondria play a role in apoptosis induction was the observation that an in vitro system for apoptosis induction required the presence of mitochondria (Newmeyer et al. 1994). The permeability transition pore (PT pore) complex (Zoratti and Szabo 1995), a protein aggregate that resides in contact sites of the inner and outer mitochondrial membrane, was subsequently identified as being responsible for apoptosis induction (Petit et al. 1995; Zamzami et al. 1995b). Multiple pharmacological stimuli have been shown to activate this complex by as yet unknown means (Petit et al. 1995; Fulda et al. 1998; Zamzami et al. 1998). The activated complex leads to the collapse of the potential over the inner mitochondrial membrane (ΔΨm), swelling of mitochondria, and the generation of oxygen radicals (Zamzami et al. 1995a; Vander Heiden et al. 1997). The subsequent release of apoptogenic caspases (Susin et al. 1999a) and a putative oxidoreductase (Susin et al. 1999b) aids in apoptosis induction.
Many genes involved in apoptosis have the dominant capacity to induce cell death upon overexpression. This feature is even conserved across species (McCarthy and Dixit 1998). It might be explained by the fact that the overexpressed proteins engage in protein–protein contacts and can thereby create the signal for apoptosis (Yang et al. 1998). Consequently, we have recently developed a screen for dominant, apoptosis-inducing genes (Grimm and Leder 1997). The screen is based on the iterative transfection of small plasmid pools of a normalized cDNA library into mammalian cells and the morphological determination of apoptosis induction. Here, we describe the characterization of adenine nucleotide translocase-1 (ANT-1), a central component of the permeability transition pore, which was identified using such a screen.
ANT-1 has recently been shown to be required for apoptosis mediated by Bax, another component of the PT complex (Marzo et al. 1998a). Conversely, ANT-1 can induce apoptosis in a Bax-dependent manner. For this, ANT-1 must be inactivated pharmacologically by the plant glucoside atractyloside, which arrests ANT-1 in a specific conformation and causes PT pore opening (Zoratti et al. 1982; Davidson and Halestrap 1987). However, in our experiments with ANT-1, such a secondary signal is not required for apoptosis induction. ANT-1's apoptosis activity does not seem to depend on its known function for ADP/ATP exchange because several transport inactive mutants could still lead to cell death. Surprisingly, we found that a very homologous protein, the ANT-2 transporter, was inactive for apoptosis induction. Furthermore, ANT-1 could not elicit a form of cell death in yeast. This is in contrast to Bax that directly interacts with ANT-1 and can dominantly induce cell death in yeast. This result suggests that ANT-1–mediated apoptosis induction depends on protein–protein interactions that are specific for mammalian cells. Since ANT-1 is activated by overexpression for apoptosis induction, it is interesting to note that this gene is already highly expressed in mitochondria. This suggests that in a normal cell it must be kept inactive by other proteins of the PT pore, which implies important stoichiometric correlations between the various components of this complex. Consistent with this, we found that cyclophilin D, another component of the PT pore, can repress ANT-1–induced apoptosis. Furthermore, our data could help to explain the observed apoptosis induction in dilated cardiomyopathy (DCM), a degenerative disease of the heart muscle that is marked by a dramatic increase of the expression level of ANT-1 and by excessive apoptosis induction. These findings are, up to now, the most direct genetic evidence that the PT pore can signal apoptosis, and may lead to the molecular elucidation of how this complex can be activated for apoptosis induction.
Materials and Methods
Plasmids
The hemagglutinin (HA) tag vector (pHA) for the expression of HA fusion proteins was constructed by annealing oligos coding for the HA epitope (YPYDVPDYA) and inserting them between the NotI and XbaI site into the pcDNA3 vector (Invitrogen Corp.). The correct sequence was verified by sequencing. The point mutants of ANT-1 were engineered by recombinant PCR using suitable primers. All amino acid changes were verified by sequencing. Deletion mutagenesis of ANT-1 was achieved with PCR using the same enzyme. For all the PCR reactions, Pwo, a thermostable polymerase with proofreading function (Roche Diagnostics) was employed with 22 cycles and 200 ng of template. All deletion and point mutants of ANT-1 contained the same Kozak sequence and were inserted into the HA tag vector (pHA) with BamHI and NotI. ANT-2 was amplified from a mouse kidney cDNA library and cyclophilin D from a 293T library, both by using specific primers. The correct sequence was verified by sequencing. The eukaryotic expression vector for baculovirus p35 has been described (Clem and Miller 1994). Reverse transcriptase–PCR was used to clone the mouse ANT-2 (5′ primer, 5′-TCAGGATCCTTTCAACATGACAGATGC-3′; and 3′ primer, 5′-TAGTGCATGCGGCCGCCTGTGTATTTCTTGATCTCATC-3′) and the human cyclophilin D (5′ primer, 5′-ATCGGATCCATTAGCCATGGTCAACCC-3′; and 3′ primer, 5′-TAGTCGGTGCGGCCGCCTTCGAGTTGTCCACAGTC-3′). Both were inserted into the pHA vector.
Cell Cultivation and Transfection
Human embryonic kidney cells (293T) (Graham et al. 1977; Pear et al. 1993) and BHK cells were kept in DME medium supplemented with 5% FCS (Sigma Chemical Co.); all other cell lines were cultivated in DME and 10% FCS with the exception of PC3 cells, which were held in RPMI with 10% FCS. All transfections were done by calcium phosphate coprecipitation as described (Roussel et al. 1984), except for PC3 cells which were transfected with Dosper (Roche Diagnostics) and 3T3 cells by using SuperFect (Qiagen). When the green fluorescent protein (GFP) was cotransfected, the indicated amount of pLantern vector (GIBCO BRL) was added to the transfection mix.
Isolation of Apoptosis-inducing Genes
mRNA was isolated from 10-wk-old CD1 mice and normalized as described (Grimm and Leder 1997). The cDNA was inserted into a modified pcDNA3 vector in which the neomycin resistance gene had been deleted (Grimm and Leder 1997). The screen for dominant apoptosis inducers was performed essentially as described (Grimm and Leder 1997). A novel 96-well DNA isolation method allowed a considerably higher throughput. Human 293T cells were used as a read-out cell line since this would allow the isolation of genes whose signals are conserved across species. Apoptosis induction was verified by transfecting the isolated, untagged ANT-1 into mouse L929 and 3T3 cells, the hamster BHK cell line, and human HeLa, MCF-7, and PC3 cells.
Immunoblotting
For detecting cytochrome c, 293T cells were transfected with an expression vector for ANT-1. After 18 h, the cells were harvested and the cytoplasmic extracts were prepared as described (Ferrari et al. 1998). Protein samples of 30 μg were loaded on SDS–polyacrylamide gels (10%), and then electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (Amersham). Western blots were probed with a monoclonal cytochrome c antibody (PharMingen) and anti-mouse HRP-conjugated antibody before enhanced chemiluminescence (ECL)–based detection (Amersham). For the poly-ADP-ribose polymerase (PARP) Western blot, nuclear fractions were obtained by differential centrifugation as described previously (Schreiber et al. 1989). Aliquots of 50 μg protein were subjected to SDS-PAGE (8% polyacrylamide) and blotted onto PVDF membranes. PARP and cleavage products were detected using a polyclonal serum directed against full-length PARP (Roche Diagnostics), anti-rabbit HRP-conjugated antibody (Amersham) and the ECL system. For the detection of HA-tagged ANT isoforms, mitochondria were isolated by differential centrifugation as described previously (Gawaz et al. 1990). 35-μg protein samples were separated on 10% polyacrylamide gels and blotted onto PVDF membranes. Detection was performed using a rat mAb raised against the influenza virus HA peptide (Roche Diagnostics) and the ECL system (Amersham).
Yeast Methods
Standard yeast methods were applied (Ausubel et al. 1991). For the inducible expression of Bax and ANT-1, the cDNAs were subcloned into pYESTrp2 (Invitrogen Corp.) in which the B42 fusion moiety was removed.
For induction of the expression constructs, EGY 48 yeast cells (MATα trp1 ura3 his3 leu2::plexAop6-leu2) were grown for 8 h in YC medium with 20% galactose and 20% raffinose instead of glucose. The yeast cells were harvested and protoplasts were formed by enzymatic digestion of the cell wall as described (Gawaz et al. 1990). The protein extracts were isolated as described above. Protein samples were separated on 10% polyacrylamide gels and transferred onto PVDF membranes. Western blots were probed with a polyclonal Bax antibody (Santa Cruz Biotechnology) or ANT-1 polyclonal serum (provided by M. Klingenberg, Munich, Germany) and anti-rabbit HRP-conjugated antibody before ECL-based detection (Amersham). The anti–Bax antibody recognizes a conserved sequence between mouse and human; the anti–ANT-1 serum detects yeast ANT-1 and also cross-reacts with the mammalian protein. A mouse mAb (Research Diagnostics) was used to detect the cytochrome oxidase subunit I.
Apoptosis Detection
Low molecular weight DNA from apoptotic cells was isolated and detected as described (Grimm and Leder 1997). Apoptosis induction was measured by an ELISA (Roche Diagnostics) specific for nucleosomal DNA fragments that are released during apoptosis. The recommendations of the manufacturer were followed. Equal transfection efficiencies were monitored by cotransfecting an expression plasmid for GFP. Other apoptosis quantifications were performed by flow cytometry (Bitzer et al. 1999). A cotransfected GFP expression plasmid was used to assess the transfection efficiency. The apoptotic population was put in relation to the percentage of GFP-positive cells. The apoptotic background of the vector control was subtracted to obtain the specific apoptosis induction. Each condition was tested in at least three independent experiments.
Mitochondrial Membrane Gradient Detection
HeLa cells were used for the investigation of the membrane potential of mitochondria because they display a better integrity of internal structures after fixation than 293T cells. After transfecting an expression vector for ANT-1 together with a plasmid for GFP, cells were loaded with Mitotracker RedCMXRos (Molecular Probes, Inc.) according to the suggestions of the manufacturer. Images were documented using a Zeiss Axioplan fluorescence microscope. RedCMXRos fluorescence was exited at 546 nm and emission was imaged at >590 nm. GFP fluorescence was exited at 450–490 nm, and emission was monitored at 515–565 nm.
Results
ANT-1 Induces Cell Death
Using our screen for dominant, apoptosis-inducing genes, we isolated a cDNA whose expression elicited an especially fast and strong apoptosis response in 293T cells. Sequencing revealed that the gene encoded ANT-1, the ADP/ATP translocator protein of the PT pore (Le Quoc and Le Quoc 1988; Halestrap 1991). ANT was active in a wide variety of cell types; every cell line tested so far underwent apoptosis after stimulation by ANT-1 (such as L929, 3T3, HeLa, BHK, MCF-7, and PC3). Fig. 1 shows the phenotype of control- and ANT-1–transfected HEK 293T cells. Whereas the vector-transfected cells displayed a normal morphology, ANT-1–expressing cells showed a constricted cytoplasm and blebbing of the plasma membrane.
Figure 1.

ANT-1 expression leads to phenotypic apoptosis induction. The empty vector or an expression plasmid for ANT-1 were transiently transfected into 293T cells. After 16 h, phase-contrast pictures were taken at a 200-fold magnification.
To assess the temporal relation between the expression of ANT-1 and apoptosis induction, we transfected 293T cells with ANT-1 and investigated apoptosis induction at different time points. Fig. 2 A shows an increase in cell death from 5 to over 50% after 20 h. This correlated with the accumulation of the HA-tagged ANT-1 protein specifically in mitochondrial fractions of the cells (Fig. 2 B). Cytochrome c oxidase was used as a marker protein for the efficient separation of cytoplasmic and mitochondrial protein fractions.
Figure 2.
Time course of apoptosis induction and protein expression after ANT-1 transfection. (A) Temporal increase of apoptosis induced by ANT-1 expression. 1 μg of the HA-tagged ANT-1 plasmid was transfected into 293T cells for each condition. Quantitative FACS analysis of sub-G1–positive cells was performed at the indicated time points. The specific apoptosis induction above background is shown as a percentage of apoptotic cells relative to all transfected cells. The means and the SDs are given for each experiment. (B) Transfected ANT-1 progressively accumulates in mitochondria. Cytoplasmic and mitochondrial cell fractions were prepared from aliquots taken from the samples under A. 35 μg of each were subjected to SDS-PAGE and subsequent immunoblot analysis using an anti–HA tag antibody to specifically detect the transfected ANT-1. The membrane was stripped and reprobed with an mAb against subunit I of cytochrome c oxidase (COX I) to demonstrate the purity of the mitochondrial preparations.


Since ANT-1 is a component of the PT pore whose activation leads to a dissipation of the proton gradient ΔΨm over the inner mitochondrial membrane (Le Quoc and Le Quoc 1988; Halestrap 1991), we tested ANT-1–transfected cells for this membrane gradient. To this end, we cotransfected ANT-1 and an expression vector for GFP into HeLa cells. The membrane gradient in transfected and, therefore, GFP-positive cells was assayed with CMXRos, a dye whose fluorescence is dependent on an intact membrane potential. Fig. 3 A shows that ANT-1– expressing cells displayed a reduced mitochondrial fluorescence with this dye. This differential staining is a very early feature of ANT-1–mediated apoptosis induction since it could be observed in cells that did not yet display an altered phenotype.
Figure 3.
Effect of ANT-1 expression on mitochondria. (A) ANT-1 overexpression leads to the collapse of the inner mitochondrial membrane potential. HeLa cells were transfected with an expression vector for GFP (2 μg) together with a control vector (Vector, 2 μg) or a plasmid for ANT-1 (ANT-1, 2 μg). After 16 h, the cells were treated with CMXRos, which stains mitochondria with an intact membrane potential. Subsequently, phase-contrast and fluorescence microscopy pictures for GFP and CMXRos activity were taken. (B) ANT-1 induces the release of cytochrome c from mitochondria. 293T cells were transfected with a control vector or an expression vector for ANT-1. Cytoplasmic extracts were prepared and assayed for the presence of cytochrome c (cyt c) by a Western blot. Molecular mass standards are given on the left. Equal loading of the gel is indicated by an upper unspecific band with equal intensity.


One of the consequences of mitochondrial damage is the release of cytochrome c from the inner mitochondrial membrane space (Kluck et al. 1997; Yang et al. 1997). Therefore, we assayed the cytoplasm of ANT-1–transfected cells for the presence of cytochrome c. Using a Western blot, we could detect this protein in the cytoplasmic fraction of ANT-1–transfected cells (Fig. 3 B). The released cytochrome c can bind Apaf-1, a mammalian homologue of the Caenorhabditis elegans gene ced-4 that leads to the aggregation and activation of downstream caspases (Srinivasula et al. 1998). A substrate for caspases is ICAD (inhibitor of caspase-activated DNase [CAD]) (Sakahira et al. 1998). Its degradation leads to the activation of CAD and the internucleosomal degradation of the DNA, a well-known biochemical sign of apoptosis. To investigate the status of the DNA, we transfected 293T cells with a control vector or ANT-1 and isolated the low molecular weight DNA. Only ANT-1–transfected cells displayed the typical DNA ladder (Fig. 4 A). We also investigated the status of PARP, one of the best known caspase substrates, in ANT-1–transfected cells. A Western blot for PARP (Fig. 4 B) revealed that ANT-1–transfected cells degraded PARP and generated the expected protein fragments. Since we have also observed apoptosis induction in MCF-7 cells (data not shown), a cell line that does not express caspase-3 (Janicke et al. 1998), we conclude that ANT-1–induced apoptosis is not dependent on this particular isoenzyme. According to a recent report (Marzo et al. 1998a), Bax, a member of the Bcl-2 gene family, interacts directly with ANT-1 and is also known to induce apoptosis (Oltvai et al. 1993). However, caspase activation does not seem to be a necessary event for Bax-induced cell death (Xiang et al. 1996). Therefore, we wanted to know whether caspase activation is required for ANT-1 apoptosis induction. To address this, we cotransfected ANT-1 and an expression vector for p35, a broad-range caspase inhibitor from baculovirus (Clem and Miller 1994). A quantitative apoptosis assay revealed a sixfold reduction in apoptosis induction when p35 was cotransfected (Fig. 4 C). These results show that ANT-1 leads to all phenotypic and biochemical alterations associated with apoptosis.
Figure 4.
Effect of ANT-1 on protein and DNA degradation. (A) ANT-1 induces internucleosomal DNA cleavage. 293T cells were transfected with an empty vector or with an expression construct for ANT-1. Low molecular mass DNA was isolated from transfected cells, separated on a 2% agarose gel, and stained with ethidium bromide. (B) ANT-1 leads to the degradation of PARP. 293T cells were transfected with an empty control vector, an expression vector for the death domain protein RIP or with ANT-1. 16 h later, nuclear extracts of the transfected cells were prepared and investigated for the status of PARP in a Western blot. The generated PARP fragment is indicated by an arrow. An unspecific signal (o) served as an internal control for equal loading of the gel. (C) Inhibition of ANT-1 apoptosis by a specific inhibitor for caspases. ANT-1 (1 μg) and a control vector (10 μg) or an expression vector (10 μg) for the caspase inhibitor p35 from baculovirus were cotransfected, and the specific apoptosis induction was determined by FACS analysis. The means and the SDs are indicated (n = 3).



The NH2-terminal Half of ANT-1 Is Sufficient for Apoptosis Induction
ANT-1 was originally described as an ADP/ATP exchange factor (Riccio et al. 1975). To assess whether the transport activity of ANT-1 is required for its apoptosis induction, we generated six different point mutations, all of which have been shown to impede ANT-1's activity to transport ADP and ATP (Muller et al. 1996; Muller et al. 1997). However, upon transfection into 293T cells, we have not observed any difference relative to the wild type (WT) in their potential to induce apoptosis as measured by the release of internucleosomal DNA fragments (Fig. 5 A).
Figure 5.
Mutational analysis of ANT-1's apoptosis activity. (A) Cell death induction by point mutants of ANT-1. 1 μg of wild-type ANT-1 (ANT-1 WT) and six point mutants that have been shown to be deficient for ADP/ATP transport were transfected into 293T cells. The wild-type amino acid, its position, as well as the mutated residue are given for each mutant construct. After 16 h, apoptosis induction by the various constructs was measured by ELISA for internucleosomal DNA fragments. The DNA fragmentation as a percentage of control is given as an index for apoptosis induction. The means and the SDs are indicated (n = 3). (B) Apoptosis activity of ANT-1 deletion mutants. Wild-type ANT-1 (1 μg) or three COOH-terminal deletion mutants (1 μg) were transiently transfected into 293T cells. The last COOH-terminal residue of each deletion mutant is indicated. For the ANT-1 WT and each mutant, the structure of the generated protein is depicted schematically with the membrane indicated as a shaded area. After 16 h, the constructs were tested for apoptosis induction by a specific ELISA as described in A.


ANT-1 is a member of the mitochondrial carrier family that consists of proteins with three 100-residue repeats comprising alternating regions of hydrophilic and hydrophobic residues. As integral membrane proteins, they span the membrane six times, interrupted by short amphipathic helices that are likely to form the transport active moiety (Klingenberg and Nelson 1994). To map the domains responsible for apoptosis induction, we have created various deletion mutants of ANT-1: ANT-1Δ 201 lacks the last two transmembrane domains and the last amphipathic helix; ANT-1Δ 142 cuts the molecule in half and comprises only the first three transmembrane domains; and ANT-1Δ 102 contains only the first two membrane spanning domains. Fig. 5 B demonstrates that ANT-1Δ 201 is as efficient for apoptosis induction as WT ANT-1, whereas ANT-1Δ 142 revealed a 50% reduction of its apoptosis potential. The largest deletion, ANT-1Δ 102, was inactive for apoptosis induction.
ANT-2 Is Unable to Induce Apoptosis
The PT pore complex is supposed to function by the conversion of the specific ADP/ATP exchange transporter into an unselective pore (Zoratti and Szabo 1995). However, since over half of the ANT-1 protein is dispensable for apoptosis induction, we wanted to know whether ANT-1 induces apoptosis by forming an unspecific pore on its own. Therefore, we assessed the specificity of apoptosis induction by ANT-1. To this end, we made use of ANT-2, an over 90% identical gene to ANT-1 which is likewise ubiquitously expressed (Doerner et al. 1997a). Transfection of ANT-2 did not lead to apoptosis induction as measured by fluorescence-activated cell sorter (FACS) analysis (Fig. 6 A). The correct mitochondrial localization and efficient expression of ANT-2 was verified by Western blot (Fig. 6 B).
Figure 6.
Effect of ANT-1 and ANT-2 on transfected cells. (A) Comparison between the apoptotic capacities of ANT-1 and ANT-2. Expression plasmids for ANT-1 (1 μg) and ANT-2 (1 μg) were transfected in 293T cells. After 16 h, apoptosis induction in transfected cells was measured by FACS analysis of sub-G1–positive cells. The specific apoptosis induction above background is shown as a percentage of apoptotic cells relative to all transfected cells. The means and the SDs are indicated (n = 3). (B) Expression and localization of ANT-1 and ANT-2. 293T cells were transfected with a control vector or expression vectors for HA-tagged ANT-1 and ANT-2. Mitochondrial extracts of transfected cells were prepared and investigated for the presence of the proteins with an anti–HA antibody in a Western blot. Equal loading of the gel was verified by two unspecific upper bands.


Growth of Yeast Cells Is Not Affected by ANT-1
Bax, a member of the Bcl-2 gene family, can induce a form of cell death by overexpression in yeast cells (Greenhalf et al. 1996; Zha et al. 1996; Jurgensmeier et al. 1997), probably by activating ANT-1 and the PT pore (Greenhalf et al. 1996; Marzo et al. 1998a). Therefore, we wanted to determine whether ANT-1 has the same capacity. To address this, we subcloned ANT-1 and Bax into a galactose- and raffinose-inducible yeast vector. The induction of these genes on suitable agar plates revealed that Bax led to a reduction in the growth of these cells, indicating cell death. In contrast, ANT-1–expressing yeast cells were completely unaffected and grew as well as on the control plate (Fig. 7 A). A Western blot revealed the efficient expression of Bax and ANT-1 in yeast cells upon induction with galactose and raffinose. (Fig. 7 B)
Figure 7.
Effect of Bax and ANT-1 expression on the growth of yeast cells. (A) Bax but not ANT-1 leads to growth inhibition in yeast. Bax and ANT-1 cloned in expression vectors under the control of a galactose and raffinose-inducible promoter and the empty control vector were introduced into yeast cells. 4.5 × 104 yeast cells of three independent clones each were plated on glucose (glu)- or galactose and raffinose (gal/raf)–containing agar. The growth of the yeast cells was monitored 36 h later. (B) Control of inducible expression of ANT-1 and Bax in yeast cells. Yeast clones from A were kept in normal medium or incubated in galactose and raffinose–containing medium for 8 h. Extracts were prepared, and equal amounts (40 μg) of proteins were investigated for the expression of Bax and ANT-1 in a Western blot. The antiserum against ANT-1 does also recognize the endogenous yeast ANT-1.


Cyclophilin D Can Repress Apoptosis Induced by ANT-1
Our experiments with ANT-2 and cell death in yeast suggested that ANT-1's apoptotic activity is mediated by specific protein–protein interactions rather than by forming an active PT pore on its own. Nevertheless, to check whether ANT-1 expression activates the endogenous PT pore, we made use of bongkrekic acid, a specific inhibitor for the PT pore (Klingenberg et al. 1970). 50 μM of this compound repressed ANT-1–induced apoptosis in a quantitative apoptosis assay more than twofold (Fig. 8 A). These findings together with the previous data from the yeast experiments led us to speculate that ANT-1 might titrate out some components of the endogenous PT pore and that replenishing these proteins might repress apoptosis. Consequently, we cotransfected ANT-1 and an expression vector for cyclophilin D, a component of the PT pore on the matrix side that directly interacts with ANT-1 (Woodfield et al. 1998). Quantification of apoptosis induction revealed a more than twofold reduced cell death by ANT-1 when cyclophilin D was cotransfected (Fig. 8 B).
Figure 8.

Inhibition of ANT-1–induced apoptosis. (A) ANT-1–induced apoptosis can be repressed by bongkrekic acid, a specific inhibitor of the PT pore. An expression vector for ANT-1 (1 μg) was transfected into plates with 293T cells. One group of plates was left untreated, the other was supplied with 50 μM bongkrekic acid (BA) for 16 h after the transfection. Apoptosis was quantified as in Fig. 5. (B) ANT-1 apoptosis can be repressed by cyclophilin D, another component of the permeability transition pore. ANT-1 (1 μg) and an expression plasmid (10 μg) for cyclophilin D (Cyclo D) or a control vector (10 μg) were transfected into 293T cells, and apoptosis was determined as described in Fig. 6.
Discussion
In this study, we have described the dominant activity of ANT-1, a central component of the PT pore in mitochondria, to induce apoptosis upon overexpression. This is the first report that shows such an activity of a component of the PT pore in the inner mitochondrial membrane. These findings have important consequences for the normal state of the cell: ANT-1 is already one of the most abundant proteins in mitochondria and makes up ∼10% of all proteins in the inner mitochondrial membrane (Klingenberg 1993). This means that its dominant activity for apoptosis induction must be repressed by other proteins in a nonapoptotic cell (see below).
In addition, ANT-1 is specifically and dramatically upregulated in heart tissue of patients with DCM (Schultheiss et al. 1996; Doerner et al. 1997b). DCM is a myocardial disease of unknown etiology that is characterized by a reduced myocardial contractility and ventricular dilatation. Its estimated prevalence is 36 in 100,000 and, therefore, represents a major cause of congestive heart failure (Dec and Fuster 1994). Interestingly enough, excessive apoptosis can be observed in tissues from DCM patients (Narula et al. 1996; Yao et al. 1996; Yamamura et al. 1999). Even though different tissues with distinct ANT isoform levels might be differentially sensitive to ANT-1 expression, we would like to propose that this pathological apoptosis induction is mediated by the increase in ANT-1. Therefore, our studies might lead to an explanation and possibly to a treatment of this disease.
Our initial hypothesis for the apoptosis induction by ANT-1 was that it is was caused by its known function of shuttling ATP and ADP over the mitochondrial membrane. However, the apoptosis potential of ANT-1 seems not to be dependent on its activity to transport ADP and ATP, because a panel of point mutants that are inactive for this transport could still induce apoptosis (Fig. 5). While the transfected ANT-1 can, therefore, be impaired for nucleotide exchange, the ADP/ATP transport activity of the endogenous ANT-1 might still be relevant for apoptosis induction and be inhibited by the overexpressed ANT-1.
The activation of the PT pore for apoptosis induction is thought to be mediated by the PT pore becoming an unspecific pore and releasing apoptogenic proteins and oxygen radicals (Marzo et al. 1998b). Isolated ANT-1 has been shown to form an unspecific, Ca2+-dependent pore (Brustovetsky and Klingenberg 1996; Ruck et al. 1998). Furthermore, ANT-1 can be activated for apoptosis induction by atractyloside, a plant glucoside that binds to ANT-1, stabilizes a specific conformation, and favors pore opening (Zoratti et al. 1982; Davidson and Halestrap 1987). Therefore, our second hypothesis was that overexpressed ANT-1 forms such an unspecific pore and thereby reproduces an activated PT pore. However, several lines of evidence suggest that overexpressed ANT-1 does not form an unspecific pore. First, deletion studies with ANT-1 showed that the COOH-terminal half of the molecule, including two transmembrane domains, is dispensable for apoptosis induction (Fig. 5 B). It is hard to imagine how such a truncated molecule could still form a membrane channel. Second, ANT-2 was inactive for apoptosis induction (Fig. 6). Since this protein is over 90% identical to ANT-1, it should also be capable of assembling into an unspecific pore. Thirdly, if ANT-1 overexpression was sufficient to form an unspecific channel, one would expect that ANT-1 could induce cell death also in yeast. However, our results show that ANT-1 was inactive in yeast (Fig. 7).
Therefore, we would like to propose a model (Fig. 9) in which ANT-1, rather than forming an activated PT pore by itself, stimulates the endogenous PT pore for apoptosis induction. This is achieved by titrating out inhibitors of this complex that activate the PT pore and induce apoptosis. One of these inhibitors could be cyclophilin D, which we have shown to be able to repress ANT-1–induced apoptosis (Fig. 8 B). Alternatively, cyclophilin D might also mask an interaction domain in ANT-1 for another protein that is a repressor for the PT pore. Therefore, this protein is not titrated out any more and the PT pore is not activated.
Figure 9.
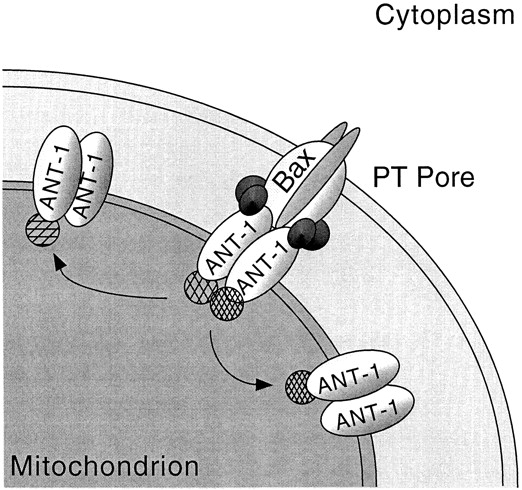
Hypothetical model for ANT-1's apoptosis-inducing activity. The PT pore complex is depicted with its different components in the outer and inner mitochondrial membrane. According to the model, ANT-1 can interact with proteins that repress the PT pore (indicated as hatched circles) and with proteins that facilitate apoptosis (indicated in dark shading). Overexpressed ANT-1 proteins (shown on the right and left) do not form an activated PT pore on their own, but rather titrate out the repressors of the endogenous PT pore that activate the PT pore. For this activation, additional proteins are needed that are not titrated out by hyperexpressed ANT-1 because they need additional interaction partners for their affinity to ANT-1. See text for a more detailed discussion of this model.
This interpretation of our results suggests that the stoichiometry of the proteins in the PT pore complex is an important factor for its inactive state. The inner mitochondrial membrane would, therefore, mirror the condition in the outer membrane where the relative abundance of pro- and anti-apoptotic proteins (e.g., the Bcl-2 family members) determines apoptosis induction (Korsmeyer 1995). According to this model, ANT-1, instead of forming an unspecific pore, would mediate apoptosis by specific protein–protein interactions. Different affinities for these proteins could, therefore, account for ANT-2's inactivity to induce apoptosis.
Other proteins in our model would facilitate apoptosis through ANT-1. Their existence is postulated because isolated ANT proteins require the presence of Ca2+ and, therefore, cannot function as an unspecific pore on their own (Brustovetsky and Klingenberg 1996; Ruck et al. 1998). These activators might not be titrated out by ANT-1 because their affinity to ANT-1 is dependent on the interaction with other proteins like, for example, Bax. Our data from yeast might be explained by assuming that the interaction of ANT-1 with the inhibitors is not conserved between yeast and mammals, and that, therefore, these inhibitors are not titrated out. Alternatively, or in addition, the inducer proteins in the mammalian complex are not present in yeast or their interaction is regulated differently.
Based on previous studies with Bax (Oltvai et al. 1993) and on this report about ANT-1, both proteins can induce apoptosis upon overexpression. It was further shown that they interact directly, and that the activation of ANT-1 for apoptosis by the plant glucoside atractyloside required the presence of Bax and vice versa (Marzo et al. 1998a). However, there are interesting differences in their capacity for apoptosis induction: Bax can induce a form of cell death in yeast, whereas ANT-1 is inactive (Fig. 7). As no Bcl-2–like gene and, therefore, no Bax homologue is found in the yeast genome, Bax might represent ANT-1's missing interaction partner in yeast. The association between Bax and ANT has been shown to be mediated by residues 105–156 of ANT (Marzo et al. 1998a). These sequences are also required for ANT-1 apoptosis induction (Fig. 5). This direct Bax–ANT interaction might suggest that Bax is activated by ANT-1 expression. However, Bax-induced apoptosis does not seem to rely on caspase activation, even though this protease family is activated when Bax is overexpressed (Xiang et al. 1996). ANT-1, on the other hand, requires caspase activation as shown through the inhibition by baculovirus p35 (Fig. 4 C). Furthermore, and most importantly, even though ANT-2 has been shown to interact with Bax (Marzo et al. 1998a), it is unable to induce apoptosis (Fig. 6). In addition, Bax might not be available for interaction with the transfected ANT-1 in the cell because the endogenous ANT-1 is already a very abundant protein and might complex all endogenous Bax. For these reasons, we believe that the transfected ANT-1 does not activate the endogenous Bax, and that overexpressed ANT-1 induces apoptosis in a different way than atractyloside-activated ANT. Taken together, ANT-1's apoptosis induction as described in this report shows how a protein that performs the essential biological function to facilitate the presence of ATP in the cytoplasm can also, under certain conditions such as in DCM, turn into a protein that elicits a deadly signal for the cell.
Acknowledgments
We would like to thank U. Cramer and F. Neudecker for excellent technical assistance, Dr. M. Klingenberg for the anti–ANT-1 serum, Dr. J.A. Duine for providing bongkrekic acid, A. Kohlhuber for suggestions concerning the manuscript, and the Bavarian Government and Roche Diagnostics for their support.
Footnotes
M.K.A. Bauer and A. Schubert contributed equally to this work.
Abbreviations used in this paper: ANT-1, adenine nucleotide translocase-1; CAD, caspase-activated DNase; DCM, dilated cardiomyopathy; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; HA, hemagglutinin; ICAD, inhibitor of caspase-activated DNase; PARP, poly-ADP-ribose polymerase; PT pore, permeability transition pore.
References
- Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith A., Struhl K. Current Protocols in Molecular Biology. Wiley Interscience; New York: 1991. [Google Scholar]
- Bitzer M., Prinz F., Bauer M., Spiegel M., Neubert W.J., Gregor M., Schulze-Osthoff K., Lauer U. Sendai virus infection induces apoptosis through activation of caspase-8 (FLICE) and caspase-3 (CPP32) J. Virol. 1999;73:702–708. doi: 10.1128/jvi.73.1.702-708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N., Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ . Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- Clem R.J., Miller L.K. Control of programmed cell death by the baculovirus genes p35 and iap. Mol. Cell. Biol. 1994;14:5212–5222. doi: 10.1128/mcb.14.8.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.M., Halestrap A.P. Liver mitochondrial pyro-phosphate concentration is increased by Ca2+ and regulates the intramitochondrial volume and adenine nucleotide content. Biochem. J. 1987;246:715–723. doi: 10.1042/bj2460715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dec G.W., Fuster V. Idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1994;331:1564–1575. doi: 10.1056/NEJM199412083312307. [DOI] [PubMed] [Google Scholar]
- Doerner A., Pauschinger M., Badorff A., Noutsias M., Giessen S., Schulze K., Bilger J., Rauch U., Schultheiss H.P. Tissue-specific transcription pattern of the adenine nucleotide translocase isoforms in humans FEBS (Fed. Eur. Biochem. Soc.) Lett. 414 1997. 258 262a [DOI] [PubMed] [Google Scholar]
- Doerner A., Schulze K., Rauch U., Schultheiss H.P. Adenine nucleotide translocator in dilated cardiomyopathypathophysiological alterations in expression and function Mol. Cell Biochem 174 1997. 261 269b [PubMed] [Google Scholar]
- Ferrari D., Stepczynska A., Los M., Wesselborg S., Schulze-Osthoff K. Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95- and anticancer drug-induced apoptosis. J. Exp. Med. 1998;188:979–984. doi: 10.1084/jem.188.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S., Scaffidi C., Susin S.A., Krammer P.H., Kroemer G., Peter M.E., Debatin K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998;273:33942–33948. doi: 10.1074/jbc.273.51.33942. [DOI] [PubMed] [Google Scholar]
- Gawaz M., Douglas M.G., Klingenberg M. Structure-function studies of adenine nucleotide transport in mitochondria. II. Biochemical analysis of distinct AAC1 and AAC2 proteins in yeast. J. Biol. Chem. 1990;265:14202–14208. [PubMed] [Google Scholar]
- Graham F.L., Smiley J., Russell W.C., Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Greenhalf W., Stephan C., Chaudhuri B. Role of mitochondria and C-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in Saccharomyces cerevisiae . FEBS (Fed. Eur. Biochem. Soc.) Lett. 1996;380:169–175. doi: 10.1016/0014-5793(96)00044-0. [DOI] [PubMed] [Google Scholar]
- Grimm S., Leder P. An apoptosis-inducing isoform of neu differentiation factor (NDF) identified using a novel screen for dominant, apoptosis-inducing genes. J. Exp. Med. 1997;185:1137–1142. doi: 10.1084/jem.185.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A.P. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of low pH against chemical and hypoxic cell damage. Biochem. J. 1991;278:715–719. doi: 10.1042/bj2780715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke R.U., Sprengart M.L., Wati M.R., Porter A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Krajewski S., Armstrong R.C., Wilson G.M., Oltersdorf T., Fritz L.C., Reed J.C., Ottilie S. Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe . Mol. Biol. Cell. 1997;8:325–339. doi: 10.1091/mbc.8.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. Dialectics in carrier researchthe ADP/ATP carrier and the uncoupling protein. J. Bioenerg. Biomembr. 1993;25:447–457. doi: 10.1007/BF01108402. [DOI] [PubMed] [Google Scholar]
- Klingenberg M., Nelson D.R. Structure-function relationships of the ADP/ATP carrier. Biochim. Biophys. Acta. 1994;1187:241–244. doi: 10.1016/0005-2728(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Klingenberg M., Grebe K., Heldt H.W. On the inhibition of the adenine nucleotide translocation by bongkrekic acid. Biochem. Biophys. Res. Commun. 1970;39:344–351. doi: 10.1016/0006-291x(70)90582-6. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Korsmeyer S.J. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- Le Quoc K., Le Quoc D. Involvement of the ADP/ATP carrier in calcium-induced perturbations of the mitochondrial inner membrane permeabilityimportance of the orientation of the nucleotide binding site. Arch Biochem. Biophys. 1988;265:249–257. doi: 10.1016/0003-9861(88)90125-7. [DOI] [PubMed] [Google Scholar]
- Loo D.T., Copani A., Pike C.J., Whittemore E.R., Walencewicz A.J., Cotman C.W. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jurgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis Science 281 1998. 2027 2031a [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Susin S.A., Beutner G., Brdiczka D., Remy R., Xie Z.H., Reed J.C., Kroemer G. The permeability transition pore complexa target for apoptosis regulation by caspases and bcl-2-related proteins J. Exp. Med. 187 1998. 1261 1271b [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy J.V., Dixit V.M. Apoptosis induced by Drosophila reaper and grim in a human system. Attenuation by inhibitor of apoptosis proteins (cIAPs) J. Biol. Chem. 1998;273:24009–24015. doi: 10.1074/jbc.273.37.24009. [DOI] [PubMed] [Google Scholar]
- McGill G. Apoptosis in tumorigenesis and cancer therapy. Front. Biosci. 1997;2:353–379. doi: 10.2741/a197. [DOI] [PubMed] [Google Scholar]
- Muller V., Basset G., Nelson D.R., Klingenberg M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations of oxidative phosphorylation and AAC expression. Biochemistry. 1996;35:16132–16143. doi: 10.1021/bi960667r. [DOI] [PubMed] [Google Scholar]
- Muller V., Heidkamper D., Nelson D.R., Klingenberg M. Mutagenesis of some positive and negative residues occurring in repeat triad residues in the ADP/ATP carrier from yeast. Biochemistry. 1997;36:16008–16018. doi: 10.1021/bi971867l. [DOI] [PubMed] [Google Scholar]
- Narula J., Haider N., Virmani R., DiSalvo T.G., Kolodgie F.D., Hajjar R.J., Schmidt U., Semigran M.J., Dec G.W., Khaw B.A. Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- Newmeyer D.D., Farschon D.M., Reed J.C. Cell-free apoptosis in Xenopus egg extractsinhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Oltvai Z.N., Milliman C.L., Korsmeyer S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit P.X., Lecoeur H., Zorn E., Dauguet C., Mignotte B., Gougeon M.L. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 1995;130:157–167. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio P., Aquila H., Klingenberg M. Purification of the carboxy-atractylate binding protein from mitochondria. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1975;56:133–138. doi: 10.1016/0014-5793(75)80127-x. [DOI] [PubMed] [Google Scholar]
- Roussel M.F., Rettenmier C.W., Look A.T., Sherr C.J. Cell surface expression of v-fms-coded glycoproteins is required for transformation. Mol. Cell. Biol. 1984;4:1999–2009. doi: 10.1128/mcb.4.10.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruck A., Dolder M., Wallimann T., Brdiczka D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1998;426:97–101. doi: 10.1016/s0014-5793(98)00317-2. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Schreiber E., Matthias P., Muller M.M., Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss H.-P., Schulze K., Dörner A. Significance of the adenine nucleotide translocator in the pathogenesis of viral heart disease. Mol. Cell. Biochem. 1996;163:319–327. doi: 10.1007/BF00408672. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Ahmad M., Fernandes-Alnemri T., Alnemri E.S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prevost M.C., Alzari P.M., Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process J. Exp. Med. 189 1999. 381 394a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor Nature 397 1999. 441 446b [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandel N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- Woodfield K., Ruck A., Brdiczka D., Halestrap A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., Chao D.T., Korsmeyer S.J. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T., Nakamura H., Yamamoto T., Umemoto S., Fujii T., Kobayashi N., Matsuzaki M. Fas expression and apoptosis correlate with cardiac dysfunction in patients with dilated cardiomyopathy. Jpn. Circ. J. 1999;63:149–154. doi: 10.1253/jcj.63.149. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yang X., Chang H.Y., Baltimore D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science. 1998;281:1355–1357. doi: 10.1126/science.281.5381.1355. [DOI] [PubMed] [Google Scholar]
- Yao M., Keogh A., Spratt P., dos Remedios C.G., Kiessling P.C. Elevated DNase I levels in human idiopathic dilated cardiomyopathyan indicator of apoptosis? J. Mol. Cell Cardiol. 1996;28:95–101. doi: 10.1006/jmcc.1996.0009. [DOI] [PubMed] [Google Scholar]
- Zamzami N., Marchetti P., Castedo M., Decaudin D., Macho A., Hirsch T., Susin S.A., Petit P.X., Mignotte B., Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death J. Exp. Med. 182 1995. 367 377a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Marchetti P., Castedo M., Zanin C., Vayssiere J.L., Petit P.X., Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo J. Exp. Med. 181 1995. 1661 1672b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Marzo I., Susin S.A., Brenner C., Larochette N., Marchetti P., Reed J., Kofler R., Kroemer G. The thiol crosslinking agent diamide overcomes the apoptosis-inhibitory effect of Bcl-2 by enforcing mitochondrial permeability transition. Oncogene. 1998;16:1055–1063. doi: 10.1038/sj.onc.1201864. [DOI] [PubMed] [Google Scholar]
- Zha H., Fisk H.A., Yaffe M.P., Mahajan N., Herman B., Reed J.C. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell. Biol. 1996;16:6494–6508. doi: 10.1128/mcb.16.11.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M., Szabo I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Zoratti M., Pietrobon D., Azzone G.F. On the relationship between the rate of ATP synthesis and H+ electrochemical gradient in rat-liver mitochondria. Eur. J. Biochem. 1982;126:443–451. doi: 10.1111/j.1432-1033.1982.tb06800.x. [DOI] [PubMed] [Google Scholar]