

Abstract
Overexpression is the most common abnormality of receptor tyrosine kinases (RTKs) in human tumors. It is presumed that overexpression leads to constitutive activation of RTKs, but the mechanism of that activation has been uncertain. Here we show that overexpression of the Met RTK allows activation of the receptor by cell attachment and that this form of activation can be tumorigenic. Transgenic mice that overexpressed Met in hepatocytes developed hepatocellular carcinoma (HCC), one of the human tumors in which Met has been implicated previously. The tumorigenic Met was activated by cell attachment rather than by ligand. Inactivation of the transgene led to regression of even highly advanced tumors, apparently mediated by apoptosis and cessation of cellular proliferation. These results reveal a previously unappreciated mechanism by which the tumorigenic action of RTKs can be mediated, provide evidence that Met may play a role in both the genesis and maintenance of HCC, and suggest that Met may be a beneficial therapeutic target in tumors that overexpress the receptor.
Keywords: Met, receptor tyrosine kinase signaling, tumorigenesis, transgenic mouse, cell adhesion
Introduction
Growth factor receptors that mediate proliferative signals are often hyperactive during tumor progression. Besides ligand-induced activation of receptors, cell adhesion to the extracellular matrix can activate several receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor, fibroblast growth factor receptor, and PDGF receptor (for reviews see Juliano 1996; Giancotti and Ruoslahti 1999; Schwartz and Baron 1999). Integrins and soluble ligands can synergistically stimulate receptor activity and signaling to promote cell growth, survival, and differentiation in vitro analyses. However, the role of adhesion-mediated activation of RTKs in tumorigenesis in vivo remains to be elucidated. We have shown previously that the majority of the Met RTK can be activated in response to cell attachment independent of the ligand (Wang et al. 1996). This provides a useful tool to analyze the receptor activation and biological consequences after cell adhesion.
The MET protooncogene was first encountered in a chromosomal translocation that had generated the TRP-MET oncogene in cultured tumor cells treated with a chemical carcinogen (Cooper et al. 1984; Park et al. 1987). The gene encodes a 170-kD protein (p170met), Met, that is processed to generate the cell surface receptor Met (p190met), composed of a glycosylated extracellular α subunit (p50met) and a transmembrane β subunit (p140met) (Gonzatti-Haces et al. 1988; Giordano et al. 1989). The β subunit possesses protein-tyrosine kinase activity and a “docking site” for cell-signaling molecules (Naldini et al. 1991a; Ponzetto et al. 1994). The ligand for Met is a growth factor known as both scatter factor and hepatocyte growth factor (HGF) (Bottaro et al. 1991; Naldini et al. 1991c), which elicits multiple biological responses, including proliferation, migration, invasion, and morphogenesis (Bardelli and Comoglio 1997; Vande Woude et al. 1997; Birchmeier and Gherardi 1998).
MET has been implicated in various human cancers. Cells transformed by MET are tumorigenic in experimental animals (Bardelli and Comoglio 1997; Vande Woude et al. 1997). Mutant alleles of MET have been found in renal, hepatocellular, and squamous cell carcinomas of humans (Schmidt et al. 1997; Olivero et al. 1999; Park et al. 1999; Di Renzo et al. 2000). Transgenic mice expressing activated mutant forms of Met develop tumors (Liang et al. 1996; Jeffers et al. 1998). Moreover, the gene is overexpressed in a variety of epithelial tumors of humans, including those of liver, prostate, colon, breast, and skin (Stuart et al. 2000). Despite this evidence, the role of MET in the genesis of human tumors remains uncertain. In particular, Met is overexpressed in some epithelial tumors while the ligand is absent (Rusciano et al. 1995, Rusciano et al. 1996; Hiscox et al. 1997; Ueki et al. 1997). This discrepancy diminishes the possibility that autocrine or paracrine activation of the Met receptor contributed to the genesis of the epithelial tumors.
Here we provide the first evidence that wild-type MET can be tumorigenic in vivo. Since HGF is not expressed or activated in normal liver and often absent from the tissue of hepatocellular carcinoma (HCC) (Kinoshita et al. 1989; D'Errico et al. 1996; Noguchi et al. 1996; Ueki et al. 1997), HGF-independent activation of Met might play a role in the genesis of liver cancer. We created transgenic mice that expressed human MET in hepatocytes under the control of tetracycline. Although human Met (hMet) cannot respond to murine HGF (Bhargava et al. 1992; Rong et al. 1992), it was enzymatically active in the hepatocytes, and the activity was dependent on cell adherence, but not HGF. The transgenic mice developed HCC that regressed when the transgene was inactivated. These results indicate that overexpression of Met may play a role in the genesis and maintenance of human HCC, provide an explanation for why HGF need not be present in tumors that overexpress Met, and suggest that cell adherence can be an alternative activation mechanism for RTKs relevant to human cancers.
Materials and Methods
Cells
The B16F1 and B16F10 murine melanoma cell lines were obtained from the Division of Cancer Treatment Tumor Bank, National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD). The A549 human lung carcinoma cells, HeLa human cervical epithelioid carcinoma cell line, 293 transformed human embryonic kidney cells, HT144 human melanoma, HT29 human colon adenocarcinoma, HCT116 human colon carcinoma cell lines, A431 human epidermoid carcinoma cells, and NCI-H441 human lung papillary adenocarcinoma cells were from American Type Culture Collection. Bovine brain capillary endothelial cells (BBE) were provided by R.I. Weiner (University of California at San Francisco). Human umbilical vein endothelial cells and normal human epidermal melanocytes (NHEMs) were obtained from Clonetics. Cells were maintained according to the providers' instructions.
Transfection
HeLa cells expressing tTA (CLONTECH Laboratories, Inc.) were cotransfected with a plasmid containing the murine Met driven by the tetracycline-responsive control element and a plasmid containing the hygromycin resistance gene as a selectable marker. Stable cells expressing murine Met were selected with Hygromycin B at 200 μg/ml. HeLa cells transiently expressing Trk/Met were obtained by transfecting HeLa cells expressing tTA with a plasmid containing trk/met driven by a tetracycline-responsive element (TRE) and cells were harvested 48 h after transfection. The murine Met cDNA was a gift from Dr. G. Vande Woude (Van Andel Research Institute, Grand Rapids, MI). The Trk/Met cDNA was a gift from Dr. Walter Birchmeier (Max Delbruck Center for Molecular Medicine, Berlin, Germany).
Cell Attachment and HGF Stimulation
Detached cells were released from plates by treating with PBS containing 0.04% EDTA, washed in PBS, and cell pellets were loosened by tapping the tubes before lysis. Attached cells were plated on dishes and incubated in PBS for 15 min at 37°C before lysis on the plates. For HGF stimulation, 40 ng/ml human recombinant HGF (R&D Systems) was added to the cells for 15 min at 37°C in serum-free medium. For attached cells, HGF was added onto the plates. For detached cells, HGF was added into the tube and the tube was inverted every 2 min to ensure the cells were kept in suspension.
Cell Surface Protein Labeling with Biotin
Cells were labeled with biotin (Pierce Chemical Co.) 0.5 mg/ml in PBS for 15 min on ice, and the reactions were stopped with 50 mM ice-cold NH4Cl/PBS for 10 min on ice. Cells then were lysed, and Met was immunoprecipitated with anti-Met antibodies. The immunocomplexes were resolved in SDS-PAGE and visualized with horseradish peroxidase–conjugated Streptavidin.
Immunoprecipitation and Immunoblotting
Antiphosphotyrosine monoclonal antibody 4G10 was produced from the 4G10 hybridoma provided by D. Morrison (National Cancer Institute, Frederick, MD). Anti-hMet and anti–murine Met (mMet) antibodies, both rabbit polyclonal antipeptide antibodies, were purchased from Santa Cruz Biotechnology, Inc.
Cell lines were starved in serum-free media supplemented with 1% bovine serum albumin for 18–24 h, and primary cells were starved in media containing 0.5% serum overnight before experiments. Isolated hepatocytes were analyzed without serum starvation. Lysis of cells and immunoblotting were performed as described (Wang et al. 1996). Where required, cells were first detached from dishes with PBS containing 0.04% EDTA before lysis.
Cell lysates were cleared by centrifugation at 14,000 g for 10 min, incubated with antibodies for 2 h, and then with protein G–Sepharose beads for 1 h at 4°C. The beads were washed three times with lysis buffer. Immune complexes were boiled in Laemmli's sample buffer for electrophoresis.
Transgenic Mice
A 4.5-kb XhoI and HindIII fragment of human MET cDNA, a gift of Dr. G. Vande Woude,was blunt-ended and inserted into pUHG 10-3 plasmid, a gift from H. Bujard (European Molecular Biology Laboratory, Heidelberg, Germany), at the XbaI site. The 6.5-kb XhoI and AseI DNA fragments containing the TRE, hCMV minimal promoter with heptamerized upstream tet-operators, human MET coding sequence, and the rabbit beta-globin intron and Poly (A) sequence were used for microinjection (see Fig. 4 A). Mice were derived from the FVB/N strain according to standard methods (Hogan et al. 1994). TRE-MET mice were screened by PCR analysis using primers derived from the TRE sequence (5′-GTCGAGTAGGCGTGTACG-3′) and the human MET sequence (5′-GAATGACATTCTGGATGGGTG-3′). LAP-tTA mice were a gift from Dr. Herman Bujard (European Molecular Biology Laboratory, Heidelberg, Germany) (Kistner et al. 1996), and the line used in this study was bred to FVB/N predominant background in Bruce Counklin's lab at the University of California at San Francisco. The PCR primers used for genotyping these mice were described by Redfern et al. 1999. Doxycycline was administered either in water at 0.2 mg/ml (Sigma-Aldrich) or in chow diet (Bio-Serv).
Figure 4.

Targeted expression of hMet in mouse hepatocytes. (a) TRE-MET transgene construct. (b) Transgenic hMet is expressed specifically and conditionally in hepatocytes. Immunohistochemistry of liver sections using antibodies to hMet. Left, liver from LAP-tTA mouse. Middle, liver of a littermate doubly transgenic for LAP-tTA and TRE-MET. Inset, Met staining at higher magnification. Right, liver from a double transgenic mouse treated with doxycycline (Dox).
Hepatocyte Isolation
Mouse hepatocytes were isolated by collagenase perfusion following techniques developed originally for the rat (Bissell et al. 1973). Livers were prepared for digestion by retrograde perfusion with DME/F-12 containing 0.01% (wt/vol) EGTA (5 ml/min); after 5 min, the perfusion medium was changed to DME/F-12 containing 0.1% collagenase (wt/vol). Collagenase perfusion continued in a nonrecirculating fashion for 20 min. At the end of the perfusion, livers were removed, minced with scissors, and filtered through cotton gauze. Crude cell isolates were suspended in 50 mL DME/F-12 containing 0.1 mg DNase I. The cells were pelleted by centrifugation at 50 g, and washed twice to remove debris and mesenchymal cells. Purified isolates contained 90% hepatocytes.
Histology
Tissues were fixed in 10% buffered formalin, and 5 μm paraffin sections were stained with hematoxylin and eosin.
Immunohistochemistry Detection of hMet
Formalin-fixed paraffin sections were deparaffinized before staining. Sections were treated with citrate buffer for 10 min at 95°C for antigen retrieval. The endogenous peroxidase activity was blocked by a 5-min treatment with 5% H202/PBS. Sections were incubated with 10% goat serum for 4 h, then with rabbit anti-hMet antibodies at 4°C overnight. The primary antibody was detected by using the Elite ABC rabbit kit (Vector Laboratories). The enzymatic activity of horseradish peroxidase was detected by DAB staining (Vector Laboratories).
Detection of Cell Proliferation and Apoptosis
Mice were injected intraperitoneally with 10 mg/ml BrdU (Boehringer) at doses of 0.01 ml/g 2 h before time of killing. Tissues were fixed and processed for paraffin sections. Slides were immunostained with an antibody to BrdU using a BrdU staining kit (Zymed Laboratories). In situ apoptotic cells were detected using the ApopTag Fluorescein apoptosis detection kit (Intergen) according to the manufacturer's instructions.
Results
Activation of Met by Cell Attachment
We first explored the cellular context in which Met might be activated by attachment, using the appearance of phosphotyrosine in Met as an indicator of receptor activation (Fig. 1; Ferracini et al. 1991). In NHEMs, Met could be activated by HGF irrespective of cell attachment, but not by cell attachment alone. In contrast, HGF failed to activate Met in NCI-H441 cells derived from a papillary carcinoma of the human lung. Instead, attachment activated Met in these cells. It thus appeared that activation of Met by cell attachment might be limited to a certain subset of cells.
Figure 1.

Cell attachment activates Met in tumor cells but not in normal cells. Immunoprecipitation and immunoblotting analyses of Met phosphorylation in NHEMs and in human lung carcinoma cells (NCI-H441). NHEMs (lanes 1–4) and NCI-H441 (lanes 5–8) were treated differentially by cell attachment or HGF as described in Materials and Methods. NHEM and NCI-H441 cells were detached from culture dishes with EDTA and held in suspension before being lysed (lanes 3, 4, 7, and 8) or lysed on the plates (lanes 1, 2, 5, and 6). Cells were incubated in the presence (lanes 2, 4, 6, and 8) or absence (lanes 1, 3, 5, and 7) of HGF for 15 min, lysed, and immunoprecipitated (IP) with antibodies to hMet. The immunocomplexes were resolved by SDS-PAGE under reducing conditions and subjected to immunoblotting with antibodies to either phosphotyrosine or hMet as indicated.
We pursued this suspicion by extending the analysis to other normal and tumor cells (Table ). The results revealed three categories of cells: those in which Met could be activated only by HGF (normal cells and tumor cells expressing approximately twice the normal amount of Met); those in which Met could be activated by either HGF or cell attachment (tumor cells expressing intermediate amounts of Met); and those in which Met could be activated only by cell attachment (tumor cells expressing abundant Met).
Table 1.
Cell Attachment–mediated Phosphorylation of Met Occurs in Tumor Cells Overexpressing the Receptor
| Met phosphorylation | |||||
|---|---|---|---|---|---|
| Cells | Det −HGF | Det +HGF | Att −HGF | Att +HGF | Met level |
| Primary cells | |||||
| NHEM | − | + | − | + | 1 |
| HUVEC | − | + | − | + | 1 |
| BBE | − | + | − | + | 1 |
| Cell lines | |||||
| A549 | − | + | − | + | 2 |
| 293 | − | + | − | + | 2 |
| HeLa | − | + | − | + | 2 |
| HT29 | − | + | + | + | 5 |
| HCT116 | − | + | + | + | 5 |
| A431 | − | + | + | + | 5 |
| NCI-H441 | − | − | + | + | >10 |
| B16 F1 | − | − | + | + | >10 |
| B16 F10 | − | − | + | + | >10 |
HUVEC, human umbilical vein endothelial cells.
Overexpression of Normal Met Is Sufficient to Permit Activation by Cell Attachment and Initiation of an Intracellular Signaling Cascade
Mutations can cause ligand-independent activation of cell surface receptors (Bishop 1991; Giordano et al. 1992). Therefore, we explored the possibility that the cells in which Met could be activated by attachment might carry mutant alleles of MET. We isolated and sequenced four cDNAs for Met derived from NCI-H441 cells. These cDNAs were each cloned from separate reverse transcription PCRs to ensure their independent origin. Although silent mutations were evident in these cDNA clones, none of the cDNAs contained mutations that could encode a mutant protein when compared with the published sequence of wild-type Met (data not shown; Park et al. 1987; Ponzetto et al. 1991). One of the silent mutations was present in two of the four clones sequenced. These polymorphisms suggest that the mRNAs were transcribed from different alleles. We conclude that both alleles of the MET gene are wild-types, and therefore the Met protein in these cells is wild-type.
We also asked whether the overexpressed Met might be abnormally processed or localized. Cell surface proteins were labeled with biotin and examined by electrophoresis in both native and reducing conditions. The samples of Met obtained from NCI-H441 and A549 cells were labeled equally well. Under reducing conditions, the α and β subunits from both sources separated as expected during electrophoresis, whereas under native conditions they remained associated with one another (Fig. 2 a). We conclude that the abundant Met produced in NCI-H441 cells is processed normally and transported to the cell surface.
Figure 2.

Met in NCI-H441 cells is normal and can elicit signaling in response to cell attachment. (a) Both α and β subunits of Met in NCI-H441 cells are expressed on the cell surface. Cell surface proteins of NCI-H441 cells (lanes 2 and 2′) and the control A459 cells (lanes 1 and 1′), in which Met does not respond to cell attachment, were labeled with biotin and lysed. Met was immunoprecipitated (IP) with antibodies to hMet and resolved under reducing (lanes 1 and 2) or nonreducing (lanes 1′ and 2′) conditions. Both P140met and P190met were detected with Streptavidin–horseradish peroxidase. (b) Activation of Met by cell adhesion elicits Grb2 recruitment to the cell surface. NCI-H441 cells were lysed while in suspension (Det) or while attached (Att) and immunoprecipitated with antibodies to hMet. The immunocomplexes were resolved by SDS-PAGE and immunoblotted with antibodies to phosphotyrosine, Grb2, or hMet.
To assess directly the effect of overexpressing wild-type Met in tumor cells, we used transfection to express mMet in HeLa cells. Although the endogenous hMet in HeLa cells was not activated by cell attachment (Table and Fig. 3 a), the abundantly expressed mMet was so activated (Fig. 3 a). We conclude that abundant overexpression of otherwise normal Met can permit activation of the receptor by cell attachment.
Figure 3.
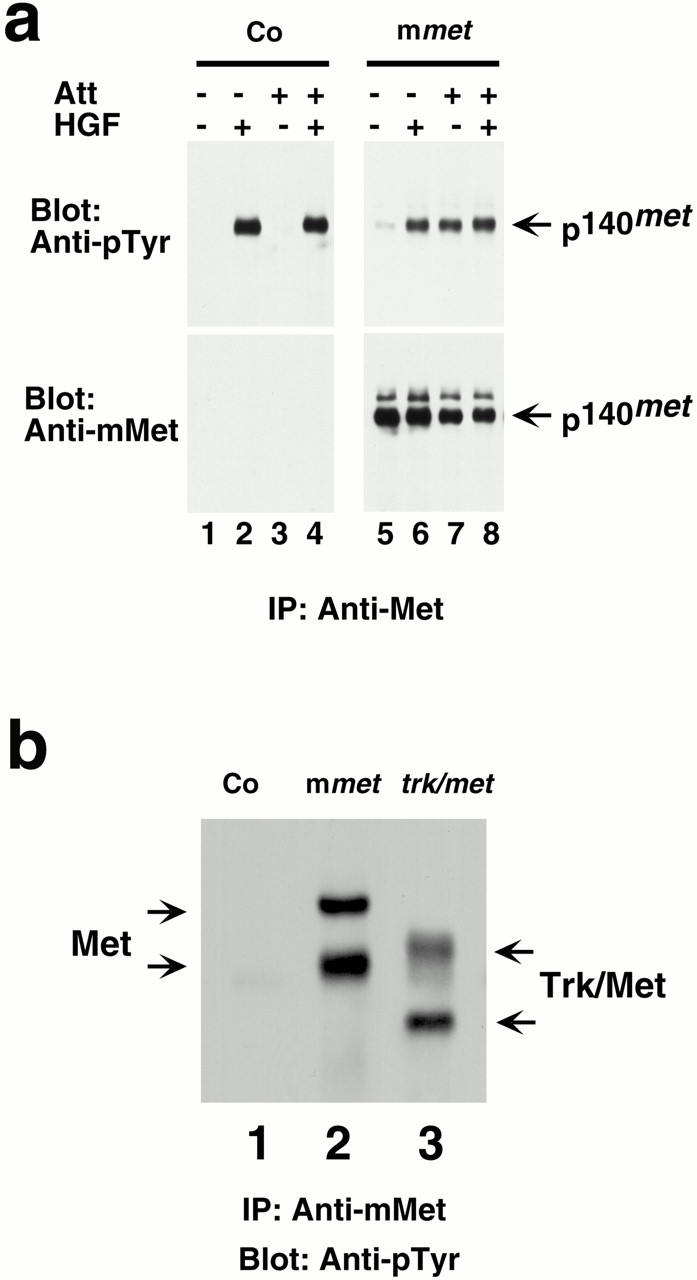
Overexpression of Met enables receptor activation by cell attachment. (a) Wild-type Met responds to cell attachment when overexpressed. HeLa cells were stably transfected with a plasmid containing mMet (lanes 5–8) or with the vector alone as a control (Co; lanes 1–4) and examined for Met phosphorylation in response to HGF or cell attachment. Cells in suspension (lanes 1, 2, 5, and 6) or on plates (lanes 3, 4, 7, and 8) were treated with either HGF (lanes 2, 4, 6, and 8) or control media (lanes 1, 3, 5, and 7). Cell lysates were immunoprecipitated (IP) with antibodies to mMet. Immunocomplexes were resolved by SDS-PAGE and immunoblotted with antibodies either to phosphotyrosine or to mMet. (b) Trk/Met hybrid protein responds to cell attachment when overexpressed. HeLa cells were transiently transfected with vector alone (lane 1) or vector containing wild-type murine met (lane 2) or trk/met (lane 3). Cells were lysed on plates, and cell lysates were immunoprecipitated with antibodies to mMet. Immunocomplexes were resolved by SDS-PAGE and immunoblotted with antibodies to phosphotyrosine.
We then explored which portion of Met might be required for activation by attachment. We examined a hybrid cDNA encoding the extracellular domain of the nerve growth factor receptor, Trk, and the membrane-spanning and intracellular domains of the Met receptor (trk/met) (Weidner et al. 1993). When expressed ectopically in HeLa cells, the hybrid protein (Trk/Met) could be activated by cell attachment (Fig. 3 b). Thus, the extracellular domain containing the ligand-binding sides of the receptor may have little bearing on activation by attachment.
The signal initiated by activation of Met is transduced by a downstream cascade of biochemical events (Naldini et al. 1991b; Ponzetto et al. 1994; Royal and Park 1995; Weidner et al. 1996; Boccaccio et al. 1998). Among the earliest of these events is the recruitment of Grb2 protein to a binding domain on Met (Ponzetto et al. 1994). We found that activation of Met by cell attachment increased the amount of Grb2 that could be coprecipitated with Met (Fig. 2 b). Thus, it is likely that activation of Met by attachment initiates a cascade of intracellular signaling.
Conditional Expression of hMet in Hepatocytes of Transgenic Mice
The preceding results raised the possibility that overexpression of Met might lead to activation of the receptor by cell attachment and subsequent tumorigenesis. Although Met is overexpressed in human HCC, HGF is frequently absent, suggesting that Met can be activated by a ligand-independent mechanism in this disease (Ueki et al. 1997). To test this hypothesis, we expressed human MET in hepatocytes and examined tumor development in mice. By using human MET in the transgene, we precluded response to intrinsic mouse HGF (Bhargava et al. 1992; Rong et al. 1992). Thus, any activation of the transgenic receptor was likely to arise from cell attachment within liver tissue.
We first generated mice in which transgenic human MET is under the control of the TRE (Kistner et al. 1996; Fig. 4 a).
MET is controlled by a TRE (Kistner et al. 1996), which allows the inducible expression of the transgene (Fig. 4 a). The TRE-MET mice were then crossed with LAP-tTA mice, in which expression of the tetracycline transactivator protein is under the control of the promoter for the liver-specific liver-activating protein gene (Kistner et al. 1996). The cross produced offspring that were transgenic for both TRE-MET and LAP-tTA. The doubly transgenic mice should express hMet in the liver, and the expression should be suppressed by the administration of doxycycline.
Examination by immunohistochemistry revealed abundant expression of hMet in hepatocytes of the doubly transgenic mice, but not in controls (Fig. 4 b). Human Met was expressed in hepatocytes (Fig. 4 b, middle panel) of the normal liver parenchyma (Fig. 4 b, middle, black arrow) as well as hyperplastic nodules (Fig. 4 b, middle, arrowheads; Fig. 5, c–e). Staining for the protein was especially pronounced at the margins of cells (Fig. 4 b, inset), as expected for a cell surface receptor. Expression was not apparent in other types of cells in the hepatic tissue (Fig. 4 b, middle, purple arrow), nor in other tissues examined, including lung, spleen, kidney, and pancreas (data not shown). Expression of hMet could be repressed by feeding the transgenic mice the tetracycline analogue doxycycline (Fig. 4 b, right).
Figure 5.

Expression of hMet in mouse hepatocytes induces HCC. (a and b) Survival and tumor incidences of TRE-MET mice in LAP-tTA background. For lines 1 and 2, doxycycline (Dox) was administered throughout pregnancy and after birth until pups were 4 wk old. For lines 3 and 4, doxycycline treatment was started when tumor-bearing mice were moribund. (c–f) Histopathology of tumor progression. (c) A 2-mo-old animal developed rare foci (black arrow around central veins). (d) By 4 mo of age, abnormal hepatocytes around the central vein grew into larger hyperplastic nodules (black arrowheads). (e) At 6 mo of age, less differentiated dysplatic nodules (white arrowheads) arise within hyperplastic liver parenchyma, resembling the progression of human HCC. (f) Diffuse trabecular growth pattern typical of HCC developed in the same liver that developed dysplatic foci in panel d.
Mice Expressing Transgenic hMet Develop HCC
Having established transgenic mice that overexpressed hMet in hepatocytes, we followed four lines of these mice for the appearance of HCC. Mice of lines 1 and 2 were born with enlarged and fatty livers, and died within 2 mo postpartum (Fig. 5 a, and data not shown). Details will be published elsewhere. The early deaths could be prevented by feeding the mating parents and newborn pups doxycycline to repress expression of the transgene. Doxycycline was then withdrawn at 4 wk of age. Under these circumstances, the mice appeared normal until 10 mo of age, when they began to die (Fig. 5 a). Approximately 85% of the deaths were accompanied by HCC, as determined by weekly palpation and eventual autopsies (Fig. 5 b and below). The incidence of HCC by 1 yr of age was >60% (Fig. 5 b).
In contrast, mice of lines 3 and 4 were healthy at birth, but began to die at 4 mo postpartum (Fig. 5 a). Like in lines 1 and 2, ∼85% of the deaths could be attributed to HCC, which appeared as an abdominal mass at 6 mo of age or later. The incidence of HCC was again 60% by 1 yr of age (Fig. 5 b).
About 15% of deaths occurred without evidence of tumor at autopsy in all MET transgenic lines when Met expression was turned on in adult animals. These deaths were often associated with abnormal gross and histological pathology of the liver. The animals were usually cachectic and jaundiced before deaths. These observations suggest that the mice died of a liver disease, the nature of which will require further analysis. We have not detected abnormalities in other organs except occasional pale kidneys in moribund tumor-bearing mice.
We monitored the development of tumors by histological analysis (Fig. 5, c–f). By 60 wk of age, small foci of hyperplasia appeared, often surrounding the central vein of the hepatic lobule (Fig. 5 c). Typically, a couple of these foci were detected per section, suggesting that dozens of these foci were present throughout the liver. These foci of hyperplasia had become bigger, increased in number by a factor of five, and uniformly distributed throughout the liver by 4 mo of age (Fig. 5 d). The cells in these foci were still recognizable as hepatocytes, but had developed foamy cytoplasm containing fat deposits that could be identified by staining with oil red-O (data not shown). These pathological changes are characteristic of the hyperplasia that typically precedes the development of HCC in humans (Ferrell et al. 1993).
Zones of progression to malignancy became apparent within the fields of hyperplasia by 6 mo of age (Fig. 5e and Fig. f). Within these zones, cells were poorly organized and less well differentiated than normal, resembling the dysplastic precursors of HCC in the human liver (Fig. 5 e; Ferrell et al. 1993). Individual livers contained multiple dysplastic foci, suggesting that tumor progression was occurring independently in multiple clonal lineages. In addition, the surrounding liver also showed atypical cytologic changes affecting hepatocytes. These included cellular and nuclear enlargement, as well as disorganization of cell plate and lobular structures. With further progression over time, the malignant foci enlarged and developed the trabeculae typical of advanced HCC (Fig. 5 f). The trabeculae were frequently separated from one another by blood-filled spaces, lined by endothelial cells, and reminiscent of widened hepatic sinusoids. Areas of necrosis were also common in the fully developed tumors. We conclude that the transgenic mice had developed HCC. The penetrance of tumorigenesis was >60% (Fig. 5 b). The area of this typical HCC was usually found in only one or two lobes of the liver, suggesting that a subset of hyperplastic foci proceed to HCC.
Activation of Transgenic Met by Cell Attachment
Our in vitro experiments show that Met can be activated when tumor cells are adherent. Since hepatocytes are adherent in liver tissues, it seemed likely that hMet from the tumor could be activated. We confirmed this expectation by using Western blotting to analyze lysates of the transgenic liver tumor (Fig. 6 a). The analysis revealed that hMet was phosphorylated on tyrosine and, thus, presumably enzymatically active. In contrast, the endogenous mMet in control liver contained no detectable phosphotyrosine (Fig. 6, lane 2′).
Figure 6.

Activation of transgenic hMet is dependent on cell attachment. (a) Transgenic hMet was phosphorylated in livers. Livers from mice transgenic for both TRE-MET and LAP-tTA (lane 1) or LAP-tTA as control (lanes 2 and 2′) were lysed and immunoprecipitated (IP) with antibodies to hMet (lanes 1 and 2) or to murine Met (lane 2′) and immunoblotted with the antibodies indicated. (b) Activation of the transgenic hMet requires cell attachment. Hepatocytes isolated from a tumor-bearing double transgenic mouse were lysed as single cell suspension (lanes 1 and 2) or after attachment to collagen-coated plates (lanes 3 and 4). Before lysing, cells were treated in the absence (lane 1 and 3) or presence of human HGF (lanes 2 and 4). Cell lysates were immunoprecipitated with antibodies to hMet and immunoblotted with antibodies to either phosphotyrosine or hMet.
To confirm that the phosphorylation of the transgenic hMet in liver was dependent on cell attachment, we analyzed preparations of hepatocytes isolated from tumor-bearing mice (Fig. 6 b). Activation of the hMet was diminished when hepatocytes were dissociated from liver and maintained in suspension. However, activation of the receptor increased substantially when these isolated hepatocytes were adherent to collagen-coated plates. Moreover, the hMet did not respond to stimulation by human HGF stimulation. We conclude that cell attachment may have led to the activation of the transgenic hMet.
Sustained Expression of Met Is Required for the Maintenance of HCC
Transgenic Met activated by cell attachment apparently triggered the genesis of HCC. To determine whether Met might also be required for maintenance of the tumor, we treated 11 moribund mice of line 3 and 13 of line 4 with doxycycline to repress expression of the transgene (see above). Within 1 mo, 21 of these mice had regained their health and lost the abdominal swelling demonstrative of tumor mass (Fig. 7, a and b). One mouse from line 3 and two from line 4 died within 1 wk of doxycycline treatment. Six mice treated with doxycycline for 4 mo have remained healthy throughout.
Figure 7.

Inactivation of Met results in the sustained regression of HCC. (a and b) Doxycycline dramatically reduces tumor burden within 1 mo. A moribund tumor-bearing mouse (lower) with enlarged abdomen before doxycycline treatment and the control littermate (upper; panel a). Substantial shrinkage of the tumor was evident after 1-mo treatment with doxycycline (b). (c–f) Gross pathology of HCC regression. Moribund tumor-bearing mice together with their wild-type littermates received doxycycline in their diet. Normal adult liver (c), HCC-containing liver before doxycycline treatment (d), HCC-containing liver after 20 d of doxycycline treatment (e), and liver of a previously HCC-bearing mouse after 4 mo of doxycycline treatment (f). The white arrows indicate normal liver tissue and black arrows point to the involuting residual mass. (g–j) Histology of liver with regressed tumors. Light microscopy of hematoxylin and eosin–stained liver sections from a previously tumor-bearing mouse treated for 4 mo with doxycycline. The majority of liver appeared normal (g), although a residual nodule was evident (h–j). The center of the residual mass was composed mainly of necrotic and calcified tissues (h). The border of normal liver (upper) and the involuted residue (lower) is marked by the white arrowheads in panel i. The border at higher magnification showed extensive infiltration of inflammatory cells (j). The arrows point to neutrophils and the arrowheads point to brown pigmented macrophages.
We also examined the macroscopic appearance of livers in tumor-bearing mice at various points after initiation of treatment with doxycycline (Fig. 7, c–f). The tumors were unchanged after 3 d of treatment (data not shown). However, after 20 d of treatment the size of the tumorous livers had diminished and apparently normal tissue had reappeared (Fig. 7 e). After 4 mo of treatment, the livers had returned almost entirely to normal, with only a small residue of abnormality (Fig. 7 f).
Histological analysis was used to examine livers from mice that had been treated for 4 mo with doxycycline (Fig. 7, g–j). The bulk of the liver appeared microscopically normal (Fig. 7 g). The residual nodule of abnormal tissue could be identified by a transition from normal tissue to an area of necrosis, scarring, calcification, and inflammatory reaction (Fig. 7h and Fig. i), the last mostly composed of neutrophils and macrophages (Fig. 7 j). We conclude that inactivation of the human MET transgene led to full regression of the hepatic tumors with reconstitution of normal tissue architecture.
Regression of HCC Was Accompanied by Apoptosis and Diminished Cellular Proliferation
We explored two possible explanations for regression of the tumors: arrest of the cell cycle and apoptosis. The proliferative activity of tissue was examined by labeling with BrdU. Numerous cells were proliferating in the liver tumors elicited by Met (Fig. 8 a). But within 3 d of instituting treatment with doxycycline, proliferation was virtually undetectable (Fig. 8 b), much as is found in a normal liver (data not shown). The absence of proliferation persisted in the remaining scar tissue when doxycycline treatment had been extended to either 20 d or 4 mo (data not shown). We conclude that removal of the stimulus from the transgenic hMet led to prompt cessation of cellular proliferation, even in the cells of advanced malignancies.
Figure 8.

Apoptosis and cessation of proliferation were involved in tumor regression. Liver paraffin sections from a tumor-bearing mouse (a and c) and a tumor-bearing mouse after 3 d of doxycycline treatment (b and d) were stained for BrdU (a and b) or fragmented DNA (c and d). Cells that have incorporated BrdU stained brown. Apoptotic cells were detected by TUNEL assay, and the fragmented DNA was labeled with fluorescein.
Using the terminal deoxynucleotidyl transferase–mediated dUTP nick end labelling (TUNEL) assay, we detected no apoptosis in either normal liver (data not shown) or fully developed tumors (Fig. 8 c). Apoptosis became abundant in tumor tissues after 3 d of doxycycline treatment (Fig. 8 d), but was no longer evident in the remaining scar tissue after either 20 d or 4 mo of treatment (data not shown). We conclude that apoptosis made an acute contribution to tumor regression, but diminished in significance as the tumor was replaced with normal tissue.
Discussion
The MET Protooncogene Can Contribute to the Genesis of HCC
HCC is among the most common cancers in the world and its incidence is rising in the United States (Okuda 2000). The majority of untreated individuals survive for <1 yr. Liver transplantation is the only effective treatment. Infection with hepatitis viruses B and C, chronic alcoholism, and exposure to aflatoxin have all been implicated in the causation of HCC (Okuda 2000).
Little is known about the molecular pathogenesis of HCC. One clue has been the observation that the protooncogene MET is frequently overexpressed in HCC (Prat et al. 1991; D'Errico et al. 1996; Noguchi et al. 1996; Ueki et al. 1997). Other evidence for the tumorigenicity of overactive MET has come from studies with transformation of cells in vitro (Iyer et al. 1990; Rong et al. 1992; Bardelli and Comoglio 1997; Giordano et al. 1997; Vande Woude et al. 1997), the finding of mutant alleles of MET in human cancers (Schmidt et al. 1997; Olivero et al. 1999; Park et al. 1999; Di Renzo et al. 2000), and the occurrence of mammary tumors in transgenic mice that express mutant versions of Met with constitutive activity using the metallothionein 1 promoter (Liang et al. 1996; Jeffers et al. 1998). Here we demonstrate a novel activation mechanism that does not rely on mutation of Met and demonstrate for the first time that overexpression of wild-type Met in hepatocytes of mice enables ligand-independent activation of the receptor that leads to HCC with a high penetrance.
Activation of the Met Receptor in the Absence of Ligand
The ligand for the Met receptor is not produced in normal liver and is often absent from the tissue of HCC (Kinoshita et al. 1989; D'Errico et al. 1996; Noguchi et al. 1996; Ueki et al. 1997). These observations diminish the prospect for autocrine or paracrine activation of Met, thus calling into doubt any role for the receptor in the genesis of HCC. We can now offer an alternative possibility. Overexpression of Met can lead to activation of the receptor by cell adherence, eliminating the requirement for ligand stimulation. In accord with this view, we were able to elicit HCC by overexpressing hMet in the hepatocytes of mice. The transgenic receptor was constitutively active and this activity receded when hepatocytes were dissociated into suspensions of single cells. The activity of Met could not be attributed to the endogenous ligand, because the transgene encodes the human version of Met, which cannot respond to murine HGF (Bhargava et al. 1992; Rong et al. 1992). Moreover, hMet in hepatocytes that were derived from HCC-bearing transgenic mice did not respond to recombinant human HGF. Therefore, we conclude that the transgenic Met was activated by cell adherence in a ligand-independent fashion.
The mechanism by which cell attachment activates Met remains obscure, but appears to be dependent on overexpression. Overexpression of Met potentiates the receptor activation by cell adherence and eliminates the requirement for ligand stimulation (Fig. 3 and Fig. 6; Wang, R., and J.M. Bishop, unpublished results). Therefore, we were able to elicit HCC by overexpressing hMet in the hepatocytes of mouse liver. Previous work failed to produce HCC in mice expressing a transgene of wild-type Met (Jeffers et al. 1998). Our findings suggest an explanation for that result. The level of Met expression may have been insufficient to permit activation of the receptor by cell adherence, and this form of activation may be the only means by which Met can be activated in HCC pathogenesis. However, genetic backgrounds of the mice differed in these two studies, and this might also explain the different outcomes.
We could not explain the activation of Met either by mutations or by abnormal processing or localization of the protein. In addition, we doubt that the extracellular domain of Met plays a role in the activation, because replacement of this domain with its unrelated counterpart from the Trk receptor had no effect on activation by cell attachment. The cytoplasmic domain of Met is obviously essential to activation, because it carries the enzymatic activity of the receptor. But membrane anchorage may also be required for activation by cell attachment, and such anchorage would be provided by the Trk component of the hybrid protein. The receptors for PDGF and macrophage-stimulating protein can also be activated by cell adhesion independent of the ligand (Sundberg and Rubin 1996; Danilkovitch-Miagkova et al. 2000). This activation is apparently mediated by signaling arising from cell surface integrins, but we and others have shown previously that such signaling may not explain the ligand-independent activation of Met (Rusciano et al. 1996; Wang et al. 1996).
We have attempted to address this problem in several ways, and the results support our hypothesis. But it is still possible that a mechanism other than adhesion might account for the results. One such possibility would be cross-talk between Met and another receptor. We have sought evidence for this without success.
The in vitro studies performed to date have detected activation of Met by cell attachment only in certain tumor cells. We have also shown that the activation of Met by cell attachment occurred in the hepatocytes isolated from tumor-bearing transgenic mice. It is possible that secondary events during tumor development may also contribute to adhesion-mediated activation of Met. A further puzzle is represented by the observation that Met, which can be activated by cell attachment in some tumor cell lines and in the hepatocytes from HCC-containing transgenic livers, is accompanied by refractoriness to HGF. We can presently offer no explanation for this observation.
The Role of Met in the Maintenance of HCC
It has not been clear whether genetic lesions responsible for initiation of tumorigenesis would eventually cease to play a role as the tumor progressed to malignancy (Hanahan and Weinberg 2000). Recent work has shown that sustained activity of an oncogene is required for maintenance of tumors elicited by myc, ras, and bcr/abl (Chin and DePinho 2000). We now show that tumors elicited by transgenic MET will regress if the transgene is inactivated, even at advanced stages of tumor progression. This finding raises the possibility that the Met receptor may be a beneficial target for the treatment of HCC and other malignancies, particularly when the receptor is overexpressed in tumor tissue.
There are at least three possible explanations for the tumor regression described here. First, malignant cells might differentiate into normal hepatocytes when the MET transgene is inactivated. This mechanism may explain the regression of hematopoietic malignancies when the expression of transgenic MYC is repressed (Felsher and Bishop 1999), but we presently have no evidence that it contributes to the regression of HCC elicited by transgenic Met.
Second, Met is among the numerous cell surface receptors whose activity can elicit and sustain the cell division cycle (Stuart et al. 2000). Thus, the repression the transgenic Met expression might lead to proliferative arrest on the part of the malignant hepatocytes. We present evidence that this is indeed the case.
Third, apoptosis of tumor cells could contribute to the reduction in tumor mass. Our data demonstrated the rapid onset of apoptosis in tumor tissue soon after the MET transgene had been inactivated, but apoptosis diminished once tumor regression was well advanced. The apoptotic response may be held in check in tumors by Met itself (Amicone et al. 1997), but it would be unleashed when the activity of Met is withdrawn, and diminishes as tumor tissue is replaced by the regrowth of normal hepatocytes.
Acknowledgments
We thank Zena Werb, D. Montgomery Bissell, and members of the Bishop laboratory for helpful suggestions; D. Montgomery Bissell, Caroline Damsky, Ken Field, and Nancy Hong for critical reading of the manuscript; Hermann Bujard for LAP-tTA mice and UHG 10-3 plasmid; George Vande Woude for human met cDNA; and Walter Bircherier for the Trk/met cDNA.
We also thank help from the Cell and Tissue Biology Core facility at the University of California at San Francisco Liver Center, supported by funds from the National Institutes of Health (5P30DK26743). This work was supported by postdoctoral fellowships from the Leukemia Society of America and American Heart Association to R. Wang, and funds from the National Institutes of Health (CA44338) and the G.W. Hooper Research Foundation.
Footnotes
Abbreviations used in this paper: HCC, hepatocellular carcinoma; HGF, hepatocyte growth factor; NHEM, normal human epidermal melanocyte; RTK, receptor tyrosine kinase; TRE, tetracycline-responsive element.
References
- Amicone L., Spagnoli F.M., Spath G., Giordano S., Tommasini C., Bernardini S., De Luca V., Della Rocca C., Weiss M.C., Comoglio P.M., Tripodi M. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 1997;16:495–503. doi: 10.1093/emboj/16.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli A., Comoglio P.M. Scatter factor receptors are key players in a unique multistep program leading to invasive growth. Ciba Found. Symp. 1997;212:133–144. doi: 10.1002/9780470515457.ch9. [DOI] [PubMed] [Google Scholar]
- Bhargava M., Joseph A., Knesel J., Halaban R., Li Y., Pang S., Goldberg I., Setter E., Donovan M.A., Zarnegar R. Scatter factor and hepatocyte growth factoractivities, properties, and mechanism. Cell Growth Differ. 1992;3:11–20. [PubMed] [Google Scholar]
- Birchmeier C., Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- Bishop J.M. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- Bissell D.M., Hammaker L.E., Meyer U.A. Parenchymal cells from adult rat liver in nonproliferating monolayer culture. I. Functional studies. J. Cell Biol. 1973;59:722–734. doi: 10.1083/jcb.59.3.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio C., Ando M., Tamagnone L., Bardelli A., Michieli P., Battistini C., Comoglio P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–288. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- Bottaro D.P., Rubin J.S., Faletto D.L., Chan A.M., Kmiecik T.E., Vande Woude G.F., Aaronson S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Chin L., DePinho R.A. Flipping the oncogene switchillumination of tumor maintenance and regression. Trends Genet. 2000;16:147–150. doi: 10.1016/s0168-9525(99)01968-x. [DOI] [PubMed] [Google Scholar]
- Cooper C.S., Park M., Blair D.G., Tainsky M.A., Huebner K., Croce C.M., Vande Woude G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- Danilkovitch-Miagkova A., Angeloni D., Skeel A., Donley S., Lerman M., Leonard E.J. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. J. Biol. Chem. 2000;275:14783–14786. doi: 10.1074/jbc.C000028200. [DOI] [PubMed] [Google Scholar]
- D'Errico A., Fiorentino M., Ponzetto A., Daikuhara Y., Tsubouchi H., Brechot C., Scoazec J.Y., Grigioni W.F. Liver hepatocyte growth factor does not always correlate with hepatocellular proliferation in human liver lesionsits specific receptor c-met does. Hepatology. 1996;24:60–64. doi: 10.1002/hep.510240112. [DOI] [PubMed] [Google Scholar]
- Di Renzo M.F., Olivero M., Martone T., Maffe A., Maggiora P., Stefani A.D., Valente G., Giordano S., Cortesina G., Comoglio P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19:1547–1555. doi: 10.1038/sj.onc.1203455. [DOI] [PubMed] [Google Scholar]
- Felsher D.W., Bishop J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracini R., Longati P., Naldini L., Vigna E., Comoglio P.M. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J. Biol. Chem. 1991;266:19558–19564. [PubMed] [Google Scholar]
- Ferrell L.D., Crawford J.M., Dhillon A.P., Scheuer P.J., Nakanuma Y. Proposal for standardized criteria for the diagnosis of benign, borderline, and malignant hepatocellular lesions arising in chronic advanced liver disease. Am. J. Surg. Pathol. 1993;17:1113–1123. doi: 10.1097/00000478-199311000-00004. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Giordano S., Di Renzo M.F., Narsimhan R.P., Cooper C.S., Rosa C., Comoglio P.M. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene. 1989;4:1383–1388. [PubMed] [Google Scholar]
- Giordano S., di Renzo M.F., Olivero M., Mondino A., Zhen Z., Medico E., Comoglio P.M. The c-met/HGF receptor in human tumours. Eur. J. Cancer Prev. 1992;3:45–49. doi: 10.1097/00008469-199210003-00007. [DOI] [PubMed] [Google Scholar]
- Giordano S., Bardelli A., Zhen Z., Menard S., Ponzetto C., Comoglio P.M. A point mutation in the MET oncogene abrogates metastasis without affecting transformation. Proc. Natl. Acad. Sci. USA. 1997;94:13868–13872. doi: 10.1073/pnas.94.25.13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzatti-Haces M., Seth A., Park M., Copeland T., Oroszlan S., Vande Woude G.F. Characterization of the TPR-MET oncogene p65 and the MET protooncogene p140 protein-tyrosine kinases. Proc. Natl. Acad. Sci. USA. 1988;85:21–25. doi: 10.1073/pnas.85.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hiscox S.E., Hallett M.B., Puntis M.C., Nakamura T., Jiang W.G. Expression of the HGF/SF receptor, c-met, and its ligand in human colorectal cancers. Cancer Invest. 1997;15:513–521. doi: 10.3109/07357909709047592. [DOI] [PubMed] [Google Scholar]
- Hogan B., Beddington R., Costantini F., Lacy E. Manipulating the Mouse EmbryoA Laboratory Manual 1994. Cold Spring Harbor Press; Cold Spring Harbor, NY: pp. 497 pp [Google Scholar]
- Iyer A., Kmiecik T.E., Park M., Daar I., Blair D., Dunn K.J., Sutrave P., Ihle J.N., Bodescot M., Vande Woude G.F. Structure, tissue-specific expression, and transforming activity of the mouse met protooncogene. Cell Growth Differ. 1990;1:87–95. [PubMed] [Google Scholar]
- Jeffers M., Fiscella M., Webb C.P., Anver M., Koochekpour S., Vande Woude G.F. The mutationally activated Met receptor mediates motility and metastasis. Proc. Natl. Acad. Sci. USA. 1998;95:14417–14422. doi: 10.1073/pnas.95.24.14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano R. Cooperation between soluble factors and integrin-mediated cell anchorage in the control of cell growth and differentiation. Bioessays. 1996;18:911–917. doi: 10.1002/bies.950181110. [DOI] [PubMed] [Google Scholar]
- Kinoshita T., Tashiro K., Nakamura T. Marked increase of HGF mRNA in non-parenchymal liver cells of rats treated with hepatotoxins. Biochem. Biophys. Res. Commun. 1989;165:1229–1234. doi: 10.1016/0006-291x(89)92733-2. [DOI] [PubMed] [Google Scholar]
- Kistner A., Gossen M., Zimmermann F., Jerecic J., Ullmer C., Lubbert H., Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T.J., Reid A.E., Xavier R., Cardiff R.D., Wang T.C. Transgenic expression of tpr-met oncogene leads to development of mammary hyperplasia and tumors. J. Clin. Invest. 1996;97:2872–2877. doi: 10.1172/JCI118744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L., Vigna E., Ferracini R., Longati P., Gandino L., Prat M., Comoglio P.M. The tyrosine kinase encoded by the MET proto-oncogene is activated by autophosphorylation Mol. Cell. Biol 11 1991. 1793 1803a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L., Vigna E., Narsimhan R.P., Gaudino G., Zarnegar R., Michalopoulos G.K., Comoglio P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET Oncogene 6 1991. 501 504b [PubMed] [Google Scholar]
- Naldini L., Weidner K.M., Vigna E., Gaudino G., Bardelli A., Ponzetto C., Narsimhan R.P., Hartmann G., Zarnegar R., Michalopoulos G.K. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor EMBO J 10 1991. 2867 2878c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi O., Enomoto N., Ikeda T., Kobayashi F., Marumo F., Sato C. Gene expressions of c-met and hepatocyte growth factor in chronic liver disease and hepatocellular carcinoma. J. Hepatol. 1996;24:286–292. doi: 10.1016/s0168-8278(96)80006-7. [DOI] [PubMed] [Google Scholar]
- Okuda K. Hepatocellular carcinoma. J. Hepatol. 2000;32:225–237. doi: 10.1016/s0168-8278(00)80428-6. [DOI] [PubMed] [Google Scholar]
- Olivero M., Valente G., Bardelli A., Longati P., Ferrero N., Cracco C., Terrone C., Rocca-Rossetti S., Comoglio P.M., Di Renzo M.F. Novel mutation in the ATP-binding site of the MET oncogene tyrosine kinase in a HPRCC family. Int. J. Cancer. 1999;82:640–643. doi: 10.1002/(sici)1097-0215(19990827)82:5<640::aid-ijc4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Park M., Dean M., Kaul K., Braun M.J., Gonda M.A., Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc. Natl. Acad. Sci. USA. 1987;84:6379–6383. doi: 10.1073/pnas.84.18.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W.S., Dong S.M., Kim S.Y., Na E.Y., Shin M.S., Pi J.H., Kim B.J., Bae J.H., Hong Y.K., Lee K.S. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999;59:307–310. [PubMed] [Google Scholar]
- Ponzetto C., Giordano S., Peverali F., Della Valle G., Abate M.L., Vaula G., Comoglio P.M. c-met is amplified but not mutated in a cell line with an activated met tyrosine kinase. Oncogene. 1991;6:553–559. [PubMed] [Google Scholar]
- Ponzetto C., Bardelli A., Zhen Z., Maina F., dalla Zonca P., Giordano S., Graziani A., Panayotou G., Comoglio P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77:261–271. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Prat M., Narsimhan R.P., Crepaldi T., Nicotra M.R., Natali P.G., Comoglio P.M. The receptor encoded by the human c-MET oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int. J. Cancer. 1991;49:323–328. doi: 10.1002/ijc.2910490302. [DOI] [PubMed] [Google Scholar]
- Redfern C.H., Coward P., Degtyarev M.Y., Lee E.K., Kwa A.T., Hennighausen L., Bujard H., Fishman G.I., Conklin B.R. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat. Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- Rong S., Bodescot M., Blair D., Dunn J., Nakamura T., Mizuno K., Park M., Chan A., Aaronson S., Vande Woude G.F. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol. Cell. Biol. 1992;12:5152–5158. doi: 10.1128/mcb.12.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal I., Park M. Hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J. Biol. Chem. 1995;270:27780–27787. doi: 10.1074/jbc.270.46.27780. [DOI] [PubMed] [Google Scholar]
- Rusciano D., Lorenzoni P., Burger M.M. Expression of constitutively activated hepatocyte growth factor/scatter factor receptor (c-met) in B16 melanoma cells selected for enhanced liver colonization. Oncogene. 1995;11:1979–1987. [PubMed] [Google Scholar]
- Rusciano D., Lorenzoni P., Burger M.M. Constitutive activation of c-Met in liver metastatic B16 melanoma cells depends on both substrate adhesion and cell density and is regulated by a cytosolic tyrosine phosphatase activity. J. Biol. Chem. 1996;271:20763–20769. doi: 10.1074/jbc.271.34.20763. [DOI] [PubMed] [Google Scholar]
- Schmidt L., Duh F.M., Chen F., Kishida T., Glenn G., Choyke P., Scherer S.W., Zhuang Z., Lubensky I., Dean M. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- Schwartz M.A., Baron V. Interactions between mitogenic stimuli, or, a thousand and one connections. Curr. Opin. Cell Biol. 1999;11:197–202. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- Stuart K.A., Riordan S.M., Lidder S., Crostella L., Williams R., Skouteris G.G. Hepatocyte growth factor/scatter factor-induced intracellular signalling. Int. J. Exp. Pathol. 2000;81:17–30. doi: 10.1046/j.1365-2613.2000.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg C., Rubin K. Stimulation of beta1 integrins on fibroblasts induces PDGF independent tyrosine phosphorylation of PDGF beta-receptors. J. Cell Biol. 1996;132:741–752. doi: 10.1083/jcb.132.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki T., Fujimoto J., Suzuki T., Yamamoto H., Okamoto E. Expression of hepatocyte growth factor and its receptor, the c-met proto-oncogene, in hepatocellular carcinoma. Hepatology. 1997;25:619–623. doi: 10.1002/hep.510250321. [DOI] [PubMed] [Google Scholar]
- Vande Woude G.F., Jeffers M., Cortner J., Alvord G., Tsarfaty I., Resau J. Met-HGF/SFtumorigenesis, invasion and metastasis. Ciba Found. Symp. 1997;212:119–130. doi: 10.1002/9780470515457.ch8. [DOI] [PubMed] [Google Scholar]
- Wang R., Kobayashi R., Bishop J.M. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. USA. 1996;93:8425–8430. doi: 10.1073/pnas.93.16.8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner K.M., Hartmann G., Naldini L., Comoglio P.M., Sachs M., Fonatsch C., Rieder H., Birchmeier W. Molecular characteristics of HGF-SF and its role in cell motility and invasion. EXS. 1993;65:311–328. [PubMed] [Google Scholar]
- Weidner K.M., Di Cesare S., Sachs M., Brinkmann V., Behrens J., Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]