

Summary
KcsA is a homotetrameric 68-kDa membrane-associated potassium channel which selectively gates the flux of potassium ions across the membrane. The channel is known to undergo a pH-dependent open-to-closed transition. Here we describe an NMR study of the monomeric subunit of the channel (KcsAM), solubilized in SDS micelles. Chemical shift, solvent exchange, backbone 15N relaxation and residual dipolar coupling (RDC) data show the TM1 helix to remain intact, but the TM2 helix contains a distinct kink, which is subject to concentration-independent but pH-dependent conformational exchange on a microsecond time scale. The kink region, centered at G99, was previously implicated in gating of the tetrameric KcsA channel. An RDC-based model of KcsAM at acidic pH orients TM1 and the two helical segments of the kinked TM2 in a configuration reminiscent of the open conformation of the channel. Thus, the transition between states appears to be an inherent capability of the monomer, with the tetrameric assembly exerting a modulatory effect upon the transition which gives the channel its physiological gating profile.
Keywords: membrane proteins, potassium channel, NMR, RDC, solvent exchange
1. Introduction
The KcsA potassium channel is a 68 kDa homotetrameric membrane protein charged with selective conduction of K+ ions across the cell membrane [1]. The structures of KcsA and homologous channels have been solved by X-ray crystallography [2], and KcsA-toxin complex structures have been determined using solid-state [3] and high-resolution NMR [4]. Each of the four KcsA subunits consists of two transmembrane (TM1 and TM2) helices and a helical pore domain. A 5-residue segment (residues 75TVGY79G) between the pore and TM2 helices forms the selectivity filter, whose backbone carbonyl atoms form a coordination cage which allows K+ ions exclusively to pass through the channel [2]. Further studies established that the cytoplasmic N- and C-terminal domains, removed for crystallization purposes, contain helical segments as well [5]. Channel gating has been the subject of extensive research, and molecular explanations for gating have focused on two KcsA domains. One is the selectivity filter, where the K+-dependent backbone conformation of the filter and pH-dependent changes in the pore helix have been shown to influence the open-to-closed transition [6–8]. The other is located at the convergence of the four inner TM2 helices on the cytoplasmic side of the channel, where a pH-sensitive “switch” has been shown [9–11]. A comparison between KcsA structures representing the open and closed states demonstrates that channel opening involves a conformational change in the TM2 helix which lines the ion pore [12]. It is well established that in liposomes this inner helix adopts the closed conformation when exposed to basic (≥ 7) pH values and the open conformation at acidic (≤ 5) pH values [13, 14].
High-resolution NMR has established itself as a valuable method in studying the structure [15–18] and dynamics [19] of membrane associated proteins. Large membrane proteins still challenge solution NMR due to the size of the micelle-protein assembly and difficulties in preparing homogeneous samples [17, 20]. Nevertheless, NMR does present the unique advantage of studying proteins under conditions that are close to their natural environment, and also is applicable to the study of low-affinity complexes [21, 22] as well as unfolded or partially unfolded proteins [23, 24]. Previously we demonstrated the potential of solution NMR for obtaining both structural data [25] as well as quantitative information on rate and amplitudes of backbone motions taking place on a ps-ns time scale [26] for KcsA. Here we focus on the 17-kDa monomeric subunit of KcsA (KcsAM) with the intent of gleaning additional structural information by studying the basic building block of the channel. We have acquired chemical shifts, solvent exchange, backbone 15N relaxation, and RDC data for KcsAM solubilized in SDS micelles. The transmembrane helical domains of tetrameric KcsA are preserved in KcsAM, but the PORE helix becomes labile and fully exposed to solvent. The TM2 helix is kinked and contains a 6-residue segment that undergoes relatively rapid conformational exchange that is pH-dependent but independent of sample concentration, suggesting a correlation with the pH-induced open-to-closed transition in tetrameric KcsA, which hinges upon this segment. In negatively charged, radially compressed gels the two TM helices are encapsulated in a single micelle and align as a single body. An RDC-based model of KcsAM at acidic pH orients TM1 and the two helical segments of the kinked TM2 in a configuration reminiscent of the open conformation of the channel. The NMR data suggest that the monomer has the inherent capability to sample the conformations of both the open and closed KcsA states, with the tetrameric assembly modulating this equilibrium to maintain the desired kinetics of ion transport.
2. Methods
NMR samples
The N-terminal deletion mutant KcsA potassium channel (residues 16–160) was expressed, isotopically labeled and purified as previously described [25, 27]. Monomeric KcsA (KcsAM) was prepared by incubating the KcsA tetramer (KcsATET) in 50mM formate buffer pH 3.0 containing 150 mM SDS, at 55 °C for 2 hours, followed by purification on a size-exclusion column equilibrated with the buffer of choice. Fractions containing KcsAM were concentrated down to 280–330 μL and placed in a Shigemi cell. Typical samples contained 0.5–0.6 mM KcsAM solubilized in SDS, with the SDS:KcsAM molar ratio maintained at 300–400:1, and were buffered with 25 mM formate at pH 4.2, 25 mM MES at pH 6.0, or 20 mM Tris at pH 8.0.
For RDC measurements KcsAM was aligned in polyacrylamide (PA) stretched gels as described in previous studies [28–31]. Briefly, the gel was polymerized from a solution containing 4.9% (w/v) acrylamide (AA), 0.1% (w/v) 2-(acrylamido)2-methyl-1-propanesulfonic acid (AMPS), 0.17% (w/v) bis(acrylamide) (BIS), 0.1% (w/v) ammonium persulfate, 0.1% (v/v) tetramethylethylenediamine (TEMED) in 100mM Tris buffer at pH 8.0. The total concentration of acrylamide monomers and crosslinker was 5.2%. The solution before polymerization was cast in a 5.7 mm diameter cylinder and allowed to polymerize for at least 4 h. After several washes over a period of 72 h the gel was incubated at 37 °C for 36–48 h until dehydrated. 270μl of the KcsAM sample was soaked into the gel and inserted into a 4.2 mm open-ended NMR tube (http://newera-spectro.com/) as described previously [30], affording an axial stretching factor of ~1.8. Aligned KcsAM samples were stable over several measurement weeks at 323 K. Small changes in the alignment tensor were monitored and compensated by periodically measuring the DNH coupling for several residues in a short 2D-experiment.
NMR measurements
All NMR measurements were conducted at 323 K on a DRX600 Bruker spectrometer using a cryogenic triple-resonance probehead equipped with z-axis pulsed field gradients. For backbone assignment, triple resonance HNCO, HN(CA)CO spectra [32] and a ct-HNCA spectrum with a 28 ms constant-time evolution period for 13Cα [33] were acquired for 2H,13C,15N-labeled KcsAM. These experiments contained a TROSY readout block optimized for water suppression on cryoprobes [26, 34]. Assignment was assisted by recording 3D 15N-separated NOE-HMQC and HMQC-NOE-HMQC experiments at 800 MHz. Solvent exchange rates of backbone amide protons were estimated by monitoring the disappearance of cross-peaks in tr-HSQC spectra acquired at intervals of 8–10 minutes after exposing the sample to D2O. Measurements of KcsAM 15N backbone relaxation rates at 323 K were based upon previously described experiments [26]. Spin-locked transverse relaxation rates R2* were derived from a measurement of R1ρ using a spin-lock field of 1.8 kHz and an appropriate correction for offset effects [35]. Residual dipolar couplings were measured for KcsAM at pH 4.2 using previously reported methods for obtaining the one-bond DNH [36], DNC′ [37] and DCC′ [38] couplings. Typically, each tr-HNCO or tr-HNCO-based experiment was acquired for 12–16 h and tr-HN(CA)CO and tr-ct-HNCA experiments were acquired for 24–36 h to obtain sufficient signal-to-noise (>100:1). Detailed acquisition parameters are presented in the Supplementary Material.
Data analysis
Spectra were processed and analyzed using the NMRPipe/NMRDraw suite containing appropriate peak-picking and spectral-analysis routines [39]. Fittings of RDCs to elements of the KcsAM structure and the tetrameric KcsA structures were performed using the DC program included in the NMRPipe software package. Due to the relatively low resolution of the MthK structure (PDB accession code 1LNQ, representing the open conformation of the channel), its coordinates were regularized before performing the RDC fitting. Briefly, using the high-resolution KcsA structure (PDB accession code 1K4C) as a template, the X-PLOR-NIH software [40] was used to position helices taken from the 1K4C structure in such a manner that they provided a best fit to the corresponding backbone atoms of 1LNQ, such as to improve the local geometry of the lower resolution structure. Modeling of the KcsAM-SDS assembly used an in-house rigid-body grid search algorithm to optimize the overall RDC fit to the KcsAM structure.
CD measurements
CD spectra of 15 μM samples of KcsAM in 15 mM SDS and in various buffers were recorded on a JASCO J-810 spectropolarimeter using a 0.1-cm path length cell at 50° C. Quantitative evaluation of secondary structure from the CD spectrum (for data in the 200–260 nm range) was carried out using the program CDNN version 2.1 (G. Bohm, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany).
Light scattering measurements
Light scattering data were obtained using an analytical Superdex-200 column 10/30 (GE Healthcare) with in-line multiangle light scattering (DAWN EOS, Wyatt Technology, Inc.) and refractive index detectors (OPTILAB DSP, Wyatt Technology, Inc.). Samples of 125 μL containing 1–2 mg/ml of either KcsATET in 25 mM MES (pH 6) or KcsAM in 20 mM Tris (pH 8) as well as 20 mM SDS were applied to the pre-equilibrated S200 column running at a flow rate of 0.5 mL/min at room temperature and eluted with the same buffer. Data were processed using the Astra V 5.1.3.0 software.
3. Results
pH-induced destabilization of tetrameric KcsA
KcsAM was prepared in a straightforward manner by exposing SDS-solubilized KcsATET to acidic conditions (pH < 4) at 55 °C for 1–2 h. KcsAM samples which were returned to ambient and neutral conditions before purification were found to contain a significant fraction of KcsA oligomers. However, after purification on a size-exclusion column, KcsAM was stable at all conditions and exhibited no oligomerization tendencies. The non-reversable nature of this dissociation process may be related to the removal of an endogenous biomolecule which has been reported to co-purify with KcsATET [41], but which is removed during purification of KcsAM. Formation of the KcsA monomer and its purity were verified using light-scattering measurements, which estimated the size of the KcsAM-containing SDS micelle to be 35 kDa, compared to the previously estimated size of ~120 kDa for the analogous KcsATET-containing micelle [25]. As expected from geometric considerations, the number of SDS molecules per KcsA subunit increases when comparing the monomer (ca 60) to the tetramer (ca 45).
We note that KcsATET exhibits long-term stability when solubilized in SDS at pH > 7, as well as in a mild detergent such as dodecyl maltoside at acidic conditions [1]. Therefore, the tetramer is destabilized not only by the denaturing effects of SDS, but also by disruption of pH-dependent inter-subunit contacts. Residue D80 immediately following the selectivity filter has been shown to be vital to tetrameric stability [42], most likely due to its participation in an inter-subunit salt bridge. As carboxylate pKa values would be elevated in the presence of the negatively charged SDS, protonation of this residue provides a possible explanation for the observed destabilization of KcsATET.
Dissociation- and pH-induced changes in KcsAM secondary structure
Despite significant overlap in its 1H,15N fingerprint region, the 17-kDa KcsAM solubilized in SDS micelles affords sufficiently resolved triple resonance spectra at 600 MHz and 323 K. Backbone resonance assignment was based upon intra- and inter-residual C′ and Cα connectivities and assisted by 15N-separated NOESY experiments. Assignments were performed for KcsAM at pH values of 4.2, 6.0 and 8.0. Depending on measurement conditions, full assignments were obtained for 135–138 of 142 non-proline residues. Local secondary structure of KcsAM was determined using the well-established correlation between backbone 13C chemical shifts and torsion angles (Figures 1–2). The four helical domains present in KcsATET [2] are observed to also be present in KcsAM: TM1 (residues 27–50), the pore helix (PORE, residues 62–74), TM2 (residues 86–115) and a helical segment in the cytoplasmic C-terminal domain (C-TER, residues 140–155). We conclude that KcsAM preserves a significant measure of the structural character present in KcsATET even when inter-subunit contacts have been abolished.
Figure 1. Secondary structure in KcsAM vs KcsATET.
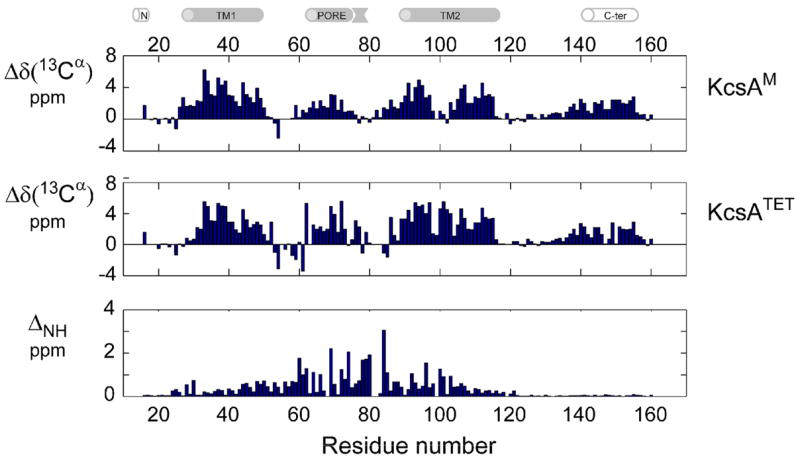
Backbone chemical shifts of KcsAM were measured to determine secondary structure elements of the channel subunit and contrasted with the homotetrameric channel assembly. Secondary 13Cα shifts (corrected for 2H isotope effects) of (top) 0.6 mM [2H,13C,15N]-labeled KcsAM and (middle) 0.8 mM (monomeric concentration) [2H,13C,15N]-labeled KcsATET , both measured in 200 mM SDS and 25 mM MES pH 6.0 at 600 MHz and 323 K. Similar behavior is observed for the 13C′ secondary chemical shifts. (Bottom) Shown is a comparison of 1H and 15N chemical shifts between the above samples of KcsAM and KcsATET along the KcsA sequence. ΔNH is defined as sqrt[(ΔδH)2 + (ΔδN/5)2], with ΔδH and ΔδN representing the respective changes in 1H and 15N chemical shifts, and the uncertainty in ηNH is estimated to be ≤ 0.02 ppm. For non-proline residues, missing bars indicate unassigned residues for one of the KcsA forms. Note that ΔNH for residues 121–160 is ≤ 0.1 ppm. The cartoon above the figure delineates the positions of secondary structure elements as found in the KcsA crystal structure [2] and a previous NMR study [25].
Figure 2. pH-induced changes in KcsAM secondary structure.
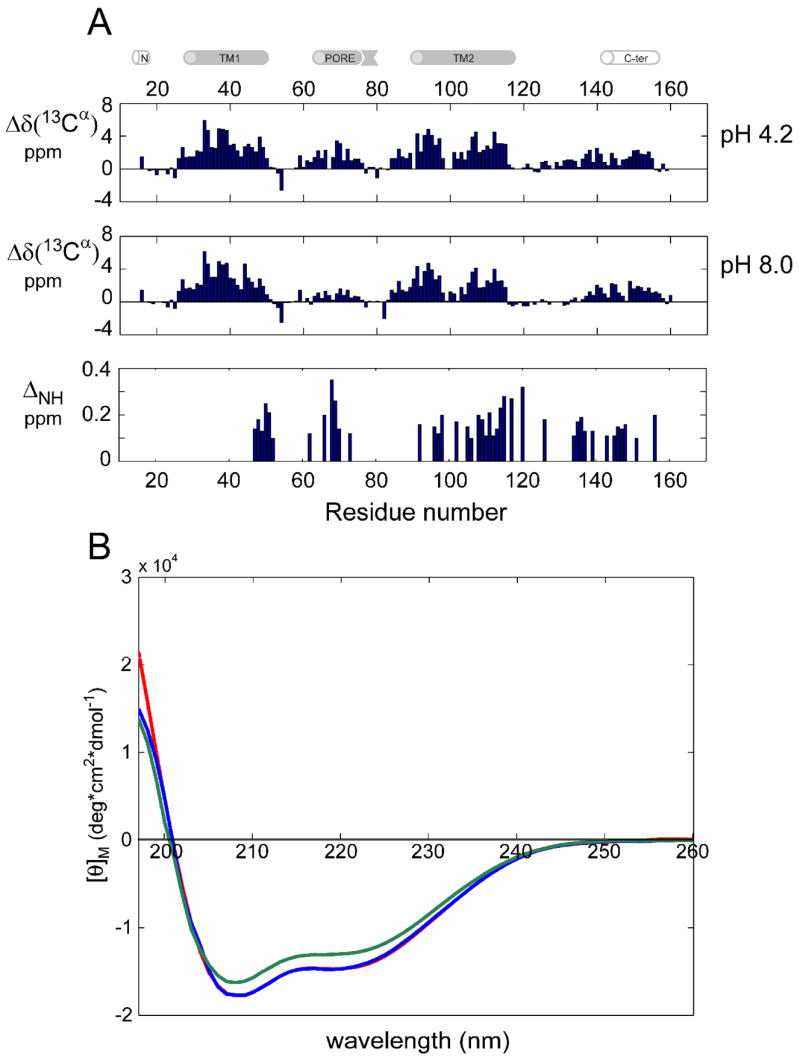
(A) Secondary 13Cα shifts (corrected for 2H isotope effects) measured at 600 MHz and 323 K of [2H,13C,15N]-labeled KcsAM in 200 mM SDS and (top) 25 mM sodium formate pH 4.2, and (middle) 20 mM Tris pH 8.0. Similar behavior is observed for the 13C′ secondary chemical shifts. (Bottom) Comparison of 1H and 15N chemical shifts between KcsAM samples at pH values of 4.2 and 8.0, with ΔNH defined as in Figure 1. For clarity, bars are shown only for residues with ΔNH ≥ 0.1 ppm. The cartoon above the figure delineates the positions of secondary structure elements as found in the KcsA crystal structure [2] and a previous NMR study [25]. (B) CD spectra of 15 μM samples of KcsAM in 15 mM SDS at various pH values. Curves obtained in 25 mM sodium formate pH 4.2, 25 mM MES pH 6.0, and 20 mM Tris pH 8.0 are shown in red, blue (practically overlapping) and green, respectively. Predicted helical contents are ~51% of residues for pH 4.2 and 6.0, compared to only 45% for pH 8.0.
A comparison between KcsAM and KcsATET structural elements reveals a change in TM2, which in KcsAM contains a 6-residue segment, 99GITSF104G (TM2K) exhibiting secondary chemical shifts incompatible with a regular α-helical conformation (Figure 1a), suggesting the presence of a kink in this transmembrane helix. The change in TM2 is particularly marked when compared to TM1, for which KcsAM and KcsATET chemical shifts are very similar. Other significant changes in secondary chemical shifts occur in the PORE helix. Based upon our previous findings [25], this strongly indicates that, in contrast to the TM domains, in KcsAM PORE is exposed to solvent, without close interactions with TM1, TM2, or the SDS micelle. Indeed, the PORE helix lies at the interface between subunits in KcsATET and is expected to be most affected by dissociation. The KcsAM C-TER domain exhibits virtually no changes in chemical shifts when compared to KcsATET (Figure 1b). This is consistent with our previous observation [26] that in SDS micelles the four C-TER domains of KcsATET are not involved in inter-subunit interactions.
pH-dependence of KcsAM chemical shifts and secondary structure is observed for the C-terminal end of TM1 (residues 47–52), PORE (65–72), TM2 (96–120) and residues preceding the C-TER helix (133–140) (Figure 2a). The PORE and C-TER chemical shift changes indicate an increase in helicity when KcsAM is transferred from pH 8 to pH 4. Circular dichroism measurements concur with these findings, reflecting a 13% increase in helicity when comparing KcsAM at these two pH values (Figure 2b). A longer C-TER helix was observed at pH 4 in another study featuring KcsATET embedded in liposomes [5]. Although in both cases residues with acidic side-chains are present, the change in PORE is remarkable as it contains only a single such residue (E71).
Solvent exchange of backbone amide sites in KcsAM
KcsATET backbone amide sites of channel residues embedded in the membrane –including the helices TM1 (residues 35–50), PORE (65–74) and TM2 (88–111) and the selectivity filter (75–79) – were highly protected from exchange with solvent, exhibiting exchange rates of kex < 10−7 sec−1 at 323 K and pH 8 [25]. Solvent exchange rates for KcsAM were measured at pH 4.2 by monitoring the disappearance of amide proton signals for lyophilized KcsAM in D2O solution (Figure 3). The hydrophobic core of KcsAM is, as expected, more exposed to exchange than KcsATET, although strong protection from hydrogen exchange is observed for micelle-buried domains. Most strongly protected were residues at the core of TM1 (residues 36–40), the only region exhibiting tetramer-like behavior, which could not be back-exchanged after D2O- expression and observed in KcsAM spectra without exposure to pH 8 and 323 K. Relative exchange rates can be summarized as
Figure 3. Solvent exchange of backbone 1HN protons in KcsAM.
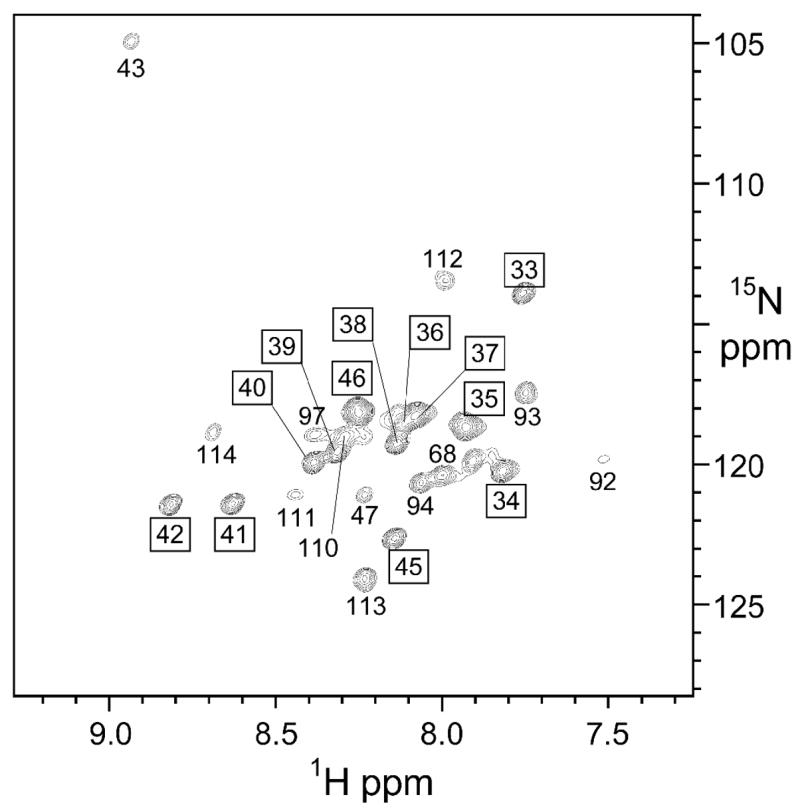
Rates of exchange with solvent for KcsAM backbone amide protons were estimated by lyophilizing a 0.5 mM KcsAM sample in 200 mM SDS and 25 mM formate buffer, pH 4.2, and reconstituting in 99.9% D2O. Shown is a [1H,15N] tr-HSQC spectrum recorded 1 h after the sample was placed in a 600 MHz spectrometer at 323 K for the D2O sample. The total experiment time was 2 h. Visible peaks are labeled by residue number, with boxed labels showing no significant (<5%) change when compared to an identical spectrum recorded 7 hours later, and regular labels indicating peaks which were not visible or appreciably weakened in the second spectrum. Based upon these results, exchange rates at 323 K and pH 4.2 can be estimated as kex≤ 10−6 s−1 for the former and 10−6 ≤ kex≤ 10−4 s−1 for the latter.
Most protected are the three helices TM1 (kex < 10−6 s−1, pH 4 and 323 K), TM2N (~10−5 s−1) and TM2C (~10−5 s−1), all values referring to central residues of these helices. Thus these helices are placed firmly within the SDS micelle. As expected from surface accessibility considerations, the PORE helix and KcsA selectivity filter, involved in extensive inter-subunit contacts in KcsATET, are no longer buried within the micelle, exhibiting faster solvent exchange. These results fully agree with the chemical shift data (Figure 1) as well.
Backbone dynamics of KcsAM: TM2K undergoes conformational exchange
Relaxation rates for the backbone amide 15N nuclei in KcsAM were measured at pH 6.0 using tr-HNCO-based two-point calculations as previously described for KcsATET [26]. The 3D tr-HNCO experiments applied to KcsAM at 323 K afford sufficient signal-to-noise and resolution, yielding full sets of relaxation rates ( R1, R2, 15N-{1H}-NOE and the cross-correlated relaxation ηxy) for 132 of the 142 observable amide sites. These measurements are summarized in Figure 4. Relaxation parameters are fully consistent with the four helical domains delineated by backbone chemical shifts. Relaxation behavior clearly divides these helical domains into two groups. With the exception of residues in the kinked domain TM2K, TM1 and TM2 exhibit average values of R1 ≈ 1.2 s−1, R2 ≈ 13 s−1, 15N-{1H}-NOE ≈ 0.65, and ηxy 9 ≈ s−1, whereas PORE and C-TER yield R1 = 1.55 s−1, R2 = 8 s−1, 15N-{1H}-NOE = 0.4, and ηxy = 6 s−1. The latter helix extends between residues 141–154 at pH 6.0. Results are consistent with the TM1 and TM2 helices being buried within the SDS micelle, increasing their effective correlation times, and PORE and C-TER being exposed to solvent, with little or no direct interaction with SDS micelles. Similar conclusions are derived using spectral density [43] and model-free [44] analyses (Figure S1 in the Supplementary Material). Micelle-buried domains, solvent-exposed helices, and unstructured segments are characterized by generalized order parameter S2 values of 0.8–0.9, 0.6–0.7, and 0.4–0.6, respectively.
Figure 4. 15N backbone relaxation for KcsAM.

15N backbone relaxation rates were measured at 600 MHz and 323 K for a 0.5 mM sample of [2H,13C,15N]-labeled KcsAM in 200 mM SDS and 25 mM MES pH 6.0. Spin-locked transverse relaxation R2 * values were derived from a measurement of R1ρ against a spin-lock field of 1.8 kHz with an appropriate correction for offset effects [35]. Relaxation rates for residues 36–40 in the TM1 helix were not measured for this sample due to insufficient back-exchange of solvent protons at these sample conditions. Measurements repeated at pH 8.0 (data not shown) establish that relaxation rates are uniform throughout the TM1 helix. The cartoon above the figure delineates the positions of secondary structure elements as found in the KcsA crystal structure [2] and a previous NMR study [25].
The kinked TM2K segment (residues 98–105) lies between the TM2N (residues 89–97) and TM2C helices (residues 106–118). In the tr-HNCO spectrum, TM2K residues give rise to weaker peaks when compared to other TM residues, and residues G99 and I100 were unobservable (Figure 5a). 15N-{1H}-NOE values suggest this segment is more flexible than its neighboring helices. Several R2/ ηxy ratios of residues in this segment are higher than expected for a domain with only fast internal dynamics [45], indicating that slower (microsecond) backbone motions contribute to backbone 15N transverse relaxation in this domain (Figure 5b). Furthermore, peaks of these residues broadened beyond detection at lower (303–308 K) temperatures, exhibited a field-dependent line-width, and their positions were sensitive to sample pH. We conclude that relaxation in this segment is affected by conformational exchange on an intermediate, microsecond time scale. Linewidths in this segment did not exhibit a dependence upon KcsAM concentration, effectively ruling out the possibility that the broadening effect could be related to transient dimerization of KcsAM. The conformational changes observed for this domain therefore occur within the KcsA monomer itself.
Figure 5. Conformational exchange on the intermediate time scale in the TM2 domain.
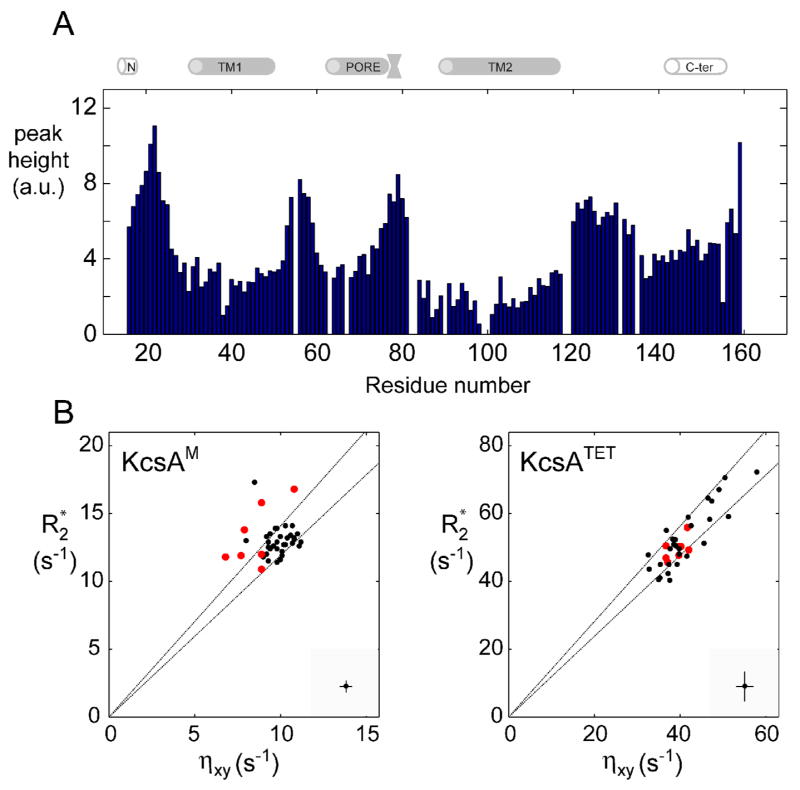
(A) Intensity of tr-HNCO peaks (arbitrary units) along the KcsAM sequence. Spectrum was recorded at 600 MHz and 323 K for a 0.5 mM [2H,13C,15N]-labeled KcsAM in 200 mM SDS and 25 mM formate buffer at pH 4.2. The cartoon above the figure delineates the positions of secondary structure elements as found in the KcsA crystal structure [2] and a previous NMR study [25]. (B) A comparison of 1H, 15N-cross-correlated (ηxy) and transverse (R2*) relaxation rates (as described in Figure 4) for micelle-embedded residues at 600 MHz and 323 K for (left) a 0.5 mM sample of [2H,13C,15N]-labeled KcsAM in 200 mM SDS and 25 mM MES pH 6.0, and (right) a similar sample containing 0.6 mM of KcsATET. In the interest of clarity, typical errors in both parameters are shown at the bottom right corner of each diagram. The two lines indicate the upper and lower limits of the expected scatter of ηxy vs. R2 value assuming an average value of −170 ppm and a spread of ±30 ppm for the 15N chemical shift anisotropy. Values obtained for detectable residues in the TM2 helix segment 98AGITSFGLV107T are shown in red. Elevated R2/ ηxy ratios (points above the upper line) are characteristic of an exchange contribution to transverse relaxation. Note that KcsATETηxy and R2 values exhibit a wider spread due to the rotational diffusion anisotropy of the KcsA-loaded micelle and the inclusion of amide sites from the PORE helix, which is the least collinear with the four-fold symmetry axis.
Aligning KcsAM in polyacrylamide gels
We set out to reveal additional structural features of KcsAM in SDS micelles using measurements of residual dipolar couplings (RDCs) in a weakly aligning environment. RDCs are well-suited for determining the relative orientation of the TM1, TM2N and TM2C helices within the micelle, and the very similar alignment strengths observed for these three helical elements confirms that they are enclosed in a single micelle. The PORE and C-TER domains show much weaker alignment and apparently reorient somewhat independently of the SDS micelle.
Preparation of aligned samples of micelle-solubilized proteins affording reliable RDC values remains technically challenging. The most generally successful approach has been the incorporation of membrane proteins into compressed polyacrylamide gels [20, 46, 47]. KcsAM presented an additional difficulty in that the TM2 domain required sample conditions which minimized intermediate conformational exchange in order to be observed. RDCs were best measured for KcsAM in a weakly negatively charged gel, which minimizes attractive forces between the gel and the SDS micelle. Such gels are expected to exert two aligning forces on the oblate SDS-KcsAM mixed micelle, a steric influence due to the radial compression and an electrostatic influence of the charged acrylamide monomer. As expected, stronger alignment was obtained in gels with similar acrylamide densities and compression factors as their negative character increased. However, lower pH values also induced a marked increase in the degree of alignment of a given sample. This effect was most pronounced for couplings in the C-TER helix, which was found to align parallel to the static magnetic field (vide infra), but also clearly observed for the micelle-embedded helices, for which the degree of alignment was doubled between samples at pH 8.0 and 4.2. For C-TER, this can be accounted for by considering the electrostatic character of this helix. With a pI value of ~7 (and >8 for the entire cytoplasmic tail), a decrease in pH increases the positive surface charge of this domain and thereby the aligning effect of the negatively charged gel upon it. We expect that such pH-sensitive helices may be generally applicable as a tunable aligning factor for samples of detergent-solubilized membrane proteins in polyacrylamide gels.
An RDC-based model for the SDS-KcsAM mixed micelle
Optimal results for measurement of backbone RDCs for KcsAM were obtained for a gel containing a 1:50 negative-to-neutral acrylamide monomer ratio and in formate buffer pH 4.2, as under these conditions suitable alignment parameters were obtained and conformational broadening in the TM2 domain was least pronounced. Three types of backbone couplings, DNH, DNC′ and DCαC′, were measured for KcsA using tr-HNCO pulse sequences in order to properly sample bond-vector orientational space, and ambiguous couplings due to spectral overlap were discarded. Overall, 90 RDCs were measured for residues of the TM1 (residues 30–48), TM2N (residues 89–97) and TM2C (residues 106–118) helices. Couplings of terminal residues in each helix were excluded to avoid fraying effects. Intrinsically smaller couplings (such as DNCα and 2DHNC′) could not be measured with sufficient accuracy for deuterated KcsAM. Despite the relatively sparse data – having measured three couplings per residue in a single alignment medium – meaningful structural conclusions could be reached.
Relatively uniform and negative (i.e. | 1JNH+1DNH| ~ 120–125 Hz) RDCs were observed for TM1 NH bond-vectors, and uniform and positive (i.e. | 1 JNH+1DNH | ~ 78–85 Hz) RDCs were observed for C-TER NH bond-vectors (Figure 6). This indicates that the TM1 helix is oriented roughly perpendicular to the magnetic field (or parallel to the unique micellar short axis); notably, this orientation was constant in all polyacrylamide samples and over varying pH values. In contrast, the C-TER helix is oriented parallel to the magnetic field (or perpendicular to the unique micellar axis). When fitting the RDCs measured for each of the two longer helices TM1 and TM2C to the corresponding helices in the prior X-ray structure, alignment tensors of quite similar magnitude and rhombicity are obtained, supporting the assumption that the KcsA transmembrane helices are aligning like a single particle. Despite the smaller number of couplings available for the shorter TM2N helix, which makes accurate definition of its alignment tensor more difficult, RDCs for TM2N are consistent with this finding.
Figure 6. Measurement of DNH residual dipolar couplings for KcsAM.
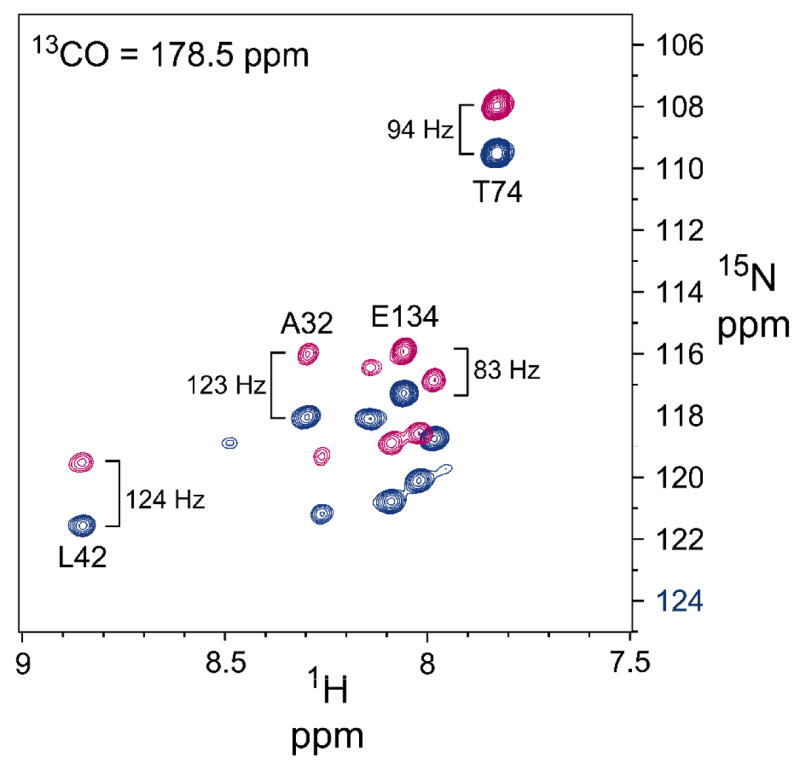
DNH couplings were measured for a sample aligned in charged polyacrylamide gel (see text) containing 0.5 mM [2H,13C,15N]-labeled KcsAM in 200 mM SDS and 25 mM formate buffer at pH 4.2, acquired at 600 MHz and 323 K. The downfield (blue) and upfield (magenta) resonances of the 1H-coupled backbone 15N nuclei were obtained from an interleaved 3D tr-HNCO-based experiment as previously described [36]. Shown is a 1H,15N plane obtained for a 13CO frequency of 178.5 ppm, demonstrating the importance of 13CO-separation for obtaining well-resolved couplings. The |1 JNH+ 1 DNH| splittings of selected backbone 1H-15N sites are indicated, showing the opposite behavior of the TM1 (residues A32 and A42) and C-TER (residue E134) domains.
RDC data for these three helical domains were used to model the SDS-KcsAM assembly. Each domain was represented by an ideal helix, since the independent fittings of helices to the RDC data were better when using ideal coordinates, in agreement with previous findings [48]. In addition, the three helices were assumed to share a common alignment tensor. The three-helix model was refined across all possible orientations to obtain the optimal fit for experimental RDC measurements while fixing all backbone torsion angles within the helical segments. Due to the inherent four-fold structural degeneracy of RDC structural data [49], this process yielded sixteen possible models for the KcsAM transmembrane arrangement of the three helices (Figure 7), with an alignment tensor of Da = 14.0 Hz, R = 0.43 and a Q-factor of 0.35. In the absence of any inter-helical translational information, these models were created assuming that 1) the 6-residue TM2K linker limits the relative positioning of the TM2N and TM2C helices, and 2) the solvent-exposed helical linker connecting TM1 and TM2N cannot traverse the micelle, i.e. the C-terminal end of TM1 and the N-terminal end of TM2N are located on the same side of the micelle. Inspection of these sixteen structures shows that only two of them create compact structures which, once embedded in the SDS micelle, are compatible with KcsAM backbone relaxation and light-scattering data.
Figure 7. RDC-based models for the structure of SDS-solubilized KcsAM.
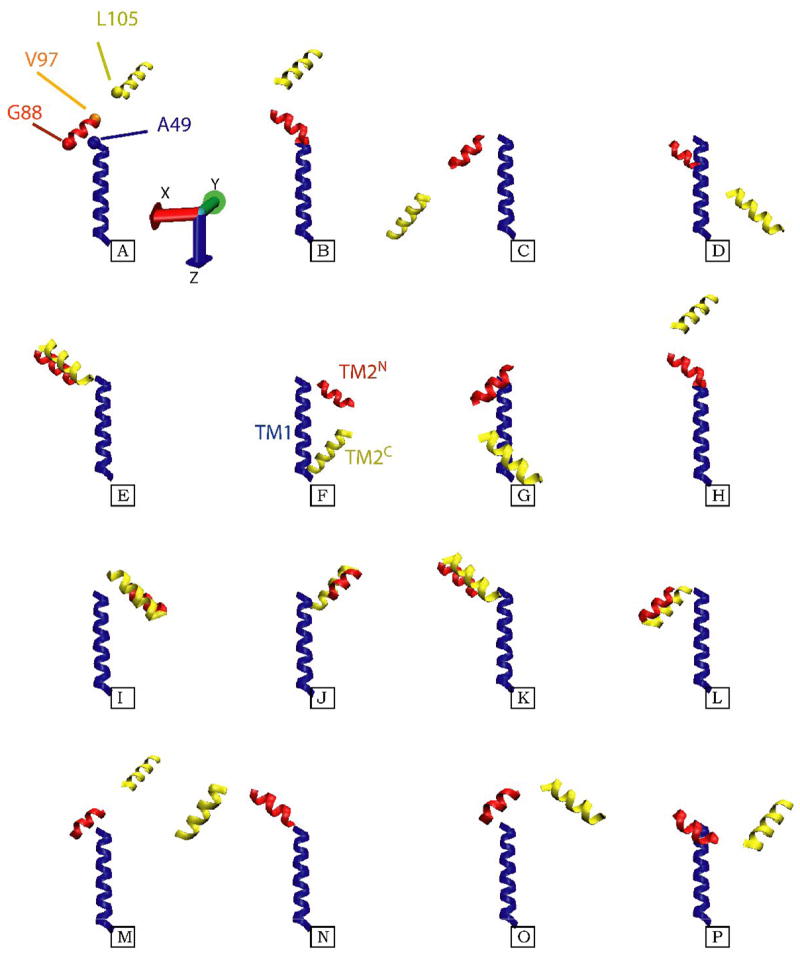
Shown are the sixteen different models for the three TM helical segments of KcsAM, TM1 (blue), TM2N (red), and TM2C (yellow), which are compatible with the acquired RDC data. The TM1 helix is almost co-linear with the z-axis of the alignment frame, and its coordinates are maintained constant in all models. For the upper left structure, the positions of Cα atoms for residues L49, G88, V97, and L105 at the inter-helical ‘joints’ are shown as blue, red, orange and yellow spheres, respectively. Since the data provides no constraints on the translational relation between the three helical fragments, the models were created as follows: 1) The short six-residue linker (TM2K) which connects the two TM2 fragments was appended to each of them in a helical conformation, and the two virtual T101 Cα atoms were superimposed; 2) Since TM1 and TM2N are linked by a solvent-exposed helical segment, residues L49 and G88 have been located on the same side of the SDS micelle. The G88 Cα atom was placed in a plane perpendicular to TM1 and containing the L49 Cα atom, and 7 Å away from the TM1 helical axis. While the position of TM2 is still ill-defined in this procedure, the models shown aim to minimize the radius of gyration and create a relatively compact structure compatible with backbone 15N relaxation and light-scattering data. Of the sixteen possibilities, structures F and G provide the most compact arrangement of the helices, and the former is depicted in the cartoon in Figure 8.
The structure of the KcsA subunit within the channel tetramer could not account for the RDC data (Figure 8a). This result was not changed when using KcsA coordinates that had been regularized to correspond to ideal helices. In contrast, in both models which form a compact structure (models F and G in Figure 7) the TM2 helix is kinked and the TM2N and TM2C helices form a ~110° angle (Figure 8b). Figure 8c suggests a plausible arrangement of the helices within the micelle based upon these results. However, in the absence of distance constraints between the various helices this must be treated as a strictly qualitative model.
Figure 8. Model of SDS-solubilized KcsAM.
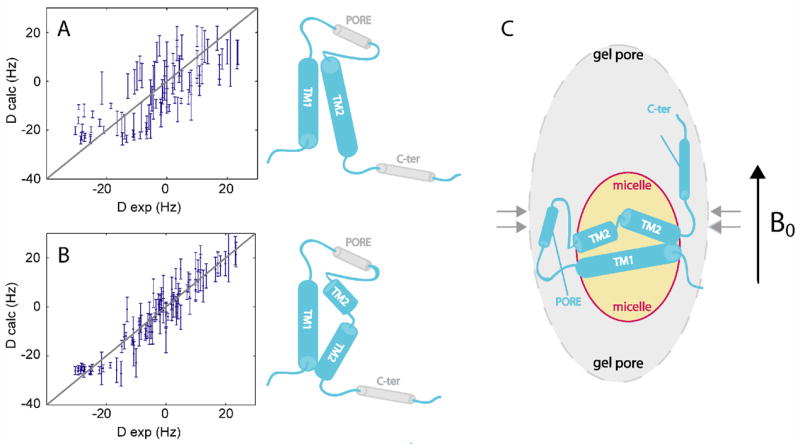
RDCs measured for the micelle-embedded helices of KcsAM were used to model this domain. (A) All 90 RDCs (normalized to DNH values) fitted to the structure of the monomeric subunit within the KcsATET structure (upper cartoon). (B) Same as (A), except the TM2 helix was allowed to bend at the kinked segment to optimize the fit between experimental and calculated RDC values, leading to a reduction in Q-factor from 0.67 to 0.35. The resulting model is shown (lower cartoon). (C) The RDC-based model of KcsAM embedded in an SDS micelle and aligned in a prolate polyacrylamide gel cavity.
Discussion
Our study attempts to deconvolute intra- and inter-subunit structural factors affecting biological activity of the KcsA potassium channel, specifically its pH-induced switch between the open and closed channel states. This approach also holds two additional benefits, namely the orientation-sensitive RDCs can be measured at higher accuracy for KcsAM, and in the absence of a KcsATET backbone assignments at acidic pH it may offer a glimpse of the KcsA open state by revealing the elements that remain unperturbed during a transition as dramatic as the switch from tetramer to monomer.
Comparing the global structures of KcsATET and KcsAM, the most significant changes are seen in regions involved in inter-subunit packing contacts, the PORE helix and the selectivity filter, which together encompass the five largest chemical shift changes in Figure 1. A similar conclusion is derived from the remarkable dissociation-induced change in protection from solvent exchange observed for these domains. The effects of dissociation upon other structural elements, however, are smaller, and generally speaking the TM1, TM2 and C-TER helices structural elements are preserved. The RDC data is consistent with the assumption that the two TM domains are embedded in a single micelle and tumble as a single body. Although it is possible that similar alignment tensors reflect partially independent tumbling of two helix-containing micelles of roughly the same size, this hypothesis is not supported by the data at hand. Light-scattering measurements are consistent with a relatively compact assembly and not a dumbbell-shaped molecule with a flexible linker. Furthermore, backbone relaxation data affords an estimated molecular weight of 30–35 kDa, which would not be reconcilable with the larger radius of gyration expected for a two-micelle entity. In addition, exchange at a slow rate observed for TM2K suggests that the two KcsAM conformations are stabilized, presumably via an interaction with TM1. Finding the two TM domains within the same micelle suggests potential similarities in the behavior of the micellar domains of KcsAM and KcsATET.
A significant finding of this study is the kinked nature and relative flexibility of the TM2 helix, manifesting itself in reduced secondary chemical shifts, increased solvent exchange rates, and pH-dependent conformational broadening of linewidths in the TM2K and TM2C domains. This result is particularly striking when comparing it to the TM1 helix, for which chemical shifts and RDCs were completely independent of sample conditions, and tetramer-like hydrogen exchange protection factors were preserved. The TM2K region corresponds to a conserved channel ‘hinge’ which allows the four TM2 helices to retract in a shutter-like manner, leading to opening of the channel [12]. Notably, the linewidth of the conserved Gly residue (G99), previously implicated in the gating-associated bending of TM2 [12] is most affected by the transition and is virtually undetectable at all sample conditions.
It is difficult to correlate the RDC-predicted orientation of the micelle-enclosed helices with a physiological state of the channel. A comparison with the analogous domains of MthK, a potassium channel structure representing the open conformation of KcsA, affords rather different bends in the TM2 domain. Whereas the MthK TM2C segment bends ~30° away from the TM2 helical director, for KcsAM the ‘splaying’ is wider, bending ~70° away; the orientation between TM1 and the TM2N segment is different as well. Notably, the fit of RDC data to the MthK structure is just as unsatisfactory as is the fit to the original KcsATET structure depicted in Figure 8a (data not shown). Since the TM2 helix is longer than the TM1 helix, the possibility that this increased bending reflects a curvature effect of the SDS micelle on the KcsAM structure can not be excluded with certainty. However, the pH-dependence of the frequencies (Figure 2) and linewidths of TM2 tr-HSQC peaks argues for a pH-dependent conformation, not simply a micelle-curvature induced bending. Presumably, the RDC-deduced conformation is stabilized at lower pH, whereas at higher pH values a second state is sufficiently populated to cause significant line-broadening. In summary, while this study stops far short of a KcsAM structure, it hints at a pH-induced population shift inherent to the channel subunit itself, which may be correlated with the open-to-closed pH-induced changes in KcsATET.
Two differences stand out when comparing our findings to a recent NMR study which characterized the tetrameric KcsA pH-dependent transition [11]. The apparent pKa value of the transition was higher (~6 vs 4.5) for KcsAM in SDS micelles. This may be attributed to the effects of the charged SDS environment upon the pKa of negatively charged side-chains, which are plausible participants in the gating transition. We also find a faster rate (~2000 vs 150 s−1) of conformational exchange compared to the above KcsATET study. It is possible that the faster rate represents the inherent flexibility of the channel subunit, which in the intact channel is modulated by the tetrameric assembly to afford the desired kinetics of channel opening. This would also be consistent with the observed and expected increase in flexibility observed for the transmembrane domain of the monomeric species. However, it is also likely that the difference in rate is impacted by the different detergent used in that study.
Additional pH-induced effects upon KcsAM structure are the increase in helical content at acidic pH observed for PORE and C-TER. Regarding the former, a network of hydrogen bonds involving the PORE helix has been implicated in channel gating at the level of the selectivity filter [8]. In KcsAM, the PORE helix maintains only its local structure, but loses its global interactions with the selectivity filter (residues 75–79) and neighboring residues (D80). Therefore, pH-induced changes in PORE observed in this study are independent of its surroundings, and may reflect an inherent sensitivity to pH. Changes in the C-TER domain are of particular interest because there is currently little if any structural evidence accounting for the role of this cytoplasmic domain (residues 125–160) in channel gating; notably, this domain is absent in all potassium channel crystal structures. Available data point to a helical structure for this domain both in SDS and in liposomes, and formation of a 4-helical bundle under appropriate conditions has been hypothesized [5, 25]. It also has been assumed that changes in cytoplasmic pH mimic the effect of a cellular regulator of the potassium channel. The elongation of the C-TER helix observed in KcsAM upon lowering the pH, possibly related to the presence of several acidic moieties in this segment, may be involved in a gating-related structural change of this domain.
Supplementary Material
Acknowledgments
We thank Dr. Alex Grishaev for assistance with the RDC fitting algorithms and Annie Aniana for technical assistance in sample preparation. J. H. C. acknowledges the support of a long-term EMBO fellowship. This work was supported by the Intramural Research Program of the NIDDK, NIH, and by the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heginbotham L, Odessey E, Miller C. Tetrameric stoichiometry of a prokaryotic K+ channel. Biochemistry. 1997;36:10335–10342. doi: 10.1021/bi970988i. [DOI] [PubMed] [Google Scholar]
- 2.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, Mackinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 3.Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 4.Yu LP, Sun CH, Song DY, Shen JW, Xu N, Gunasekera A, Hajduk PJ, Olejniczak ET. Nuclear magnetic resonance studies of a potassium channel-charybdotoxin complex. Biochemistry. 2005;44:15834–15841. doi: 10.1021/bi051656d. [DOI] [PubMed] [Google Scholar]
- 5.Cortes DM, Cuello LG, Perozo E. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen Physiol. 2001;117:165–180. doi: 10.1085/jgp.117.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry on ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 7.Berneche S, Roux B. A gate in the selectivity filter of potassium channels. Structure. 2005;13:591–600. doi: 10.1016/j.str.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 8.Cordero-Morales JF, Cuello LG, Zhao Y, Jogini V, Cortes DM, Roux B, Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Mol Struc Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 9.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, Mackinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 10.Kelly BL, Gross A. Potassium channel gating observed with site-directed mass tagging. Nat Struc Biol. 2003;10:280–284. doi: 10.1038/nsb908. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi K, Takahashi H, Kawano S, Shimada I. Identification and characterization of the slowly exchanging pH-dependent conformational rearrangement in KcsA. J Biol Chem. 2007;282:15179–15186. doi: 10.1074/jbc.M608264200. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, Mackinnon R. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 13.Cuello LG, Romero JG, Cortes DM, Perozo E. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 1998;37:3229–3236. doi: 10.1021/bi972997x. [DOI] [PubMed] [Google Scholar]
- 14.Heginbotham L, LeMasurier M, Kolmakova-Partensky L, Miller C. Single streptomyces lividans K+ channels: functional asymmetries sidedness of proton activation. J Gen Physiol. 1999;114:551–560. doi: 10.1085/jgp.114.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez C, Wuthrich K. NMR solution structure determination of membrane proteins reconstituted in detergent micelles. FEBS Lett. 2003;555:144–150. doi: 10.1016/s0014-5793(03)01155-4. [DOI] [PubMed] [Google Scholar]
- 16.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Page RC, Moore JD, Nguyen HB, Sharma M, Chase R, Gao FP, Mobley CK, Sanders CR, Ma L, Sonnichsen FD, Lee S, Howell SC, Opella SJ, Cross TA. Comprehensive evaluation of solution nuclear magnetic resonance spectroscopy sample preparation for helical integral membrane proteins. J Struct Funct Genomics. 2006;7:51–64. doi: 10.1007/s10969-006-9009-9. [DOI] [PubMed] [Google Scholar]
- 18.Rooslid TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 19.Tamm LK, Abildgaard F, Arora A, Blad H, Bushweller JH. Structure, dynamics and function of the outer membrane protein A and influenza hemagglutinin fusion domain in detergent micelles by solution NMR. FEBS Lett. 2003;555:139–143. doi: 10.1016/s0014-5793(03)01127-x. [DOI] [PubMed] [Google Scholar]
- 20.Sanders CR, Sonnichsen FD. Solution NMR of membrane proteins: practice and challenges. Magn Res Chem. 2006;44:S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 21.Zuiderweg ERP. Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry. 2003;41:1–7. doi: 10.1021/bi011870b. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi K, Wagner G. NMR studies of protein interactions. Curr Opin Struct Biol. 2006;16:109–117. doi: 10.1016/j.sbi.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Wimmer J, Peti W, Schwalbe H. Motional properties of unfolded ubiquitin: a model for a random coil protein. J Biomol NMR. 2006;35:175–186. doi: 10.1007/s10858-006-9026-9. [DOI] [PubMed] [Google Scholar]
- 24.Gebel EB, Shortle D. Characterization of denatured proteins using residual dipolar couplings. Methods Mol Biol. 2007;350:39–48. doi: 10.1385/1-59745-189-4:39. [DOI] [PubMed] [Google Scholar]
- 25.Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Prot Sci. 2006;15:684–698. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chill JH, Louis JM, Baber J, Bax A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J Biomol NMR. 2006;36:123–136. doi: 10.1007/s10858-006-9071-4. [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi K, Yokogawa M, Matsuda T, Sugai M, Kawano S, Kohno T, Nakamura H, Takahashi H, Shimada I. Structural basis of the KcsA K+ channel and agitoxin2 pore-blocking toxin interaction by using the transferred cross-saturation method. Structure(Camb) 2003;11:1381–1392. doi: 10.1016/j.str.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Tycko R, Blanco FJ, Ishii Y. Alignment of biopolymers in strained gels: A new way to create detectable dipole-dipole couplings in high-resolution biomolecular NMR. J Am Chem Soc. 2000;122:9340–9341. [Google Scholar]
- 29.Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. Solution NMR of proteins within polyacrylamide gels: diffusional properties and residual alignment of by mechanical stress or embedding of oriented purple membranes. J Biomol NMR. 2000;18:303–309. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]
- 30.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 31.Meier S, Haussinger D, Grzesiek S. Charged acrylamide copolymer gels as media for weak alignment. J Biomol NMR. 2002;24:351–356. doi: 10.1023/a:1021609207024. [DOI] [PubMed] [Google Scholar]
- 32.Salzmann M, Wider G, Pervushin K, Senn H, Wuthrich K. TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J Am Chem Soc. 1999;121:844–848. [Google Scholar]
- 33.Salzmann M, Pervushin K, Senn H, Wuthrich K. 13C-constant-time [15N,1H]-trosy-HNCA for sequential assignments of large proteins. J Biomol NMR. 1999;14:85–88. doi: 10.1023/a:1008346931993. [DOI] [PubMed] [Google Scholar]
- 34.Pervushin K, Wider G, Wuthrich K. Single transition-to-single transition polarization transfer (ST2-PT) in [15N,1H]-TROSY. J Biomol NMR. 1998;12:345–348. doi: 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- 35.Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton N. Protein NMR Spectroscopy: Principles and Practice. Academic Press; San Diego, CA, USA: 1996. p. 283. [Google Scholar]
- 36.Kontaxis G, Clore GM, Bax A. Evaluation of cross-correlation effects and measurement of one-bond couplings in proteins with short transverse relaxation times. J Magn Res. 2000;143:184–196. doi: 10.1006/jmre.1999.1979. [DOI] [PubMed] [Google Scholar]
- 37.Chou JJ, Delaglio F, Bax A. Measurement of one-bond 15N-13C′ dipolar couplings in medium sized proteins. J Biomol NMR. 2000;18:101–105. doi: 10.1023/a:1008358318863. [DOI] [PubMed] [Google Scholar]
- 38.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Meth Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Mag Res. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 41.Valiyaveetil FI, Zhou Y, Mackinnon R. Lipids in the structure, folding and function of the KcsA K+ channel. Biochemistry. 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- 42.Choi H, Heginbotham L. Functional influence of the pore helix glutamate in the KcsA K+ channel. Biophys J. 2004;86:2137–2144. doi: 10.1016/S0006-3495(04)74273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farrow NA, Zhang O, Szabo A, Torchia DA, Kay LE. Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 44.Cole R, Loria JP. FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;13:289–302. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 45.Fushman D, Cowburn D. Model-independent analysis of 15N chemical shift anisotropy from NMR relaxation data: ubiquitin as a test example. J Am Chem Soc. 1998;120:7109–7110. [Google Scholar]
- 46.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 47.Jones DH, Opella SJ. Weak alignment of membrane proteins in stressed polyacrylamide gels. J Mag Res. 2004;171:258–269. doi: 10.1016/j.jmr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 48.Mesleh MF, Lee S, Veglia G, Thiriot DS, Marassi FM, Opella SJ. Dipolar waves map the structure and topology of helices in membrane proteins. J Am Chem Soc. 2003;125:8928–8935. doi: 10.1021/ja034211q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Hashimi HM, Valafar H, Terrell M, Zartler ER, Eidsness MK, Prestegard JH. Variation of molecular alignment as a means of resolving orientational ambiguities in protein structures from dipolar couplings. J Mag Res. 2000;143:402–406. doi: 10.1006/jmre.2000.2049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.