

Abstract
Protein disulfide isomerase (PDI) interacts with secretory proteins, irrespective of their thiol content, late during translocation into the ER; thus, PDI may be part of the quality control machinery in the ER. We used yeast pdi1 mutants with deletions in the putative peptide binding region of the molecule to investigate its role in the recognition of misfolded secretory proteins in the ER and their export to the cytosol for degradation. Our pdi1 deletion mutants are deficient in the export of a misfolded cysteine-free secretory protein across the ER membrane to the cytosol for degradation, but ER-to-Golgi complex transport of properly folded secretory proteins is only marginally affected. We demonstrate by chemical cross-linking that PDI specifically interacts with the misfolded secretory protein and that mutant forms of PDI have a lower affinity for this protein. In the ER of the pdi1 mutants, a higher proportion of the misfolded secretory protein remains associated with BiP, and in export-deficient sec61 mutants, the misfolded secretory protein remain bounds to PDI. We conclude that the chaperone PDI is part of the quality control machinery in the ER that recognizes terminally misfolded secretory proteins and targets them to the export channel in the ER membrane.
Keywords: protein disulfide isomerase, endoplasmic reticulum-associated degradation, endoplasmic reticulum quality control, BiP, yeast
Secretory proteins are targeted to the ER of mammalian cells and are translocated into the ER lumen through a channel formed by the Sec61 protein complex (Andrews and Johnson 1996; Hanein et al. 1996). In the ER, secretory protein folding, covalent modification, and appropriate oligomerization are prerequisites for the packaging of these proteins into ER-to-Golgi complex transport vesicles (Hurtley and Helenius 1989). Misfolded secretory proteins are recognized by the quality control machinery in the ER and reexported to the cytosol in a Sec61p-dependent fashion, indicating that protein import and export across the ER membrane may be mediated by the same channel (Wiertz et al. 1996b; Pilon et al. 1997). In the cytosol, misfolded secretory proteins are degraded by the proteasomes (Hiller et al. 1996; Werner et al. 1996; Wiertz et al. 1996a).
The mechanism of recognition of proteins destined for degradation is poorly understood (Cresswell and Hughes 1997; Suzuki et al. 1998). Chaperones that facilitate protein folding in the ER are capable of distinguishing folded and unfolded proteins (Simons et al. 1995; Hendershot et al. 1996) and are, therefore, in a good position to recognize proteins as terminally misfolded and to target them for export from the ER. Using a cell-free assay that faithfully reproduces ER export and degradation of a mutant secretory protein, McCracken and Brodsky 1996 initially demonstrated that the ER chaperone calnexin is required for export. In addition, the authors recently showed that mutations in the ER lumenal chaperone BiP also interfere with export of misfolded secretory proteins from the yeast ER (Brodsky et al. 1999).
Protein disulfide isomerase (PDI) is a major ER-resident protein with multiple functions (Gilbert 1997). As an enzyme, PDI catalyzes the formation and breakage of disulfide bonds (oxidoreductase) and rearranges preexisting disulfide bonds (isomerase; Gilbert 1997). The isomerase activity of PDI is essential in the yeast Saccharomyces cerevisiae (Laboissiere et al. 1995). Kemmink et al. 1997 analyzed the domain structure of human PDI and suggested that it consists of active (a and a′) and inactive (b and b′) thioredoxin modules (Fig. 1 a). In addition to its enzymatic activities, mammalian PDI can chaperone the refolding of disulfide-free, denatured proteins in vitro (nonenzymatic chaperone activity; Gilbert 1997), and both mammalian and yeast PDI can bind to peptides in the ER lumen (Welply et al. 1985; LaMantia et al. 1991; Klappa et al. 1997). Recently, it has been shown that mammalian PDI also binds to secretory proteins late during protein translocation into the ER and that, like BiP, purified mammalian PDI has a higher affinity for unfolded than for correctly folded proteins, irrespective of their disulfide content (Klappa et al. 1995, Klappa et al. 1997; Hendershot et al. 1996).
Figure 1.
Expression of PDI mutants Δ222–302 and Δ252–277. a, Structures of wild-type S. cerevisiae PDI and deletion mutants Δ222–302 and Δ252–277. b, Mutant PDI proteins are overexpressed. Equal amounts of wild-type and mutant microsomal protein (20 μg/lane) were resolved on a 7.5% SDS gel, and transferred to nitrocellulose. PDI wild-type and mutant proteins, and BiP were detected by incubation with the respective antibodies (1:500 for PDI, 1:1,000 for BiP) 125I-labeled protein A, and PhosphorImaging. c, A fraction of wild-type and mutant PDI proteins are underglycosylated. Cells were pulse-labeled with 35S-methionine/cysteine for 15 min at 30°C, lysed, and PDI immunoprecipitated. Immunoprecipitates were incubated in the absence or presence of endoglycosidase H for 4 h at 37°C. Immunoprecipitated proteins were resolved on a 7.5% SDS gel and detected by PhosphorImaging.



We created two deletions in the central region of yeast PDI that contains a stretch of acidic amino acids with similarity to the peptide binding region identified in mammalian PDI (LaMantia and Lennarz 1993; Noiva et al. 1993). We found that these PDI deletion mutants displayed decreased affinity for a synthetic glycosylation acceptor peptide. We subsequently investigated the effects of our mutations on forward and retrograde secretory protein transport from the ER lumen; the pdi1 deletion mutants specifically affected export of a sulfhydryl-free misfolded protein from the ER to the cytosol. While wild-type PDI efficiently recognized this thiol-free misfolded protein, the PDI mutant proteins had a significantly lower affinity for this substrate. As a consequence, misfolded secretory proteins retained in the lumen of mutant microsomes remained associated with BiP. In sec61 mutants deficient in retrograde transport from the ER, a high proportion of misfolded secretory proteins was bound to PDI. We conclude that PDI is a component of the quality control machinery in the ER that recognizes misfolded proteins and targets them for export to the cytosol via the Sec61 channel.
Materials and Methods
Strains and Growth Conditions
MLY200a (MATa ade2-1 can1-100 ura3-1 leu2-3, 112 trp1-1 his3-11, 15 pdi1::HIS3 pPDI1 URA3; LaMantia and Lennarz 1993) was used as PDI wild-type strain. Plasmids containing pdi1 deletion mutants (pRS-Δ222–302, pRS-Δ252–277) were exchanged for the plasmid containing wild-type PDI1 by plasmid shuffling on 5-fluoroorotic acid (5-FOA; Sikorski and Boeke 1991). The prc1-1 allele was introduced into these strains by crossing with W303-1C (MATα ade2-1 can1-100 ura3-1 leu2-3, 112 trp1-1 his3-11, 15 prc1-1; Knop et al. 1996a), and PEP4 replaced with URA3 using one-step gene replacement with pTS15 (Tachibana and Stevens 1992). WCG4a (MATa leu2-3, -112 ura3 his3-11, -15) and WCG4-11/22a (pre1-1 pre2-2, same as WCG4a; Hiller et al. 1996) were used for cytosol preparations. RSY1293 (MATα can1-100 leu2-3, -112 his3-11,-15 trp1-1 ura3-1 ade2-1 sec61::HIS3 [pDF40], HindIII–StyI fragment of SEC61 in pRS316), RSY1294 (as RSY1293, except [psec61-32]), and RSY1295 (as RSY1293, except [psec61-41]) were used for SEC61 wild-type and mutant microsomes. W303-1B pCT37 is MLY200 (pGAL1-PDI1) and was a kind gift from Tom Stevens (University of Oregon at Eugene, OR; Tachibana and Stevens 1992). MLY200 (pS1S2pdi1) is described in Luz and Lennarz 1998. Media were purchased from Difco Inc. Yeast cells were grown in YPDA (1% yeast extract, 2% peptone, 2% glucose, 20 mg/ml adenine sulfate), YPGA (1% yeast extract, 2% peptone, 2% galactose, 20 mg/ml adenine sulfate), or synthetic media with the appropriate additions (Sherman 1991).
Construction of pdi1 Deletion Mutants
DNA manipulations were performed according to Sambrook et al. 1989. Plasmids pUC18-PDI and pGX-PDI (pGEX3X-PDI; LaMantia and Lennarz 1993) were used as the source for the PDI1 gene. Mutagenesis and subcloning: pGX-Δ222–302 was obtained by sequential digestion of pGX-PDI with NaeI and EcoRV, followed by ligation of the 4.2-kb fragment with the 2.1-kb fragment. pUC18-PDI was digested with EcoRV and NaeI, and the 4.2-kb fragment was ligated to the 2.1-kb fragment, yielding pUC18-Δ222–302. This vector was digested with EcoRI and BamHI and the 2.2-kb cassette was ligated into the corresponding sites of pRS314, thereby producing pRS-Δ222–302. pGX-Δ252–277 was generated by PCR, using the ExSite kit from Stratagene, pGX-PDI as the template, and the mutagenic primers P12 (5′-ACC GAG TTG GCC AAA AAG AAC-3′) and P14 (5′-TTG GGC GAA AAC GGA ACC GTC-3′). To generate pRS-Δ252–277, the internal HpaI–StuI fragment from pGX-Δ252–277 was inserted into pRS-PDI that had been digested with the same enzymes.
Radiolabeling of Cells and Immunoprecipitation
Wild-type and mutant cells were grown to an OD600 of 0.5–1.0 at 30°C in minimal medium without methionine and cysteine. Cells were concentrated to OD600 = 4/ml and 500 μl aliquots were preincubated at 30°C for 15 min. Radiolabeling was initiated by the addition of [35S]Promix (1,200 Ci/mol; New England Nuclear) to 30 μCi/OD600 of cells and incubation was continued for 10 min. Chase mix was added from a 100× stock solution (0.3% cysteine, 0.4% methionine, 100 μΜ ammonium sulfate) and incubation continued for the indicated periods of time. An equal volume of ice-cold 20 mM NaN3/20 mM Tris-HCl, pH 7.5, was added to the cultures; protein extracts were prepared according to Doering and Schekman 1996; secretory precursors (CPY, invertase) were immunoprecipitated as described (Feldheim et al. 1993) and analyzed on 7.5% polyacrylamide gels. Where indicated (Fig. 1 c), samples were treated with 5 mU endoglycosidase H (Boehringer) for 4 h at 37°C before gel electrophoresis.
Immunoblotting
Microsomal proteins (20 μg) from wild-type and mutant strains were resolved on 7.5% polyacrylamide gels and transferred to nitrocellulose. Blots were incubated with a 1:500 dilution of affinity-purified anti-PDI antibody (R. Freedman, University of Kent at Canterbury, UK), a 1:500 dilution of affinity-purified anti-Sec61p antibody (M. Pilon, University of California at Berkeley), or a 1:1,000 dilution of anti-BiP antiserum (R. Schekman, University of California at Berkeley). Antibody binding was quantitated using 125I-protein A (Nycomed Amersham Inc.) and a PhosphorImager (BioRad).
Peptide Binding
A synthetic glycosylation acceptor peptide, N-benzoyl-NYT-amide, was iodinated using chloramine T as in Römisch and Schekman 1992. Wild-type or pdi1 mutant microsomes (10 μl, 100 μg) were incubated with 7.5 × 106 cpm of peptide in 100 μl B88 for 30 min at 10°C. Membranes were washed and exposed to a longwave UV light for 5 min on ice. Membranes were heated in SDS sample buffer for 5 min, proteins were resolved on 7.5% SDS gels, and peptide–cross-linking products were visualized by autoradiography. Peptide binding to PDI was quantitated using a PhosphorImager (BioRad).
Budding of ER-to-Golgi Complex Transport Vesicles
Permeabilized yeast cells from wild-type and mutant strains were prepared from cultures grown to OD600 = 1 as described (Baker et al. 1988). Cytosol was prepared from WCG4a by liquid nitrogen lysis as described in Pilon et al. 1997. Budding of vesicles from the ER of permeabilized cells containing 35S-labeled glyco-pro-α-factor (gpαf) was done at 24°C, but otherwise as described (Römisch and Schekman 1992). Radiolabeled gpαf in the vesicle and ER-fractions was quantitated by precipitation with concanavalin A (ConA) and scintillation counting.
Endoplasmic Reticulum Degradation Assay
Microsomes were prepared from cells grown to OD600 = 1 as described in Pilon et al. 1997. ER degradation of the nonglycosylated form of pro-α-factor (Δgpαf) was assayed at 24°C as in Pilon et al. 1997. In brief, 20 μl translocation reactions contained 2 μl microsomes of OD280 = 30, B88 (20 mM Hepes, pH 6.8, 150 mM potassium acetate, 5 mM magnesium acetate, 250 mM sorbitol), ATP, and a regenerating system (40 mM creatine phosphate, 0.2 mg/ml creatine phosphokinase, 1 mM ATP, 50 μΜ GDP-mannose) and 2 μl in vitro translated, 35S-labeled pΔgpαf (500,000 cpm). Translocation reactions were incubated for 50 min at 24°C, and the membranes were washed twice in B88. Membranes were resuspended in B88 containing ATP and the regenerating system and degradation reactions were started by adding cytosol to 6 mg/ml final concentration in a 20-μl/reaction final volume. Degradation reactions were incubated at 24°C for the indicated periods of time. At the end of the incubation, samples were precipitated with trichloro-acetic acid and analyzed after electrophoresis on 18% polyacrylamide, 4 M urea SDS gels with a BioRad PhosphorImager.
Cross-linking and Immunoprecipitation
Washed microsomes (20 μl, 200 μg protein) loaded with 20 μl Δgpαf translation as above were resuspended in 100 μl B88, pH 7.4 (20 mM Hepes, pH 7.4, 150 mM potassium acetate, 5 mM magnesium acetate, 250 mM sorbitol). Cytosol (100 μl at 12 mg/ml) was added and DSP (Pierce Chemical Co.) was added to 2.5 mM. Incubation was continued at 24°C for 20 min. Samples were transferred to ice, and ammonium acetate added to 400 mM to quench cross-linking. After 20 min on ice, samples were solubilized by adding SDS to 1% and heating to 65°C for 10 min. Immunoprecipitations were done with 10 μg affinity-purified anti-Sec61p antibody (M. Pilon, University of California at Berkeley), 10 μl anti-PDI serum (T. Stevens, University of Oregon at Eugene, OR) or 8 μl anti-BiP antiserum (R. Schekman, University of California at Berkeley) as described (Lyman and Schekman 1995). Cross-linked immunoprecipitated proteins were resolved on 7.5% polyacrylamide gels or cleaved by heating in sample buffer containing 100 mM DTT and resolved on 18% polyacrylamide, 4 M urea SDS gels. Where appropriate, proteins were subsequently transferred to nitrocellulose and BiP detected with a 1:1,000 dilution of anti-BiP antiserum (R. Schekman, UC Berkeley) and 125I-protein A (Nycomed Amersham Inc.). The anti-PDI antiserum from T. Stevens recognized wild-type and mutant PDI proteins equally well in denaturing immunoprecipitations. On immunoblots, however, this antiserum reacted preferentially with PDI mutant proteins. We therefore obtained affinity-purified anti-PDI antibodies from R. Freedman (University of Kent at Canterbury, UK) for the quantitative immunoblots shown in Fig. 1 b. The constituents of the Δgpαf-BiP cross-link (in Fig. 7 a) were analyzed as follows: after solubilization samples were first immunoprecipitated with a polyclonal anti–α-factor serum; the immunoprecipitated proteins were dissociated from protein A–Sepharose by heating to 90°C in 1% SDS for 5 min, followed by 1:10 dilution in buffer, and immunoprecipitation with anti-BiP antibodies. The precipitates were dissociated and cross-links reduced by heating in sample buffer containing 1% SDS, 100 mM DTT. Proteins were resolved on 7.5% SDS gels and analyzed by silverstaining to visualize BiP, and autoradiography to visualize radiolabeled Δgpαf.
Figure 7.
PDI and BiP are associated in the ER lumen and dissociate in the presence of a folding substrate. a, Wild-type and pdi1 mutant microsomes were incubated in translocation reactions containing pΔgpαf (lanes 1–3) or no translocation substrate (lanes 4–6). At the end of the translocation, DSP cross-linking was performed as described above, followed by solubilization of the membranes, and immunoprecipitation with anti-PDI antiserum. Cross-links in the immunoprecipitate were cleaved with DTT, proteins were resolved on 7.5% SDS gels, and transferred to nitrocellulose membranes. BiP was detected by immunoblotting with anti-BiP antiserum (1:1,000) followed by incubation with 125I-protein A and PhosphorImaging. b, Quantitation of the experiment shown in a.


Structure Prediction for PDI Mutants
We used secondary structure prediction and alignment between mature yeast and human PDI to determine the boundaries of the yeast PDI b and b′ domains. The secondary structure prediction was obtained using the PHD program (Rost and Sander 1993). Alignments were done with the GAP program from the GCG package (Program Manual for the Wisconsin Package, version 8. 1994. Genetics Computers Group). The proposed b domain boundaries, including the loop linking a and b, are amino acids 139–238, the boundaries for the yeast b′ domain, including the loop linking b′ to the a′ domain, are amino acids 241–378. To cross-validate these results, we generated 30 different versions of both the b and the b′ domains, in which their boundaries were gradually increased by one residue. Subsequently, these new domains were aligned with the corresponding domains of human PDI using the GAP program. The results from this analysis are consistent with the yeast b and b′ domain boundaries outlined above.
Next, we mapped the Δ252–277 PDI deletion into the human thioredoxin structure using our b′ domain definition, and mapped the Δ222–302 PDI deletion into two thioredoxin structures using our b and b′ domain definitions, assuming that both yeast b and b′ domains have a thioredoxin fold like the corresponding domains in human PDI (Kemmink et al. 1997). The two thioredoxin structures were docked manually, using the QUANTA package (Molecular Modeling Software Package, 1992). Gross steric overlaps were eliminated by using a 3.8 Å cutoff distance between Cα atom pairs. The docking orientation shown was chosen on the basis that the bonded ends of the two molecules should be close, that the potential b and b′ domain active sites (by analogy with that of thioredoxin) and the free ends of each domain should not be occluded, and that both domains share a reasonable amount of buried surface.
Results
Analysis of PDI Deletion Mutants Δ222–302 and Δ252–277
PDI has both enzymatic (oxidoreductase, disulfide isomerase) and, so far, poorly defined nonenzymatic (chaperone) functions (Gilbert 1997). Both yeast and mammalian PDI also bind to small peptides in the ER lumen (Welply et al. 1985; LaMantia et al. 1991; Klappa et al. 1997). The physiological significance of this interaction is unclear, but the peptide binding domain of mammalian PDI is required for its chaperone function (Dai and Wang 1997). We created two deletions in the central region of yeast PDI that shows homology to the mammalian peptide binding domain (LaMantia and Lennarz 1993) and investigated the effects of these deletions on secretory protein transport from the ER. The structure of pdi1 deletions Δ252–277 and Δ222–302 is illustrated in Fig. 1 a. Mutations were created as described in Materials and Methods and exchanged for wild-type PDI by plasmid shuffling of MLY200a on 5-FOA (Sikorski and Boeke 1991). In rich medium at 30°C, both mutant strains had longer generation times than the wild-type (Δ222–302, 125 min; Δ252–277, 150 min; wild-type, 100 min). We quantified the steady state expression levels of wild-type and mutant PDI proteins by immunoblotting and found that both mutant forms are overexpressed relative to wild-type PDI (2.1× for Δ222–302, 2.2× for Δ252–277; Fig. 1 b). This overexpression of mutant PDI does not lead to the induction of the unfolded protein response, as illustrated by the identical levels of BiP in wild-type and pdi1 mutant cells (Fig. 1 b). Wild-type and mutant PDI proteins migrate as doublets or triplets of bands on SDS gels (Fig. 1 b) that collapse to single bands after endoglycosidase H digestion (Fig. 1 c). Mild underglycosylation of PDI has been observed previously (Ng et al. 1996) and has no effect on PDI functions, per se.
Effect of PDI Deletions on Peptide Binding
Peptide binding to PDI was first observed using small acceptor peptides containing the recognition site for oligosaccharyl transferase (Noiva et al. 1993). To investigate the effect of the deletions in yeast PDI on its capability to bind to small peptides, we prepared microsomes from wild-type and pdi1 mutant strains. Membranes were incubated for 30 min at 10°C in the presence of iodinated N-benzoyl-NYT-amide, a synthetic substrate of oligosaccharyl transferase. Membranes were washed and irradiated with long-wave UV light for 5 min on ice to photo–cross-link the peptide to PDI. Radiolabeled proteins were resolved by SDS gel electrophoresis and visualized by autoradiography. The amount of 125I-benzoyl-NYT-amide bound to PDI was quantified by PhosphorImage analysis. As illustrated in Fig. 2, peptide binding to Δ252–277 PDI was reduced to ∼38% of wild-type, and Δ222–302 PDI showed 14% of peptide binding, compared with wild-type. Comparable peptide binding activity was found with a glycosylation acceptor peptide containing a p-azidobenzoyl-substituted lysyl residue (not shown). Our data indicate that amino acids 252–277 do not encompass the sole peptide binding site of PDI, but suggest that the principal peptide binding domain of yeast PDI is contained within amino acids 222–302.
Figure 2.

Peptide binding to PDI mutants Δ252–277 and Δ222–302 is reduced, compared with wild-type. 125I-labeled glycosylation acceptor peptide, N-benzoyl-NYT-amide, was translocated into PDI wild-type or mutant microsomes as described in Materials and Methods and cross-linked with longwave UV light. All samples were analyzed in duplicate. Proteins were resolved on 7.5% SDS gels and peptide–cross-linking products were visualized by autoradiography. Peptide binding to PDI was quantitated using a PhosphorImager (BioRad).
Effects of PDI Deletions on Secretory Protein Transport from the Endoplasmic Reticulum
The essential function of yeast PDI is the isomerization of disulfide bonds in the ER (Laboissiere et al. 1995). Mutations in the enzymatically active sites of PDI (CGHC; Fig. 1 a) lead to the retention of disulfide-containing secretory proteins in the ER, but do not affect proteins without disulfide bonds (LaMantia and Lennarz 1993; Holst et al. 1997). The physiological substrates for the chaperone function of PDI are not known. We investigated the effects of the pdi1 deletion mutants Δ252–277 and Δ222–302 on the transport of several proteins from the ER in intact yeast cells and in a permeabilized cell-based in vitro system.
The vacuolar protease carboxypeptidase Y (CPY) is synthesized as a prepro-protein whose signal sequence is cleaved after translocation into the ER (Feldheim et al. 1993). In the ER lumen, pro-CPY is N-glycosylated to the p1 form, and its five disulfide bonds are formed in a PDI-dependent fashion (Tachibana and Stevens 1992; Endrizzi et al. 1994). Correct disulfide bonding is a prerequisite for packaging of CPY into ER-to-Golgi transport vesicles (Tachibana and Stevens 1992), and in mutants deficient in the enzymatic activities of PDI p1CPY is retained in the ER (LaMantia and Lennarz 1993; Holst et al. 1997). In the Golgi complex p1CPY is converted to p2CPY by the addition of outer-chain mannose residues, and upon arrival in the vacuole, p2CPY is proteolytically cleaved to mature CPY (mCPY; Feldheim et al. 1993). To study the effects of our pdi1 deletion mutants on the maturation of CPY, wild-type and pdi1 mutant cells were pulse-labeled with 35S-methionine/cysteine, followed by immunoprecipitation of CPY after various chase periods. Within the 10-min pulse in wild-type PDI1 cells, 58% of CPY reached the vacuole and was converted to mCPY (Fig. 3 a, PDI panel, 0 min chase). The remaining ER (p1) and Golgi (p2) forms were rapidly transported to the vacuole and only mCPY was detected after 30 min of chase (Fig. 3 a). In the Δ222–302 mutant, a slightly lower proportion of CPY reached the vacuole within the labeling period (36%; Fig. 3 a, 0 min chase, Δ222–302 panel), but subsequent transport and maturation of CPY proceeded with kinetics that were comparable to wild-type (Fig. 3 a, PDI vs. Δ222–302). In Δ252–277, 31% of CPY matured within the 10-min pulse. The subsequent transport of p1 and p2CPY to the vacuole and concomitant disappearance of the p1 and p2 bands proceeded with wild-type kinetics, there was no accumulation of the ER-specific p1 form of CPY, and only mCPY was immunoprecipitated after 30 min of chase (Fig. 3 a, Δ252–277 vs. PDI). By contrast, an isogenic strain expressing a mutant PDI in which both cysteines in the NH2-terminal active site had been replaced by serines (S1S2 PDI; Luz and Lennarz 1998), contained mostly p1CPY after a 10-min pulse and displayed a significant delay in maturing CPY (Fig. 3 a, S1S2 PDI). We conclude that PDI deletions Δ252–277 and Δ222–302 do not lead to the retention of disulfide-bonded secretory proteins in the ER, indicating that both mutants have significant enzymatic activities.
Figure 3.
Effects of deletions in PDI on transport through the secretory pathway. a, Wild-type and mutant cells were pulse-labeled for 10 min at 30°C with 35S-methionine/cysteine, followed by chase incubations for the indicated periods of time. At each time point, cells were lysed and CPY immunoprecipitated. Proteins were resolved on a 7.5% SDS gel and visualized by PhosphorImaging. b, SICs were prepared from wild-type cells and PDI deletion mutants. In vitro translated, radiolabeled ppαf was translocated into the ER of these SICs, and cells were incubated in the presence of ATP, an ATP-regenerating system, and 120 μg/25 μl reaction yeast cytosol for the indicated periods of time at 24°C. Budded vesicles containing gpαf were separated from SICs by differential centrifugation; radiolabeled, glycosylated gpαf was isolated from vesicle and SIC fractions by ConA precipitation, and quantified by scintillation counting.


The periplasmic enzyme invertase contains two cysteine residues that do not form a disulfide bond (Jämsä et al. 1994). Invertase is translated as a preprotein that is signal-cleaved and core-glycosylated after translocation into the ER lumen (Feldheim et al. 1993). In the Golgi apparatus, the core-oligosaccharides are elongated heterogeneously to produce the extended outer-chain oligosaccharide (Feldheim et al. 1993). Invertase transport through the secretory pathway was monitored after derepression for 3 h in 0.1% glucose. Cultures were labeled for 10 min and chased for up to 60 min before cell lysis and invertase immunoprecipitation. As in the case of CPY, we found no significant difference in the maturation of invertase in the pdi1 deletion mutants versus wild-type, and no specific accumulation of the core-glycosylated ER form of invertase (data not shown). These results indicate that the overexpression of mutant forms of PDI had no nonspecific negative effects on export of proteins from the ER.
We used a well-characterized in vitro system based on permeabilized yeast cells to assess export of a cysteine-free secretory protein from the ER of wild-type and pdi1 mutant cells. The mating pheromone precursor prepro-α-factor (ppαf) was radiolabeled by in vitro translation in the presence of 35S-methionine and translocated into the ER of permeabilized cells. Upon translocation ppαf is signal-cleaved and glycosylated (gpαf; Römisch and Schekman 1992). In the presence of cytosol and ATP at a physiological temperature vesicles containing gpαf bud off the ER and can be separated from the semi-intact cells (SICs) by differential centrifugation (Baker et al. 1988). The amounts of gpαf in the vesicle and ER fractions were quantified by lectin precipitation with ConA-Sepharose and scintillation counting as described in Römisch and Schekman 1992. As illustrated in Fig. 3 b the kinetics and maximal level of vesicle budding from the ER was indistinguishable in wild-type and Δ222–302 SICs. While maximal export was lower in Δ252–277 (Fig. 3 b, ▪), the initial kinetics of export were identical in both pdi1 mutants and the wild-type (t1/2 = 5 min for each mutant strain, t1/2 = 6 min for wild-type) indicating that there was no specific retention of a cysteine-free secretory protein in the ER of the mutants in vitro.
PDI Deletion Mutants Are Defective in Misfolded Secretory Protein Export to the Cytosol
Misfolded secretory proteins are recognized by a poorly defined quality control machinery in the ER and exported back to the cytosol for degradation by the proteasomes (for review, see Cresswell and Hughes 1997). In the past, we have used a cell-free assay system developed by McCracken and Brodsky 1996 to demonstrate that a mutant form of the secretory protein ppαf, which has its three glycosylation sites mutated (Δgpαf), is transported from the ER lumen to cytosol through a channel containing Sec61p, the core component of the protein translocation channel that is also responsible for protein import into the ER (Pilon et al. 1997). Here, we used this in vitro assay to ask whether membranes derived from our pdi1 deletion mutants were deficient in the export of Δgpαf to the cytosol for degradation. Signal-sequence containing pΔgpαf was translated in vitro and translocated posttranslationally into wild-type and mutant microsomes for 50 min at 24°C, resulting in signal-cleaved Δgpαf in the lumen of the washed membranes (Fig. 4 a, t = 0). Also present in these membranes is the nonspecifically membrane-associated precursor pgpαf that is resistant to washes, but partially accessible to exogenously added protease or cytosolic proteasomes (Fig. 4 a, compare lower mobility bands in t = 0 vs. t = 60-min samples). After translocation was completed, the membranes were washed and incubated with ATP and 6 mg/ml wild-type or proteasome–mutant (pre1 pre2) cytosol at 24°C for the indicated periods of time. As shown in Fig. 4 a, export and degradation of Δgpαf in wild-type microsomes is dependent on the presence of ATP, cytosol, and functional proteasomes in the cytosol (McCracken and Brodsky 1996; Pilon et al. 1997). In PDI1 wild-type microsomes, Δgpαf was exported and degraded with a half-life of 25 min (Fig. 4b and Fig. c). In contrast, Δgpαf was exported from Δ222–302 membranes with a half-life of >60 min (Fig. 4b and Fig. c), and there was no export of Δgpαf from Δ252–277 membranes within the 60 min of incubation time (Fig. 4b and Fig. c). Intact yeast cells expressing pΔgpαf release 40% of the amount of α-factor secreted by wild-type α cells, indicating that retention of the mutant precursor in the ER is not complete (Caplan et al. 1991). Thus, the half-life of Δgpαf in intact cells is determined by both its rate of packaging into ER-to-Golgi complex transport vesicles and its rate of transport to the cytosol and degradation. In good agreement with Caplan et al. 1991, we found in pulse–chase experiments that the half-life of the intracellular Δgpαf in PDI1 wild-type cells was 11 min. The active site mutant S1S2 PDI did not affect the half-life of Δgpαf in intact yeast cells (t1/2 = 10 min). In Δ222–302 cells, however, the half-life of Δgpαf increased to 22 min, and in Δ252–277, its half-life was more than threefold increased (37 min). The kinetics of the release of mature α-factor, however, were identical in wild-type and all pdi1 mutant cells. These data suggest that the observed delay in Δgpαf degradation in our cell-free assay faithfully reflects the situation in intact yeast cells. Export and degradation of Δgpαf was not affected in microsomes containing the active site mutant S1S2 PDI (Fig. 4b and Fig. c), suggesting that reduced enzymatic activities in the deletion mutants were not the primary cause for their failure to export Δgpαf. Our data demonstrate that deletions in the central region of PDI strongly affect the export of a thiol-free misfolded secretory protein from the ER for degradation in the cytosol.
Figure 4.
PDI deletion mutants are defective in export of misfolded secretory proteins from the ER to the cytosol for degradation. a, Export and degradation are dependent on ATP and cytosolic proteasomes. Mutant pΔgpαf was translocated into wild-type microsomes for 50 min at 24°C. Membranes were washed and incubated in the presence or absence of ATP, an ATP-regenerating system, and 6 mg/ml wild-type or proteasome mutant (pre1 pre2) yeast cytosol for the indicated periods of time at 24°C. At each time point, proteins were precipitated with trichloro-acetic acid, and resolved on an 18% polyacrylamide gel containing 4 M urea. Radiolabeled protein was visualized and quantified by PhosphorImaging. A fraction of pΔgpαf (upper band) remains aggregated on the cytosolic face on the microsomes, cannot be removed by buffer washes, and is partially susceptible to proteolysis by cytosolic proteasomes. b, Microsomes were prepared from wild-type cells and pdi1 mutants. Mutant pΔgpαf was translocated into wild-type and mutant microsomes and membranes were incubated in the presence of ATP, an ATP-regenerating system, and 6 mg/ml wild-type yeast cytosol for the indicated periods of time at 24°C. Samples were analyzed as for a. c, The Δgpαf bands from two (wild-type) or four (mutants) degradation experiments performed as in b were quantified using a PhosphorImager (BioRad). Variation at each time point was <10% for wild-type and <5% for the mutants. d, PDI1 wild-type and mutant cells expressing prc1-1 were pulse-labeled for 10 min, chased, and CPY* immunoprecipitated and analyzed as described for CPY in Fig. 3 a. Note that in Δ222–302 and 252–277, an increased proportion of CPY* is transported to the Golgi complex (p2CPY*). p2CPY* is not secreted, and is rapidly degraded in PEP4 wild-type cells, but relatively stable in pep4::URA3 cells (shown here).





In addition, we investigated the effects of deletions in the PDI peptide binding site on export from the ER and degradation of another mutant secretory protein, CPY*, which contains multiple N-linked oligosaccharides and disulfide bonds (Knop et al. 1996a,Knop et al. 1996b). We performed pulse–chase experiments in wild-type and pdi1 mutant cells expressing prc1-1, which encodes CPY*, and found that, in contrast to Δgpαf, CPY* required the enzymatic activity of PDI for export to the cytosol (Fig. 4 d; half-life of CPY* in PDI1 wild-type cells, 14 min; in S1S2 PDI cells, 60 min). Deletions in the peptide binding site of PDI caused increases in half-life to 21 min in Δ222–302, and 28 min in 252–277, respectively (Fig. 4 d). These comparatively moderate defects suggest that additional components of the quality control machinery in the ER are rate-limiting for export of CPY* to the cytosol for degradation.
PDI Interacts Specifically with a Cysteine-free Misfolded Secretory Protein Destined for Export
Secretory proteins translocating into the ER interact initially with the chaperone BiP, which promotes both translocation and early steps of secretory protein folding (Sanders et al. 1992; Simons et al. 1995; Hendershot et al. 1996). Late during translocation and/or after translocation is complete, PDI binds to cysteine-containing secretory proteins and promotes correct disulfide bonding (Bulleid and Freedman 1988). PDI also interacts with thiol-free secretory proteins late in translocation indicative of its chaperone activity (Klappa et al. 1995). We used the amino-group reactive, thiol-cleavable cross-linker dithiobis-(succinimidyl)-propionate (DSP) to examine whether PDI can distinguish between the cysteine-free wild-type and mutant forms of secretory pro-α factor (gpαf and Δgpαf). After 50-min translocation reactions in the presence of ATP, an ATP-regenerating system, and radiolabeled pΔgpαf or ppαf, we treated PDI wild-type microsomes with DSP. Mutant Δgpαf is devoid of oligosaccharyl side chains and yeast PDI is one of the few highly glycosylated ER-resident lumenal proteins. Therefore, we initially subjected cross-linked proteins in Δgpαf-containing membranes to lectin precipitation with ConA-Sepharose and resolved cross-linked proteins on nonreducing 7.5% SDS gels. We lectin-precipitated one major cross-linking product of ∼85 kD and two minor higher molecular weight bands (Fig. 5 a, lane 2) whose identity as PDI cross-linked to Δgpαf was confirmed by immunoprecipitation with anti-PDI antiserum (Fig. 5 a, lane 3, PDIxΔgpαf). The cross-linked Δgpαf in these bands accounts for ∼7% of the Δgpαf in the sample. Anti-ppαf antiserum and anti-PDI antibodies quantitatively precipitated the 85-kD cross-linking product (not shown; and Fig. 5 a, lanes 6 and 7). Unglycosylated, signal-cleaved Δgpαf migrates at ∼19 kD on SDS polyacrylamide gels, and full-length, fully glycosylated PDI migrates at 65 kD, thus the observed 85-kD cross-linking product is the correct size for one molecule of PDI cross-linked to one molecule of Δgpαf. The higher molecular weight smear that is evident in the lectin-precipitate did not contain PDI, indicating that PDI, in contrast to the chaperone BiP, does not interact with misfolded secretory proteins that are part of large aggregates (Fig. 5 a, lanes 2 vs. 3, and Fig. 6 a). The major cross-linking product of PDI and wild-type gpαf migrated at 95 kD, due to the three oligosaccharyl side chains in gpαf (Fig. 5 a, lane 5). Its intensity was eightfold lower than that of the PDI cross-linking product containing mutant Δgpαf (Fig. 5 a, compare PDIxΔgpαf, lane 2, to PDIxgpαf, lane 5). We conclude that PDI is a chaperone that can distinguish between wild-type and misfolded secretory proteins irrespective of their thiol-content.
Figure 5.
PDI specifically interacts with a misfolded, cysteine-free secretory protein. a, Wild-type PDI preferentially binds to Δgpαf. Radiolabeled pΔgpaf (lanes 1–3) or ppαf (lanes 4 and 5) were translocated into wild-type microsomes. Directly after termination of the translocation reaction, DSP cross-linking was performed as described in Materials and Methods, followed by solubilization of the membranes, and precipitation of glycoproteins with ConA-Sepharose or immunoprecipitation with anti-PDI antiserum. Cross-linked proteins were resolved on nonreducing 7.5% SDS gels and visualized by PhosphorImaging. The positions of major cross-linking products are indicated (PDIxΔgpαf and PDIxgpαf). Note that wild-type gpαf is a glycoprotein, thus, both monomeric gpαf (•, lanes 4 and 5) and gpαf dimers (*, lane 5) bind to ConA-Sepharose, whereas Δgpαf contains no oligosaccharyl side chains and, therefore, only binds to ConA-Sepharose if cross-linked to the glycoprotein PDI (lane 2). b, Radiolabeled pΔgpαf was translocated into wild-type and pdi1 mutant microsomes and cross-linked with DSP as described. Cross-linking products of Δgpαf and wild-type or mutant PDI proteins were detected by precipitation with ConA-Sepharose.


Figure 6.
Deletions in PDI result in increased association of misfolded secretory proteins with BiP. a, BiP binds to monomeric and aggregated misfolded secretory proteins. Left, Radiolabeled pΔgpαf (lanes 1 and 2) or ppαf (lanes 3 and 4) were translocated into wild-type microsomes, followed by DSP cross-linking, solubilization, and immunoprecipitation with anti-BiP antibodies. BiP–cross-linked proteins were resolved on nonreducing 7.5% SDS gels and visualized by PhosphorImaging. Note that a fraction of monomeric and dimeric gpαf (•, lanes 3 and 4; *, lane 4) and dimeric Δgpaf (••, lane 2) precipitated nonspecifically with protein A–Sepharose. Right, To reveal the individual constituents of the BiPxΔgpαf cross-linking product, we performed sequential immunoprecipitations of the cross-linked material first with anti-ppαf, then with anti-BiP antiserum. Cross-links were cleaved by incubation in sample buffer containing 100 mM DTT, proteins were resolved on a 7.5% SDS gel, and visualized by silverstaining (lanes 1–3) and autoradiography (lanes 4–6). b, Deletions in PDI result in increased association of misfolded secretory proteins with BiP. Radiolabeled pΔgpαf was translocated into wild-type or pdi1 mutant microsomes as indicated, followed by DSP cross-linking, solubilization, and immunoprecipitation with anti-BiP antibodies (lanes 1–3), or incubation with protein A-Sepharose only (lanes 4–6). To facilitate quantitation of BiP-associated Δgpαf, cross-links were cleaved with DTT before SDS-PAGE on 18% 4 M urea gels.



Next, we asked whether deletions in the central region of PDI had any effects on its ability to interact with misfolded Δgpαf. Cross-linking was performed in wild-type and pdi1 mutant microsomes containing Δgpαf as described, and cross-linked proteins were analyzed by ConA precipitation, rather than immunoprecipitation, to ensure quantitative precipitation. We found that the interaction of Δ222–302 with Δgpαf was substantially lower than that of wild-type PDI with Δgpαf (Fig. 5 b, lanes 1 vs. 2), and the cross-linking product of Δ252–277 and Δgpαf was barely detectable (Fig. 5 b, lane 3). Quantitation of the cross-linking products revealed that the intensity of Δ222–302xΔgpαf was 43%, and Δ252–277xgpαf was ∼24% of wild-type. Taking into account the higher expression levels of mutant vs. wild-type PDI (Fig. 1 b), these percentages are equivalent to a fivefold lower affinity of Δ222–302 and a ninefold lower affinity of Δ252–277 for Δgpαf compared with wild-type. These levels of interaction are in the same range as the amount of wild-type gpαf cross-linked to wild-type PDI. We could not detect any interaction of the PDI mutant proteins with wild-type gpαf, however (data not shown), indicating that the PDI deletion mutants can still differentiate between misfolded and wild-type secretory proteins.
Deletions in PDI Cause Increased Association of Misfolded Secretory Proteins with BiP
During translocation into the ER, secretory proteins interact initially with the chaperone BiP (Sanders et al. 1992). BiP also promotes protein folding in the ER lumen (Simons et al. 1995; Hendershot et al. 1996). Whereas wild-type proteins bind BiP only transiently, misfolded proteins often have prolonged interactions with BiP, resulting in a higher percentage of misfolded proteins associated with BiP at any given time point (Ng et al. 1992; Jämsä et al. 1994; Schmitz et al. 1995; Hendershot et al. 1996). We performed DSP cross-linking experiments in wild-type microsomes containing either wild-type gpαf or mutant Δgpαf, followed by immunoprecipitation with anti-BiP antiserum to reveal the relative amounts of these secretory proteins associated with BiP in the ER lumen. As shown in Fig. 6 a, we observed no significant association of BiP with wild-type gpαf (Fig. 6 a, lane 4), whereas a large fraction (10%) of mutant Δgpαf present in the sample could be cross-linked to BiP (Fig. 6 a, lane 2). In contrast to PDI, BiP also interacted with Δgpαf that was part of high molecular weight aggregates (compare Fig. 5 a, lane 3, to Fig. 6 a, lane 2).
To reveal the individual constituents of the BiPxΔgpαf cross-linking product, we performed sequential immunoprecipitations of the cross-linked material first with anti-ppαf, then with anti-BiP antiserum; cross-links were cleaved by incubation in sample buffer containing 100 mM DTT, proteins resolved on a 7.5% SDS gel, visualized by silverstaining (Fig. 6 a, right, lanes 1–3) and autoradiography (Fig. 6 a, right, lanes 4–6). The principal components of the radiolabeled cross-linking product immunoprecipitated with anti-BiP antibodies shown in Fig. 6 a, right, lane 2, were indeed BiP (Fig. 6 a, right, lane 2) and Δgpαf (Fig. 6 a, right, lane 5).
As BiP interaction precedes PDI interaction with secretory proteins in the ER (Klappa et al. 1995), we subsequently analyzed whether deletions in PDI had any effects on the amount of mutant Δgpαf associated with BiP. Cross-linking with DSP in microsomes was performed as described, and followed by anti-BiP immunoprecipitation. To facilitate quantitation of BiP-associated Δgpαf after immunoprecipitation, we cleaved the cross-linked proteins by reduction with dithiotreitol and resolved Δgpαf on 18% acrylamide gels containing 4 M urea. As shown in Fig. 6 b, increased amounts of Δgpαf were associated with BiP in the PDI deletion mutants compared with wild-type (1.4× in Δ222–302, 2.5× in Δ252–277). These findings suggest that PDI is required to remove terminally misfolded Δgpαf from BiP, or that in the ER, Δgpαf is at equilibrium between binding to BiP and to PDI.
PDI and BiP Are Associated with Each Other and Dissociate in the Presence of a Folding Substrate
Next, we asked whether PDI and BiP interacted with each other directly, and whether their interaction was affected by the presence of newly translocated secretory proteins in the ER lumen. To this end, we translocated pΔgpαf into PDI1 wild-type and mutant microsomes, or incubated the membranes in the absence of secretory precursors, but in the presence of ATP and an ATP-regenerating system for 50 min at 24°C. At the end of the translocation reaction, we cross-linked proteins with DSP as described earlier, lysed the membranes, and immunoprecipitated with anti-PDI antiserum. Cross-linked immunoprecipitates were cleaved with DTT. We resolved PDI and coprecipitated proteins on 7.5% SDS gels, transferred the proteins to nitrocellulose, and immunoblotted for BiP. Bound antibody was revealed by incubation with 125I-labeled protein A followed by PhosphorImaging. As shown in Fig. 7 a, a fraction of wild-type PDI was associated with BiP in the absence of Δgpαf (Fig. 7 a, lane 4). In the presence of newly translocated Δgpαf, BiP and PDI dissociated, and BiP no longer coimmunoprecipitated with anti-PDI antibodies (Fig. 7 a, lanes 4 vs. 1, 10.9-fold reduction; Fig. 7 b). Our in vitro translations routinely contain ∼100 nM pΔgpαf. Assuming a 50% translocation efficiency and taking into account that yeast microsomes, in contrast to microsomes from mammalian secretory tissues, are a mixture of cellular membranes containing <50% ER-derived vesicles, we calculated a 15–20× molar excess of BiP/PDI over Δgpαf in the ER lumen after translocation. The near quantitative dissociation of BiP and PDI in the presence of this substoichiometric amount of Δgpαf suggests that each Δgpαf molecule interacts with multiple BiP/PDI dimers during translocation. Decreasing the pΔgpαf concentration in the translocation reaction to 50 or 25% resulted in partial dissociation of BiP from wild-type PDI, proportional to the amount of Δgpαf present in the ER (not shown). In the absence of a folding substrate, an increased amount of BiP (2.7×) immunoprecipitated with the deletion mutant Δ222–302, compared with wild-type PDI (Fig. 7 a, lanes 5 vs. 4; Fig. 7 b). Δ222–302 is overexpressed 2.1-fold over wild-type PDI (Fig. 1 b), thus, the amount of BiP coprecipitating with this mutant protein was roughly proportional to the amount of mutant PDI present in the ER. Like BiP and wild-type PDI, BiP and PDI Δ222–302 dissociated in the presence of Δgpαf (Fig. 7 a, lanes 5 vs. 2, 4.7-fold decrease; Fig. 7 b), but compared with wild-type PDI, a larger proportion of BiP remained bound to Δ222–302, even in the presence of gpαf (Fig. 7 a, lane 2; Fig. 7 b). Mild overexpression of wild-type PDI1 from the GAL1 promoter likewise resulted in a proportional increase in the amount of BiP associated with PDI (not shown), suggesting that the higher levels of BiP coprecipitating with PDI Δ222–302 reflect the higher concentration of PDI Δ222–302 protein in the ER lumen, and not an increased affinity of this PDI mutant for BiP. Although PDI Δ252–277 is also overexpressed compared with wild-type (2.2-fold; Fig. 1 b), the amount of BiP coimmunoprecipitating with this mutant protein was lower than that coprecipitating with wild-type PDI in the absence of Δgpαf (Fig. 7 a, lanes 6 vs. 4). This indicates that the affinity of Δ252–277 for BiP is reduced ∼2.8-fold compared with wild-type. Furthermore, only a small fraction of PDI Δ252–277 dissociated from BiP in the presence of Δgpαf (Fig. 7 a, lanes 6 vs. 3, 1.3-fold reduction; Fig. 6 b). Our experiments demonstrate that PDI and BiP are bound to each other in the ER lumen and dissociate in the presence of a newly translocated folding substrate. As all translocating secretory proteins are unfolded, dissociation of BiP and PDI should be triggered by newly translocated proteins, irrespective of whether they will be able to fold correctly or not. We indeed found that both Δgpαf and wild-type gpαf caused dissociation of BiP and PDI (not shown). While PDI Δ222–302 displayed a normal dynamic interaction with BiP, PDI Δ252–277 had a reduced affinity for BiP in the absence of a folding substrate, and did not dissociate from BiP appropriately in the presence of folding substrate. It remains to be investigated whether the dynamic interaction of BiP and PDI is a prerequisite for misfolded secretory protein export from the ER, or whether the more severe export defect in Δ252–277 microsomes solely is due to the low affinity of the mutant PDI for misfolded secretory proteins.
Misfolded Secretory Proteins Remain Associated with PDI in Export-deficient sec61 Mutant Microsomes
Transport of misfolded secretory proteins from the ER lumen to the cytosol is dependent on an ER-membrane protein, Sec61p, which is also the principal component of the translocation channel mediating protein import into the ER (Wiertz et al. 1996b; Pilon et al. 1997). Previously, we have described two mutants in SEC61, sec61-32 and sec61-41, which are cold-sensitive for protein import into the ER and defective for misfolded protein export from the ER to the cytosol (Pilon et al. 1997). We argued that in sec61-32 and sec61-41 microsomes, Δgpαf should accumulate at a stage just before export, and if PDI interaction was required at this point, an increased amount of Δgpαf should be associated with PDI in sec61 mutant microsomes. We translocated pΔgpαf into SEC61 wild-type and mutant microsomes prepared from cells grown at the permissive temperature (30°C) as described, cross-linked proteins with DSP, and immunoprecipitated from the lysed membranes with anti-PDI and anti-BiP antibodies. Cross-links were cleaved by incubating the immunoprecipitates in SDS sample buffer containing 100 mM DTT, and proteins resolved on 18% polyacrylamide gels containing 4 M urea. Radiolabeled Δgpαf associated with PDI or BiP was visualized by autoradiography and quantitated using a PhosphorImager. As shown in Fig. 8, in sec61-32 microsomes, which are entirely deficient for protein export to the cytosol (Pilon et al. 1997), there is a dramatic increase in Δgpαf associated with PDI relative to wild-type microsomes (Fig. 8, lane 5 vs. lane 4; 5.1×). In sec61-41 microsomes, which display a partial misfolded protein export defect (Pilon et al. 1997), we observed a 3.2-fold increase in the amount of Δgpαf associated with PDI (Fig. 8, lane 6 vs. lane 4). In both sec61 mutants, we also found moderately increased amounts of Δgpαf bound to BiP (Fig. 8, lanes 11 and 12 vs. lane 10; 2.1× and 1.3×).
Figure 8.

In export-deficient sec61 mutant microsomes, misfolded secretory proteins remain associated with PDI. Radiolabeled pΔgpαf was translocated into SEC61 wild-type and mutant microsomes as described in Materials and Methods and cross-linked with DSP. PDI– and BiP–cross-linked proteins were immunoprecipitated with the respective antibodies. Cross-links were cleaved with DTT, proteins resolved on 18% polyacrylamide gels containing 4 M urea, and radiolabeled Δgpαf associated with PDI or BiP was visualized by autoradiography and quantified using a PhosphorImager. Relative amounts of Δgpαf associated with PDI and BiP are indicated with and without adjustment for increased levels of BiP and PDI due to the unfolded protein response induction in the sec61 mutant microsomes (1.7× for sec61-32; 1.4× for sec61-41).
Accumulation of misfolded proteins in the ER induces the unfolded protein response that leads to an increased synthesis of ER-resident proteins (Sidrauski et al. 1998). The amount of Sec61p in sec61-32 and sec61-41 microsomes is identical to wild-type (Pilon et al. 1997). Quantitative immunoblotting for BiP and PDI, however, revealed that the levels of both proteins were moderately elevated in the ER of the export-deficient sec61 mutants (1.7× in sec61-32; 1.4× in sec61-41). If we factor in the elevated amounts of BiP and PDI into our quantitation of Δgpαf associated with both chaperones, we find that Δgpαf in export-deficient sec61 mutant microsomes accumulated primarily at PDI (Fig. 8, 3.0× for sec61-32; 2.3× for sec61-41). Our data suggest that misfolded secretory proteins destined for export to the cytosol interact sequentially with BiP, PDI, and Sec61p. We were unable to detect a significant direct association of PDI and Sec61p that may indicate that additional ER-resident proteins are required for the transfer of Δgpαf from PDI to the export channel.
Discussion
We have provided genetic and biochemical evidence for a role of PDI in recognition of a misfolded cysteine-free secretory protein in the ER lumen and its export to the cytosol for degradation. Our data define an important novel function of PDI in addition to its established enzymatic activities. Membranes prepared from two new deletion mutants in PDI1, Δ222–302 and Δ252–277, were deficient in a cell-free assay for export of a misfolded secretory protein from the ER to the cytosol for degradation by the proteasomes. The export substrate in this assay system, a mutant form of the yeast pheromone precursor prepro-α factor, Δgpαf, does not contain any cysteine residues, thus all our observations are independent of the enzymatic functions of PDI.
Chaperone-like properties of purified PDI, such as prevention of unfolded protein aggregation, promotion of folding of denatured substrates, and binding to peptides have been described previously (Welply et al. 1985; LaMantia et al. 1991; Dai and Wang 1997; Gilbert 1997; Klappa et al. 1997). The physiological substrates of the chaperone PDI were unknown until now, however, and the importance the chaperone activity of PDI in the cell was not understood. Noiva and coworkers mapped the peptide binding site of mammalian PDI to an acidic region within the COOH-terminal 50 amino acids of the protein, a region distinct from the enzymatically active sites (Fig. 1 a; Noiva et al. 1993). A purified truncation mutant of mammalian PDI, from which this peptide binding region has been deleted, no longer promotes the folding of denatured substrate proteins (Dai and Wang 1997). The central part of S. cerevisiae PDI (amino acids 252–277) is rich in acidic amino acids and shows weak homology to the peptide binding region of mammalian PDI (LaMantia and Lennarz 1993). Therefore, we created deletion mutants in yeast PDI centering around this region to investigate the physiological role of the chaperone function of PDI in yeast. Cells harboring these deletion mutations were viable, and cell growth and transport of proteins through the secretory pathway was only moderately affected (Fig. 3). Neither of our deletion mutants specifically retained secretory proteins in the ER, indicating that both PDI mutant proteins had significant enzymatic activity, and that neither interfered nonspecifically with secretory protein packaging into ER-to-Golgi complex transport vesicles (Fig. 3).
Export from the ER for degradation of disulfide-containing substrate proteins is redox potential dependent, suggesting that disulfide bonds need to be reduced before export (Young et al. 1993; Wilson et al. 1995; Tortorella et al. 1998). Here, we show that mutations in the enzymatically active sites of PDI retard the export and degradation of disulfide-containing CPY*, but not of thiol-free Δgpαf (Fig. 4), suggesting that PDI is responsible for the reduction of disulfide bonds in degradation substrates before export.
CPY* and Δgpαf differ not only with respect to disulfide content: the removal of the N-glycosylation acceptor sites from wild-type α-factor precursor by mutagenesis causes degradation of the resulting Δgpαf (McCracken and Brodsky 1996). In contrast, the presence and the appropriate trimming of the N-linked oligosaccharides of CPY* are essential for its degradation (Knop et al. 1996b; Jakob et al. 1998). Based on these data, Jakob and colleagues 1998 suggested that lectins recognizing specific N-linked oligosaccharide structures may be rate-limiting for export of CPY* to the cytosol. Therefore, it is not surprising that the chaperone function of PDI is not strictly required for CPY* degradation (Fig. 4). Finally, deletion of the gene encoding the ER membrane protein Der1p results in dramatic stabilization of CPY* in the ER (Knop et al. 1996a), but causes only a twofold increase of the half-life of Δgpαf (K. Römisch, unpublished results). Taken together, these data suggest that different subsets of quality control machinery in the ER recognize a variety of features in folding proteins, and subsequently trigger their export either into anterograde transport vesicles or back across the ER membrane to the cytosol.
Predicted Domain Rearrangements in the PDI Deletion Mutants
Although both mutant PDI proteins were overexpressed (Fig. 1 b), this did not lead to the induction of the unfolded protein response in the mutant cells as indicated by the comparable levels of BiP expression in wild-type and mutant cells (Fig. 1 b), suggesting that these mutant forms of PDI are not grossly misfolded. In all our experiments, however, the pdi1 mutant carrying the smaller deletion (Δ252–277) displayed stronger functional defects than the mutant with the larger deletion (Δ222–302; Fig. 4, Fig. 5 b, 6 b, and 7). Human PDI consists of two enzymatically active (a and a′) and two inactive (b and b′) thioredoxin modules that are arranged in the order a-b-b′-a′ (Kemmink et al. 1997). The boundaries of the yeast a and a′ domains have been identified by multiple sequence alignment (Kanai et al. 1998). We determined the domain boundaries of the yeast PDI b and b′ domains by secondary structure prediction and alignment of human and yeast PDI, as described in Materials and Methods (Fig. 9 a). Using these domain boundaries, we mapped the yeast PDI b and b′ domains into the structure of human thioredoxin and determined the location of our deletions Δ252–277 and Δ222–302 with respect to these PDI domains (Fig. 9 a). We then graphically analyzed the potential changes in packing between the newly generated interfaces in the deletion mutants. As shown in Fig. 9 b in red, Δ222–302 removes the last α helix from the b domain, and the first four helices and three beta strands from the b′ domain. In this deletion mutant, the COOH-terminal end of the truncated b domain is physically close to, and on the same side of the molecule as the new NH2-terminal residue of the truncated b′ domain. This suggests that the deletion of amino acids 222–302 causes loss of a substantial fraction of the b′ domain, but does not cause a significant domain rearrangement (Fig. 9 b). Deletion of amino acids 252–277 removes the second half of the first helix, the first beta sheet, and the first half of the second helix from the b′ domain (Fig. 9, a and c). The distance between the new terminal ends is large (>27 Å; Fig. 9 c), and the ends are located on opposite faces of the molecule (Fig. 9 c). Therefore, this deletion is predicted to introduce a rotation of the COOH-terminal part of PDI (truncated b′ domain and a′ domain) with respect to the NH2-terminal part (a and b domains, Fig. 9 a).
Figure 9.
Predicted structure of PDI deletion mutants. a, Domain structure of mature yeast PDI and relative positions of deletions. Each rectangle in PDI corresponds to a thioredoxin fold. Domain boundaries are indicated and were determined as described in Materials and Methods. The domain boundaries for b and b′ include the loops linking them to domains a and a′, respectively. Regions shaded in gray correspond to deleted amino acids in the b and b′ domains. b, Predicted structure of Δ222–302. The deleted amino acids were mapped into two thioredoxin structures using the b and b′ domain definitions and assuming the b′ domain has a thioredoxin fold. The two thioredoxin structures were docked manually, as described in Materials and Methods. Deleted residues are in red and remaining residues are in green. The figure shown on the bottom is identical to the one shown on top, but tilted 90° towards the viewer. c, Predicted structure of Δ252–277. Deleted residues (red) were mapped into the thioredoxin structure using the b′ domain boundary definitions described in the text. These figures were obtained using the QUANTA package (molecular modeling software package, 1992).


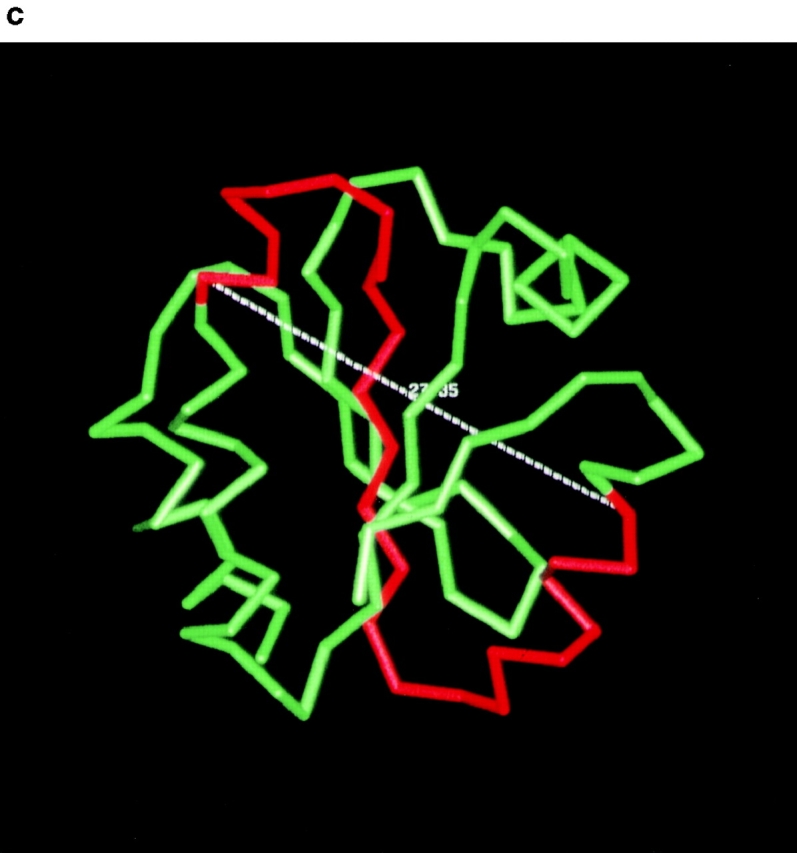
The substantial domain rearrangement in PDI Δ252–277, but not in PDI Δ222–302, is the most likely explanation for the stronger functional defects displayed by Δ252–277, with respect to misfolded secretory protein binding and export, and its failure to interact dynamically with BiP (Fig. 4 b and c, 5 b, 6 b, and 7). While our work was in progress, Klappa et al. 1998 published a paper characterizing the misfolded protein and peptide binding domains of human PDI. The authors found that the human PDI b′ domain is essential, but not sufficient, for the binding of misfolded proteins, and that other domains contribute to protein binding (Klappa et al. 1998). Our data suggest that the orientation of the PDI thioredoxin modules, with respect to each other, is an important factor for the efficient interaction of PDI with other proteins (folding substrates and BiP), and that deletion of >50% of the yeast b′ domain in Δ222–302 has a less deleterious effect than the domain rearrangement in Δ252–277. Klappa and colleagues also defined the b′ domain as the principal binding site for small peptides to human PDI (Klappa et al. 1998). In good agreement with their observation, we found that the binding of a small glycosylation acceptor peptide to our yeast PDI deletion mutants was inversely proportional to the fraction of the b′ domain deleted, i.e., Δ252–277 displayed reduced, but still significant, peptide binding activity, compared to wild-type (38%; Fig. 2), whereas peptide binding to Δ222–302 was substantially weaker (13% of wild-type; Fig. 2).
Interaction and Cooperation of Chaperones in the ER Lumen
Transient interactions of PDI with cysteine-free secretory proteins late during translocation into the ER have been observed previously (Klappa et al. 1995; Oliver et al. 1997). In addition, Klappa et al. 1997 demonstrated that misfolded, but not native, proteins competed with the binding of purified PDI to peptides (Klappa et al. 1997). This competition was independent of the thiol-content of the competing proteins, and suggested that PDI can distinguish between folded and misfolded proteins, like the chaperone BiP. Our cross-linking data confirm this notion (Fig. 5 and Fig. 6, a). We found, however, that in contrast to BiP, PDI does not interact with misfolded secretory proteins that are part of large aggregates (compare Fig. 5 a, lane 3, and 6 a, lane 2), but rather binds misfolded proteins in their monomeric form only (Fig. 5 a, lane 3). One of the functions of BiP may be the dissociation of single molecules from protein aggregates. After dissociation, the unfolded protein could productively interact with PDI and other chaperones and ultimately reach its native conformation.
Both of the PDI deletion mutants had significantly lower affinity for misfolded secretory proteins (Fig. 5 b) and resulted in an increase of misfolded secretory protein associated with BiP (Fig. 6 b). Therefore, we propose an equilibrium of folding substrates binding to and dissociating from BiP and a second equilibrium reaction coupled to the first of binding to and dissociation from PDI. Under these circumstances, a decreased affinity of PDI for the misfolded protein would lead to the observed increase of substrate bound to BiP (Fig. 6 b).
In the absence of a newly translocated secretory protein (i.e., a folding substrate), a fraction of BiP and PDI were associated with each other in wild-type cells (Fig. 7 a, lane 4). Upon translocation of a secretory protein into the ER, BiP and PDI dissociated and no longer coimmunoprecipitated (Fig. 7 a, lane 1, and b). PDI Δ222–302 behaved similarly, but the higher protein levels and lower substrate affinity of the mutant PDI led to a higher level of PDI–BiP complexes, even in the presence of substrate (Fig. 7 a, lanes 2 vs. 5, and b). In contrast, PDI Δ252–277 displayed decreased affinities for both BiP and secretory proteins, thus the levels of PDI–BiP complexes in these mutant strains were low and refractory to the presence of newly translocated secretory proteins (Fig. 7 a, lanes 3 vs. 6, and b). Cross-linking in our experiments was performed for 20 min at 24°C in the absence of ATP. If we cross-linked in the presence of ATP, we found that a higher proportion of BiP coprecipitated with PDI in the presence of Δgpαf in both wild-type and Δ222–302 microsomes, but not Δ252–277 microsomes (data not shown). Thus, the reassociation of BiP and PDI after completion of folding and substrate release was ATP-dependent. Folding of secretory proteins chaperoned by BiP requires ATP and cycles of association and dissociation of BiP and its folding substrates (Hendershot et al. 1996). ATP binding to BiP causes a conformational change in the protein that leads to release of its folding substrate (Wei et al. 1995). PDI can subsequently bind to BiP in the ATP-bound, substrate-free state (Fig. 7 a, lane 4; and data not shown). This complex formation between chaperones in the ER may have evolved to ensure concerted action in the presence of folding substrates and to prevent unproductive folding attempts in their absence.
Gating the Export Channel
Hamman and colleagues recently showed that BiP is identical with the lumenal permeability barrier of the protein import channel in the ER membrane, and that formation of a tight seal by BiP is dependent on the presence of nucleotide (ATP or ADP; Hamman et al. 1998). We and others have demonstrated previously that the core components of this protein-conducting channel, Sec61p, is also required for misfolded protein export from the ER (Wiertz et al. 1996b; Pilon et al. 1997). Therefore, it is likely that the protein import and export channels in the ER membrane are identical and that the protein conducting channels responsible for export also are sealed by BiP on the lumenal side. Opening of the lumenal channel gate during protein import is dependent upon, and directly mediated by, a functional signal sequence in the incoming secretory protein (Jungnickel and Rapoport 1995; Plath et al. 1998). The nature of the export signal and the mechanism by which the export channel is opened from the ER lumen are unknown so far. Our data suggest a possible mechanism: misfolded secretory proteins may be targeted for export in their PDI-bound form. PDI can interact with nucleotide-bound BiP (Fig. 7, and data not shown), the form of BiP that seals the protein translocation channel (Hamman et al. 1998). Thus, the PDI-export substrate complex may bind to BiP at the export channel and cause BiP dissociation from the lumenal side of the channel. This would allow insertion of the misfolded protein into the channel, and ultimately result in its export to the cytosol for degradation. In microsomes derived from sec61-32 and sec61-41 mutants that are defective in misfolded secretory protein export, misfolded Δgpαf accumulates at the lumenal face of the export sites (Pilon et al. 1997) and binds to PDI (Fig. 8), suggesting that PDI is indeed involved in targeting of Δgpαf to the export channel. Other components of the quality control machinery in the ER may similarly bind to their misfolded substrate proteins and target them to the Sec61 channel.
A Model for Initiation of Misfolded Protein Export from the ER to the Cytosol
We propose the following series of events during and after pΔgpαf entry into the ER. Unfolded secretory precursor in the ER lumen interacts with BiP–PDI complexes and causes their rapid dissociation (Fig. 10, a and b, step 1). Signal-cleaved Δgpαf subsequently oscillates between free and chaperone-bound states (Fig. 10, a and b, step 2, a and b). In the model we have only depicted BiP and PDI, but, depending on the folding substrate, there may be any number of other chaperones involved, such as calnexin/calreticulin, immunophilins, or Grp94 (Hammond and Helenius 1994; McCracken and Brodsky 1996; Muresan and Arvan 1997; Davis et al. 1998). In the case of wild-type secretory proteins, folding is fast and efficient, and the equilibrium reactions favor the free over the chaperone-bound states of the secretory protein (Fig. 10 a, step 2, a and b). The secretory protein becomes progressively more folded, which leads to a decrease of its residence time on the chaperones until it no longer interacts with BiP and PDI (Fig. 10 a, step 3), and it is packaged into ER-to-Golgi complex transport vesicles (Fig. 10 a, step 4). In contrast, mutant secretory proteins are preferentially bound to chaperones to prevent protein aggregation (Fig. 10 b, step 2 a* and b*). In some way yet to be determined, possibly by exposure of specific hydrophobic residues, PDI recognizes Δgpαf as terminally misfolded, removes it from the folding reactions (Fig. 10 b, step 3*), and targets it for export to the cytosol and degradation (Fig. 10 b, step 4*). In both scenarios, free BiP and PDI reassociate after substrate release. Other chaperones complexed to misfolded substrate proteins may be able to trigger channel opening in a similar fashion.
Figure 10.

Postulated sequence of events after secretory protein translocation into the ER for wild-type and misfolded proteins (see Discussion).
Acknowledgments
We thank Tom Stevens (University of Oregon at Eugene) for strains and anti-PDI antibodies, Howard Riezman (Biozentrum, Basel, Switzerland) for pTS15, Dieter Wolf (University of Stuttgart, Germany) for the prc1-1 allele, Robert Freedman (University of Kent at Canterbury, UK) for anti-PDI antibodies and helpful comments on the manuscript, Randy Schekman (University of California at Berkeley) for anti-CPY, anti-BiP, and anti-Sec61p antibodies, and an anonymous reviewer for ensuring a high degree of quality control.
K. Römisch is a Senior Fellow of The Wellcome Trust (042216). W.J. Lennarz acknowledges the National Institutes of Health for support (GM33184).
Footnotes
Abbreviations used in this paper: Δgpαf, mutant glycosylation site-free pro-α factor; BiP, heavy chain binding protein; CPY, carboxypeptidase Y; CPY*, mutant CPY; ConA, concanavalin A; DSP, dithiobis-(succinimidyl)-propionate; gpαf, glycopro-α factor; mCPY, mature CPY; PDI, protein disulfide isomerase; SICs, semi-intact cells.
References
- Andrews D.W., Johnson A.E. The transloconmore than a hole in the membrane? Trends Biochem. Sci. 1996;21:365–369. [PubMed] [Google Scholar]
- Baker D., Hicke L., Rexach M., Schleyer M., Schekman R. Reconstitution of SEC gene product dependent intercompartmental protein transport. Cell. 1988;54:335–344. doi: 10.1016/0092-8674(88)90196-1. [DOI] [PubMed] [Google Scholar]
- Brodsky J.L., Werner E.D., Dubas M.E., Goeckeler J.L., Kruse K.B., McCracken A.A. The requirement for molecular chaperones during endoplasmic reticulum associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- Bulleid N.J., Freedman R.B. Defective cotranslational formation of disulphide-bonds in protein disulphide-isomerase-deficient microsomes. Nature. 1988;335:649–651. doi: 10.1038/335649a0. [DOI] [PubMed] [Google Scholar]
- Caplan S., Green R., Rocco J., Kurjan J. Glycosylation and structure of the yeast MFα1 α-factor precursor is important for efficient transport through the secretory pathway. J. Bact. 1991;173:627–635. doi: 10.1128/jb.173.2.627-635.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresswell P., Hughes E.A. Protein degradationthe ins and outs of the matter. Curr. Biol. 1997;7:R552–R555. doi: 10.1016/s0960-9822(06)00279-x. [DOI] [PubMed] [Google Scholar]
- Dai Y., Wang C. A mutant truncated protein disulfide isomerase with no chaperone activity. J. Biol. Chem. 1997;272:27572–27576. doi: 10.1074/jbc.272.44.27572. [DOI] [PubMed] [Google Scholar]
- Davis E.C., Broekelmann T.J., Ozawa Y., Mecham R.P. Identification of tropoelastin as a ligand for the 65-kD FK506-binding protein, FKBP65, in the secretory pathway. J. Cell Biol. 1998;140:295–303. doi: 10.1083/jcb.140.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering T.L., Schekman R. GPI anchor attachment is required for Gas1p transport from the endoplasmic reticulum in COP II vesicles. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:182–191. [PMC free article] [PubMed] [Google Scholar]
- Endrizzi J.A., Breddam K., Remingtion S.J. 2.8-Å structure of yeast serine carboxypeptidase. Biochemistry. 1994;33:11106–11120. doi: 10.1021/bi00203a007. [DOI] [PubMed] [Google Scholar]
- Feldheim D., Yoshimura K., Admon A., Schekman R. Structural and functional characterization of Sec66p, a new subunit of the polypeptide translocation apparatus in the yeast endoplasmic reticulum. Mol. Biol. Cell. 1993;4:931–939. doi: 10.1091/mbc.4.9.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert H.F. Protein disulfide isomerase and assisted protein folding. J. Biol. Chem. 1997;272:29399–29402. doi: 10.1074/jbc.272.47.29399. [DOI] [PubMed] [Google Scholar]
- Hamman B.D., Hendershot L.M., Johnson A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell. 1998;92:747–758. doi: 10.1016/s0092-8674(00)81403-8. [DOI] [PubMed] [Google Scholar]
- Hammond C., Helenius A. Folding of VSV G proteinsequential interaction with BiP and calnexin. Science. 1994;266:456–458. doi: 10.1126/science.7939687. [DOI] [PubMed] [Google Scholar]
- Hanein D., Matlack K.E.S., Jungnickel B., Plath K., Kalies K.-U., Miller K.R., Rapoport T.A., Akey C.W. Oligomeric rings of the Sec61p complex induced by ligands required for protein translocation. Cell. 1996;87:721–732. doi: 10.1016/s0092-8674(00)81391-4. [DOI] [PubMed] [Google Scholar]
- Hendershot L., Wei J., Gaut J., Melnick J., Aviel S., Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc. Natl. Acad. Sci. USA. 1996;93:5269–5274. doi: 10.1073/pnas.93.11.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M.M., Finger A., Schweiger M., Wolf D. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin–proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- Holst B., Tachibana C., Winther J.R. Active site mutations in yeast protein disulfide isomerase cause dithiothreitol sensitivity and a reduced rate of proteins folding in the endoplasmic reticulum. J. Cell Biol. 1997;138:1229–1238. doi: 10.1083/jcb.138.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtley S.M., Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- Jakob C.A., Burda P., Roth J., Aebi M. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämsä E., Simonen M., Makarow M. Selective retention of secretory proteins in the yeast endoplasmic reticulum by treatment of cells with a reducing agent. Yeast. 1994;10:355–370. doi: 10.1002/yea.320100308. [DOI] [PubMed] [Google Scholar]
- Jungnickel B., Rapoport T.A. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 1995;82:261–270. doi: 10.1016/0092-8674(95)90313-5. [DOI] [PubMed] [Google Scholar]
- Kanai S., Toh H., Hayano T., Kikuchi M. Molecular evolution of the domain structure of protein disulfide isomerases. J. Mol. Evol. 1998;47:200–210. doi: 10.1007/pl00006377. [DOI] [PubMed] [Google Scholar]
- Kemmink J., Darby N.J., Dijkstra K., Nilges M., Creighton T.E. The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr. Biol. 1997;7:239–245. doi: 10.1016/s0960-9822(06)00119-9. [DOI] [PubMed] [Google Scholar]
- Klappa P., Freedman R.B., Zimmermann R. Protein disulphide isomerase and a lumenal cyclophilin-type peptidyl prolyl cis-trans isomerase are in transient contact with secretory proteins during late stages of translocation. Eur. J. Biochem. 1995;232:755–764. [PubMed] [Google Scholar]
- Klappa P., Hawkins H.C., Freedman R.B. Interactions between protein disulphide isomerase and peptides. Eur. J. Biochem. 1997;248:37–42. doi: 10.1111/j.1432-1033.1997.t01-1-00037.x. [DOI] [PubMed] [Google Scholar]
- Klappa P., Ruddock L.W., Darby N.J., Freedman R.B. The b′ domain provides the principal peptide binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Finger A., Braun T., Hellmuth K., Wolf D. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast EMBO (Eur. Mol. Biol. Organ.) J. 15 1996. 753 763a [PMC free article] [PubMed] [Google Scholar]
- Knop M., Hauser N., Wolf D.H. N-glycosylation affects endoplasmic reticulum degradation of a mutated derivative of carboxypeptidase yscY in yeast Yeast. 12 1996. 1229 1238b [DOI] [PubMed] [Google Scholar]
- Laboissiere M.C.A., Sturley S.L., Raines R.T. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J. Biol. Chem. 1995;270:28006–28009. doi: 10.1074/jbc.270.47.28006. [DOI] [PubMed] [Google Scholar]
- LaMantia M.L., Lennarz W.J. The essential function of yeast protein disulfide isomerase does not reside in its isomerase activity. Cell. 1993;74:899–908. doi: 10.1016/0092-8674(93)90469-7. [DOI] [PubMed] [Google Scholar]
- LaMantia M.L., Miura T., Tachikawa H., Kaplan H.A., Lennarz W.J., Mizunaga T. Glycosylation site binding protein and protein disulfide isomerase are identical and essential for cell viability in yeast. Proc. Natl. Acad. Sci. USA. 1991;88:4453–4457. doi: 10.1073/pnas.88.10.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz J.M., Lennarz W.J. The non-active site cysteine residues of yeast protein disulfide isomerase are not required for cell viability. Biochem. Biophys. Res. Com. 1998;248:621–627. doi: 10.1006/bbrc.1998.8992. [DOI] [PubMed] [Google Scholar]
- Lyman S.K., Schekman R. Interaction between BiP and Sec63p is required for the completion of protein translocation into the ER of Saccharomyces cerevisiae . J. Cell Biol. 1995;131:1163–1171. doi: 10.1083/jcb.131.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken A.A., Brodsky J.L. Assembly of ER-associated degradation in vitrodependence on cytosol, calnexin, and ATP. J. Cell Biol. 1996;132:291–298. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z., Arvan P. Thyroglobulin transport along the secretory pathway. J. Biol. Chem. 1997;272:26095–26102. doi: 10.1074/jbc.272.42.26095. [DOI] [PubMed] [Google Scholar]
- Ng D.T.W., Watowich S.S., Lamb R.A. Analysis in vivo of GRP78-BiP/substrate interactions and their role in the induction of the GRP78-BiP gene. Mol. Biol. Cell. 1992;3:143–155. doi: 10.1091/mbc.3.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D.T.W., Brown J.D., Walter P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell. Biol. 1996;134:269–278. doi: 10.1083/jcb.134.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noiva R., Freedman R.B., Lennarz W.J. Peptide binding to protein disulfide isomerase occurs at a site distinct from the active sites. J. Biol. Chem. 1993;268:19210–19217. [PubMed] [Google Scholar]
- Oliver J.D., van der Wal F.J., Bulleid N.J., High S. Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science. 1997;275:86–88. doi: 10.1126/science.275.5296.86. [DOI] [PubMed] [Google Scholar]
- Pilon M., Schekman R., Römisch K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K., Mothes W., Wilkinson B., Stirling C.J., Rapoport T.A. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 1998;94:795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- Römisch K., Schekman R. Distinct processes mediate glycoprotein and glycopeptide export from the endoplasmic reticulum in S. cerevisiae . Proc. Natl. Acad. Sci. USA. 1992;89:7227–7231. doi: 10.1073/pnas.89.15.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B., Sander C. Prediction of protein structure at better than 70% accuracy. J. Mol. Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular CloningA Laboratory Manual. Cold Spring Harbor Laboratories Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Sanders S.L., Whitfield K.M., Vogel J.P., Rose M.D., Schekman R. Sec61p and BiP directly facilitate polypeptide translocation into the ER. Cell. 1992;69:353–365. doi: 10.1016/0092-8674(92)90415-9. [DOI] [PubMed] [Google Scholar]
- Schmitz A., Maintz M., Kehle T., Herzog V. In vivo iodination of a misfolded proinsulin reveals colocalized signals for BiP binding and for degradation in the ER. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:1091–1098. doi: 10.1002/j.1460-2075.1995.tb07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sidrauski C., Chapman R., Walter P. The unfolded protein responsean intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S., Boeke J.D. In vitro mutagenesis and plasmid shufflingfrom cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- Simons J.F., Ferro-Novick S., Rose M.D., Helenius A. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J. Cell Biol. 1995;130:41–49. doi: 10.1083/jcb.130.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Yan Q., Lennarz W.J. Complex, two-way traffic of molecules across the membrane of the endoplasmic reticulum. J. Biol. Chem. 1998;273:10083–10086. doi: 10.1074/jbc.273.17.10083. [DOI] [PubMed] [Google Scholar]
- Tachibana C., Stevens T.H. The yeast EUG1 gene encodes an endoplasmic reticulum protein that is functionally related to protein disulfide isomerase. Mol. Cell. Biol. 1992;12:4601–4611. doi: 10.1128/mcb.12.10.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella D., Story C.M., Huppa J.B., Wiertz E.J.H.J., Jones T.R., Ploegh H.L. Dislocation of type I membrane proteins from the ER to the cytosol is sensitive to changes in redox potential. J. Cell Biol. 1998;142:365–376. doi: 10.1083/jcb.142.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Gaut J.R., Hendershot L.M. In vitro dissociation of BiP-peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. J. Biol. Chem. 1995;270:26677–26682. doi: 10.1074/jbc.270.44.26677. [DOI] [PubMed] [Google Scholar]
- Welply J.K., Shenbagamurthi P., Naider F., Park H.R., Lennarz W.J. Active-site directed photoaffinity labeling and partial characterization of oligosaccharyltransferase. J. Biol. Chem. 1985;260:6459–6465. [PubMed] [Google Scholar]
- Werner E.D., Brodsky J.L., McCracken A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradationan unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz E.J.H.J., Jones T.R., Sun L., Bogyo M., Geuze H.J., Ploegh H.L. The human cytomegalovirus US11 gene product dislocates MHC class I chains from the endoplasmic reticulum to the cytosol Cell. 84 1996. 769 779a [DOI] [PubMed] [Google Scholar]
- Wiertz E.J.H.J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T.R., Rapoport T.A., Ploegh H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction Nature. 384 1996. 432 438b [DOI] [PubMed] [Google Scholar]
- Wilson R., Allen A.J., Oliver J., Brookman J.L., High S., Bulleid N.J. The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem. J. 1995;307:679–687. doi: 10.1042/bj3070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J., Kane P.L., Exley M., Wileman T. Regulation of selective protein degradation in the endoplasmic reticulum by redox potential. J. Biol. Chem. 1993;268:19810–19818. [PubMed] [Google Scholar]