

Abstract
Nuclear RNA transcription is repressed when eukaryotic cells enter mitosis. Here, we found that the derepression of ribosomal gene (rDNA) transcription that normally takes place in telophase may be induced in prometaphase, metaphase, and anaphase mitotic HeLa cells, and therefore appears not to be dependent on completion of mitosis. We demonstrate for the first time that in vivo inhibition of the cdc2– cyclin B kinase activity is sufficient to give rise to okadaic acid–sensitive dephosphorylation of the mitotically phosphorylated forms of components of the rDNA transcription machinery, and consequently to restore rDNA transcription in mitotic cells. These results, showing that during mitosis the rDNA transcription machinery is maintained repressed by the cdc2–cyclin B kinase activity, provide an in vivo demonstration of the cell cycle–dependent regulation of rDNA transcription. Interestingly in mitotic cells, the newly synthesized 47S precursor ribosomal RNA (pre-rRNA) is not processed into the mature rRNAs, indicating that rDNA transcription and pre-rRNA processing may be uncoupled. Moreover this suggests that inhibition of the cdc2– cyclin B kinase is not sufficient to activate the 47S pre-rRNA processing machinery and/or to induce its relocalization at the level of newly synthesized 47S pre-rRNA. This in vivo approach provides new possibilities to investigate the correlation between pre-rRNA synthesis and pre-rRNA processing when the nucleolus reforms.
Keywords: ribosomal DNA transcription, cdc2–, cyclin B kinase, ribosomal RNA processing, roscovitine, mitosis
Introduction
As higher eukaryote cells enter mitosis, transcription is repressed. Silencing of ribosomal gene (rDNA) transcription occurs from prophase ( Prescott and Bender 1962; Gébrane-Younès et al. 1997) to telophase ( Prescott and Bender 1962; Roussel et al. 1996; and for review see Thiry and Goessens 1996). This appears to be a general feature, since only slight variability of this timing can be observed depending on cells. Concurrently, precursor ribosomal RNA (pre-rRNA) processing is arrested at the entrance to and restored at the exit from mitosis ( Fan and Penman 1971). The timing of these events suggests a link with cell cycle controls that could be mediated by the cdc2–cyclin B kinase pathway. Moreover, recent results obtained in vitro have shown that mitotic silencing of rDNA transcription is most probably due to cdc2–cyclin B kinase–directed phosphorylation ( Heix et al. 1998; Kuhn et al. 1998), as reported previously for RNA polymerase (RNA pol) II– and III–dependent transcription (for review see Gottesfeld and Forbes 1997). However, it remains to be elucidated how mitotic cells maintain repression of rDNA transcription in vivo, how they activate transcription at the end of mitosis, and in particular if this activation may be induced in cells without completion of mitosis. Moreover it would be interesting to know if resumption of rDNA transcription gives rise to pre-rRNA processing.
The rDNA transcription machinery remains assembled during mitosis. The different complexes and factors (RNA pol I, upstream binding factor [UBF], TATA-binding protein [TBP]-containing factor SL1) that are sufficient to promote rDNA transcription in vitro (for review see Moss and Stefanovsky 1995) remain localized with rDNAs on chromosomes ( Roussel et al. 1993, Roussel et al. 1996; Zatsepina et al. 1993; Weisenberger and Scheer 1995; Jordan et al. 1996; Gébrane-Younès et al. 1997). In addition, the transcription termination factor TTF-1, which facilitates initiation and mediates termination of rDNA transcription ( Grummt et al. 1986; Längst et al. 1997, Längst et al. 1998; Sander and Grummt 1997), is also colocalized with the mitotic rDNA machinery ( Sirri et al. 1999). Moreover, complexes engaged in transcription seem to be present on chromosomes, since they can be reactivated in vitro by agents blocking new initiation events ( Matsui et al. 1979; Matsui and Sandberg 1985). Considering these observations, it may be proposed that the mitotic inactivation of rDNA transcription occurs at the level of transcription elongation ( Weisenberger and Scheer 1995). However, mitotic phosphorylation impairs the interaction of SL1 with UBF, suggesting that phosphorylation might prevent preinitiation complex formation and shut down rDNA transcription at mitosis ( Heix et al. 1998). Models proposed for repression of RNA pol II– and III–dependent transcription during mitosis include phosphorylation that would induce inactivation and displacement of transcription factors (for review see Gottesfeld and Forbes 1997). In particular, the majority of RNA pol II–specific TBP is detached from the chromatin in mitosis ( Segil et al. 1996). Conversely, the TBP-containing factor SL1 is not dispersed during mitosis, whereas it is inactivated by cdc2–cyclin B kinase–directed phosphorylation ( Heix et al. 1998; Kuhn et al. 1998).
Since the rDNA transcription machinery remains associated with rDNAs during mitosis and its repression is most probably under the control of the cdc2–cyclin B kinase pathway, we anticipated that inactivation of this kinase activity would make it possible to reinduce rDNA transcription on mitotic chromosomes. Our results show that cdc2–cyclin B kinase activity is indispensable to maintain repression of rDNA transcription during mitosis. The inhibition of the cdc2–cyclin B kinase induces resumption of rDNA transcription in colchicine-arrested or metaphase synchronized mitotic HeLa cells in a manner dependent on an okadaic acid–sensitive phosphatase. Interestingly, the newly synthesized 47S pre-rRNA is not processed and therefore accumulates.
Materials and Methods
Cell Culture and Synchronization
HeLa cells were cultured in MEM (Sigma Chemical Co.) supplemented with 10% FCS. For mitosis synchronization, HeLa cells were exposed to 2 mM thymidine for 16 h and then resuspended in fresh medium supplemented with 24 μM 2′-deoxycytidine and allowed to grow for 9 h. Thymidine (2 mM) was added again for 16 h, causing cells to accumulate near the G1/S boundary. The mitotic cells evaluated to represent 90% of the total cells were collected at 11 h after release from the double thymidine block. HeLa cells were also blocked in mitosis (prometaphase) by colchicine treatment (0.02 μg/ml for 14 h). Mitotic cells were harvested by mechanical shock.
Kinase Inhibitor Treatments
A highly selective inhibitor, roscovitine (BIOMOL Research Laboratories) was used to inhibit cdc2–cyclin B kinase. Colchicine-arrested mitotic HeLa cells were treated or not with okadaic acid (0.5 μM) or actinomycin D (0.05 μg/ml) for 1 h. Thereafter, 150 μM roscovitine was added for 30 min. Mitotic HeLa cells obtained by double thymidine block were treated with taxol (5 μg/ml) and 150 μM roscovitine for 30 min.
Antibodies and Probes
The human autoimmune sera with specificity against UBF (A17) and TTF-1 (P21) have been described ( Roussel et al. 1993; Sirri et al. 1999). The rabbit polyclonal antiphosphohistone H1 antibodies and the MPM-2 detection kit were from Upstate Biotechnology. The goat polyclonal anti–TBP-associated factor for RNA pol I (TAFI110) (C-18) antibodies and rabbit polyclonal anti-TBP (N-12) antibodies were from Santa Cruz Biotechnology, Inc. The mouse monoclonal anti–β-tubulin antibody was from Zymed Laboratories, Inc. Texas red–conjugated secondary antibodies specific for human and rabbit IgGs, FITC-conjugated goat anti–mouse antibodies, peroxidase-conjugated secondary antibodies specific for human IgAs and human IgGs were obtained from Jackson ImmunoResearch Laboratories, Inc. Peroxidase-conjugated secondary antibodies specific for rabbit IgGs and goat IgGs were obtained from Amersham France and Sigma Chemical Co., respectively.
The 5′-external transcribed spacer (ETS) leader rDNA probe was an EcoRI-SapI fragment of human rDNA (nucleotide −512/+420) prepared from pBES ( Wilson et al. 1982), and the 5′-ETS core rDNA probe (pBSS; Wilson et al. 1982) was a SalI-SalI fragment of human rDNA (nucleotide +693/+2921).
Immunoblotting
For protein analysis, the cells were resuspended in SDS-PAGE sample buffer ( Laemmli 1970), sonicated, boiled for 5 min, and centrifuged. The supernatant corresponding to the same number of cells was loaded into the gel and proteins were run on a 12% polyacrylamide (acrylamide/bis-acrylamide, 30:0.2)-SDS gel and electrotransferred to nitrocellulose membranes (Protran, Schleicher & Schuell). Membranes were blotted with antibodies to UBF, TTF-1, TBP, TAFI110, and phosphohistone H1 as described previously ( Sirri et al. 1999).
Assay of RNA Pol Activity In Situ and Immunofluorescence Labeling
The cells were washed in PBS and transferred onto poly-l-lysine–coated glass slides. The assay of in situ RNA pol activity was performed as described previously ( Roussel et al. 1996) in conditions set up to reveal RNA pol I and RNA pol II transcription ( Moore and Ringertz 1973). Bromo-uridine 5′-triphosphate (BrUTP) incorporation was detected by immunofluorescence labeling using a mouse monoclonal anti-BrdU antibody (Boehringer Mannheim) revealed by FITC-conjugated goat anti–mouse antibodies. UBF was simultaneously detected using serum A17 ( Roussel et al. 1993) and revealed by Texas red–conjugated goat anti–human antibodies. Phosphorylated histone H1 was detected with rabbit polyclonal antiphosphohistone H1 antibodies followed by Texas red–conjugated goat anti–rabbit antibodies. DNA was visualized with 4,6-diamidino-2-phenylindole (DAPI).
Fluorescent microscopy was performed using a CCD camera Leitz DMRB. Images were then assembled using Adobe Photoshop and printed directly from the computer on a printer (ColorEase PS Printer; Eastman-Kodak Co.).
Metabolic Labeling and Northern Blot Analysis
For RNA analysis, colchicine-arrested mitotic HeLa cells were metabolically labeled or not for 3 h with [32P]orthophosphate at a final concentration of 250 μCi/ml (ICN Biomedicals) in phosphate-free MEM. During metabolic labeling, the cells were treated or not with okadaic acid (0.5 μM) or actinomycin D (0.05 μg/ml) for 1 h. Roscovitine (150 μM) was then added for 30 min. Total RNAs were isolated using the kit, RNA NOW (Biogentex). The 32P-labeled RNAs were separated by electrophoresis on 1% agarose formaldehyde gels. The RNAs were transferred to positively charged membranes (Boehringer Mannheim). After UV cross-linking, autoradiographies were performed with a PhosphorImager (Molecular Dynamics, Inc.). The size of the RNAs was determined by comparison to an RNA ladder (GIBCO BRL). In parallel, the unlabeled RNAs were run in 0.75% agarose formaldehyde gels and transferred to positively charged membranes. The 5′-ETS core rDNA and 5′-ETS leader rDNA probes were labeled with [α32P]dCTP by nick translation (GIBCO BRL). The membranes were prehybridized for 5 h at 42°C in buffer containing 50% formamide, 5× SSPE, 10× Denhardt's solution, 0.1% SDS, and 50 μg/ml salmon sperm DNA ( Maniatis et al. 1982). Hybridization with the radio-labeled probes was carried out in the same buffer for 24 h at 42°C. After hybridization, the membranes were washed twice for 10 min each in 2× SSC and 0.1% SDS at 65°C, and finally once for 5 min in 0.1× SSC and 0.1% SDS at 65°C. Autoradiographies were performed with a PhosphorImager.
Results
Mitotic Repression of Transcription Is Abolished by Inhibition of the cdc2–cyclin B Kinase Pathway
Global inhibition of transcription occurring during mitosis in higher eukaryotes appears at the G2/M transition phase when the cdc2–cyclin B kinase is activated. To determine whether the cdc2–cyclin B kinase pathway is responsible in vivo for the mitotic inhibition of transcription, in situ transcription assays were carried out on colchicine-arrested mitotic HeLa cells treated or not with roscovitine, a highly selective inhibitor of cyclin-dependent kinases ( De Azevedo et al. 1997; Meijer et al. 1997). Several cyclin-dependent kinases including cdc2–cyclin B and cdk2–cyclin A or E are very sensitive to roscovitine (IC50 < 7 μM) when tested in vitro. However, because cdk2–cyclin E and cdk2–cyclin A are active at the G1/S transition and during S phase, respectively, the cdc2–cyclin B kinase is the only known kinase inhibited by roscovitine when mitotically synchronized cells are treated with this inhibitor. The inhibition of the cdc2–cyclin B kinase after roscovitine treatment was verified by analysis of the level of the hyperphosphorylated form of histone H1, a well-characterized substrate for cdc2–cyclin B kinase. As expected, no transcription activity was observed by in situ transcription assays set up to reveal both RNA pol I and RNA pol II transcription in colchicine-arrested mitotic HeLa cells ( Fig. 1 b) and the hyperphosphorylated form of histone H1 was detected ( Fig. 1 c). More interestingly, when cells were treated with roscovitine for 30 min before being processed for in situ transcription assays, transcription activity was detected in ∼60% of the cells as determined by the incorporation of BrUTP appearing in spots in association with chromosomes ( Fig. 1 e). The inhibition of the cdc2–cyclin B kinase after roscovitine treatment was demonstrated by the low level of the hyperphosphorylated form of histone H1 in a large proportion of cells (compare Fig. 1c and Fig. f). By analyzing simultaneously both the level of phosphohistone H1 and BrUTP incorporation, we could relate the inhibition of the cdc2–cyclin B kinase to the resumption of transcription activity. Conversely, cells showing a high level of the hyperphosphorylated form of histone H1 did not exhibit any transcription activity ( Fig. 1e and Fig. f, arrowheads). The resumption of transcription in mitotic cells after roscovitine treatment was dependent on an okadaic acid–sensitive phosphatase as shown in Fig. 1 h. Indeed, neither dephosphorylation of histone H1 (compare Fig. 1f and Fig. i) nor restoration of transcription activity was observed when cells were treated with both roscovitine and okadaic acid before being processed for in situ transcription assays (compare Fig. 1e and Fig. h).
Figure 1.
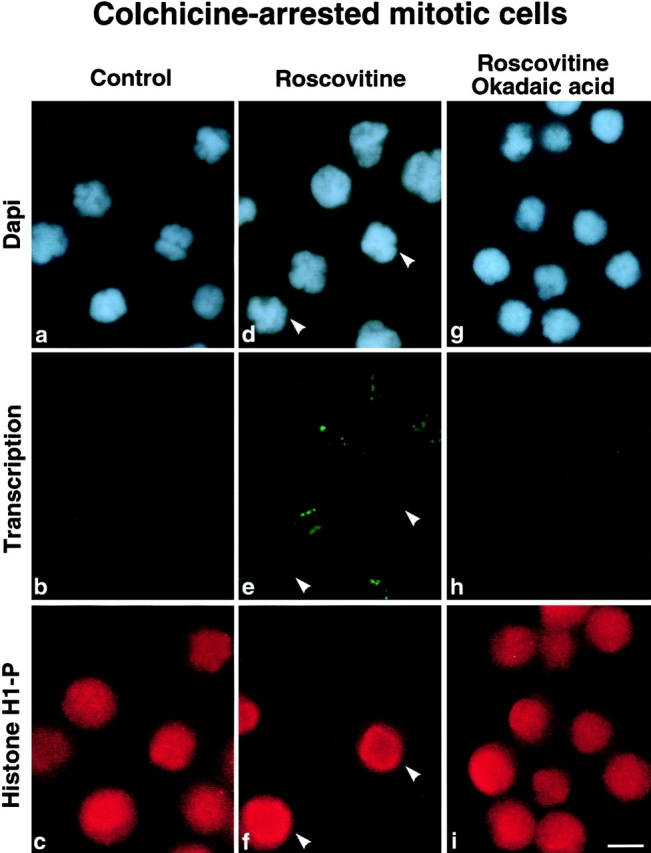
Mitotic repression of transcription is abolished by roscovitine treatment. HeLa cells blocked in mitosis by colchicine treatment (0.02 μg/ml for 14 h) were treated (d–f) or not (a–c) with 150 μM roscovitine, a specific inhibitor of cdc2–cyclin B kinase, for 30 min. Mitotic cells were also treated with 0.5 μM okadaic acid for 1 h before addition of 150 μM roscovitine for 30 min (g–i). Transcription activity was revealed by the detection of in situ incorporation of BrUTP in conditions favoring both RNA pol I and RNA pol II activity (b, e, and h). Simultaneously, the inhibition of cdc2–cyclin B kinase was verified by the detection of the hyperphosphorylated form of histone H1 (c, f, and i). The chromosomes were stained with DAPI (a, d, and g). In control cells (a–c) no transcription activity was detected (b) and histone H1 was hyperphosphorylated (c). In roscovitine-treated cells (d–f) transcription activity was detected (e) in mitotic cells negative for hyperphosphorylated histone H1 (f). Arrowheads indicate cells positive for phosphorylation of histone H1 (f) and negative for transcription activity (e). In mitotic cells, additionally pretreated with okadaic acid (g–i), neither dephosphorylation of histone H1 (i) nor transcription activity (h) was detected. The inhibition of cdc2–cyclin B kinase abolished mitotic repression of transcription dependent on an okadaic acid–sensitive phosphatase. Bar, 10 μm.
Inhibition of the cdc2–cyclin B Kinase Pathway Specifically Restores rDNA Transcription in Mitotic Cells
Inhibition of the cdc2–cyclin B kinase pathway by roscovitine treatment in colchicine-arrested mitotic HeLa cells leads rapidly to the recovery of transcription activity in a major proportion of the cells as demonstrated by in situ BrUTP incorporation. The fluorescent patterns observed in the same cells for BrUTP incorporation and for UBF showed that both labelings were superimposable ( Fig. 2e and Fig. f). The localization of UBF, and consequently of the rDNA transcription machinery, and the detection of transcription activity made it possible to observe that after inhibition of the cdc2–cyclin B kinase pathway, the transcription activity was restored at nucleolar organizer regions (NORs), i.e., in chromosomal sites where rDNAs associated with the rDNA transcription machinery clustered during mitosis. This observation suggests that the transcription activity that had resumed in colchicine-arrested mitotic cells most probably corresponded to rDNA transcription. This was confirmed by results obtained when mitotic cells were treated with a low concentration (0.05 μg/ml) of actinomycin D known to inhibit rDNA transcription in vivo. As shown in Fig. 2 h, the transcription activity observed in mitotic cells after roscovitine treatment was hypersensitive to actinomycin D. In conclusion, cdc2–cyclin B kinase activity is indispensable to maintain mitotic silencing of rDNA transcription, and its inhibition restores rDNA transcription in mitotic cells, whereas no RNA pol II–dependent transcription is detected.
Figure 2.
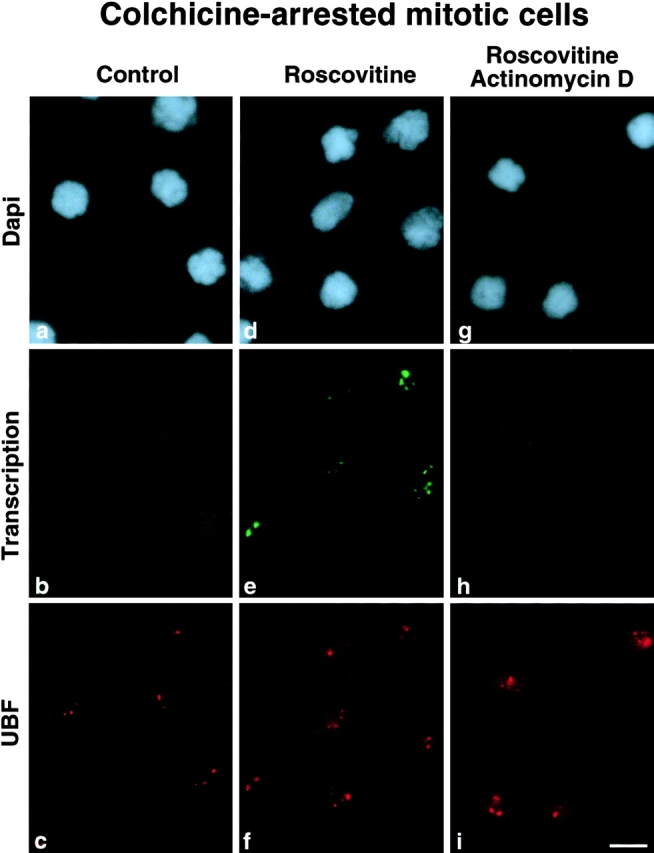
Roscovitine treatment relieves mitotic silencing of rDNA transcription. HeLa cells blocked in mitosis by colchicine treatment (0.02 μg/ml for 14 h) were treated (d–f) or not (a–c) with 150 μM roscovitine for 30 min. Mitotic cells were also treated with a low concentration of actinomycin D (0.05 μg/ml for 1 h) before roscovitine treatment (g–i). Transcription activity was detected by in situ BrUTP incorporation in conditions favoring both RNA pol I and RNA pol II activity (b, e, and h). Localization of the rDNA transcription machinery was revealed by UBF localization (c, f, and i). In roscovitine-treated cells the sites of BrUTP incorporation (e) were superimposable to the sites of localization of the rDNA transcription machinery revealed by UBF labeling (f). The actinomycin D treatment abolished this transcription activity. No incorporation of BrUTP was detected in actinomycin D–treated cells (h) as for control cells (b). Bar, 10 μm.
Restoration of rDNA Transcription in Mitotic Cells Leads to Accumulation of the Unprocessed 47S Pre-rRNA
To definitively establish that rDNA transcription is restored in roscovitine-treated mitotic cells and to determine what rRNA transcript is synthesized, colchicine-arrested mitotic HeLa cells were metabolically labeled with [32P]orthophosphate. Cellular RNAs were extracted, separated on agarose formaldehyde gels, transferred to nylon membranes, and the 32P-labeled RNA was recorded using a PhosphorImager ( Fig. 3 A). To identify the newly synthesized RNA, the same experiment was reproduced without 32P metabolic labeling, and cellular RNA was hybridized with a 32P-labeled 5′-ETS core rDNA probe recognizing both the partially processed (45S and 46S) and the unprocessed (47S) pre-rRNAs ( Fig. 3 B, lanes a′–d′), and with a 32P-labeled 5′-ETS leader rDNA probe recognizing specifically the unprocessed 47S pre-rRNA ( Fig. 3 B, lanes e′–h′). In colchicine-arrested mitotic HeLa cells ( Fig. 3 A, lanes a and a′), several RNA species appeared weakly 32P-labeled after 32P metabolic labeling ( Fig. 3 A, lane a′). This was most probably due to the presence of 2–5% of contaminating interphase cells as assessed by microscope observations (data not shown). When colchicine-arrested mitotic HeLa cells were treated with roscovitine ( Fig. 3 A, lanes b and b′), a large RNA was synthesized ( Fig. 3 A, lane b′). This RNA, whose synthesis is inhibited by 0.05 μg/ml actinomycin D ( Fig. 3 A, compare lanes b′ and c′), was identified by Northern blot analyses as pre-rRNA ( Fig. 3 B, lanes b′ and f′). As observed by in situ transcription assays ( Fig. 1), resumption of rDNA transcription triggered by roscovitine treatment is abolished if the roscovitine effects are counteracted by okadaic acid treatment ( Fig. 3 A, lane d′). It is noteworthy that the levels of 32P-labeled 28S and 18S rRNA did not vary ( Fig. 3 A, compare lanes a′ and b′). Therefore, the newly synthesized pre-rRNA did not seem to be processed into mature rRNAs. The absence of any processing of the newly synthesized pre-rRNA is supported by Northern blot analyses using the 5′-ETS core rDNA probe of pre-rRNA present in mitotic cells ( Fig. 3 B, lanes a and a′) for which rDNA transcription was restored by roscovitine treatment ( Fig. 3 B, lanes b and b′), and in mitotic cells for which rDNA transcription was simultaneously restored by roscovitine and inhibited by actinomycin D treatment ( Fig. 3 B, lanes c and c′). Indeed, the partially processed pre-rRNA known to be present in mitotic cells ( Dundr and Olson 1998; Fig. 3 B, lane a′) was actively processed and/or degraded after roscovitine treatment, and consequently, largely decreased as observed when rDNA transcription was simultaneously restored by roscovitine and inhibited by actinomycin D treatment ( Fig. 3 B, compare lanes a′ and c′). As a consequence, we can conclude that the pre-rRNA easily detectable in mitotic cells treated with roscovitine ( Fig. 3 B, lane b′) corresponds to newly synthesized pre-rRNA, which is not processed and therefore accumulates. The electrophoretic behavior of this pre-rRNA present in mitotic cells treated with roscovitine ( Fig. 3 B, lane b′) argues in favor of the absence of any processing event of the newly synthesized pre-rRNA, even of the primary processing events. Indeed this pre-rRNA migrates slightly slower than the partially processed pre-rRNA present in mitotic cells ( Fig. 3 B, compare lanes a′ and b′). This is confirmed using the 5′-ETS leader rDNA probe, which recognizes specifically the unprocessed 47S pre-rRNA ( Fig. 3 B, lane f′), and therefore we can conclude that restoration of rDNA transcription in mitotic cells leads to accumulation of the unprocessed 47S pre-rRNA. On the other hand it is interesting to note that processing and/or degradation of the partially processed pre-rRNA present in mitotic cells ( Dundr and Olson 1998; Fig. 3 B, lane a′) that occurs after roscovitine treatment ( Fig. 3 B, lane c′) is no longer observed in the presence of okadaic acid ( Fig. 3 B, lane d′).
Figure 3.

Identification of RNAs synthesized after roscovitine treatment in mitotic cells. (A) Colchicine-arrested mitotic HeLa cells were metabolically labeled with [32P]orthophosphate (250 μCi/ml). During labeling, cells were treated or not with okadaic acid (OA) or actinomycin D (AMD) for 1 h, after which roscovitine (Rosc) was added for 30 min. The RNAs corresponding to 8 × 105 cells were analyzed. (A, lanes a–d) RNAs detected in 1% agarose gel by ethidium bromide staining. (A, lanes a′–d′) RNAs detected after autoradiography. In roscovitine-treated cells (A, lane b′) an intense, slowly migrating RNA is synthesized (A, arrow) when compared with the control cells (A, lane a′). Synthesis of RNA was abolished by actinomycin D (A, lane c′) and okadaic acid treatment (A, lane d′). (B) The same experiment was reproduced without metabolic labeling and the RNAs were analyzed by Northern blot using 32P-labeled 5′-ETS core or 32P-labeled 5′-ETS leader rDNA probes. (B, lanes a–h) RNAs detected in 0.75% agarose gel by ethidium bromide staining. (B, lanes a′–d′) Blot hybridized with a 32P-labeled 5′-ETS core rDNA probe to identify the 47–45S pre-rRNAs. In extracts prepared from roscovitine-treated mitotic cells (B, lane b′) the pre-rRNA detected by the 5′-ETS core rDNA probe migrated slightly slower than in those prepared from control cells (B, lane a′) and from mitotic cells treated with both actinomycin D and roscovitine (B, lane c′) or with both okadaic acid and roscovitine (B, lane d′). When mitotic cells were treated with actinomycin D and roscovitine, the level of pre-rRNA was largely decreased (B, lane c′). (B, lanes e′–h′) Blot hybridized with a 32P-labeled 5′-ETS leader rDNA probe to specifically identify the unprocessed 47S pre-rRNA. The unprocessed 47S pre-rRNA accumulated in extracts prepared from roscovitine-treated mitotic cells (B, lane f′) contrary to those prepared from control cells (B, lane e′) and from mitotic cells treated with actinomycin D and roscovitine (B, lane g′) or with okadaic acid and roscovitine (B, lane h′).
Mitotically Phosphorylated Forms of Components of the rDNA Transcription Machinery Are Dephosphorylated by Roscovitine Treatment
Several components of the rDNA transcription machinery, TAFI110, and TBP ( Heix et al. 1998), UBF ( Klein and Grummt 1999), and more recently TTF-1 ( Sirri et al. 1999) were reported to be phosphorylated during mitosis. Except for UBF, mitotic phosphorylation was shown to modify the electrophoretic mobility of these components ( Heix et al. 1998; Sirri et al. 1999), and is therefore easily observed. As shown in Fig. 4, TTF-1, TAFI110, and TBP migrated slower in colchicine-arrested mitotic HeLa cells ( Fig. 4, lane 2) than in interphase cells ( Fig. 4, lane 1). When mitotic cells were treated for 30 min with roscovitine, phosphorylation of these components was largely reversed, as demonstrated by their electrophoretic migration (compare Fig. 4, lanes 1 and 3). The dephosphorylation, occurring when cdc2–cyclin B kinase is inhibited, implicated the activity of an okadaic acid–sensitive phosphatase as observed in Fig. 4, lane 4. Indeed, if mitotic cells were treated previously with okadaic acid, subsequent roscovitine treatment produced no effect on the phosphorylation state of these components. On the other hand, dephosphorylation is obviously not a consequence of the restoration of rDNA transcription activity. Inhibition of rDNA transcription by actinomycin D did not affect dephosphorylation from occurring when mitotic cells were treated with roscovitine ( Fig. 4, lane 5). In this series of experiments, cdc2–cyclin B kinase activity was detected by analyzing the level of the hyperphosphorylated form of histone H1. The hyperphosphorylated form of histone H1, only present in mitotic cells (compare Fig. 4, lanes 1 and 2), disappeared when mitotic cells were treated with roscovitine ( Fig. 4, lane 3) or with both roscovitine and actinomycin D ( Fig. 4, lane 5). The absence of histone H1 dephosphorylation when mitotic cells were treated with both roscovitine and okadaic acid ( Fig. 4, lane 4) is explained by the fact that okadaic acid inhibits protein phosphatase-1 (PP-1), which is required for dephosphorylation of histone H1 ( Paulson et al. 1996). Considering these results and those reported above, it seems clearly established that the mitotic repression of rDNA transcription is dependent on phosphorylation events, and most probably on phosphorylation of components of the rDNA transcription machinery. Inhibition of the cdc2–cyclin B kinase is sufficient to restore rDNA transcription activity only if no okadaic acid–sensitive phosphatase is inhibited. This observation suggests that cdc2–cyclin B kinase might repress rDNA transcription during mitosis by inhibiting an okadaic acid–sensitive phosphatase. Nevertheless, we cannot rule out the possibility that inhibition of an okadaic acid–sensitive phosphatase counteracts the roscovitine effect by increasing cdc2–cyclin B kinase activity.
Figure 4.

Mitotically phosphorylated forms of components of the rDNA transcription machinery are dephosphorylated by roscovitine treatment. Immunoblots of unsynchronized HeLa cells (lane 1) and colchicine-arrested mitotic HeLa cells (lanes 2–5) not treated with roscovitine (lane 2) or treated with roscovitine (+Ros; lane 3), with both okadaic acid and roscovitine (+OA +Ros; lane 4), or with both actinomycin D and roscovitine (+AMD +Ros; lane 5) probed with antibodies directed against different components of the rDNA transcription machinery (TTF-1, TAFI110, UBF, and TBP). Antiphosphohistone H1 (indicated by H1-circled P) antibodies were used to verify the inhibition of cdc2–cyclin B kinase by roscovitine treatment. Unsynchronized HeLa cells (lane 1) were used to identify the nonphosphorylated form of the proteins studied. Except for UBF, the other factors were phosphorylated in untreated mitotic cells (compare lanes 1 and 2). In mitotic extracts the bands corresponding to TTF-1 and TAFI110 migrated slightly slower (lane 2) than in interphase extracts (lane 1). For TBP, different phosphorylated forms were resolved in mitotic extracts (lane 2). These mitotic phosphorylations were partially reversed for TTF-1 and completely for TAFI110, TBP, and histone H1 by roscovitine treatment (compare lanes 1–3). Okadaic acid pretreatment counteracted the effect of roscovitine on mitotic phosphorylation of these proteins: only hyperphosphorylated forms were visible (lanes 3 and 4). Pretreatment with actinomycin D did not affect the dephosphorylation induced by roscovitine treatment (lane 5).
The Resumption of rDNA Transcription Activity Is Not a Consequence of Exit from Mitosis
During the cell cycle, mitotic repression of rDNA transcription is released at telophase when cells exit from mitosis. Therefore, it may be asked whether the inhibition of cdc2–cyclin B kinase in colchicine-arrested mitotic cells induces exit from mitosis, and consequently derepression of rDNA transcription, or whether cdc2–cyclin B kinase directly regulates rDNA transcription. To address this question, roscovitine treatments were reproduced using synchronized mitotic HeLa cells. The population of mitotic cells used was collected 11 h after release from a double thymidine block and treated or not with roscovitine for 30 min. As illustrated in Fig. 5, control cells ( Fig. 5, a and b) exhibited a large percentage of prometaphase and metaphase cells (∼50%), whereas among roscovitine-treated cells ( Fig. 5d and Fig. e), very few if any were found at these middle mitotic stages. Obviously the roscovitine treatment caused an accelerated and most probably abnormal progress through mitosis, and cells appeared in later mitotic stages. As shown in Fig. 5 f, by in situ transcription, most roscovitine-treated cells exhibited rDNA transcription, but it was impossible to say if resumption of rDNA transcription was directly due to inhibition of cdc2–cyclin B kinase or to accelerated progress through mitosis.
Figure 5.

Roscovitine treatment causes accelerated and abnormal progress through mitosis. Mitotic HeLa cells were collected 11 h after release from double thymidine block and treated (d–f) or not (a–c) with 150 μM roscovitine for 30 min. The cells were analyzed by phase-contrast and DAPI staining. In the control cells a large percentage (∼50%) of prometaphase and metaphase stages (a and b, arrowheads) was detected, contrary to roscovitine-treated cells in which later mitotic stages were visible (d and e). rDNA transcription activity was detected after roscovitine treatment (f). Bar, 10 μm.
To overcome this problem, a population of mitotic cells enriched in middle mitotic stages was treated ( Fig. 6, e–h) or not ( Fig. 6, a–d) with roscovitine for 30 min, and the same population of mitotic cells was treated with both roscovitine and the microtubule poison taxol for the same length of time ( Fig. 6, i–l). Cells were analyzed by DNA and β-tubulin staining to follow progress through mitosis. As reported above, roscovitine treatment accelerated progress through mitosis as shown by the disappearance of prometaphase and metaphase cells ( Fig. 6; compare Fig. 6, a–d and Fig. 6, e–h). When cells were treated with both roscovitine and taxol, the microtubule stabilization effect obtained by taxol treatment reversed the effect of roscovitine on the kinetics of mitosis. As observed when Fig. 6, a–d are compared with Fig. 6i–l, prometaphase and metaphase cells are maintained. We also analyzed derepression of rDNA transcription by in situ transcription assays ( Fig. 7g and Fig. k), and either UBF localization ( Fig. 7 h) or cdc2–cyclin B kinase activity by hyperphosphorylated histone H1 detection ( Fig. 7 l). The results obtained are similar to those obtained on colchicine-arrested mitotic cells, i.e., transcription is restored in prometaphase and metaphase cells at NORs, where the rDNA transcription machinery is localized in association with rDNAs during mitosis (compare Fig. 7g and Fig. h) only in cells for which cdc2–cyclin B kinase activity is actually inhibited as demonstrated by the disappearance of hyperphosphorylated histone H1 (compare Fig. 7k and Fig. l). Therefore, the resumption of rDNA transcription in mitotic cells is not dependent on completion of mitosis nor is it a consequence of exit from mitosis. Inhibition of cdc2–cyclin B kinase relieves mitotic silencing of rDNA transcription in metaphase HeLa cells ( Fig. 8) independently of separation of sister chromatids and chromosome decondensation. The same conclusion is derived from results obtained when MPM-2 antibody, which recognizes mitosis-specific epitopes ( Davis et al. 1983), is used on colchicine-arrested mitotic HeLa cells treated with roscovitine. No loss of reactivity with the MPM-2 antibody, characterizing exit from mitosis ( Andreassen and Margolis 1994), is observed in colchicine-arrested mitotic HeLa cells exhibiting rDNA transcription activity (data not shown).
Figure 6.
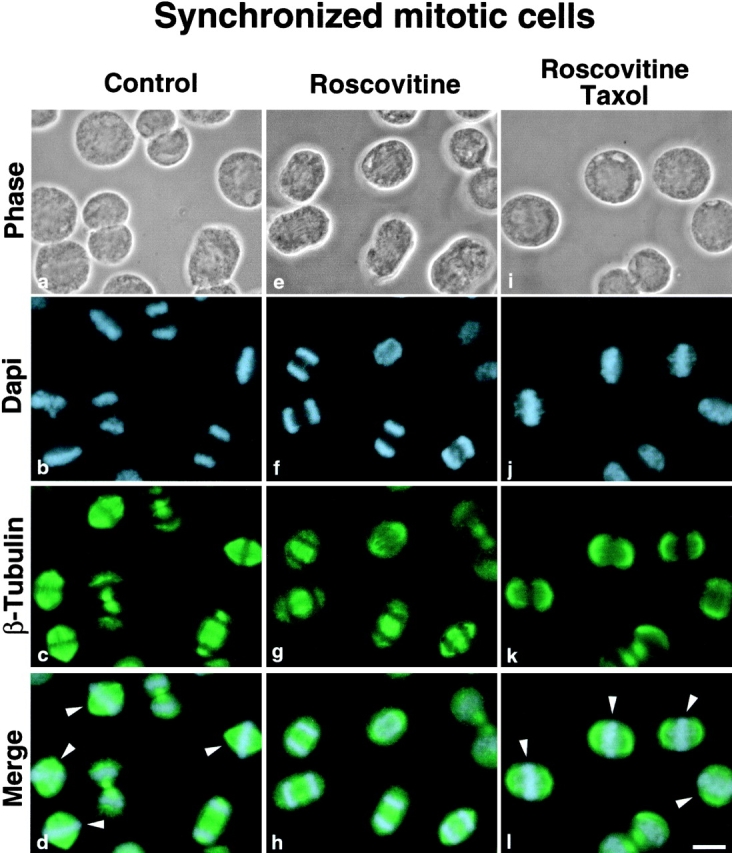
Taxol treatment reverses the effect of roscovitine on the kinetics of mitosis. HeLa cells synchronized in middle mitotic stages by a double thymidine block were not treated (a–d) or treated with roscovitine (e–h), or with both roscovitine and taxol (i–l) for 30 min. The cells were analyzed by phase-contrast (a, e, and i), DAPI staining (b, f, and j), and β-tubulin labeling (c, g, and k). (d, h, and l) Superimposition of DAPI staining and β-tubulin labeling. In roscovitine-treated cells (h), a higher percentage of late mitotic stages than in control cells (d) was observed. Conversely, addition of taxol blocked the cells at middle mitotic stages (l) by reversing the effect of roscovitine on the kinetics of mitosis. Arrowheads (d and l) indicate prometaphase and metaphase cells. Bar, 10 μm.
Figure 7.
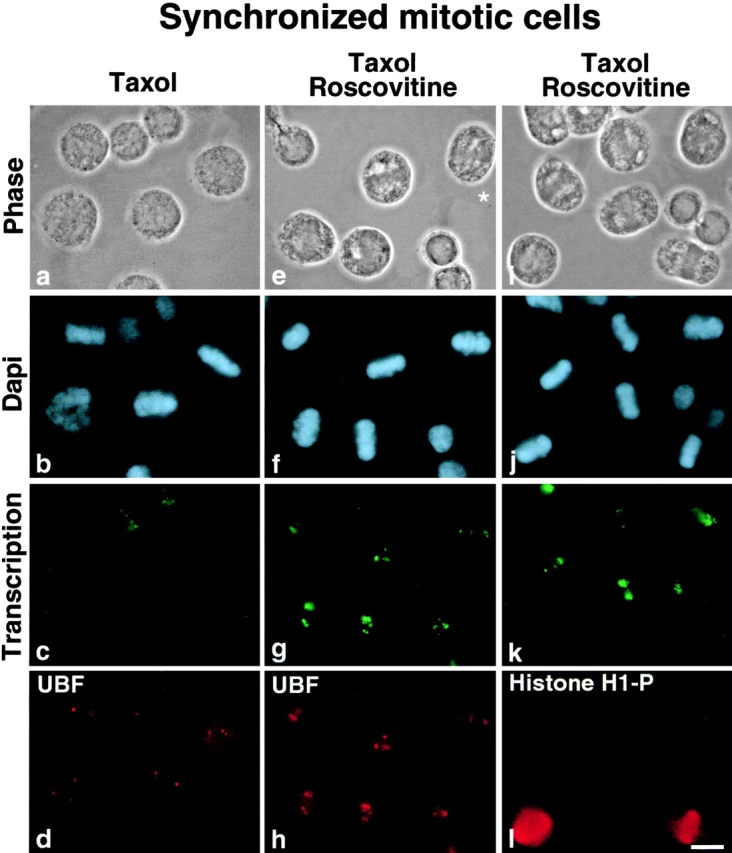
Roscovitine treatment restores rDNA transcription in metaphase cells. Mitotic HeLa cells synchronized by a double thymidine block were treated with taxol (a–d) or taxol and roscovitine (e–l) for 30 min. The cells were processed to detect the transcription activity (c, g, and k), and to label UBF (d and h) and the hyperphosphorylated form of histone H1 (l). The mitotic stages were visualized by phase-contrast (a, e, and i) and DAPI staining (b, f, and j). The roscovitine treatment was sufficient to restore transcription activity in metaphase (g and k). This transcription activity was detected at the NORs, where UBF is localized (compare g and h), only in cells in which cdc2–cyclin B kinase was actually inhibited as evidenced by the absence of the hyperphosphorylated form of histone H1 (compare k and l). Asterisk (e) indicates the cell for which enlargement is shown in Fig. 8. Bar, 10 μm.
Figure 8.
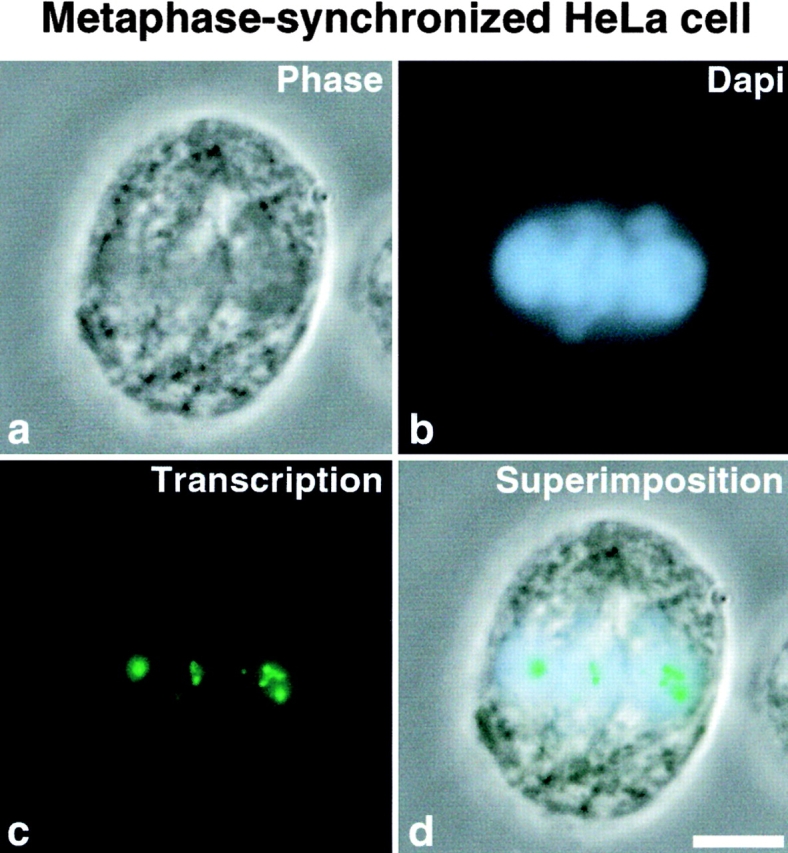
After roscovitine treatment, the sites of rDNA transcription detected by BrUTP incorporation (c) are observed in a metaphase HeLa cell (a) on the DAPI-stained chromosomes (b) aligned at the metaphase plate as shown by superimposition of the phase-contrast image, DAPI staining, and in situ transcription detection (d). Bar, 10 μm.
Discussion
Mitotic Silencing of rDNA Transcription
It is now well-established that cdc2–cyclin B kinase–directed phosphorylation plays a key role in mitotic repression of RNA pol I– ( Heix et al. 1998; Kuhn et al. 1998), RNA pol II– ( Leresche et al. 1996; Long et al. 1998), and RNA pol III–dependent ( Hartl et al. 1993; Leresche et al. 1996; Gottesfeld and Forbes 1997) transcription. Our results show that the cdc2–cyclin B kinase is not only necessary to establish rDNA transcription repression as proposed for the mitotic repression of RNA pol III transcription ( Hartl et al. 1993), but also to maintain repression from prophase to telophase. Indeed, reactivation of rDNA transcription after in vivo inhibition of the cdc2–cyclin B kinase pathway in colchicine-arrested and synchronized mitotic cells shows that cdc2–cyclin B kinase activity directly controls rDNA transcription during mitosis. We also show that the cdc2–cyclin B kinase maintains mitotic rDNA silencing, most probably by inhibiting an as-yet unidentified serine/threonine phosphatase. Indeed, inhibition of the cdc2–cyclin B kinase leads to dephosphorylation of the components of the rDNA transcription machinery specifically phosphorylated during mitosis, i.e., TBP, TAFI110, and TTF-1, and consequently to derepression of mitotic rDNA silencing only in the absence of okadaic acid. PP-1, which plays a key role for anaphase progression and exit from mitosis ( Fernandez et al. 1992) and which is required for dephosphorylation of histone H1 ( Paulson et al. 1996), seems to be a good candidate for dephosphorylation of components of the rDNA transcription machinery. This hypothesis is also supported by the fact that the PP-1 δ isoform remains associated with chromosomes throughout mitosis ( Andreassen et al. 1998). Moreover, in this work, dephosphorylation of TBP, TAFI110, and TTF-1 occurs after cdc2–cyclin B kinase inhibition in a manner dependent on an okadaic acid–sensitive phosphatase as does dephosphorylation of histone H1.
Derepression of rDNA Transcription Is Not the Consequence of Exit from Mitosis and Does Not Require Completion of Mitosis
Restoration of rDNA transcription after cdc2–cyclin B kinase inhibition might be interpreted as a consequence of exit from mitosis. Indeed, it is known that inhibition of mitotic kinases by the protein kinase inhibitor 2-aminopurine induces checkpoint override and mitotic exit in cells arrested in mitosis by inhibitors of microtubule function ( Andreassen and Margolis 1994). Moreover, as shown in this study, inhibition of cdc2–cyclin B kinase by roscovitine treatment induces rapid exit from mitosis, most probably by shunting the spindle-assembly checkpoint. Nevertheless, we have been able to verify that the reactivity with MPM-2 antibodies, which recognize mitosis-specific epitopes ( Davis et al. 1983), is maintained in colchicine-arrested mitotic cells treated with roscovitine for 30 min. Moreover, rDNA transcription may be restored in synchronized metaphase cells treated both by roscovitine and taxol. Consequently, rDNA transcription is restored in mitotic cells by inhibition of cdc2–cyclin B kinase activity. The resumption of rDNA transcription is only dependent on cdc2–cyclin B kinase activity and does not need progression of cells through late mitotic stages.
Mitotic Repression Is Released Successively for the Different RNA Pols
During the cell cycle, derepression takes place successively for the different RNA pols. Whereas RNA pol I transcription is reactivated in telophase ( Roussel et al. 1996), reactivation of RNA pol III transcription takes place later during the G1 phase ( White et al. 1995). Moreover, as shown here, inhibition of cdc2–cyclin B kinase restores rDNA transcription rapidly and uniquely. The rapid switch-on of rDNA transcription in telophase or after roscovitine treatment, i.e., when cdc2–cyclin B kinase is inactivated, is most probably made possible by the fact that, as opposed to other RNA pol–dependent transcription machineries, the rDNA transcription machinery is not released from rDNAs during mitosis. Concerning the molecular basis of mitotic repression of rDNA transcription, two somewhat contradictory mechanisms have been proposed. The first mechanism suggests that rDNA transcription is regulated at the level of transcription elongation ( Weisenberger and Scheer 1995). This is supported by biochemical experiments ( Matsui and Sandberg 1985) showing that rDNA transcription may be reactivated in vitro in the presence of heparin, i.e., an agent that blocks new initiation events, and by immunocytochemical studies ( Zatsepina et al. 1993; Weisenberger and Scheer 1995; Jordan et al. 1996; Roussel et al. 1996; Gébrane-Younès et al. 1997; Sirri et al. 1999) showing that the rDNA transcription machinery is never disengaged from rDNAs. The second mechanism proposed is that mitotic repression of rDNA transcription takes place at the level of transcription initiation. This is based on recent results obtained using a reconstituted cell-free transcription system ( Heix et al. 1998) showing that the mitotic phosphorylation of SL1 impairs the capability of SL1 to interact with UBF, and therefore prevents preinitiation complex formation. Our data do not make it possible to discriminate between these two mechanisms, but they establish that whatever the step of the rDNA transcription process affected, the mechanism that represses rDNA transcription during mitosis is reversed in vivo after inactivation of the cdc2–cyclin B kinase.
Newly Synthesized 47S Pre-rRNA Is Not Processed after cdc2–cyclin B Inhibition
In eukaryotes, the rRNAs are cotranscribed as a single large pre-rRNA that is rapidly processed into the mature rRNAs ( Allmang and Tollervey 1998). In humans, the 47S pre-rRNA is processed into the mature 18S, 5.8S, and 28S rRNAs. Interestingly, the resumption of rDNA transcription in mitotic cells does not give rise to processing of the newly synthesized 47S pre-rRNA. As reported here, when rDNA transcription is restored, the newly synthesized 47S pre-rRNA accumulates without increase of the 18S and 28S rRNA species. These results seem to indicate that when rDNA transcription is restored, it is uncoupled from 47S pre-rRNA processing. It is noteworthy that the partially processed pre-rRNA preserved during mitosis in association with pre-rRNA processing components ( Dundr and Olson 1998) appears to be actively processed after inhibition of cdc2–cyclin B kinase in colchicine-arrested mitotic HeLa cells. Our results suggest that the machinery involved in processing of the partially processed pre-rRNA is also derepressed by inhibition of cdc2–cyclin B kinase in a manner dependent on an okadaic acid–sensitive phosphatase.
With respect to the newly synthesized 47S pre-rRNA, its processing could be inhibited at the level of the primary processing events that had already taken place for the partially processed pre-rRNA ( Dundr and Olson 1998), or could be prevented by the absence of components implicated in pre-rRNA processing. Indeed, as opposed to the rDNA transcription machinery that remains associated with rDNAs at the level of NORs during mitosis, the pre-rRNA processing components appear to be excluded from NORs ( Hernandez-Verdun and Gautier 1994; Weisenberger and Scheer 1995). Pre-rRNA processing components such as fibrillarin, nucleolin, protein B23, and U3 small RNAs leave the NORs and relocalize around the chromosomes early in prophase ( Gautier et al. 1992, Gautier et al. 1994). Normally, at the end of mitosis, in telophase, these components are recruited in prenucleolar bodies ( Fomproix et al. 1998) that relocalize to transcription sites, i.e., the reforming nucleoli, with different kinetics ( Westendorf et al. 1998; Fomproix and Hernandez-Verdun 1999; Savino et al. 1999). It is presently unknown how the coordination between rDNA transcription activation and targeting of the rRNA processing machinery to sites of transcription is controlled at the end of mitosis. In this study, the accumulation of the newly synthesized 47S pre-rRNA suggests that the activity of one or several factors involved in pre-rRNA processing are not under the control of the cdc2–cyclin B kinase or that the relocalization of the pre-rRNA processing machinery is not regulated by this kinase. Further investigations are necessary to establish the fate of the pre-rRNA processing components in relation to resumption of pre-rRNA processing when rDNA transcription is restored in mitotic cells.
Acknowledgments
The authors thank J.E. Sylvester for the gift of rDNA-containing plasmids, and A.L. Haenni for critical reading of the manuscript.
This work was supported in part by grants from the Centre National de la Recherche Scientifique and the Association pour la Recherche sur le Cancer (Contracts 9143 and 5304). V. Sirri is the recipient of a grant from the Centre National de la Recherche Scientifique.
Footnotes
Abbreviations used in this paper: BrUTP, bromo-uridine 5′-triphosphate; DAPI, 4,6-diamidino-2-phenylindole; ETS, 5′-external transcribed spacer; NOR, nucleolar organizer region; PP-1, protein phosphatase-1; pre-rRNA, precursor ribosomal RNA; rDNA, ribosomal gene; RNA pol, RNA polymerase; rRNA, ribosomal RNA; SL1, promoter selectivity factor; TAFI110, TATA-binding protein–associated factor for RNA polymerase I; TBP, TATA-binding protein; TTF-1, transcription termination factor for RNA polymerase I; UBF, upstream binding factor.
References
- Allmang C., Tollervey D. The role of the 3′ external transcribed spacer in yeast pre-rRNA processing. J. Mol. Biol. 1998;278:67–78 . doi: 10.1006/jmbi.1998.1693. [DOI] [PubMed] [Google Scholar]
- Andreassen P.R., Margolis R.L. Microtubule dependency of p34cdc2 inactivation and mitotic exit in mammalian cells. J. Cell Biol. 1994;127:789–802 . doi: 10.1083/jcb.127.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen P.R., Lacroix F.B., Villa-Moruzzi E., Margolis R.L. Differential subcellular localization of protein phosphatase-1 α, γ1, and δ isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 1998;141:1207–1215 . doi: 10.1083/jcb.141.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis F.M., Tsao T.Y., Fowler S.K., Rao P.N. Monoclonal antibodies to mitotic cells. Proc. Natl. Acad. Sci. USA. 1983;80:2926–2930 . doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Azevedo W.F., Leclerc S., Meijer L., Havlicek L., Strnad M., Kim S.-H. Inhibition of cyclin-dependent kinases by purines analoguescrystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997;243:518–526 . doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Dundr M., Olson M.O.J. Partially processed pre-rRNA is preserved in association with processing components in nucleolus-derived foci during mitosis. Mol. Biol. Cell. 1998;9:2407–2422 . doi: 10.1091/mbc.9.9.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H., Penman S. Regulation of synthesis and processing of nucleolar components in metaphase-arrested cells. J. Mol. Biol. 1971;59:27–42 . doi: 10.1016/0022-2836(71)90411-6. [DOI] [PubMed] [Google Scholar]
- Fernandez A., Brautigan D.L., Lamb N.J.C. Protein phosphatase type 1 in mammalian cell mitosischromosomal localization and involvement in mitotic exit. J. Cell Biol. 1992;116:1421–1430 . doi: 10.1083/jcb.116.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomproix N., Hernandez-Verdun D. Effects of anti-PM-Scl 100 (Rrp6p exonuclease) antibodies on prenucleolar body dynamics at the end of mitosis. Exp. Cell Res. 1999;251:452–464 . doi: 10.1006/excr.1999.4578. [DOI] [PubMed] [Google Scholar]
- Fomproix N., Gébrane-Younès J., Hernandez-Verdun D. Effects of anti-fibrillarin antibodies on building of functional nucleoli at the end of mitosis. J. Cell Sci. 1998;111:359–372 . doi: 10.1242/jcs.111.3.359. [DOI] [PubMed] [Google Scholar]
- Gautier T., Robert-Nicoud M., Guilly M.-N., Hernandez-Verdun D. Relocation of nucleolar proteins around chromosomes at mitosisa study by confocal laser scanning microscopy. J. Cell Sci. 1992;102:729–737 . doi: 10.1242/jcs.102.4.729. [DOI] [PubMed] [Google Scholar]
- Gautier T., Fomproix N., Masson C., Azum-Gélade M.C., Gas N., Hernandez-Verdun D. Fate of specific nucleolar perichromosomal proteins during mitosiscellular distribution and association with U3 snoRNA. Biol. Cell. 1994;82:81–93 . doi: 10.1016/s0248-4900(94)80010-3. [DOI] [PubMed] [Google Scholar]
- Gébrane-Younès J., Fomproix N., Hernandez-Verdun D. When rDNA transcription is arrested during mitosis, UBF is still associated with non-condensed rDNA. J. Cell Sci. 1997;110:2429–2440 . doi: 10.1242/jcs.110.19.2429. [DOI] [PubMed] [Google Scholar]
- Gottesfeld J.M., Forbes D.J. Mitotic repression of the transcriptional machinery. TIBS (Trends Biochem. Sci.). 1997;22:197–202 . doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- Grummt I., Rosenbauer H., Niedermeyer I., Maier U., Ohrlein A. A repeated 18 bp sequence motif in the mouse rDNA spacer mediates binding of a nuclear factor and transcription termination. Cell. 1986;45:837–846 . doi: 10.1016/0092-8674(86)90558-1. [DOI] [PubMed] [Google Scholar]
- Hartl P., Gottesfeld J., Forbes D.J. Mitotic repression of transcription in vitro. J. Cell Biol. 1993;120:613–624 . doi: 10.1083/jcb.120.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heix J., Vente A., Voit R., Budde A., Michaelidis T.M., Grummt I. Mitotic silencing of human rRNA synthesisinactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:7373–7381 . doi: 10.1093/emboj/17.24.7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Verdun D., Gautier T. The chromosome periphery during mitosis. Bioessays. 1994;16:179–185 . doi: 10.1002/bies.950160308. [DOI] [PubMed] [Google Scholar]
- Jordan P., Mannervik M., Tora L., Carmofonseca M. In vivo evidence that TATA-binding protein SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J. Cell Biol. 1996;133:225–234 . doi: 10.1083/jcb.133.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J., Grummt I. Cell cycle-dependent regulation of RNA polymerase I transcriptionthe nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA. 1999;96:6096–6101 . doi: 10.1073/pnas.96.11.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A., Vente A., Dorée M., Grummt I. Mitotic phosphorylation of the TBP-containing factor SL1 represses ribosomal gene transcription. J. Mol. Biol. 1998;284:1–5 . doi: 10.1006/jmbi.1998.2164. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685 . doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Längst G., Blank T.A., Becker P.B., Grummt I. RNA polymerase I transcription on nucleosomal templatesthe transcription termination factor TTF-1 induces chromatin remodeling and relieves transcriptional repression. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:760–768 . doi: 10.1093/emboj/16.4.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Längst G., Becker P.B., Grummt I. TTF-1 determines the chromatin architecture of the active rDNA promoter. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3135–3143 . doi: 10.1093/emboj/17.11.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche A., Wolf V.J., Gottesfeld J.M. Repression of RNA polymerase II and III transcription during M phase of the cell cycle. Exp. Cell Res. 1996;229:282–288 . doi: 10.1006/excr.1996.0373. [DOI] [PubMed] [Google Scholar]
- Long J.J., Leresche A., Kriwacki R.W., Gottesfeld J.M. Repression of TFIIH transcriptional activity and TFIIH-associated cdk7 kinase activity at mitosis. Mol. Cell. Biol. 1998;18:1467–1476 . doi: 10.1128/mcb.18.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 9.47–9.55.
- Matsui S., Sandberg A.A. Intranuclear compartmentalization of DNA-dependent RNA polymerasesassociation of RNA polymerase I with nucleolar organizing chromosomes. Chromosoma. 1985;92:1–6 . doi: 10.1007/BF00327238. [DOI] [PubMed] [Google Scholar]
- Matsui S.-I., Weinfeld H., Sandberg A.A. Quantitative conservation of chromatin-bound RNA polymerases I and II in mitosis. J. Cell Biol. 1979;80:451–464 . doi: 10.1083/jcb.80.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L., Borgne A., Mulner O., Chong J.P.J., Blow J.J., Inagaki N., Inagaki M., Delcros J.-G., Moulinoux J.-P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997;243:527–536 . doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Moore G.P.M., Ringertz N.R. Localization of DNA-dependent RNA polymerase activities in fixed human fibroblasts by autoradiography. Exp. Cell Res. 1973;76:223–228 . doi: 10.1016/0014-4827(73)90439-4. [DOI] [PubMed] [Google Scholar]
- Moss T., Stefanovsky V.Y. Promotion and regulation of ribosomal transcription in eukaryotes by RNA polymerase I. Prog. Nucl. Acid Res. Mol. Biol. 1995;50:25–65 . doi: 10.1016/s0079-6603(08)60810-7. [DOI] [PubMed] [Google Scholar]
- Paulson J.R., Patzlaff J.S., Vallis A.J. Evidence that the endogenous histone H1 phosphatase in HeLa mitotic chromosomes is protein phosphatase 1, not protein phosphatase 2A. J. Cell Sci. 1996;109:1437–1447 . doi: 10.1242/jcs.109.6.1437. [DOI] [PubMed] [Google Scholar]
- Prescott D.M., Bender M.A. Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp. Cell Res. 1962;26:260–268 . doi: 10.1016/0014-4827(62)90176-3. [DOI] [PubMed] [Google Scholar]
- Roussel P., André C., Masson C., Géraud G., Hernandez-Verdun D. Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J. Cell Sci. 1993;104:327–337 . doi: 10.1242/jcs.104.2.327. [DOI] [PubMed] [Google Scholar]
- Roussel P., André C., Comai L., Hernandez-Verdun D. The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. J. Cell Biol. 1996;133:235–246 . doi: 10.1083/jcb.133.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander E.E., Grummt I. Oligomerization of the transcription termination factor TTF-1implications for the structural organization of ribosomal transcription units. Nucleic Acids Res. 1997;25:1142–1147 . doi: 10.1093/nar/25.6.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino T.M., Bastos R., Jansen E., Hernandez-Verdun D. The nucleolar antigen Nop52, the human homologue of the yeast ribosomal RNA processing RRP1, is recruited at late stages of nucleologenesis. J. Cell Sci. 1999;112:1889–1900 . doi: 10.1242/jcs.112.12.1889. [DOI] [PubMed] [Google Scholar]
- Segil N., Guermah M., Hoffmann A., Roeder R.G., Heintz N. Mitotic regulation of TFIIDinhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev. 1996;10:2389–2400 . doi: 10.1101/gad.10.19.2389. [DOI] [PubMed] [Google Scholar]
- Sirri V., Roussel P., Hernandez-Verdun D. The mitotically phosphorylated form of the transcription termination factor TTF-1 is associated with the repressed rDNA transcription machinery. J. Cell Sci. 1999;112:3259–3268 . doi: 10.1242/jcs.112.19.3259. [DOI] [PubMed] [Google Scholar]
- Thiry M., Goessens G. The Nucleolus during the Cell Cycle 1996. Springer-Verlag; Heidelberg: pp. 146 pp [Google Scholar]
- Weisenberger D., Scheer U. A possible mechanism for the inhibition of ribosomal RNA gene transcription during mitosis. J. Cell Biol. 1995;129:561–575 . doi: 10.1083/jcb.129.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf J.M., Konstantinov K.N., Wormsley S., Shu M.-D., Matsumoto-Taniura N., Pirollet F., Klier F.G., Gerace L., Baserga S.J. M phase phosphoprotein 10 is a human U3 small nucleolar ribonucleoprotein component. Mol. Biol. Cell. 1998;9:437–449 . doi: 10.1091/mbc.9.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Goettlieb T.M., Downes C.S., Jackson S.P. Mitotic regulation of a TaTa-binding-protein-containing complex. Mol. Cell. Biol. 1995;15:1983–1992 . doi: 10.1128/mcb.15.4.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson G.N., Szura L.L., Rushford C., Jackson D., Erickson J. Structure and variation of human ribosomal DNAthe external transcribed spacer and adjacent regions. Am. J. Hum. Genet. 1982;34:32–49 . [PMC free article] [PubMed] [Google Scholar]
- Zatsepina O.V., Voit R., Grummt I., Spring H., Semenov M.V., Trendelenburg M.F. The RNA polymerase I-specific transcription initiation factor UBF is associated with transcriptionally active and inactive ribosomal genes. Chromosoma. 1993;102:599–611. doi: 10.1007/BF00352307. [DOI] [PubMed] [Google Scholar]