

Abstract
The broad aim of this work was to explore the feasibility of using light-directed perturbation techniques to study cell locomotion. Specifically, a caged form of thymosin β4 (Tβ4) was photoactivated in a defined local region of locomoting fish scale keratocytes and the resulting perturbation of locomotion was studied. Purified Tβ4 was produced in an inactive form by “caging” with ([n-nitroveratryl]oxy)chlorocarbamate. In vitro spectrophotofluorometric assays indicated that caged Tβ4 did not change the normal actin polymerization kinetics, whereas photoactivated Tβ4 significantly inhibited actin polymerization. With an a priori knowledge of the cytoplasmic diffusion coefficient of Tβ4 as measured by fluorescence recovery after photobleaching experiments, the rapid sequestration of actin monomers by uncaged Tβ4 and the consequent reduction in the diffusional spread of the Tβ4–actin complex were predicted using Virtual Cell software (developed at the Center for Biomedical Imaging Technology, University of Connecticut Health Center). These simulations demonstrated that locally photoactivating Tβ4 in keratocytes could potentially elicit a regional locomotory response. Indeed, when caged Tβ4 was locally photoactivated at the wings of locomoting keratocytes, specific turning about the irradiated region was observed, whereas various controls were negative. Additionally, loading of exogenous Tβ4 into both keratocytes and fibroblasts caused very rapid disassembly of actin filaments and reduction of cellular contractility. Based on these results, a mechanical model is proposed for the turning behavior of keratocytes in response to photoreleased Tβ4.
Keywords: thymosin β4, caged compounds, cell locomotion, FRAP, keratocyte
Introduction
Cell movement involves the coordinated action of many biomechanical events, the complexity of which is yet to be clearly understood. The key molecular events underlying directed cell locomotion are: (a) lamellipodial protrusion (driven largely by actin polymerization), (b) formation of stable cell-substrate adhesions at the front, (c) forward translocation of the cell body, presumably powered by actomyosin contraction, (d) tail retraction as a result of contractility-induced mechanical disruption of adhesions and/or enzymatic destabilization of adhesions, and finally, (e) recycling of adhesion receptors to the front part of the cell (for reviews see Condeelis 1993; Lee et al. 1993; Lauffenburger and Horwitz 1996; Mitchison and Cramer 1996; Sheetz et al. 1999). In essence, three key supramolecular phenomena, namely protrusion, adhesion, and contractility, are regulated spatiotemporally in a precise fashion to produce cell locomotion. Thus, a true understanding of the mechanism of cell locomotion would require not only the elucidation of the molecular basis of these events, but also an in-depth knowledge of how these many local diverse molecular events are globally integrated into an overall action. The central theme of this work was to formulate a novel experimental strategy in which a perturbation is produced in defined local regions of locomoting cells and the resulting effects on locomotion are studied. Results from these experiments can be used to test the adequacy of provisional mathematical models that attempt to integrate the interplay of local molecular events into a quantitative global description of locomotion.
To elicit a motile response, cells must regulate their content of F-actin spatially and temporally by changing the steady state filament assembly. The dynamics of the actin-based cytoskeleton in motile cells are regulated by concerted actions of different actin-binding proteins, and among these capping and G-actin sequestering proteins are major players (Carlier et al. 1996; Sun et al. 1996). Thus, it is conceivable that a local perturbation in the activity of such actin-binding proteins would disrupt normal actin dynamics, thereby leading to altered locomotory behavior.
The large intracellular G-actin pool is thought to be maintained by the action of thymosin β4 (Tβ4), a member of the β-thymosin superfamily of polypeptides originally isolated from mammalian thymus (Safer and Nachmias 1994; Safer and Chowrashi 1997). The monomer-sequestering action of Tβ4 lies in its ability to bind preferentially to ATP–G-actin (1:1 stoichiometry) with micromolar affinity (Safer et al. 1990; Goldschmidt-Clermont et al. 1992; Nachmias 1993). By virtue of its binding to G-actin, Tβ4 inhibits actin polymerization in resting cells. Thus, under stimulated conditions, when profilin is activated by the hydrolysis of membrane phosphatidylinositol 4,5-bisphosphate 2, Tβ4 can also release a large pool of G-actin available for profilin to accelerate polymerization (Pantaloni and Carlier 1993). This property is functionally correlated with the abundance of Tβ4 in highly motile blood cells (Hannapel and Van Kampen 1987; Weber et al. 1992). Experimental evidence such as acceleration of dermal wound healing in response to topical application of Tβ4 (Malinda et al. 1999) and overexpression of Tβ10 (functionally similar to Tβ4) in highly motile carcinogenic cell types (Califano et al. 1998; Santelli et al. 1999; Clark et al. 2000) further exemplifies the role of Tβ4 in cell motility.
In the present work, we investigated the effect of a localized increase in the activity of Tβ4 on the overall locomotory behavior of fish scale keratocytes. Tβ4 activity was modulated by means of the photoactivation approach, which permits concentrations of intracellular mediators to be varied locally, rapidly, and repeatedly in a noninvasive manner (Adams and Tsien 1993; Marriott 1994; Bayley et al. 1998; Marriott et al. 1998). A caged form of Tβ4 was photoreleased by absorption of UV light (365 nm) in a defined local region of locomoting keratocytes. Such a light-directed perturbation of the Tβ4 level produced very specific changes in the global locomotory pattern of keratocytes. To elucidate the possible molecular mechanism underlying the specific changes in locomotion in response to photoreleased Tβ4, the immediate effects of Tβ4 on the actin cytoskeleton and cellular contractility were investigated.
Materials and Methods
Fish Actin Preparation
G-actin was prepared from muscle acetone powder of fish filets as described by Spudich and Watt 1971. The extracted G-actin was stored in G-actin buffer (2 mM Tris, pH 8.0, 0.2 mM ATP, 0.05 mM CaCl2, 0.1% β-mercaptoethanol). The concentration of G-actin was determined by spectrophotometric reading at 280 nm (extinction coefficient = 26,000 M−1cm−1) and the purity of the actin preparation was confirmed by SDS-PAGE (see Fig. 1 A).
Figure 1.

(A) SDS-PAGE of purified fish actin (right lane) after Coomasie blue staining. The presence of only one major band at the 43-kD marker position (left lane) confirms the purity of extracted actin. (B) Spectrofluorophotometric data from in vitro acrylodan actin polymerization assay under different conditions as indicated, demonstrating the loss of actin-sequestering capability of Tβ4 as a result of caging with NVOC (molar ratio of caged Tβ4/actin was equal to 4:1). Uncaging restored the biochemical activity of caged Tβ4, comparable to the efficiency of pure Tβ4. (C) Western blot of keratocyte scale extracts with anti-Tβ4 antibody (right lane). GST-Tβ4 served as a positive control for the antibody binding (left lane). (D) Immunostaining of Tβ4 in keratocytes demonstrating its diffuse intracellular distribution.
Fluorescent Labeling of Actin
Actin was specifically labeled at cysteine residues with acrylodan (Molecular Probes) according to the protocol described by Marriott et al. 1988. Essentially, β-mercaptoethanol was removed from the storage protein buffer before conjugating actin with 10× molar excess of acrylodan (stock solution prepared at 10 mM in dimethylformamide) for 2 h in the dark at room temperature. The excess, unconjugated probe was removed by dialyzing the labeled actin against G-actin buffer followed by centrifugation at 21,000 g for 20 min. The labeling ratio (dye/protein typically ranged from 1 to 2) was calculated from OD280 and OD385 measurements (actin: ε280 = 26,000 M−1cm−1; acrylodan: ε385 = 18,500 M−1cm−1).
Tβ4
For most of the applications, synthesized Tβ4 (Peptide Synthesis facility at the University of North Carolina) was used. A plasmid encoding glutathione S-transferase (GST)-Tβ4 was expressed in BL 21–competent cells, and the fusion protein was purified on a glutathione-linked affinity column using standard techniques.
Actin Polymerization Assay
Actin polymerization was initiated by adding 1/50 vol of actin polymerization buffer (2.5 M KCl, 100 mM MgCl2, 50 mM ATP) to a reaction mixture of ∼4 μM actin and 3 μM acrylodan-labeled actin in G-actin buffer. The polymerization kinetics were obtained from fluorometric recordings (λexcitation = 410 nm; λemission = 465 nm) taken at 1-min intervals for ∼45 min. Occasionally, the biochemical activities of Tβ4 and its caged form were assessed using a pyrene-labeled rabbit skeletal muscle actin assay kit (Cytoskeleton).
Caging of Tβ4
Synthesized Tβ4 was caged according to the protocol described by Marriott 1994 with minor modifications. In brief, synthesized Tβ4, reconstituted in PBS at 2 mM and adjusted to a pH of 9.0 with the addition of 1.0 M NaHCO3 (final concentration of bicarbonate was 0.1 M), was incubated with 10× molar excess of the caging compound ([n-nitroveratryl]oxy)chlorocarbamate (NVOC-Cl) (Fluka; prepared as a 100 mM stock solution in acetone) in the dark for 1 h at 4°C. The unbound NVOC was removed by centrifugation at 14,000 rpm for 5 min followed by desalting through a G-15 Sephadex spin column (Amersham Pharmacia Biotech). Final concentrations of Tβ4 and NVOC were determined using the bicinchoninic acid assay (Pierce Chemical Co.) and OD360 measurements (ε360 = 5,000 M−1cm−1 for NVOC), respectively. The labeling ratio (NVOC/Tβ4) typically ranged from 2 to 4. The caged protein was concentrated up to 8–10 mg/ml using a Speedvac for future use.
Cell Culture and Protein Loading
Primary cultures of fish epithelial keratocytes from the scales of Molly fish were prepared as described by Lee et al. 1993. In brief, scales were cultured between two 22-mm square glass coverslips in RPMI 1640 (supplemented with 10% fetal calf serum and 0.1% penicillin/streptomycin/fungizone) for 1–2 d in a humidified chamber at room temperature. Sheets of keratocytes that had migrated from the scales were first disaggregated by incubating with Ca2+- and Mg2+-free PBS for 5–10 min, washed once with PBS, and incubated with RPMI media for 30 min. Cells were washed twice with PBS before loading the caged molecule into cells by bead loading. In brief, cells were overlaid with 50 μl of the protein solution (40 μl of either 10–40 mg/ml 3,4-dihydro-3-methyl-6-nitro-2H-1,3-benzoxazin-2-one [DMNB]-caged FITC-dextran or 10 mg/ml caged Tβ4 and 10 μl of 1 mg/ml rhodamine-dextran). Glass beads of diameter 425–600 μm (Sigma-Aldrich) were then sprinkled on the cells, creating transient pores in the plasma membrane and facilitating the intracellular uptake of the molecule of interest. Bead-loaded cells were immediately washed two times with PBS and then, after allowing a recovery of 30 min in RPMI media, the coverslip with attached keratocytes was mounted face up in the well of a metal slide chamber for video microscopy.
Photoactivation and Video Microscopy
Observations of keratocytes were made at room temperature using a 60×/1.4 NA Nikon objective. Video images from a Dage MTI camera were digitally acquired at 15-s intervals on a Matrox LC imaging card, driven by a time-lapse dialogue of Metamorph software (Universal Imaging Corp.). For uncaging of caged Tβ4 and caged FITC-dextran, a 354-nm UV light generated from a He-Cd laser (Liconix) was focused onto an ∼3-μm-diameter spot on the specimen using a 100× Nikon objective. The estimated power of the laser beam after passing through the objective and the duration of illumination were equal to 10 μW (measured by a powermeter) and 100 ms, respectively. These experimental parameters were identical in all the experiments. In vitro uncaging of Tβ4 was performed in a cuvette placed directly in front of the unfocussed laser beam before adding to G-actin. The photoactivation set up is essentially the same one used for photoreleasing caged calcium ionophore or caged IP3 previously in our laboratory (Ishihara et al. 1997; Lee et al. 1999).
Immunocytochemistry and Western Blotting of Tβ4
Sheets of keratocytes were disaggregated into single cells after 1 d of culture using Ca2+- and Mg2+-free PBS as described above. Immunostaining of Tβ4 was performed using a 1:100 dilution of anti-Tβ4 polyclonal primary antibody (a gift from Dr. Helen Yin, University of Texas Southwestern Medical Center at Dallas, Dallas, TX) and 1:160 dilution of FITC-conjugated goat anti–rabbit secondary antibody (Pierce Chemical Co.) according to the protocol described by Yu et al. 1994. Immunofluorescence images were digitally acquired using a Hamamatsu cooled CCD camera.
For Western blots, cells were harvested by enzymatic digestion of fish scales with collagenase (2 mg/ml in MEM) at 37°C for 3 h. Cells were lysed with RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 2 mM EDTA, 1 mM PMSF, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 1 mM Na-pervanadate, 1 μM DTT) and ∼20 μg of lysate was run on a 10–20% gradient Tris-tricine gel (Bio-Rad Laboratories). The gel was fixed with 10% glutaraldehyde in PBS for 1 h before transferring proteins to a nitrocellulose membrane for maximum retention of Tβ4 (Lin and Morrison-Bogorad 1991; Yu et al. 1994). Western blotting was performed using a 1:125 dilution of the primary antibody (the same as was used for immunostaining) and 1:10,000 dilution of goat anti–rabbit HRP-conjugated secondary antibody (Calbiochem). The immunoreactive bands were visualized using the ECL detection system (Amersham Pharmacia Biotech).
Staining of Actin Filaments
Keratocytes coloaded with Tβ4 and rhodamine-dextran were identified from the fluorescence of the loading indicator. Staining of the loaded cell was performed in situ on the microscope stage. In brief, cells were washed with PBS once and fixed with 4% paraformaldehyde for 10 min. After two brief washes with PBS, cells were permeabilized with 1% Triton X-100 for 15 min and washed twice with PBS before being stained with Alexa-Phalloidin (1:5 dilution in PBS; Molecular Probes) for 30 min. Cells were finally washed three times with PBS before performing fluorescence microscopy with a 100× objective.
Wrinkling Assay
Wrinkling rubber films were produced by vulcanizing a drop of silicone fluid (dimethylpolysiloxane, 12,500 centistokes; Sigma-Aldrich) at the center of a 35-mm petri dish using a glow discharge apparatus (polaron sputter coater E5100, 10 mA, 5–10 s), similar to the methods adopted by Lee et al. 1994 and Oliver et al. 1995, Oliver et al. 1999. Swiss 3T3 fibroblasts expressing GFP–α-actinin (provided by Dr. Carol Otey, University of North Carolina at Chapel HIll, Chapel HIll, NC) were sparsely cultured on the top of the rubber film for 6–24 h before being microinjected with Tβ4 or PBS. For the wrinkling study with keratocytes, cell sheets were first loaded with either caged Tβ4 or caged FITC-dextran as described previously. Loaded cell sheets were disaggregated with Ca2+- and Mg2+-free PBS for 5–10 min and then trypsinized for 1–2 min before being replated on the silicone films. Uncaging of the molecules was done as described previously.
Diffusion Measurements
FITC-conjugated dextran (10 kD), Tβ4, and GST-Tβ4 were bead loaded into C3H fibroblasts. Diffusion coefficients of these molecules were measured using the FRAP technique (bleaching time, 50 ms; sampling time, 100 ms; power, 800 mW from 488-nm argon laser).
Simulation of Diffusional Spread of Photoactivated Tβ4 in Keratocytes
With the knowledge of the diffusion coefficient of Tβ4, the diffusional spread of photoreleased Tβ4 and its interaction with the intracellular G-actin pool in keratocytes were predicted by simulating the uncaging phenomenon using the Virtual Cell software (developed at the Center for Biomedical Imaging Technology, University of Connecticut Health Center; Schaff et al. 1997, Schaff et al. 2000). For simplicity, a two-dimensional model of the keratocyte with a semicircular geometry (diameter equal to 30 μm) was used (see Fig. 2 A). The following reaction equilibrium was taken into account: T + A ←Æ T-A (T, free Tβ4; A, unsequestered G-actin; T-A, Tβ4–G-actin complex). For the purpose of simulation, physiological concentrations of total intracellular [Tβ4] (= T + TA) was assumed to be 100 μM (Carlier et al. 1993). Assuming an initial baseline concentration of A equal to 50 μM, a dissociation constant (K d) of 2 μM for the Tβ4–actin interaction (Nachmias 1993) resulted in T and actin-conjugated Tβ4 (TA) equal to 2 and 98 μM, respectively, indicating that the vast majority of the intracellular Tβ4 pool is actually bound to actin monomers. Thus, the measured diffusion coefficient of Tβ4 equal to 1.2 × 10−8 cm2/s from FRAP experiments reflected predominantly the value of the conjugated form (TA). For the Virtual Cell program, the diffusion coefficients of T and A were computed from that of TA by adjusting according to the molecular size. From the known K d and off rate (2/s) of the Tβ4–actin interaction (Safer and Nachmias 1994; Safer and Chowrashi 1997), the calculated on rate of the reaction was equal to 106 M−1s−1; these rate constants were input for the purpose of simulation. Uncaging of Tβ4 was simulated at a 3-μm-diameter zone at the keratocyte wing and the simulation was run with different values of uncaged Tβ4 concentrations ranging from 200 to 2,000 μM (equal to the loading concentration of caged Tβ4). The simulation time was limited to 10 s for computational convenience.
Figure 2.

(A) Schematic representation of the model of keratocyte simulating the uncaging of Tβ4. (B) Virtual Cell simulation data of time-dependent concentration profiles of [T], [TA], and [A] along the line scan through the cell (as depicted in A) for different concentrations of uncaged Tβ4 (left, 200 μM; middle, 1,000 μM; right, 2,000 μM). The uncaged Tβ4 is indicated as a 3-μm-wide spike in the [T] curve at time t = 0. Note that although a minor component of photoreleased Tβ4 diffuses rapidly, resulting in [T] decreasing to its baseline value, much of Tβ4 rapidly binds to the neighboring actin monomers and diffuses slowly as shown by the localization of [TA] at a high concentration around the photoactivated region. Concomitantly, a dramatic decrease in the free G-actin concentration takes place.
Results
Biochemical Assessment of Caged Tβ4
The caging efficiency of Tβ4 was biochemically evaluated by its effect on the kinetics of fish actin polymerization. Fig. 1 B gives the in vitro actin polymerization kinetics from spectrophotofluorometric measurements under different conditions. In control experiments, a time-dependent increase in the polymerization of G-actin was found immediately after adding the polymerization buffer (t = 0), as evident from the increase in the fluorescence intensity. Actin polymerization reached a plateau in ∼30 min. When Tβ4 was added at four times molar excess to the G-actin, dramatic inhibition of actin polymerization occurred. Upon preincubation of G-actin with caged Tβ4 at a 1:4 molar ratio, no significant inhibition in actin polymerization with respect to the control experiment was observed after adding the polymerization buffer, indicating caged Tβ4 was inactive. By contrast, when caged Tβ4 was first uncaged in vitro in a cuvette by using an unfocussed UV laser beam and then added to G-actin, the biochemical activity of Tβ4 was restored, as demonstrated by its ability to retard the actin polymerization, similar to the inhibition observed when Tβ4 was added.
Fish Scale Keratocytes Express Tβ4
Expression of Tβ4 was detected in the Western blot of fish scale extracts (Fig. 1 C). Realizing the possibility of contamination from the lysates of cells other than keratocytes in the scales, additional immunostaining of Tβ4 in keratocytes was done. Fig. 1 D confirms the presence of Tβ4 in keratocytes. The efficacy of the antibody for immunostaining was also tested on human neutrophils and platelets which are known to express Tβ4 at a very high concentration (data not shown). Tβ4 was found to be diffusely distributed throughout the cytoplasm without any preferential localization. The higher fluorescence intensity of FITC detected in the nuclear region is most likely the result of signal integration over a larger volume.
Cytoplasmic Diffusion of Tβ4 Is Slow Enough to Cause a Local Biological Effect
The diffusion coefficients (D) of FITC-dextran (10 kD), FITC-Tβ4 (∼5 kD), and FITC-GST-Tβ4 (∼31 kD) were determined from FRAP experiments using stationary C3H fibroblasts. The choice of the fibroblast as a model system was for technical reasons, since accurate FRAP measurements are much easier if the photobleached spot remains stationary during measurement, an obvious limitation for measurements on rapidly migrating keratocytes. The average diffusion coefficients of FITC-dextran, FITC-Tβ4, and FITC-GST-Tβ4 were 1.5 × 10−8 cm2/s (n = 8 cells), 1.2 × 10−8 cm2/s (n = 16 cells), and 0.48 × 10−8 cm2/s (n = 16 cells), respectively. The modest cytoplasmic diffusion coefficients of these molecules (on the order of 10−8 cm2/s) are almost two orders of magnitude slower than their free diffusion in the aqueous solution (diffusion constant is on the order of 10−6 cm2/s; Luby-Phelps et al. 1987). To check for the applicability of these results to keratocytes, video FRAP measurements of FITC-Tβ4 were performed, where the recovery kinetics of the imaged bleached spot (data not shown) were fully consistent with the quantitative FRAP data on fibroblasts.
The diffusional spread of photoreleased Tβ4 and its interaction with the intracellular G-actin pool in keratocytes were simulated at various uncaging concentrations (200, 1,000, and 2,000 μM). Fig. 2 B shows the intracellular concentration profiles of T and TA at different time points (1, 5, and 10 s) after uncaging, as measured along a line spanning across the cell through the zone of photoactivation (Fig. 2 A). It is evident from Fig. 2 B that while the concentration of free Tβ4 ([T]) rapidly decreased to the initial baseline value of 2 μM due to diffusion, the active form of Tβ4 (complexed to G-actin, [TA]) persisted for 5–10 s at a high concentration around the localized photoactivated region (<10 μm) compared with its baseline value elsewhere in the cell. This also resulted in a dramatic local decrease in the free G-actin concentration ([A]). A time-dependent recovery of [A] (up to ∼60% of baseline value in 10 s) was observed in the photoactivated region when Tβ4 was uncaged at 200 μM. At higher values of uncaged Tβ4 (1 and 2 mM), no such recovery was observed in 10 s of simulation time. In summary, these results indicate that after local uncaging, photoreleased Tβ4 can rapidly sequester neighboring actin monomers, markedly reducing its diffusional efflux from the photoactivated region; thus a local biological effect could be elicited.
Loading Exogenous Tβ4 into Keratocytes Affects Cell Morphology
To assess the global effect of exogenous Tβ4 on cell morphology, keratocytes were either bead loaded with rhodamine-dextran alone (control) or were coloaded with pure Tβ4 at a concentration of 10 mg/ml. When loaded with rhodamine-dextran alone, keratocytes maintain an unimpaired shape during locomotion (Fig. 3 A). However, aberrant changes in cell morphology were observed under the influence of exogenously loaded Tβ4 (Fig. 3 B). The fluctuation in cell morphology was quantified by the variance of the ratio of cell area to perimeter and a significantly larger value of this parameter was observed for keratocytes loaded with Tβ4 (n = 10 cells; Fig. 3 C).
Figure 3.

Morphological changes induced by loading of exogenous Tβ4 into keratocytes. (A) In control experiments, keratocytes loaded with rhodamine-dextran alone maintain an unimpaired shape during locomotion. Time interval between two successive frames is 30 s. (B) Exogenously loaded Tβ4 causes aberrant changes in cell morphology. (C) Larger variance of cell area to perimeter for Tβ4-loaded keratocytes compared with those loaded with rhodamine-dextran (Rh-dextran) alone quantitatively confirms the observed higher fluctuations in cell morphology (variance in μm2).
Photorelease of Tβ4 at the Wings of Keratocytes Causes Specific Changes in Cell Locomotion
A total of nine control experiments were performed in which keratocytes were coloaded with DMNB-caged FITC-dextran and rhodamine-dextran, and the caged FITC-dextran was photoreleased. Fig. 4 shows the data from a typical control experiment. Before irradiation, a loaded cell (identified by fluorescence in the rhodamine channel, as shown in Fig. 4 A) did not fluoresce in the FITC channel (Fig. 4 B). After a brief (100 ms) UV irradiation at the wing of the keratocyte, the caged FITC-dextran was photoreleased, as is evident from the fluorescence in the FITC channel (Fig. 4 C). Time-lapse video microscopy of the same cell was performed in order to assess the effect of the laser and the released caging group on the locomotory behavior of keratocytes (Fig. 4, D–G). Under this irradiation condition at the wing (marked by a circle), keratocyte movement appeared unaffected as assessed from the maintenance of direction (mean degree of turning = 3 ± 2.5°) and unaltered speed of locomotion after photoactivation. The loading concentration of caged FITC-dextran was varied from 10 to 40 mg/ml in order to span the possible ranges of concentration of the leaving groups generated by photoreleasing caged Tβ4. With the known dye to dextran molar ratio (2:1), photoactivation of FITC-dextran at a loading concentration of 40 mg/ml (equivalent to 4 mM FITC-dextran) presumably releases up to 8 mM of the caging group. This is comparable to the concentration of the caging group in the case of photoreleased Tβ4 at a loading concentration of 2 mM (dye to protein ratio ranged from 2 to 4). In other control experiments, all unloaded keratocytes and those loaded with rhodamine-dextran alone, when irradiated, behaved in a similar manner (data not shown). This indicates that there were no nonspecific changes in keratocyte locomotion induced by laser irradiation, loading of rhodamine-dextran, or the released caging group.
Figure 4.

Control experiment: photoactivation of caged FITC-dextran in the wing of the keratocyte. (A) Positive rhodamine fluorescence identifies a loaded cell. (B) Before uncaging, the fluorescence in the FITC channel was negligible, as expected. (C) After photoactivation, caged FITC-dextran was uncaged successfully as demonstrated by FITC fluorescence throughout the cell. (D–G) Time-lapse microscopy of the same cell shows unaltered locomotion as a result of such photorelease (the 3-μm zone of photoactivation is indicated by a circle).
Fig. 5A–H illustrate the changes in keratocyte locomotion induced by photorelease of Tβ4 at the wing of the cell under the same irradiation conditions as used in the control experiments. When Tβ4 was uncaged locally at a small region on the wing (marked by a circle), the normal locomotory characteristics continued briefly (<1 min) before the cell appeared to become tethered at the photoactivated site. This caused the cell to turn dramatically toward the direction of photoactivation, pivoting around the irradiated region. The cell eventually deadhered from the substrate and was able to locomote, maintaining its usual persistence in direction. This turning phenomenon was observed in 19 out of a total of 23 cells we photoactivated, although there was a large variation in the degree of turning (15–103°, mean turning = 56 ± 24°; see Table ). In preliminary experiments where Tβ4 was photoreleased at the leading lamella of locomoting keratocytes, a transient furrowing of the lamella was observed in some cases (data not shown).
Figure 5.

Time-lapse video microscopy of a locomoting keratocyte after photorelease of Tβ4 at the wing (marked by a circle) showing dramatic turning of the cell towards the direction of photoactivation. B and C are the frames acquired right before and after photoactivation, respectively. Note the pivoting of the irradiated zone to the substrate as a result of photorelease of Tβ4. About 8 min after photoactivation of Tβ4, normal locomotion resumed (H). Bar, 15 μm.
Table 1.
Amount of Turning of Different Keratocytes in Response to Photoreleased Tβ4 (Four Cells Did Not Turn)
| Cell number | Amount of turning |
|---|---|
| ° | |
| 1 | 34 |
| 2 | 78 |
| 3 | 44 |
| 4 | 32 |
| 5 | 30 |
| 6 | 83 |
| 7 | 89 |
| 8 | 90 |
| 9 | 80 |
| 10 | 15 |
| 11 | 60 |
| 12 | 70 |
| 13 | 35 |
| 14 | 49 |
| 15 | 50 |
| 16 | 103 |
| 17 | 39 |
| 18 | 30 |
| 19 | 61 |
| Mean ± SD | 56 ± 24 |
Tβ4 Causes a Rapid Disassembly of Actin Filaments and Reduction of Contractility in both Swiss 3T3 Fibroblasts and Keratocytyes
To elucidate the possible molecular mechanism of the action of the step-wise increase of Tβ4 in a comparable model system, pure Tβ4 was microinjected at a high concentration (needle concentration = 10 mg/ml) into Swiss 3T3 fibroblasts expressing GFP–α-actinin that served as a molecular marker for studying the dynamics of the actin cytoskeleton. Fibroblasts were preferred over keratocytes as a model system for studying immediate cytoskeletal changes, if any, for the following reasons: (a) keratocytes are extremely difficult to microinject, and (b) with GFP–α-actinin, it was easy to visualize actin filament dynamics in real time, which is not possible with untransfectable cells like keratocytes. A dramatic disassembly of actin filaments (as evident from the disappearance of actin filaments) was observed within a minute of microinjection (Fig. 6A and Fig. B), which was consistent with earlier observations in NRK or PtK2 cells (Sanders et al. 1992; Sanger et al. 1995; Golla et al. 1997). However, the kinetics of disassembly in our experiments was faster than that observed by Sanger et al. 1995 (of the order of 10 min), which is presumably due to the differences in cell type and the actual concentration of microinjected Tβ4. In control experiments, PBS was microinjected into the fibroblasts and no change in the integrity of the actin cytoskeleton was detected (Fig. 6C and Fig. D), indicating that the actin filament disassembly in response to Tβ4 was specific.
Figure 6.

(A and B) Microinjection of Tβ4 into GFP-α-actinin–expressing Swiss 3T3 fibroblasts causes dramatic disassembly of actin filament bundles (A, before microinjection, indicated by thin arrows; B, 1 min after microinjection). Note that the actin cytoskeleton remains unaltered in the noninjected neighboring cell (indicated by thick arrow). (C and D) Control experiments: besides negligible membrane damage at the site of microinjection, the overall integrity of the actin cytoskeleton (C) is maintained after microinjection of PBS (D). The fluorescent images were obtained with a 100× objective. (E and F) Contractility of Swiss 3T3 fibroblasts, as judged by the strength of the cell-induced wrinkles on the silicone substrate (indicated by an arrow in E), is reduced within 1 min of microinjection of Tβ4 (F, note the disappearance of the wrinkles). (G and H) Control experiment: microinjection of PBS does not cause any change in the cellular contractility (G and H refer to before and after microinjection of PBS, respectively). These phase–contrast images were acquired using a 40× objective.
For assessment of the changes in cell contractility, Swiss 3T3 fibroblasts plated on the wrinkling rubber film were microinjected with either Tβ4 (at 10 mg/ml) or PBS (control experiments). Immediately after the microinjection of Tβ4, a dramatic reduction of cell contractility occurred, as is evident from a complete relaxation of the wrinkles underneath the cell body (Fig. 6E and Fig. F). In control experiments, microinjection of PBS did not change the strength of the wrinkles (Fig. 6G and Fig. H), demonstrating that the decrease in cell contractility was specific to the action of Tβ4.
The effects of Tβ4 on the actin cytoskeleton and cellular contractility as described above are not limited only to the fibroblast model system. Similar results were obtained in the case of keratocytes as well. Fig. 7 depicts the effect of exogenously loaded Tβ4 on the actin cytoskeleton of keratocytes, as judged from the phalloidin staining. Control (unloaded) keratocytes (Fig. 7 A) show a meshwork of actin filaments in the lamella (marked 1) and a parallel sarcomeric type of actin filaments in the perinuclear region (marked 2). These cytoskeletal features were clearly absent in the Tβ4-loaded keratocytes (Fig. 7 B), suggesting a fibroblast-like disruption of actin filaments induced by Tβ4. Loading of rhodamine-dextran alone did not result in such cytoskeletal changes (data not shown).
Figure 7.

Effect of exogenously loaded Tβ4 on the actin cytoskeleton of keratocytes, as judged from the phalloidin staining after the experiment. (A) Control (unloaded) keratocytes show meshwork of actin filaments in the lamella (marked 1) and parallel sarcomeric type of actin filaments in the perinuclear region (marked 2). (B) In Tβ4-loaded keratocytes, lack of these cytoskeletal features suggests Tβ4-induced actin filament depolymerization.
To evaluate the effect of Tβ4 on the contractility of keratocytes, caged Tβ4 was globally photoreleased in cells plated on silicone film by defocussing the laser beam. Keratocytes normally exhibit strong wrinkles parallel to the direction of locomotion (Oliver et al. 1999). As controls, either unloaded cells or cells loaded with caged FITC-dextran were irradiated under the same condition and the strength of the cell-induced wrinkles before and after laser irradiation were compared. Results from these experiments are shown in Fig. 8. As demonstrated in Fig. 8A and Fig. B, photoactivation of caged Tβ4 clearly reduced the strength of the wrinkles under the irradiated cell (indicated by arrow). In control experiments, neither laser irradiation alone on unloaded cells (Fig. 8C and Fig. D) nor photorelease of caged FITC-dextran (Fig. 8E and Fig. F) resulted in such weakening of the wrinkle strength. These data allow the conclusion that the reduction of cellular contractility in keratocytes was specific to the action of Tβ4 and not the result of any nonspecific effects of laser irradiation and released caging groups.
Figure 8.
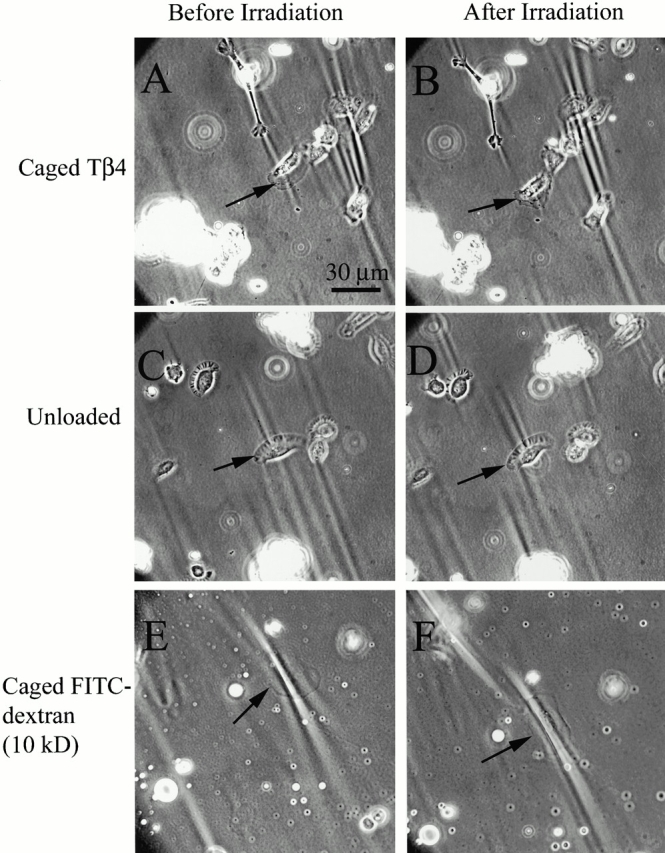
(A and B) Photorelease of caged Tβ4 into keratocyte (marked by an arrow) causes a rapid downregulation in contractility, as judged by comparing the strength of the wrinkles before (A) and after (B) photoactivation. Note that in neighboring cells which were not photoactivated the strength of the wrinkles remain unaltered. (C and D) In the unloaded cell (marked by an arrow), laser irradiation alone does not decrease the contractility (control experiment). (E and F) Photoactivation of caged FITC-dextran in keratocyte (marked by and arrow) also does not reduce the strength of the wrinkles (control experiment).
Discussion
Cell locomotion is a complex interplay of many diverse local molecular events; one of the underlying intellectual challenges in the study of locomotion is to elucidate how local molecular events are integrated to move the cell. This question can be eventually addressed by means of mathematical modeling of cell locomotion in order to keep track of diverse molecular processes. However, establishing the validity of models of any physical system requires corroborating data from experimental systems. Apart from immediate scientific insights into the cellular response of Tβ4, one of the strengths of the present work lies in the development of an experimental strategy, the results of which can eventually be used to test the accuracy of mathematical models of cell locomotion. More specifically, we have established the feasibility of using light-directed manipulation of molecules in defined regions of single motile cells to perturb the locomotion process. Such photomanipulative techniques have distinct advantages over genetic overexpression or deletion approaches, in that perturbations are made in a spatiotemporally defined fashion while avoiding genetic compensation. To the best of our knowledge, this is the first successful demonstration of the specific effects of a locally photoreleased actin-binding protein in modulating cell locomotion.
Since precisely regulated actin assembly in both time and space is crucial for cell locomotion (Condeelis 1993; Schafer and Cooper 1995), we hypothesized that a localized increase in concentration of a G-actin–sequestering protein like Tβ4 would decrease actin polymerization locally, and the resulting perturbation in cytoskeletal dynamics would in turn affect global cell locomotion. Our hypothesis, rooted in previous studies (Yu et al. 1994; Sun et al. 1996; Golla et al. 1997), was based on data from experiments in which both fibroblasts (data not shown) and keratocytes, when loaded with Tβ4 at a high concentration, underwent dramatic morphological changes (Fig. 3). In the present work, we photoactivated caged Tβ4 in defined regions of locomoting keratocytes and studied the changes in the locomotory characteristics as a result of instantaneous release of Tβ4.
A nine-residue segment (LKKTETQEK; residues 17–25) present in all isoforms of β-thymosins is homologous and was identified as an actin-binding sequence (Heintz et al. 1993). Specifically, electrostatic contacts between actin and lysine residues 18 and 19 of Tβ4 are critical for proper binding (Vancompernolle et al. 1991, Vancompernolle et al. 1992; Van Troys et al. 1996). Thus, in principle, the actin binding ability of Tβ4 can be reversibly inactivated by attaching a caging group to one or both of those two lysine residues. In our preparation, the caged Tβ4 was characterized in vitro using a fluorescence-based actin polymerization assay using acrylodan-labeled G-actin. A very large blue shift in the emission occurs when acrylodan–G-actin is incorporated into F-actin (from 492 nm for G-actin to 465 nm for F-actin). This makes it an ideal probe for studying actin polymerization. In addition, the acrylodan-conjugated actin behaves similarly to native actin with respect to polymerization kinetics and critical monomer concentration (Marriott et al. 1988). No significant inhibition in the rate of actin polymerization was observed in the presence of caged Tβ4 when compared with the control actin (Fig. 1 B), which confirms the inactivation of Tβ4 as a result of masking by the photolabile nitrobenzyl moiety. The minor differences in the kinetics could result from either some spontaneous uncaging during handling of the protein and/or from the inhibitory action of a proportion of Tβ4 that is not caged at the proper lysine residues. Upon unmasking of the active sites of Tβ4 by light-directed removal of the photolabile group, the biochemical activity was restored so that actin polymerization was inhibited in a fashion comparable to that of pure Tβ4.
A potential problem in photoreleasing proteins inside cells is the possibility of rapid diffusion of the released molecules from the site of photoactivation, thereby lowering the concentration of the protein below the threshold level of activity. This problem was encountered in studies by Walker et al. 1998, where local photorelease of caged inhibitory peptides against calmodulin and myosin light chain kinase failed to produce any detectable changes in eosinophil locomotion, whereas global photorelease immediately stopped the cells from moving. To estimate the rate of diffusion of Tβ4, FRAP experiments were performed on fibroblasts. Our FRAP data showed that the cytoplasmic diffusion rate of Tβ4 was modest and on the order of 10−8 cm2/s, compared with ∼10−6 cm2/s estimated in aqueous solution. A difference of two orders of magnitude between the diffusion constants measured in cytoplasm and aqueous solution is consistent with the idea that diffusion of molecules inside the cytoplasm is severely limited by various factors, including obstructions from filamentous structures, steric hindrance, and transient interactions with the elements of the cytomatrix (Jacobson and Wojcieszyn 1984; Luby-Phelps et al. 1987, Luby-Phelps et al. 1988; Luby-Phelps 1994). Furthermore, our Virtual Cell simulation data were consistent with the notion that rapid binding of photoreleased Tβ4 to G-actin occurs, thereby promoting the localization of the complexed Tβ4 to a considerable degree. The high concentration of the Tβ4–G-actin complex in the photoactivated region compared with its baseline value elsewhere in the cell suggests that while there may be a minor fast diffusing component, much of the Τβ4 quickly binds to the neighboring G-actin to elicit a local biological effect. The dramatic decrease in the free G-actin concentration near the photoactivated region implies that (a) a local shift in equilibrium from F-actin to G-actin would occur, thereby causing a local depolymerization of existing actin filaments, and (b) a severe retardation of existing filament growth would also occur. A full simulation of the kinetics of actin filament depolymerization is complex. This would require incorporation of the actions of other actin-binding proteins and also the potential, at high concentration, for Τβ4 to copolymerize with actin (Carlier et al. 1996; Sun et al. 1996).
Since much cytoplasmic actin monomer is bound to various actin-binding proteins, one can argue that Tβ4, when locally uncaged, may not act until actin filament turnover provides a free monomer to which Tβ4 can bind. If this were the case, a local effect would not be expected because the time required for sufficient filament depolymerization would permit free, uncaged Tβ4 to diffuse throughout the cell. We contend that local photorelease of Tβ4 can rapidly sequester the neighboring G-actin pool and in turn cause a local depolymerization of actin filaments based on the following arguments. First, G-actin sequestering proteins such as profilin and Tβ4 rapidly exchange between bound and free states, so that rapid shuttling of G-actin between these two classes of actin-binding proteins occurs (Pantaloni and Carlier 1993). Thus, it is indeed possible that a large, local increase in the Tβ4 concentration could rapidly exchange with the profilin-bound G-actin pool and, by means of this sequestering action, suppress local actin polymerization and simultaneously favor the disassembly of actin filaments. Second, contractility upon globally uncaging Tβ4 was downregulated within a minute in keratocytes. Since downregulation of contractility undoubtedly requires the diminution of myosin motor activity and the disassembly of actin filaments, we can presume that local disassembly of F-actin proceeds with similar or even more rapid kinetics. Finally, the marked asymmetry in the keratocyte geometry during cell turning in response to a local photorelease of Tβ4 is further indicative of a localized disturbance in the cytoskeletal dynamics in the cell.
When Tβ4 was photoreleased at the wings of locomoting keratocytes, a dramatic turning of cells using the irradiated zone as the pivot point was observed (Fig. 5). This turning phenomenon was specific to the action of Tβ4 since photorelease of caged FITC-dextran (Fig. 4), loading of rhodamine-dextran, and simply the laser irradiation itself failed to produce any such effects on keratocyte locomotion. In control experiments, the concentration of caged FITC-dextran was varied over a range (up to 40 mg/ml) to cover a range of released caging group comparable to that in the experiments with caged Tβ4. The underlying assumption was that the loading efficiency of the caged forms of Tβ4 (5 kD) and FITC-dextran (10 kD) into keratocytes is comparable with the bead-loading technique. Over the range of the concentration of caged FITC-dextran we tested, photoactivation did not cause any change in keratocyte locomotion. The large variability in the degree of turning in response to uncaging of Tβ4 is most likely due to cell-to-cell variability in both loading concentration of caged Tβ4 and the efficiency of uncaging. Since our simulation data suggest a rapid sequestration of actin monomers by the uncaged Tβ4 by 10 s, it is not immediately clear why it took almost 1 min for the cells to turn. It is plausible that an initial trigger to turn is delivered by the Tβ4 jump, which causes a local perturbation in actin filament dynamics and sets in motion a sequence of events leading to the cell turning. In this view, when the cell has turned the effects of Tβ4 jump could be a distant memory.
Finally, the experimental results demonstrating Tβ4-induced changes in the actin cytoskeleton and the cellular contractility provide significant insight into the molecular mechanisms underlying the cell turning phenomenon. Previous studies have shown that strong “pinching” type retraction forces are exerted at the wings of keratocytes that are, at least in part, responsible for detachment of the rear margin of the cell during locomotion (Lee et al. 1994; Oliver et al. 1995, Oliver et al. 1999; Galbraith and Sheetz 1999). These forces are presumably exerted by the molecular interaction of actomyosin parallel bundles (schematically represented in Fig. 9A and Fig. B) in the perinuclear region (demonstrated by distribution of actin and myosin II; Svitkina et al. 1997) since myosin II inhibitors dramatically reduce the pinching traction under keratocytes (Oliver et al. 1999). Furthermore, detailed analysis indicated that the propulsive tractions are exerted predominantly in the wings of keratocyte (Oliver et al. 1999; Fig. 9 B). Based on the results from the experiments studying the effects of increased Tβ4 on the actin cytoskeleton and the contractility of both keratocytes and fibroblasts, we propose the following model (Fig. 9) for the turning behavior of keratocytes after photoactivation at the wing. In order for a cell to turn, there must be an imbalance in the propulsive tractions at the wings of the keratocyte (Oliver et al. 1999). This imbalance results in a net propulsive torque that produces a turn about the pivot point. In this mechanical context, we postulate that local photorelease of caged Tβ4 depolymerizes the existing actin filaments (Fig. 9 C) and also inhibits growth of the actin filaments, leading to a downregulation of contractility that causes both propulsive and pinching tractions generated in the right wing of the keratocyte to be abrogated, as depicted schematically in Fig. 9 D. These combined, asymmetric force inhibitions produce a poorly retracting zone that becomes the pivot point. The result of this markedly asymmetric force distribution is that the cell turns clockwise powered by the now unbalanced left wing propulsive tractions acting through the indicated moment arm (Fig. 9 D) (recall that the substrate exerts a forward-directed reaction force equal and opposite to the propulsive traction, and the reaction force acting through the moment arm “d” produces the torque that causes the cell to turn).
Figure 9.

Proposed model for the turning of keratocytes in response to photoreleased Tβ4 at the wing. Cell turning is the result of a clockwise torque generated by substrate reaction to unbalanced propulsive traction (the details of the model are described in Discussion).
Our hypothesis of local depolymerization of actin filaments and diminution of filament growth rates in the photoactivated zone is also consistent with the results of other preliminary experiments, where photorelease of Tβ4 at the leading lamella of locomoting keratocytes caused a transient furrowing of the lamella in some cases, thus suggesting inhibition in lamellar protrusion as a result of reduction in actin filament growth, either by local actin filament disassembly and/or by inhibition in actin polymerization (data not shown). Future experiments involving detailed traction force mapping of keratocytes after photoactivation will provide additional insights into the molecular mechanisms of the action of photoreleased Tβ4.
Acknowledgments
We thank Dr. Carol Otey (University of North Carolina at Chapel Hill, Chapel Hill, NC) and Dr. Helen Yin (University of Texas Southwestern Medical Center at Dallas, Dallas, TX) for providing us with GFP-α-actinin–expressing Swiss 3T3 fibroblasts and an antibody to Tβ4, respectively. We also thank Jessica Cohen for her technical assistance and Dr. Elizabeth deBeus for her advice in the early stages of this project.
K. Jacobson was supported by National Institutes of Health grant GM35325 and P60-DE13079 from the National Institute for Dental and Cranial Research.
Footnotes
D. Jones' present address is Wake Forest University School of Medicine, Winston-Salem, NC 27106.
Abbreviations used in this paper: A, unsequestered G-actin; GST, glutathione S-transferase; NVOC, ([n-nitroveratryl]oxy)chlorocarbamate; T, free Tβ4; TA, actin–β4 complex; Tβ4, thymosin β4.
References
- Adams S.R., Tsien R.Y. Controlling cell chemistry with caged compounds. Annu. Rev. Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- Bayley H., Chang C.Y., Miller W.T., Niblack B., Pan P. Caged peptides and proteins by targeted chemical modification. Methods Enzymol. 1998;291:117–134. doi: 10.1016/s0076-6879(98)91010-2. [DOI] [PubMed] [Google Scholar]
- Califano D., Monaco C., Santelli G., Giuliano A., Veronese M.L., Berlingieri M.T., de Franciscis V., Berger N., Trapasso F., Santoro M., Viglietto G. Thymosin beta-10 gene overexpression correlated with highly malignant neoplastic phenotype of transformed thyroid cells in vivo and in vitro. Cancer Res. 1998;58:823–828. [PubMed] [Google Scholar]
- Carlier M.F., Jean C., Rieger K.J., Lenfant M., Pantaloni D. Modulation of interaction between G-actin and thymosin β4 by the ATP/ADP ratiopossible implication in the regulation of actin dynamics. Proc. Natl. Acad. Sci. USA. 1993;90:5034–5038. doi: 10.1073/pnas.90.11.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M.F., Didry D., Erk I., Lepault J., Van Troys L., Vandekerckhove J., Perelroizen I., Yin H., Doi Y., Pantaloni D. Tβ4 is not a simple G-actin sequestering protein and interacts with F-actin at high concentration. J. Biol. Chem. 1996;271:9231–9239. doi: 10.1074/jbc.271.16.9231. [DOI] [PubMed] [Google Scholar]
- Clark E.A., Golub T.R., Lander E.S., Hynes R. Genomic analysis of metastatic reveals an essential role for rhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edgethe formation of cell protrusions. Annu. Rev. Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Galbraith C.G., Sheetz M.P. Keratocytes pull with similar forces on their dorsal and ventral surfaces. J. Cell Biol. 1999;147:1313–1324. doi: 10.1083/jcb.147.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont P.J., Furman M.I., Wachsstock D., Safer D., Nachmias V.T., Pollard T.D. The control of actin nucleotide exchange by thymosin β4 and profilin. A potentially regulatory mechanism for actin polymerization in cells. Mol. Biol. Cell. 1992;3:1015–1024. doi: 10.1091/mbc.3.9.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golla R., Philp N., Safer D., Chintapalli J., Hoffman R., Collins L., Nachmias V.T. Co-ordinate regulation of the cytoskeleton in 3T3 cells overexpressing thymosin-β4. Cell Motil. Cytoskeleton. 1997;38:187–200. doi: 10.1002/(SICI)1097-0169(1997)38:2<187::AID-CM7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hannapel E., Van Kampen M. Determination of thymosin β4 in human blood cells and serum. J. Chromatogr. 1987;397:279–285. doi: 10.1016/s0021-9673(01)85010-x. [DOI] [PubMed] [Google Scholar]
- Heintz D., Reichart A., Mihelic M., Voelter W., Faulstich H. Use of bimanyl actin derivative (TMB-actin) for studying complexation of β-thymosins. FEBS Lett. 1993;329:9–12. doi: 10.1016/0014-5793(93)80181-s. [DOI] [PubMed] [Google Scholar]
- Ishihara A., Gee K., Schwartz S., Jacobson K., Lee J. Photoactivation of caged compounds in single, living cellsan application to the study of cell locomotion. Biotechniques. 1997;23:268–274. doi: 10.2144/97232st01. [DOI] [PubMed] [Google Scholar]
- Jacobson K., Wojcieszyn J. The translational mobility of substances within the cytoplasmic matrix. Proc. Natl. Acad. Sci. USA. 1984;81:6747–6751. doi: 10.1073/pnas.81.21.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger D.A., Horwitz A.F. Cell migrationa physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Theriot J.A., Jacobson K. Principles of locomotion for simple-shaped cells. Nature. 1993;362:167–171. doi: 10.1038/362167a0. [DOI] [PubMed] [Google Scholar]
- Lee J., Leonard M., Oliver T., Ishihara A., Jacobson K. Traction forces generated by locomoting keratocytes. J. Cell Biol. 1994;127:1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Oxford G., Johnson B., Jacobson K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature. 1999;400:382–386. doi: 10.1038/22578. [DOI] [PubMed] [Google Scholar]
- Lin S.C., Morrison-Bogorad M. Cloning and characterization of a testis-specific thymosin β10 cDNAexpression in post-meiotic male germ cells. J. Biol. Chem. 1991;266:23347–23353. [PubMed] [Google Scholar]
- Luby-Phelps K. Physical properties of cytoplasm. Curr. Opin. Cell Biol. 1994;6:3–9. doi: 10.1016/0955-0674(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Luby-Phelps K., Castle P.E., Taylor D.L., Lanni F. Hindered diffusion of inert tracer particle in the cytoplasm of mouse 3T3 cells. Proc. Natl. Acad. Sci. USA. 1987;84:4910–4913. doi: 10.1073/pnas.84.14.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K., Lanni F., Taylor D.L. The submicroscopic properties of cytoplasm as a determination of cellular function. Annu. Rev. Biophys. Biophys. Chem. 1988;17:369–396. doi: 10.1146/annurev.bb.17.060188.002101. [DOI] [PubMed] [Google Scholar]
- Malinda K.M., Sidhu G.S., Mani H., Banaudha K., Maheshwari R.K., Goldstein A.L., Kleinman H.K. Thymosin beta 4 accelerates wound healing. J. Invest. Dermatol. 1999;113:364–368. doi: 10.1046/j.1523-1747.1999.00708.x. [DOI] [PubMed] [Google Scholar]
- Marriott G. Caged protein conjugates and light-directed generation of protein activitypreparation, photoactivation, and spectroscopic characterization of caged G-actin conjugates. Biochemistry. 1994;33:9092–9097. doi: 10.1021/bi00197a010. [DOI] [PubMed] [Google Scholar]
- Marriott G., Zechel K., Jovin T.M. Spectroscopic and functional characterization of an environmentally sensitive fluorescent actin conjugate. Biochemistry. 1988;27:6214–6220. doi: 10.1021/bi00417a004. [DOI] [PubMed] [Google Scholar]
- Marriott G., Ottl J., Heidecker M., Gabriel D. Light-directed activation of protein activity from caged protein conjugates. Methods Enzymol. 1998;291:95–116. doi: 10.1016/s0076-6879(98)91009-6. [DOI] [PubMed] [Google Scholar]
- Mitchison T.J., Cramer L.P. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Nachmias V.T. Small actin-binding proteinsthe β-thymosin family. Curr. Biol. 1993;5:56–62. doi: 10.1016/s0955-0674(05)80008-0. [DOI] [PubMed] [Google Scholar]
- Oliver T., Dembo M., Jacobson K. Traction forces in cells locomoting along a substratum. Cell Motil. Cytoskeleton. 1995;31:225–240. doi: 10.1002/cm.970310306. [DOI] [PubMed] [Google Scholar]
- Oliver T., Dembo M., Jacobson K. Separation of actomyosin-generated and adhesive traction forces in tethered cells provides a physical basis for gliding and turning locomotion in keratocytes. J. Cell Biol. 1999;145:589–604. doi: 10.1083/jcb.145.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D., Carlier M.F. How profilin promotes actin assembly in the presence of Tβ4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- Safer D., Chowrashi P. β-Thymosins from marine invertebratesprimary structure and interaction with actin. Cell Motil. Cytoskeleton. 1997;38:163–171. doi: 10.1002/(SICI)1097-0169(1997)38:2<163::AID-CM5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Safer D., Nachmias V.T. Beta thymosins as actin binding peptides. Bioessays. 1994;16:4873–4879. doi: 10.1002/bies.950160814. [DOI] [PubMed] [Google Scholar]
- Safer D., Golla R., Nachmias V.T. Isolation of a 5-kilodalton actin-sequestering peptide from human blood platelets. Proc. Natl. Acad. Sci. USA. 1990;87:2536–2540. doi: 10.1073/pnas.87.7.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders M.C., Goldstein A.L., Wang Y.L. Thymosin beta 4 (Fx peptide) is a potent regulator of actin polymerization in living cells. Proc. Natl. Acad. Sci. USA. 1992;89:4678–4682. doi: 10.1073/pnas.89.10.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger J.M., Golla R., Safer D., Choi J.K., Yu K.R., Sanger J.W., Nachmias V.T. Increasing intracellular concentrations of thymosin β4 in PtK2 cellseffects on stress fibers, cytokinesis, and cell spreading. Cell Motil. Cytoskeleton. 1995;31:307–322. doi: 10.1002/cm.970310407. [DOI] [PubMed] [Google Scholar]
- Santelli G., Califano D., Chiapetta G., Vento M.T., Bartoli P.C., Zullo F., Trapasso F., Viglietto G., Fusco A. Thymosin beta-10 gene overexpression is a general event in human carcinogenesis. Am. J. Pathol. 1999;155:799–804. doi: 10.1016/s0002-9440(10)65178-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D.A., Cooper J.A. Control of actin assembly at filament ends. Annu. Rev. Cell Dev. Biol. 1995;11:497–518. doi: 10.1146/annurev.cb.11.110195.002433. [DOI] [PubMed] [Google Scholar]
- Schaff J.C., Fink C., Slepchenko B., Carson J.H., Loew L.M. A general computational framework for modeling cellular structure and function. Biophys. J. 1997;73:1135–1146. doi: 10.1016/S0006-3495(97)78146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaff J.C., Slepchenko B.M., Loew L.M. Physiological modeling with the virtual cell framework. Methods Enzymol. 2000;321:1–23. doi: 10.1016/s0076-6879(00)21184-1. [DOI] [PubMed] [Google Scholar]
- Sheetz M.P., Felsenfeld D., Galbraith C.G., Choquet D. Cell migration as a five-step cycle. Biochem. Soc. Symp. 1999;65:233–243. [PubMed] [Google Scholar]
- Spudich J.A., Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Sun H., Kwiatkowska K., Yin H.L. β-Thymosins are not simple actin monomer buffering proteinsinsights from overexpression studies. J. Biol. Chem. 1996;271:9223–9230. [PubMed] [Google Scholar]
- Svitkina T.M., Verkhovsky A.B., McQuade K.M., Borisy G.G. Analysis of the actin–myosin II system in fish epidermal keratocytesmechanism of cell body translocation. J. Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancompernolle K., Vandekerckhove J., Bubb M.R., Korn E.D. The interfaces of actin and acanthamoeba actobindin. Identification of a new actin-binding motif. J. Biol. Chem. 1991;266:15427–15431. [PubMed] [Google Scholar]
- Vancompernolle K., Goethals M., Huet C., Louvard D., Vandekerckhove J. G- to F-actin modulation by a single amino acid substitution in the actin binding site of actobindin and thymosin β4. EMBO J. 1992;11:4739–4746. doi: 10.1002/j.1460-2075.1992.tb05579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Troys M., Dewitte D., Goethals M., Carlier M.F., Vandekerckhove J., Ampe C. The actin binding site of thymosin β4 mapped by mutational analysis. EMBO J. 1996;15:201–210. [PMC free article] [PubMed] [Google Scholar]
- Walker J.W., Gilbert S.H., Drummond R.M., Yamada M., Sreekumar R., Carraway R.E., Ikebe M., Fay F.S. Signaling pathways underlying eosinophil cell motility revealed by caging peptides. Proc. Natl. Acad. Sci. USA. 1998;95:1568–1573. doi: 10.1073/pnas.95.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A., Nachmias V.T., Pennise C.R., Pring M., Safer D. Interaction of thymosin β4 with muscle and platelet actinimplications for actin sequestration in resting platelets. Biochemistry. 1992;31:6179–6185. doi: 10.1021/bi00142a002. [DOI] [PubMed] [Google Scholar]
- Yu F., Lin S., Morrison-Bogorad M., Yin H.L. Effect of thymosin β4 and β10 isoforms on actin structures in living cells. Cell Motil. Cytoskeleton. 1994;27:13–25. doi: 10.1002/cm.970270103. [DOI] [PubMed] [Google Scholar]