

Abstract
Ras is believed to stimulate invasion and growth by different effector pathways, and yet, the existence of such effectors under physiologic conditions has not been shown. Estrogen receptor (ER), on the other hand, is both anti-invasive and proliferative in human breast cancer, with mechanisms for these paradoxical actions remaining largely unknown. Our previous work showed an essential role of p38γ mitogen-activated protein kinase in Ras transformation in rat intestinal epithelial cells, and here, we show that p38γ integrates invasive antagonism between Ras and ER to increase human breast cancer invasion without affecting their proliferative activity. Ras positively regulates p38γ expression, and p38γ in turn mediates Ras nonmitogenic signaling to increase invasion. Expression of the Ras/p38γ axis, however, is trans-suppressed by ER that inhibits invasion and stimulates growth also by distinct mechanisms. Analysis of ER and its cytoplasmic localized mutant reveals that ER additionally binds to p38γ protein, leading to its specific down-regulation in the nuclear compartment. A p38γ-antagonistic activity of ER was further shown in a panel of breast cancer cell lines and was shown independent of estrogens by both ER depletion and ER expression. These results revealed that both Ras and ER use distinct pathways to regulate breast cancer growth and invasion, and that p38γ specifically integrates their antagonistic activity to stimulate cell invasion. Selective targeting of p38γ-dependent invasion pathways may be a novel strategy to control breast cancer progression.
Introduction
Two defining hallmarks of cancer are uncontrolled proliferation and increased invasion (1). Although increased proliferation is frequently associated with enhanced invasion (2), recent evidence suggests that these two events are distinct (3–6). Ras protein is involved in human malignancies through multiple processes that include increased proliferation, enhanced invasion, and altered cytoskeleton organization (2). Although studies with Ras effector domain mutants suggest a specific role of individual Ras effectors/pathways in these processes (7, 8), the existence of such effectors under physiologic conditions remains unproven. Ras protein is overexpressed in up to 60% of human breast cancer (1, 9), and dissecting Ras mitogenic signaling from its invasive activity is, therefore, critical in understanding its promoting roles in breast cancer progression.
Estrogen receptor (ERα) is a nuclear transcription factor that regulates gene expression through binding to specific estrogen-responsive elements (ERE) on a target gene promoter. ER is proliferative in mammary tissues and consequently implicated in the etiology of breast cancer (10, 11). Paradoxically, ER inhibits breast cancer in vitro invasion and in vivo metastasis (12, 13). Furthermore, higher levels of ER protein expression in human breast cancers correlate negatively with clinical metastasis but positively with the prognosis (14, 15). These results together indicate that ER may use distinct pathways to stimulate breast cancer growth and to inhibit its invasion/metastasis.
ER may regulate breast cancer progression through crosstalk with Ras/mitogen-activated protein kinase (MAPK) signaling. Activation of the Ras/extracellular signal-regulated kinase (ERK) pathway downstream of growth factor receptors such as epidermal growth factor receptor (EGFR) increases both cell proliferation and invasion (4, 16). However, forced Ras expression in ER+ breast cancer cells inhibits ER activity (17), and expression levels of EGFR and/or Ras proteins are higher in ER− and/or metastatic breast cancers (18–20). These results suggest that ER and Ras cooperate in stimulating growth but antagonize each other in regulating invasion, and dissecting their crosstalk may contribute to understanding breast cancer progression.
MAPKs consist of ERK, c-Jun NH2-terminal kinase, and p38 cascades. ERK activity is essential for Ras-induced proliferation/transformation (21, 22), whereas p38 is inhibitory to Ras activity (23–25). In response to Ras-induced invasion, however, both ERK and p38 are required in this process (26). Although these different p38 effects on Ras-induced proliferation and invasion are not understood, some p38 family members may have distinct activity in these regulations. For example, p38 isoform proteins can be either cooperative or antagonistic in regulating gene expression and cell proliferation (27, 28). Studies of p38 isoform-specific effects may contribute to understanding different Ras-p38 signaling interactions in regulating cell proliferation and invasion.
The p38 family consists of four isoforms: α, β, γ, and δ (29). p38γ, also called stress-activated protein kinase 3 or ERK6, shares about 60% identity with p38α and p38β (30, 31). In contrast to the ubiquitously expressed p38α, p38γ is only detectable in certain normal tissues (30, 31). Recent studies, however, show that p38γ protein is highly expressed in several human malignant cell lines (27, 32, 33), indicating its possible contributing role in cancer development. Indeed, our recent studies showed that Ras activates p38γ by increasing its expression in rat intestinal epithelial (IEC-6) cells, and induced p38γ, albeit nonmitogenic by itself, is required for Ras transformation (34). Specific roles of p38γ in Ras-associated human cancers, however, remain to be established. Because Ras plays critical roles in increasing breast cancer progression (2), p38γ may function as a Ras effector in this regulation.
Here, we sought to test the hypothesis that the Ras/p38γ axis may exist in human breast cancer and thereby regulate breast cancer progression in coordination with ER protein. Our results revealed that in human breast cancer, Ras dictates p38γ expression, which in turn mediates Ras nonmitogenic signaling to stimulate cell invasion. The invasive activity of the Ras/p38γ axis, however, is antagonized by ER protein through trans-repression, and ER can additionally disrupt p38γ protein through direct binding. These results suggest an invasive role of p38γ in human breast cancer through integrating Ras proinvasive and ER anti-invasive signaling.
Materials and Methods
cDNA constructs
Recombinant adenovirus vector (Ad-Vect) and vector-containing oncogenic H-Ras (Leu61, L61) or dominant-negative H-Ras (Asn17, N17) were provided by Dennis Stacey (35). Adenovirus β-gal (ad-β-galactosidase), ad-MKK6, and ad-p38γ were described elsewhere (32). Retroviral vector pLHCX (provided by Dan Mercola; ref. 36) was used to express p38γ. To deplete endogenous protein, a retroviral vector pSUPER (pSR; ref. 37) was used (target sequence for sip38γ was 5′-AAGGAGAT-CATGAAGGTGACG-3′ and for siERα was 5′-AAGAGGAGGGAGAATGTT-GAA-3′) using a luciferase gene sequence as a control (5′-CGTACGCGG-AATACTTCGA-3′; ref. 34). Human ER cDNA and ERE luciferase reporter (ERE-Luc, four ERE repeats upstream of a luciferase gene) were provided by David Shapiro (38). ER site–directed mutagenesis (T311A) was carried out by a PCR-based technique using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA) with primer sequences as previously published (39). All mutations generated were confirmed by DNA digestion with restriction enzymes and DNA sequencing.
Cell culture, reagents, and Tet-inducible ER expression
MEM and other reagents for cell culture were purchased from Life Technologies (Gaithersburg, MD). Protein-Sepharose G beads and Cy3- and FITC-labeled second antibodies were obtained from Zymed (South San Francisco, CA). p38 isoform–specific antibodies were described previously (27, 34), and their specificity was further confirmed by antibodies from BD Clontech (Palo Alto, CA). ERK1/2 and ER antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), whereas phosphorylated p38 (p-p38) and phosphorylated-ERK (p-ERK) antibodies were from Cell Signaling (Beverly, MA). Mouse pan-Ras antibody was purchased from Oncogene Research Products (San Diego, CA). Human breast cancer cell lines [ER+: MCF-7 and 47D; ER−: MDA-MB-231 (231) and MDA-MB-468 (468)] were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in MEM containing 10% fetal bovine serum (FBS) and antibiotics at 37°C with 5% CO2. The Tet-on expression system (T-Rex) was purchased from Invitrogen (San Diego, CA) and used to express human ER or ER/T311A in 231 cells as previously described (40). To evaluate effects of estrogens, cells were cultured with 10% charcoal-treated serum in phenol-free medium for 48 hours and then treated with 10 nmol/L estrogens for additional 24 hours.
Transfection, viral infection, reporter assays, and small interfering RNA experiments
ERE-Luc was transiently transfected by calcium phosphate into Tet-ER cells, which were then incubated with and without Tet for an additional 24 hours before the reporter assay using dual luciferase kit (41). For adenoviral infection, cells were infected with ad-vector or ad-L61/ad-N17 Ras in no serum medium for 4 hours and cultured in normal medium for 24 hours for protein expression and migration assays. In the case of retroviral infection, pSUPER (pSR-Lucif, pSR-sip38γ, and pSR-siER) and pLHCX (Vector and p38γ; refs. 24, 34) were transfected into Phoenix-Ampo retrovirus packaging cells (ATCC), and supernatants were used to infect target cells. Typically, cells were analyzed for protein or RNA expression at 24 to 48 hours.
Invasion, cell growth, and data analysis
Invasion assays were carried out using the BioCoat Matrigel Invasion Chamber (BD Biosciences, Bedford, MA) according to the manufacturer’s instruction. Briefly, 2.5 × 105 cells in serum-free medium were plated into each chamber, which was placed into the six-well plate containing 20% FBS and incubated for 22 hours, at 37°C, 5% CO2. Tetracycline (Tet) was added 2 hours later to both inserts and the bottom well to induce ER expression. For infection, cells are replated for invasion assay 24 hours after removal of virus-containing medium. Following incubation, the noninvaded cells on the upper surface of membrane were removed, and the invaded cells on the lower surface were washed, fixed, and stained with crystal violet. The number of invaded cells from 13 to 20 representative fields was counted manually under a phase-contrast microscope, normalized to the respective control. To further verify these results, some crystal-stained attached cells were solubilized with 10% acetic acid and quantitated at 600 nm (42). For cell growth assay, cells were plated with or without Tet and pulsed labeled with [3H]thymidine, and the incorporated radioactivity was determined as previously described (41). All results were analyzed by Student’s t test for statistically significant difference.
Immunostaining, immunoprecipitation, glutathione S-transferase pulldown, and cell fractionation/Western and Northern blot assays
For immunostaining, cells were plated on coverslips and fixed in 3.7% formaldehyde. After permeabilized in a buffer containing 0.5% Triton X-100 and 0.5% NP40, cells were blocked in 3% bovine serum albumin in PBS. A mouse monoclonal anti-flag (M2, Sigma, St. Louis, MO) and anti-ER (F10) antibody at 1:100 were used for flag-p38γ and ER staining, as previously described (34, 40). For immunoprecipitation, cell lysates were incubated with a goat p38γ antibody, and the precipitates were examined for ER protein by Western blot. For glutathione S-transferase (GST) pulldown assay, GST, GST-ER and GST-ER/T311A proteins were expressed in Escherichia coli BL21 and purified using reduced glutathione-agarose beads (23).
To examine protein expression, cells were directly lysed in 1× loading buffer. After heating, samples were separated on SDS-PAGE and transferred to a nitrocellulose membrane for detection of the molecule of interest using enhanced chemiluminescence kit. For cell fractionations, cytoplasmic and nuclear fractions were prepared as previously described (43). For Northern blot, total RNA was prepared by TRIzol (Invitrogen) and separated by agarose gel. p38γ and p38α probe was prepared from their corresponding human cDNAs in pcDNA3 vector by enzymatic digestion and/or PCR (34). The probe was labeled with [32P]dCTP using High Primer kit (Roche Molecular Biochemical, Indianapolis, IN). The Northern blots were standardized for equal RAN loading using the UV fluorescence of the 18S rRNA band from the same membrane.
Results
Ras positively regulates p38γ expression independent of ERK and/or p38 phosphorylation, whereas ER trans-suppresses Ras/p38γ in human breast cancer
Previous studies have shown that both transient and stable Ras expressions induce p38γ protein expression in IEC-6 cells (34). To determine whether Ras regulates p38γ expression in human breast cancer, adenovirus-mediated gene delivery was used to express oncogenic H-Ras (L61) and the dominant-negative mutant (N17; ref. 35) in ER− 231 human breast cancer cells. Following infections, cells were analyzed for protein expression and phosphorylation. Consistent with the positive regulatory effect in IEC-6 cells (34), L61 increases whereas N17 decreases p38γ but not p38α protein expression compared with the control β-gal infection (Fig. 1A and B). In contrast to the p38γ induction in IEC-6 cells, Ras-regulating p38γ protein expression does not associate with significant alterations in ERK and/or p38 phosphorylations (Fig. 1A and B), in agreement with a recent observation of Ras activity dissociating with ERK phosphorylation in human breast cancer cells (44). These results reveal a determinant role of Ras activity in p38γ protein expression in human breast cancer independent of ERK/p38 phosphorylation and indicate a possible role of p38γ in Ras regulating breast cancer progression.
Figure 1.
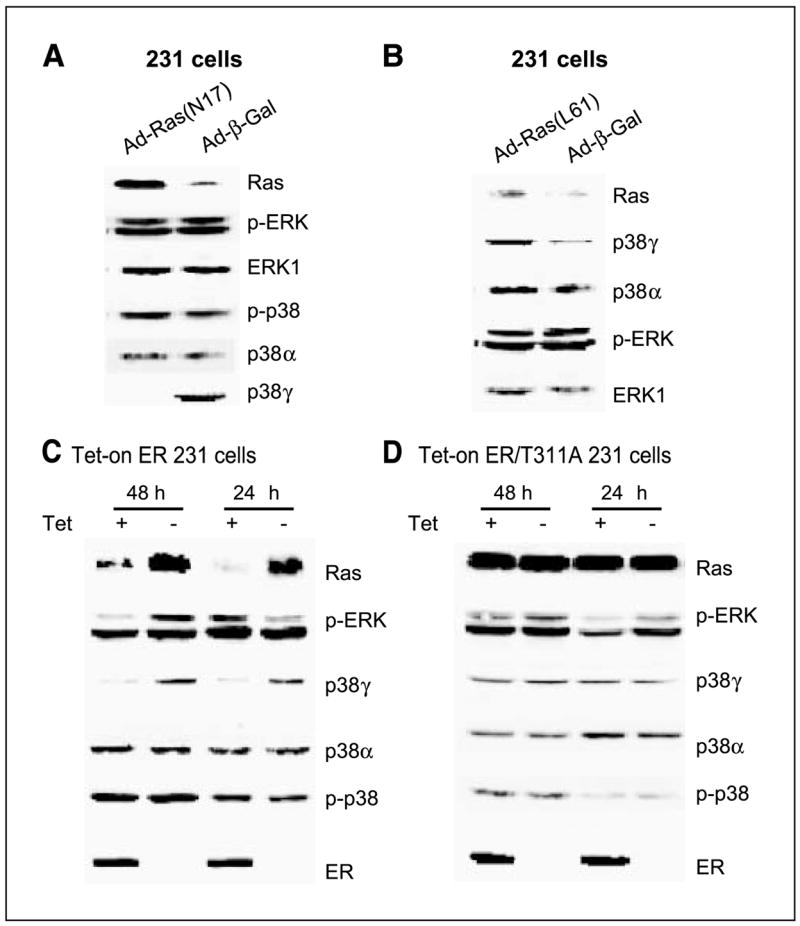
Ras positively regulates p38γ protein expression independent of ERK phosphorylation, whereas both Ras and p38γ protein expression is trans-suppressed by ER in human breast cancer cells. A, Ras inhibition suppresses p38γ expression without affecting ERK/p38 phosphorylation. ER− 231 cells were infected with control adenovirus (Ad-β-gal) or virus expressing dominant-negative Ras (Ad-N17) for 4 hours and incubated for an additional 24 hours before analyzed for protein expression by Western blot. B, Ras activation induces p38γ expression without stimulating ERK phosphorylation. Cells were infected with Ad-β-gal or virus expressing oncogenic H-Ras (Ad-L61) as in (A) and examined for protein expression. C and D, ER inhibits Ras/p38γ protein expression dependent of its transcription activity. Cells were cultured with and without Tet for the indicated time to induce wild-type (C) and the mutant ER expression (D), and their effects on Ras/p38γ protein expression were examined by Western. Representative from three separate experiments.
Ras is known to antagonize endogenous ER activity in MCF-7 human breast cancer cells (17, 45). To explore whether exogenous ER conversely inhibits endogenous Ras activity, consequently leading to down-regulation of p38γ protein expression, a Tet-on system was used to express ER protein by including its transcription-deficient mutant (T311A) for comparison (39, 40). As expected, Tet-induced ER inhibits Ras protein expression (Fig. 1C). This effect requires ER transcription activity, as levels of Ras proteins remain constant with and without ER/T311A expression (Fig. 1D). Strikingly, ER down-regulating Ras protein expression does not result in persistent alterations in p-ERK and/or p-p38 proteins. Rather, it leads to a significant and sustained decrease in p38γ protein expression (Fig. 1C and D). This effect is specific, as expression levels of p38α protein remain constant with and without ER expression. Additional experiments with p-p38 immunoprecipitation and p38γ/p38α Western blotting showed that there was no detectable p38γ phosphorylation in the absence and presence of ER expression (data not shown). These results, by both Ras expression and inhibition, revealed a unique signaling axis from Ras to p38γ through regulating protein expression in human breast cancer. Because of the ER inhibitory role, the Ras/p38γ axis may only act to regulate cell invasion/proliferation in ER− breast cancer.
p38γ transmits Ras nonmitogenic signaling to promote ER− breast cancer invasion
The Ras determinant role in p38γ protein expression prompted us next to explore if p38γ acts as a Ras effector to execute its functions in regulating breast cancer invasion and growth. To assess roles of endogenous Ras in these regulations, ER− 231 cells were infected with Ad-β-gal or Ad-Ras (N17), and its effects on cell invasion/growth were determined. Functional aspects of endogenous p38γ in these processes, on the other hand, were analyzed by small interfering RNA–mediated p38γ depletion (34). As expected, inhibition of Ras activity by N17 substantially decreases Matrigel cell invasion and DNA synthesis (P < 0.001 for both cases; Fig. 2A–C), indicating that Ras activity is required for both processes (26, 46). In contrast to the Ras inhibition, p38γ depletion only inhibits invasion (P < 0.01) without affecting thymidine incorporation (P > 0.05, Fig. 2A–C), suggesting a specific role of p38γ in stimulating cell invasion. These results indicate that although p38γ is downstream of Ras, they have common and distinct roles in regulating breast cancer invasion and growth.
Figure 2.
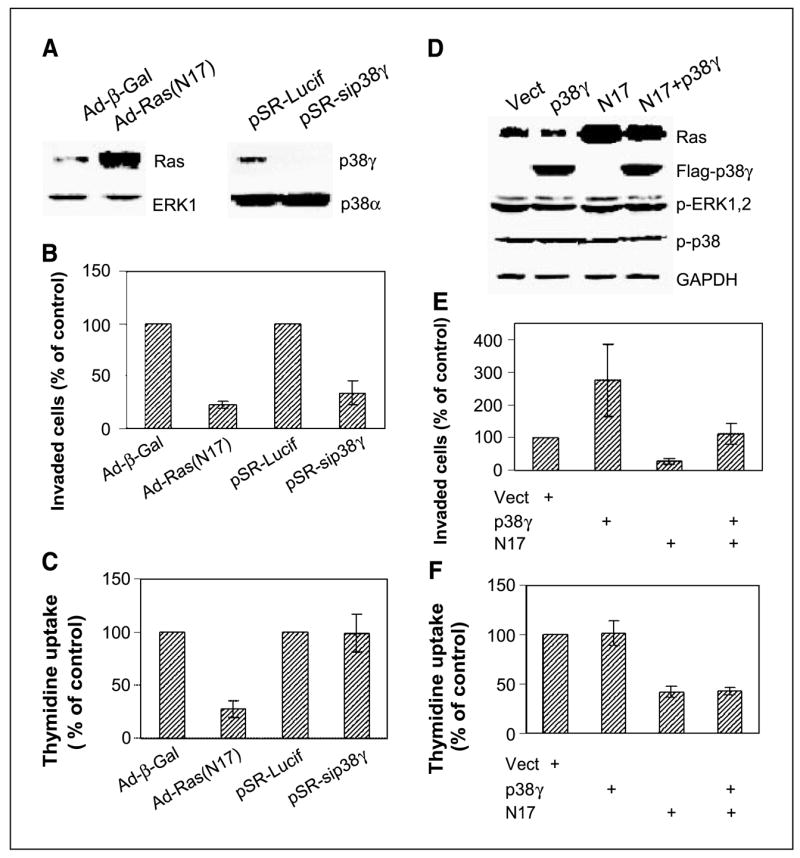
Endogenous Ras activity is required for both proliferation and invasion, whereas p38γ only transmits nonmitogenic Ras signaling to stimulate invasion. A, adenovirus-mediated N17 expression and small interfering RNA–induced p38γ depletion. Cells were infected with either adenovirus (Ad-β-gal or Ad-N17) or retrovirus (pSR-Lucif, control retrovirus; pSR-sip38γ, small interfering RNA retrovirus) and incubated for 24 hours before Western analysis. B, both Ras inhibition and p38γ depletion inhibit cell invasion. Cells were plated for invasion assay after infection, and invaded cells were counted and normalized to respective controls. Columns, mean of 15 fields (P < 0.01 for N17 and sip38γ versus their respective controls); bars, SD. Similar results were obtained from additional two experiments. C, Endogenous Ras but not p38γ is required for cell proliferation. Cells were infected as above, and cell proliferation was estimated by thymidine incorporation. Columns, mean of three experiments (P < 0.05 only for Ad-N17 versus Ad-Vect); bars, SD. D, N17 and p38γ overexpression. ER− 231 cells were infected either with adenovirus (Vect, N17) and/or pLHCX retrovirus (Vect, p38γ) and analyzed for protein expression 24 hours later. E, p38γ overexpression rescues N17-mediated invasion inhibition. Cells were coinfected with Ad-N17 with and without pLHCX-p38γ and assessed for cell invasion as described above. Columns, mean of 13 fields (P < 0.01 for p38γ versus Vect, N17 versus Vect, and for N17 versus p38γ + N17); bars, SD. Similar results are obtained from one additional experiment. F, high levels of p38γ protein expression do not overcome N17-induced growth inhibition. Columns, mean of three separate experiments (P < 0.05 for N17 versus Vect, but P > 0.05 for p38γ versus Vect and N17 + p38γ versus N17); bars, SD.
Because N17 inhibits both p38γ expression and cell invasion, whereas p38γ is proinvasive, the p38γ down-regulation may be required for N17 inhibiting invasion. If this is the case, p38γ overexpression should rescue N17-induced invasion-suppression. To this end, p38γ protein is overexpressed by a retroviral vector pLHCX in Ad-N17–infected cells, and its effects on cell invasion and DNA synthesis were determined. Consistent with the inhibitory effect of p38γ depletion, higher levels of p38γ protein increase invasion 2.8-fold (P < 0.01) without increasing thymidine uptake (Fig. 2D–F). More importantly, p38γ overexpression almost completely overcomes the N17-induced inhibition of cell invasion (P < 0.01) without affecting DNA synthesis (P > 0.05), indicating that p38γ only transmits the nonmitogenic Ras signaling to stimulate invasion. By both depletion and overexpression with and without Ras inhibition, these experiments show that p38γ is necessary and sufficient for breast cancer invasion through transmitting Ras nonmitogenic invasive signaling.
ER regulates breast cancer growth and invasion by distinct pathways
The above results showed that Ras stimulates invasion and DNA synthesis by p38γ-dependent and p38γ-independent mechanisms, indicating Ras using distinct pathways to increase breast cancer invasion and growth. Because ER has well-established activities in breast cancer progression (47, 48), we wished next to determine whether ER also regulates invasion and growth through distinct signaling. Previous studies have shown that ER suppresses breast cancer invasion by estrogen-dependent and estrogen-independent mechanisms (13), whereas estrogen can inhibit its own receptor expression (11). Consequently, experiments were done in normal complete medium without estrogen addition to assess effects of ER protein expression.
Results in Fig. 3A and B showed that Tet induces a similar level of ER and ER/T311A protein expression, but ER transcription activity (ERE-Luc, ref. 38) decreases about 90% (P < 0.01) in mutant ER expressed cells. These results are consistent with the previous observation in endometrial adenocarcinoma cells (39). ER expression inhibited invasion >80% (P < 0.01), whereas the mutant ER had no substantial effects (P > 0.05, Fig. 3C, left). A similar result was also obtained when crystal violet–stained cells were quantitated at 600 nm (ref. 42; Fig. 3C, right). Although ER has well-established invasion-inhibitory activity (12, 13), these results suggest that ER may do so through transcriptional regulations. In contrast to the effects on cell invasion, ER and ER/T311A increased DNA synthesis similarly (P > 0.05; Fig. 3D), indicating that ER regulates invasion and DNA synthesis by distinct mechanisms. These results together show that both Ras and ER regulate breast cancer growth and invasion by distinct signal transduction pathways.
Figure 3.

ER inhibits invasion and stimulates growth by distinct mechanisms. A, ER protein expression in Tet-on cells. Tet-on 231 cells were incubated with and without Tet for 24 hours and examined for ER and ER/T311A protein expression. B, Thr311 is required for ER transcription activity. Cells were transfected with ERE-Luc, incubated with and without Tet for 24 hours, and assessed for luciferase activity. Columns, mean of five independent experiments; bars, SD. C, ER requires its transcription activity to inhibit invasion. Cells were cultured ± Tet for 22 hours in the invasion chamber for the assay (see Materials and Methods). Columns, mean from 16 fields in one experiment; bars, SD. Similar results were obtained in two additional experiments. The absorption at A 600 nm (OD600 ) is mean of three separate experiments. D, ER does not require its transcription activity to stimulate DNA synthesis. Cells were cultured for 24 hours with or without Tet and cell growth was estimated by thymidine incorporation.
ER requires its nuclear localization to bind to p38γ protein, leading to its specific down-regulation in the nuclear compartment
Although antagonistic effects between ER and Ras have been described (17, 45), ER-suppressing p38γ activity is novel and warrants further investigations. We sought, therefore, to further explore mechanistic aspects of signaling interactions between ER and p38γ. Previous studies have shown that ER/T311A is predominantly localized in cytoplasm compared with nuclear wild-type ER protein (39), whereas p38γ is both in the nucleus and the cytoplasm where it functionally interacts with other proteins (49). We wish next to determine if ER and ER/T311A are differently localized in human breast cancer cells, and whether their distributions affect p38γ localization through direct binding, thereby additionally contributing to the p38γ antagonizing activity.
Consistent with previous findings (39), immunostaining analyses showed that ER is mostly localized in the nucleus, whereas ER/T311A is predominantly in the area surrounding the nuclei after a 24-hour incubation with Tet (Fig. 4A). p38γ signals are detected in the nucleus and cytoplasm in ER− cells. In ER-expressed cells, however, the nuclear p38γ signal seems negatively correlating with the ER intensity, indicating that nuclear ER is able to disrupt nuclear p38γ protein, leading to its down-regulation in the nuclear compartment. This conclusion is further supported by the observation in ER/T311A cells in which the mutant ER is mostly outside the nucleus and in which there is no decrease in nuclear p38γ signals (Fig. 4A).
Figure 4.
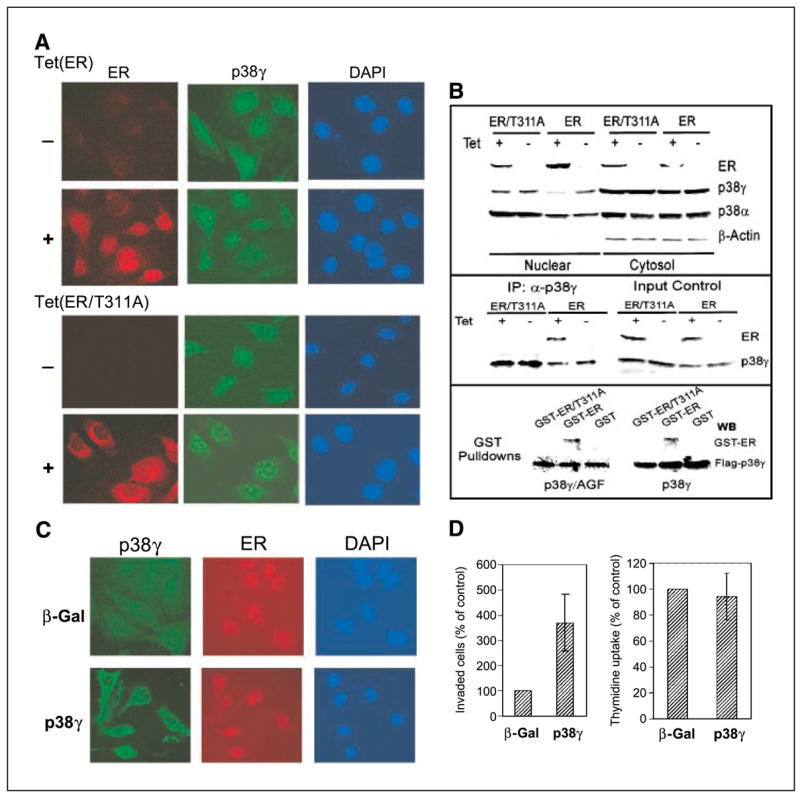
ER antagonizes nuclear p38γ activity through direct binding. A, ER is nuclear while ER/T311A is predominantly cytoplasmic. Cells were incubated for 24 hours (±Tet) and double stained for ER (detected with Cy3-labeled second antibody) and for p38γ expression (detected with FITC-labeled second antibody) with 4′,6-diamidino-2-phenylindole (DAPI) staining as a control for nuclear signals. B, ER disrupts nuclear p38γ protein through Thr311-dependent binding. Cells were prepared for nuclear and cytoplasmic fractions, which were analyzed for protein expression (top). p38γ binding activity of ER was assessed by immunoprecipitation with a p38γ antibody followed by Western analyses (middle). For pulldown assays (bottom), the purified GST, GST-ER, and GST-ER/T311A proteins were incubated with equal amounts of lysates prepared from 293T cells transfected with flag-p38γ and flag-p38γ/AGF, and the precipitates were analyzed for p38γ-bound ER protein. C, overexpressed p38γ is cytoplasmic in ER+ cells. ER+ 231 cells (Tet-on cells incubated with Tet for 24 hours) were infected with Ad-Vect (Vect) or Ad-p38γ for 24 hours and stained with anti-flag antibody for transfected flag-p38γ. D, higher levels of p38γ proteins increase invasion (right) but not DNA synthesis (left) in ER+ breast cancer cells. Tet-on 231 cells were plated for invasion and thymidine incorporation after infection. Right, columns, mean from 18 fields of one representative experiment; bars, SD. Similar results were obtained from one additional experiment. Left, columns, mean of four experiments; bars, SD.
To further confirm the decreased nuclear p38γ protein concentration in response to ER expression, cellular fractionation and Western blotting experiments were done (43). Cytoplasmic actin (50) and both cytoplasmic and nuclear p38α (51) serve as controls in this experiment. Results in Fig. 4B (top) showed that wild-type ER is mostly nuclear, whereas its mutant seems to be more abundant in the cytoplasmic fractions, generally consistent with the immunostaining results. Endogenous ER protein was also previously found to be both cytosolic and nuclear in ER+ MCF-7 cells by cell fractionation experiments (52). Of great interest, nuclear p38γ protein is decreased upon ER induction, whereas its levels in cytoplasm remain constant. Because the mutant did not have this effect, Thr311-directed ER nuclear localization is apparently required for its antagonizing activity against p38γ protein, leading to its specific down-regulation in the nuclear compartment.
To determine whether there is a physical interaction between ER and p38γ, thereby leading to p38γ down-regulation, immunoprecipitation and Western analyses were done. Results in Fig. 4B (middle) show that p38γ binds to ER but not ER/T311A, although both proteins are expressed to a similar level from the input control, indicating ER requiring Thr311 to bind to p38γ protein. To further show whether p38γ requires its phosphorylation to bind to ER, flag-tagged p38γ and its nonphosphorable mutant (p38γ/AGF) were expressed in 293T cells. Lysates derived were incubated with bacterially expressed GST, GST-ER, GST-ER/T311A proteins, and flag precipitates were examined for bound ER proteins. Results in Fig. 4B (bottom) show that ER, but not ER/T311A, binds to both p38γ and p38γ/AGF proteins, indicating that the direct ER-p38γ binding requires ER Thr311 but not p38γ phosphorylation. Together with the immunostaining and cell fractionation analysis, these results show that ER requires its nuclear localization to bind and thereby to disrupt nuclear p38γ protein.
p38γ overexpression increases invasion but not DNA synthesis in ER+ breast cancer cells
Results of ER-inhibiting p38γ expression and p38γ-stimulating ER− cell invasion suggest that p38γ may act downstream of ER to increase invasion. If this is the case, forced p38γ expression in ER+ cells should overcome this genetic restriction and lead to an invasive response. To relate invasion-regulatory effects with cellular localizations, flag-p38γ was overexpressed in ER+ cells (Tet-on), and its cellular distributions and activities on invasion were determined. Results in Fig. 4C show that in contrast to the endogenous p38γ protein, overexpressed p38γ is mostly in the cytoplasm, an effect similar to a recent observation in which overexpressed MK2 leads to its own relocalization (53). Although mechanisms are unclear at present, the cytoplasmic localization of exogenous p38γ proteins in ER expressed cells further supports the notion that ER antagonizes nuclear p38γ activity, leading to a reduced nuclear p38γ concentration even under overexpressed conditions. In the cytoplasm, transfected p38γ may have escaped the nuclear ER targeting as a result of their different localizations. As expected, overexpressed p38γ led to an increased invasion (3.7-fold) but not thymidine incorporation in these ER+ cells (Fig. 4D). These results, thus, further establish the p38γ-antagonizing activity of ER within the nuclear compartment and reveal that elevation of p38γ protein concentrations by forced expression can also increase ER+ cell invasion.
ER negatively regulates p38γ gene expression in a panel of human breast cancer cell lines
ER-inhibiting p38γ protein expression dependent of ERE activity suggests that this regulation may be transcriptional. We would like next to determine if ER antagonizes p38γ at RNA levels to further establish the trans-suppression mechanism. Total RNA in this case was prepared from Tet-on cells and analyzed for p38γ RNA expression (41) by including p38α as a control. As shown in Fig. 5A (left), Tet-induced ER decreases p38γ but not p38α RNA expression, thereby consolidating our conclusion of ER trans-suppressing p38γ expression. To show if this trans-suppression is a general phenomenon, a panel of natural ER+ and ER− breast cancer cell lines were analyzed for p38γ RNA expression. ER protein expression has been previously confirmed in ER+ MCF-7 and T47D but not in ER− 231 and 468 human breast cancer cells by Western analyses (40). Consistent with Tet-induced ER expression, higher levels of p38γ RNA were detected in ER− than in ER+ cells (Fig. 5A, right), ruling out cell type–specific effects of the p38γ antagonistic activity.
Figure 5.
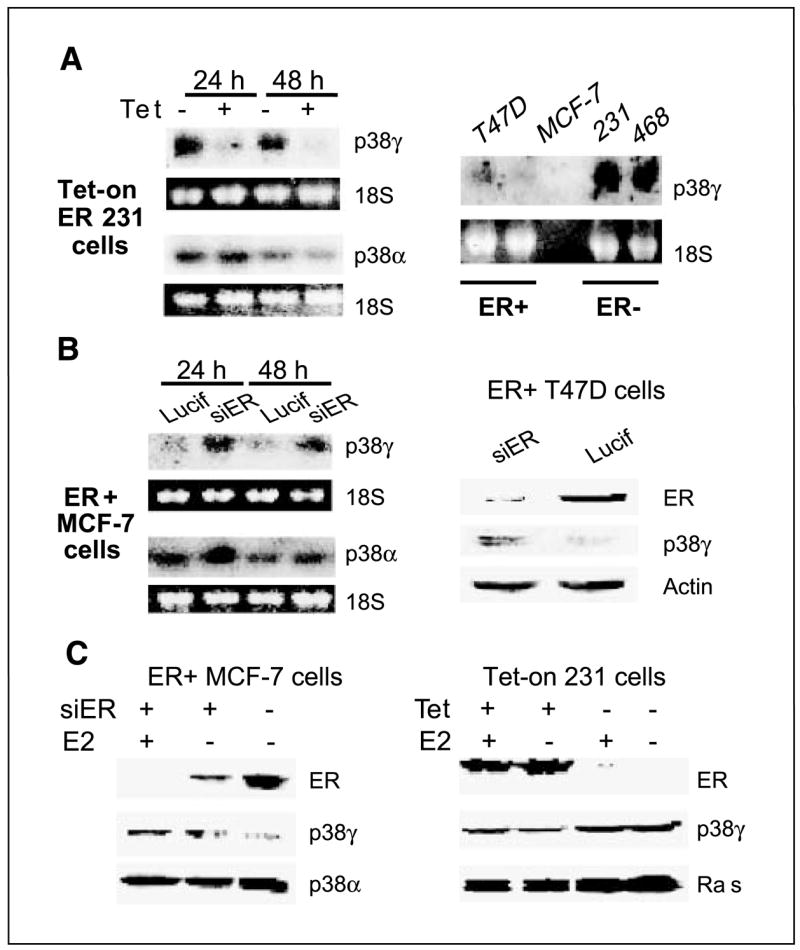
ER negatively regulates p38γ expression in a panel of human breast cancer cell lines. A, ER+ phenotype correlates with lower levels of p38γ RNA expression. Total RNA was prepared, separated, and transferred to a nitrocellulose membrane that was hybridized with a specific radioactive-labeled probe for p38γ expression. B, ER depletion increases p38γ expression. ER+ cells were infected with pSR-Lucif (Lucif) or pSR-siER (siER) for 24 hours and examined for p38γ RNA (left) or protein (right) expression. C, ER negatively regulates p38γ expression by ligand-independent mechanisms. Cells in steroid-depleted medium were incubated for 24 hours (±10 nmol/L E2) after the pSR infection (left) or in the presence or absence of Tet (right) and analyzed by Western analysis for protein expression.
To determine whether endogenous ER is also inhibitory to p38γ expression, ER+ human breast cancer cells were depleted of ER proteins by retroviral-mediated gene silencing (34), and its effects on p38γ expression were determined. Results in Fig. 5B show that ER depletion increases p38γ expression, further establishing ER-inhibitory role in p38γ expression. To assess if ligand can further enhance the trans-repression, estrogens were added after the retroviral infection and p38γ protein contents were determined. Similar to the RNA up-regulation, ER depletion leads to an increase in p38γ protein expression (Fig. 5C, left). Although estrogen treatment can further lower ER protein concentrations as a result of its well-known ER-depleting activity in MCF-7 cells (11), it fails to further increase p38γ protein expression, indicating a ligand-independent p38γ antagonistic activity of ER protein. Furthermore, addition of estrogens to steroid-depleted culture did not further increase the ER suppressive effects on Ras/p38γ protein expression in Tet-on 231 cells (Fig. 5C, right). These results together show that ER trans-suppresses p38γ and/or Ras by ligand-independent mechanisms. Because loss of ER expression frequently occurs in late-stage breast cancer (54), demonstration of a p38γ antagonistic activity of ER independent of ligand further highlights the role of p38γ proinvasive activity in breast cancer progression.
Discussion
Uncontrolled proliferation and increased invasion are key characteristics of human cancer, and dissecting these two events is consequently critical for understanding and controlling malignant progression (1). Here, we showed that Ras increases breast cancer invasion and DNA synthesis by p38γ-dependent and p38γ-independent pathways. A specific role of p38γ in increasing invasion independent of growth was shown by both its depletion and overexpression. Moreover, we provide evidence indicating that ER also inhibits invasion and increases DNA synthesis by distinct pathways, where p38γ is specifically involved in regulating cell invasion. These results together show that there exist distinct signal transduction pathways for both Ras and ER to regulate breast cancer invasion and growth in which p38γ specifically integrates their antagonistic activity to stimulate invasion (Fig. 6). Because invasion is a major characteristic of tumor but not normal cells (1), inhibiting p38γ-dependent invasive pathways may be a novel strategy to control breast cancer progression.
Figure 6.

An experimental model illustrates that ER and Ras regulate breast cancer progression through distinct invasive and proliferative pathways where p38γ only integrates their antagonistic activity to increase invasion. ER requires transcription activity to suppress Ras/p38γ expression and to bind p38γ protein, whereas it increases DNA synthesis by ERE-independent mechanisms. Ras, on the other hand, increases invasion and DNA synthesis through p38γ-dependent and p38γ-independent pathways in ER− cells. Although loss of ER expression may reduce cell growth, resultant Ras activation will provide mitogenic signaling, leading to increased DNA synthesis [and possible cell proliferation (?)]. Induced p38γ following ER inactivation and/or Ras activation will specifically mediate Ras nonmitogenic signaling to stimulate invasion. Through these two mechanisms, Ras/p38γ activation will lead to an increased breast cancer progression. This model suggests a feasibility to individually target invasive and proliferative signal transduction pathways in human breast cancers.
Our results presented herein have revealed a unique Ras/p38γ axis that acts to regulate breast cancer progression in coordination with ER protein. Here are the major characteristics of the Ras/p38γ axis: (a) Ras, by both its activation and inhibition, primarily signals to regulate p38γ expression independent of ERK/p38 phosphorylation; (b) Ras and its effector p38γ have common and individual roles in stimulating invasion and growth; and finally, (c) the Ras/p38γ axis is trans-repressed by ER and p38γ is additionally down-regulated by ER in the nuclear compartment through direct binding. These results together suggest that p38γ may serve as an integrating point for Ras proinvasive and ER anti-invasive activity through programmed protein expressions. In ER+ cells, invasion signals may be genetically silent in part through ER-induced suppression of Ras/p38γ expression. In response to loss of ER expression during breast cancer progression, Ras/p38γ will be activated by increased expression. The activated Ras will increase both proliferation and invasion, whereas the induced p38γ will only confer proinvasive signaling, leading to increased breast cancer progression (Fig. 6).
The central mechanism by which ER antagonizes the Ras/p38γ activity is likely through its nuclear localization that integrates transcriptional and posttranscriptional regulatory processes. This was shown by the fact that inhibition of ER nuclear localization through Thr311 mutation both reduces ERE-dependent transcription and disrupts its p38γ binding activity. These results are consistent with the notion that nuclear localization is essential for activities of nuclear receptors (55). Although a decreased ERE by ER/T311A correlating with the loss of p38γ binding activity suggests a role of this interaction in ER transcription, a decreased p38γ expression in ER+ breast cancer cells may argue against its physiologic roles in regulating ER nuclear localization and ER activity. Additional studies, however, are needed to explore mechanisms by which the ER direct binding leads to a p38γ down-regulation in the nuclear compartments.
Our results may offer an explanation about paradoxical ER effects in stimulating growth and in inhibiting invasion, which may otherwise antagonize each other in regulating breast cancer progression. Although loss of ER expression may reduce breast cancer growth, the resultant Ras activation can compensate for this effect by a proliferative response through increased expression (Fig. 6). This theory is supported by the observation of higher levels of Ras protein expression in ER− and/or metastatic human breast cancers (19, 44, 54, 56, 57). Because ER inactivation and Ras activation frequently occur in late stage of the disease (54, 56), these results may suggest a stage-specific role of ER and Ras in promoting breast cancer progression through a coordinative signaling integration.
It is well accepted that Ras uses distinct effector pathways to induce malignant transformation/progression (2, 46, 58). Our studies here are the first to show the existence of such pathways under physiologic conditions in which induced p38γ transmits Ras nonmitogenic signaling to amplify its invasion signals without affecting its proliferative activity. This specific pathway was first suggested by positive regulation of p38γ expression through Ras activation and inactivation, then confirmed by ER inhibiting both Ras and p38γ expression, and finally proven by p38γ-mediated rescue of N17-induced invasion inhibition. Although ERK and/or p38 phosphorylations are involved in regulating breast cancer invasion (26, 42), these effects could be secondary to altered cell proliferation. Here, we show that Ras dictates p38γ expression without affecting ERK/p38 phosphorylation in human breast cancer, and p38γ in turn increases invasion without affecting Ras mitogenic activity. These results together suggest that p38γ act as a Ras effector to stimulate invasion in response to ER inactivation/Ras activation.
Acknowledgments
Grant support: NIH grant 2R01 CA91576 and Department of Veterans Affairs Merit Review (G. Chen).
We thank Melanine Weytkow for assistance in some experiments and Drs. Jiahuai Han (The Scripps Research Institute, La Jolla, CA), Dennis Stacey (The Cleveland Clinic Foundation, Cleveland, OH), David Shapiro (University of Illinois, Urbana, IL), and Dan Mercola (Sidney Kimmel Cancer Center, San Diego, CA) for providing reagents.
References
- 1.Hanahan D, Weiberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004;14:105–14. doi: 10.1016/j.semcancer.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Gao CF, Xie Q, Su YL, et al. Proliferation and invasion: plasticity in tumor cells. Pro Natl Acad Sci U S A. 2005;102:10528–33. doi: 10.1073/pnas.0504367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol. 2002;4:487–94. doi: 10.1038/ncb807. [DOI] [PubMed] [Google Scholar]
- 5.Huntington JT, Shields JM, Der CJ, et al. Over-expression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells. Role of b-raf mutation and fibroblast growth factor signaling. J Biol Chem. 2004;279:33168–76. doi: 10.1074/jbc.M405102200. [DOI] [PubMed] [Google Scholar]
- 6.Scott LA, Vass JK, Parkinson EK, Gillespie DAF, Winnie JN, Ozanne BW. Invasion of normal human fibroblasts induced by v-Fos is independent of proliferation, immortalization, and the tumor suppressors p16INK4a and p53. Mol Cell Biol. 2004;24:1540–59. doi: 10.1128/MCB.24.4.1540-1559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–2. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 8.Roderiguez-Viciana P, Warne PH, Khwaja A, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–67. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 9.Downward J. Targeting Ras signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 10.Fujita T, Kobayashi Y, Wada O, et al. Full activation of estrogen receptor a activation function-1 induces proliferation of breast cancer cells. J Biol Chem. 2003;278:26704–14. doi: 10.1074/jbc.M301031200. [DOI] [PubMed] [Google Scholar]
- 11.Pinzone JJ, Stevenson H, Stroble J, Berg PE. Molecular and cellular determinants of estrogen receptor α expression. Mol Cell Biol. 2004;24:4605–12. doi: 10.1128/MCB.24.11.4605-4612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia M, Deroco D, Freiss G, Rochefort H. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. Pro Natl Acad Sci U S A. 1992;89:11538–42. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platet N, Cunat S, Chalbos D, Rochefort H, Garcia M. Unliganded and liganded estrogen receptors protect against cancer invasion via different mechanisms. Mol Endocrinol. 2000;14:999–1009. doi: 10.1210/mend.14.7.0492. [DOI] [PubMed] [Google Scholar]
- 14.Clark R, Thompson EW, Leonessa F, et al. Hormone resistance, invasiveness and metastatic potential in breast cancer. Breast Cancer Res Treat. 1993;24:227–39. doi: 10.1007/BF01833263. [DOI] [PubMed] [Google Scholar]
- 15.Van’t Veer LJ, Dai H, Van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 16.Janda E, Lehmann K, Killisch I, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasid A, Lippman ME, Papageorge AG, Lowy DR, Gelmann EP. Transfection of v-rasH DNA into MCF-7 human breast cancer cells bypasses dependence on estrogen for tumorigenicity. Science. 1985;228:725–8. doi: 10.1126/science.4039465. [DOI] [PubMed] [Google Scholar]
- 18.deFazio A, Chiew YE, McEvoy M, Watts CK, Sutherland RL. Antisense estrogen receptor RNA expression increases epidermal growth factor receptor gene expression in breast cancer cells. Cell Growth Differ. 1997;8:903–11. [PubMed] [Google Scholar]
- 19.Rundle A, Tang D, Brand-Rauf P, et al. Association between the ras p21 oncoprotein in blood samples and breast cancer. Cancer Lett. 2002;185:71–8. doi: 10.1016/s0304-3835(02)00236-7. [DOI] [PubMed] [Google Scholar]
- 20.Ozer E, Sis B, Ozen E, Sakizli M, Canda T, Sarioglu S. BRCA1, C-erbB-2, and H-ras gene expression in young women with breast cancer. An immunohistochemical study. App Imm Mol Morphol. 2000;8:12–8. doi: 10.1097/00129039-200003000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Mansour S, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–70. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 22.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–52. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 23.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback to Ras proliferative signaling. J Biol Chem. 2000;275:38973–80. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 24.Qi X, Tang J, Pramanik R, et al. p38 MAPK activation selectively induces cell death in K-ras mutated human colon cancer cells through regulation of vitamin D receptor. J Biol Chem. 2004;279:22138–44. doi: 10.1074/jbc.M313964200. [DOI] [PubMed] [Google Scholar]
- 25.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 26.Kim MS, Lee EJ, Kim HRC, Moon A. p38 kinase is a key molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–61. [PubMed] [Google Scholar]
- 27.Pramanik R, Qi X, Borowicz S, et al. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun: the determinant role of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003;278:4831–9. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Ma R, Flavell R, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38α and p38δ MAPK isoforms by TGF-β1 in murine mesangial cells. J Biol Chem. 2002;277:42257–62. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 29.Ono K, Han J. The p38 signal transduction pathway. Activation and function Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 30.Lechner C, Zahalka MA, Giot J, Moller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Pro Natl Acad Sci U S A. 1996;93:4355–9. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38g: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228:334–40. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, McGowan CH, Zhao M, et al. Involvement of the MKK6-38γ cascade in γ-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–52. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdollahi T, Robertson NM, Abdollahi A, Litwack G. Identification of interleukin 8 as an inhibitor of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in the ovarian carcinoma cell line OVCAR3. Cancer Res. 2003;63:4521–6. [PubMed] [Google Scholar]
- 34.Tang J, Qi X, Mercola D, Han J, Chen G. Essential role of p38γ in K-Ras transformation independent of phosphorylation. J Biol Chem. 2005;280:23910–7. doi: 10.1074/jbc.M500699200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu J, Lee JH, Harwalka JA, Oh-Siskovic S, Stacey DW, Golubic M. Adenovirus-mediated gene transfer of dominant negative Ha-Ras inhibits proliferation of primary meningioma cells. Neurosurgery. 1999;44:579–88. doi: 10.1097/00006123-199903000-00080. [DOI] [PubMed] [Google Scholar]
- 36.Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278:20582–92. doi: 10.1074/jbc.M210992200. [DOI] [PubMed] [Google Scholar]
- 37.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 38.Zhang CC, Shapiro DJ. Activation of the p38 mitogen-activated protein kinase pathway by estrogen or by 4-hydroxytamoxifen is coupled to estrogen receptor-induced apoptosis. J Biol Chem. 2000;275:479–86. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Bai W. Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002;22:5835–45. doi: 10.1128/MCB.22.16.5835-5845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qi X, Borowicz S, Pramanik R, Schultz RM, Han J, Chen G. Estrogen receptor inhibits c-Jun-dependent stress-induced cell death by binding and modifying c-Jun activity in human breast cancer cells. J Biol Chem. 2004;279:6769–77. doi: 10.1074/jbc.M311492200. [DOI] [PubMed] [Google Scholar]
- 41.Qi X, Pramank R, Wang J, et al. The p38 and JNK pathways cooperate to trans-activate vitamin D receptor via AP-1 and sensitize human breast cancer cells to vitamin D3-induced growth inhibition. J Biol Chem. 2002;277:25884–92. doi: 10.1074/jbc.M203039200. [DOI] [PubMed] [Google Scholar]
- 42.Huang S, New L, Pan Z, Han J, Nemerow GR. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38α mitogen-activated protein kinase activity. J Biol Chem. 2000;275:12266–72. doi: 10.1074/jbc.275.16.12266. [DOI] [PubMed] [Google Scholar]
- 43.Asada M, Yamada T, Ichijo H, et al. Apoptosis inhibitory activity of cytoplasmic p21cip/WAF1 in monocytic differentiation. EMBO J. 1999;8:1223–34. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eckert LB, Repasky GA, Ulku AS, et al. Involvement of Ras activation in human breast cancer signaling, invasion, and anoikis. Cancer Res. 2004;64:4585–92. doi: 10.1158/0008-5472.CAN-04-0396. [DOI] [PubMed] [Google Scholar]
- 45.Sommers CL, Papageorge AG, Wilding G, Gelmann EP. Growth properties and tumorigenesis of MCF-7 cells transfected with isogenic mutants of rasH. Cancer Res. 1990;50:67–71. [PubMed] [Google Scholar]
- 46.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 47.Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogen and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51:55–67. doi: 10.1016/j.critrevonc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Oesterreich S, Zhang P, Guler RL, et al. Re-expression of estrogen receptor α in estrogen α-negative MCF-7 cells restores both estrogen and insulin-like growth factor-mediated signaling and growth. Cancer Res. 2001;61:5771–7. [PubMed] [Google Scholar]
- 49.Sabio G, Simon J, Arthur C, et al. p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–45. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawrence T, Beblen M, Liu GY, Nizet V, Karin M. IKKα limits macrophage NF-kB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 51.New L, Jiang Y, Han J. Regulation of PRAK subcellular location by p38 MAP kinases. Mol Biol Cell. 2003;14:2603–16. doi: 10.1091/mbc.E02-08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piotrowicz RS, Ding L, Maher P, Levin EG. Inhibition of cell migration by 24-kDa fibroblast growth factor-2 is dependent upon the estrogen receptor. J Biol Chem. 2001;276:3963–70. doi: 10.1074/jbc.M004868200. [DOI] [PubMed] [Google Scholar]
- 53.Seternes O, Mikalsen T, Johansen B, et al. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004;23:4780–91. doi: 10.1038/sj.emboj.7600489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000;62:51–62. doi: 10.1023/a:1006491619920. [DOI] [PubMed] [Google Scholar]
- 55.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–72. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 56.Lundy J, Grimson R, Mishriki Y, Chao S, Ovarez S, Fromowitz F. Elevated ras oncogene expression correlates with lymph node metastases in breast cancer patients. J Clin Oncol. 1986;4:1321–5. doi: 10.1200/JCO.1986.4.9.1321. [DOI] [PubMed] [Google Scholar]
- 57.Clair T, Miller W, Cho-Chung Y. Prognostic significance of the expression of the ras protein with a molecular weight of 21,000 by human breast cancer. Cancer Res. 1987;47:5290–3. [PubMed] [Google Scholar]
- 58.Malumbres M, Barbacid M. Ras oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:7–13. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]