

Abstract
Phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) on serine 51 integrates general translation repression with activation of stress-inducible genes such as ATF4, CHOP, and BiP in the unfolded protein response. We sought to identify new genes active in this phospho-eIF2α–dependent signaling pathway by screening a library of recombinant retroviruses for clones that inhibit the expression of a CHOP::GFP reporter. A retrovirus encoding the COOH terminus of growth arrest and DNA damage gene (GADD)34, also known as MYD116 (Fornace, A.J., D.W. Neibert, M.C. Hollander, J.D. Luethy, M. Papathanasiou, J. Fragoli, and N.J. Holbrook. 1989. Mol. Cell. Biol. 9:4196–4203; Lord K.A., B. Hoffman-Lieberman, and D.A. Lieberman. 1990. Nucleic Acid Res. 18:2823), was isolated and found to attenuate CHOP (also known as GADD153) activation by both protein malfolding in the endoplasmic reticulum, and amino acid deprivation. Despite normal activity of the cognate stress-inducible eIF2α kinases PERK (also known as PEK) and GCN2, phospho-eIF2α levels were markedly diminished in GADD34-overexpressing cells. GADD34 formed a complex with the catalytic subunit of protein phosphatase 1 (PP1c) that specifically promoted the dephosphorylation of eIF2α in vitro. Mutations that interfered with the interaction with PP1c prevented the dephosphorylation of eIF2α and blocked attenuation of CHOP by GADD34. Expression of GADD34 is stress dependent, and was absent in PERK−/− and GCN2−/− cells. These findings implicate GADD34-mediated dephosphorylation of eIF2α in a negative feedback loop that inhibits stress-induced gene expression, and that might promote recovery from translational inhibition in the unfolded protein response.
Keywords: endoplasmic reticulum, translation, gene expression regulation, signal transduction, molecular cloning
Introduction
Translation initiation is inhibited in cells exposed to different stressful conditions. The phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) plays an important role in this stereotyped response, and is mediated by distinct kinases that are activated by specific stress signals. The interferon-inducible, double-stranded RNA–activated kinase (PKR) phosphorylates eIF2α in response to viral infection (Clemens and Elia 1997; Samuel et al. 1997). Other kinases, such as PERK and general control nonrepressed– (GCN) 2, phosphorylate eIF2α in cells experiencing stress from protein malfolding in the ER (Harding et al. 1999, Harding et al. 2000b; Sood et al. 2000a) or amino acid deprivation (Berlanga et al. 1999; Harding et al. 2000a; Sood et al. 2000b), respectively. Toxins like arsenite, that are believed to modify protein structure, are also associated with high levels of eIF2α phosphorylation, but the responsible kinase(s) has not been identified (Brostrom and Brostrom 1998).
When phosphorylated on serine 51, eIF2α binds to and inhibits the guanine nucleotide exchange factor, eIF2B. The latter is required for the formation of the eukaryotic translational preinitiation complexes, and its sequestration in an inactive complex with phosphorylated eIF2α inhibits the initiation step of protein synthesis. In the case of viral infection, the physiological significance of PKR-induced eIF2α phosphorylation is revealed by the observation that nearly all successful viruses have adapted a mechanism for circumventing the activity of this upstream kinase or the downstream response (Clemens 1996). The significance of translational control during the unfolded protein response (UPR) is revealed by the phenotype of PERK−/− cells that are markedly impaired in their ability to survive exposure to conditions that promote protein malfolding in the ER (Harding et al. 2000b). In the absence of PERK, unmitigated protein synthesis leads to critical levels of ER stress, promoting programmed cell death (Harding et al. 2000b).
We have recently discovered that in addition to its role in regulating protein synthesis, eIF2α phosphorylation is also required for stress-induced gene expression. Cells lacking the upstream kinases PERK or GCN2 are impaired in the induction of the C/EBP homologous protein (CHOP) and immunoglobulin binding protein of B cells (BiP) genes by cognate stress signals (Harding et al. 2000a). Paradoxically, eIF2α phosphorylation and the reduced formation of translational preinitiation complexes activate the translation of a transcription factor, ATF4 (Harding et al. 2000a), which accumulates under stress and activates CHOP (Fawcett et al. 1999; Harding et al. 2000a). CHOP is a downstream transcription factor that binds to and activates the promoter of target genes that are believed to play a role in programmed cell death and tissue regeneration (Wang et al. 1998a; Zinszner et al. 1998). BiP is an ER chaperone whose transcription is positively regulated by the UPR through at least three independent signaling pathways (Mori 2000), one of which involves the ER stress-inducible eIF2α kinase PERK (Harding et al. 2000a, Figure 1A therein). Thus, phosphorylation of eIF2α on serine 51 integrates transcriptional and translational responses in mammalian cells. We tentatively refer to this pathway as the integrated stress response.
We sought to identify new components of the integrated stress response by screening for genes or gene fragments that, when expressed ectopically in either their sense or antisense orientation, would block the response. We report here on the isolation of one such genetic suppressor element (GSE) of the integrated stress response that encodes the COOH terminus of the stress-inducible growth arrest and DNA damage gene (GADD)34 protein. Our studies suggest that GADD34 participates in a negative feedback loop that attenuates signaling in the integrated stress response.
Materials and Methods
Identifying GSEs That Impair CHOP::GFP Activation
CHO-K1 cells were stably transformed with a CHOP::GFP reporter plasmid. The plasmid was constructed by fusing an 8.5-kb 5′ murine CHOP gene fragment, whose 3′ end is at the PmlI site in exon 3, nine nucleotides 5′ of the CHOP coding region, to enhanced green fluorescent protein (GFP) (CLONTECH Laboratories, Inc.) and termination sequences from the SV-40 virus (Wang et al. 1998b). A clone of CHOP::GFP cells was selected for low basal GFP activity and high inducibility by tunicamycin and amino acid starvation, and was used in all subsequent studies. It is referred to as the parental line.
A random primed cDNA library from CHO-K1 cells was constructed in the retroviral plasmid pBabe Puro® (Morgenstern and Land 1990). To increase representation of genes that may impact on the integrated stress response, we pooled mRNA from untreated, tunicamycin-treated, and thapsigargin-treated cells. The polylinker of the preretroviral plasmid was adapted to incorporate an AUG initiation codon in all three reading frames upstream of the site of insertion of the cDNAs, and a UAG stop codon in all three reading frames downstream of the cDNA insert (Gudkov and Roninson 1997).
Retroviruses were packed into vesicular stomatitis virus glycoprotein (VSV-G) envelope pseudotyped viruses (Landau and Littman 1992), and retroviral pools of 106 individual recombinants were used to infect CHOP::GFP cells. FACS® selection for cells that had acquired retroviruses containing GSEs of CHOP::GFP was carried out by treating pools of cells with tunicamycin (1.75 μg/ml for 3 h), and 16 h later sorting the 1% dullest cells. Cells recovered from the sorting procedure were expanded and selected for acquisition of the Puro® marker by culture in Puromycin (5 μg/ml for 7 d). Cells were subjected to three successive cycles of selection by the aforementioned criteria. After the third sort, cells were plated at clonal dilution, and the clones were tested for CHOP::GFP activity under stress.
Cloning, Validation, and Functional Characterization of the A1 GSE, GADD34, and Mutant Derivatives
Genomic DNA was prepared from clones defective in CHOP::GFP expression, and the cDNA insert was recovered by PCR of the integrated retrovirus. The insert was ligated into the parental preretroviral plasmid, packaged into infectious particles, and transduced into parental CHOP::GFP cells. Reconstitution of the defect in CHOP::GFP expression was taken as evidence for the presence of a GSE in the retroviral clone.
The cDNA insert was also introduced into a pCDNA3 plasmid (Invitrogen) in which the Neo® marker was replaced by the human CD2 cell surface marker, creating pCDNA3-CD2. This plasmid was used in transient transfection (Lipofectamine Plus; Life Technologies) to transduce the GSE into parental CHOP::GFP cells. A full-length GADD34 EST (GenBank/EMBL/DDBJ accession no. AW320574) was obtained (Incyte), and a combination of PCR-based manipulations and restriction digests were used to introduce a FLAG epitope tag at the NH2 terminus, and to create the deletion mutants and the point mutations indicated in the text.
The eIF2α cDNA was a gift of R. Schneider (New York University, New York, NY). The S51D mutation was introduced by PCR mutagenesis, and the wild-type and mutant cDNA were introduced into the pCDNA3-CD2 plasmid.
Transfected cells were treated with tunicamycin (Calbiochem) or cultured in media lacking amino acids to activate CHOP::GFP. Treated cells were recovered by trypsinization, stained with a Tricolor anti–human CD2 antibody (Caltag Laboratories) to report on transfected cells, and analyzed by dual channel FACS® analysis for the GFP and anti–CD2-Tricolor signals.
Immunoprecipitation, Immunoblotting, and Detection of CHOP, eIF2α, PERK, GCN2, ATF4, and PP1c
Cell treatment, lysate preparation, immunoprecipitation, and immunoblotting followed procedures described previously (Harding et al. 1999, Harding et al. 2000a,Harding et al. 2000b). Tunicamycin and thapsigargin were purchased from Calbiochem-Novabiochem and Sigma-Aldrich, respectively. The antisera for detecting total content of eIF2α and eIF2α phosphorylated on serine 51 have been described previously (Scorsone et al. 1987; DeGracia et al. 1997). The antisera reactive with total PERK and GCN2, and the activated, phosphorylated forms of the proteins have been described previously (Harding et al. 1999, Harding et al. 2000a,Harding et al. 2000b; Bertolotti et al. 2000). As described previously, CHOP (Wang et al. 1996) and ATF4 (Vallejo et al. 1993) were detected by immunoblot. GADD34, tagged at the NH2 terminus with a FLAG epitope, was immunoprecipitated from cell lysates prepared in 1% Triton X-100–containing buffer (Harding et al. 2000b) using an anti-FLAG monoclonal antibody (Eastman Kodak Co.). Coprecipitating PP1 was detected by immunoblot using a rabbit anti–human PP1c antiserum (Santa Cruz Biotechnology, Inc.) at a dilution of 1:200.
In Vitro Dephosphorylation of eIF2α
Bacterially expressed GST-PERK was used to phosphorylate eIF2α in vitro in rabbit reticulocyte lysate as described previously (Harding et al. 1999). After gel filtration to remove the unincorporated [γ-32P]ATP, 1-μl aliquots of the radiolabeled proteins in the reticulocyte lysate were placed in a 10-μl dephosphorylation reaction (dephosphorylation buffer: 20 mM Tris-HCl, pH 7.4, 50 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 0.8 mM ATP; He et al. 1997), 5 μl of CHO cell lysate with a protein content of 1 mg/ml was added, and the reaction was incubated at 30°C for the specified time. The reaction was terminated by boiling the sample in an equal volume of 2% SDS loading buffer, and the radiolabeled material was resolved on a 10% SDS-PAGE that was exposed to autoradiography. Cell lysates were obtained by extraction in a buffer containing 0.5% Triton X-100, 50 mM NaCl, 20 mM Tris-HCl, pH 7.4, 10% glycerol, 0.1 mM EDTA, and protease inhibitors.
Northern Blot Analysis
The PERK−/− and GCN2−/− cells have been described previously (Harding et al. 2000a,Harding et al. 2000b). Total RNA from untreated and stressed cells was fractionated on MOPS-formaldehyde gels, transferred to nylon filters, and hybridized to the CHOP, BiP, glutaraldehyde 3-phosphate dehydrogenase (GAPDH), the insert of murine GADD34 EST, or A1 retroviral insert, and scanned in a Molecular Dynamics PhosphorImager.
Results
Isolating a GSE That Interferes with CHOP Expression in Stressed Cells
We have described previously a stable CHO cell line carrying a CHOP::GFP transgene that faithfully reports on the activity of the endogenous CHOP gene; the reporter is tightly repressed in unstressed cells, and is strongly activated by the UPR or amino acid deprivation (Fig. 1 A, and Wang et al. 1998b). In an effort to identify genes that regulate CHOP activation by stress, we constructed a retroviral library of cDNA made from ER-stressed CHO cells, and screened it for clones that inhibited the activation of the CHOP::GFP reporter. The recombinant retroviruses were created with an in-frame AUG for translation of the inserted cDNA in all three frames. This design encourages the expression of GSEs that can be truncated dominant negative alleles of genes that activate the pathway, gain-of-function mutations in genes that suppress the pathway, or antisense hypomorphic alleles of activators of the pathway, among other possibilities (Gudkov and Roninson 1997).
Figure 1.
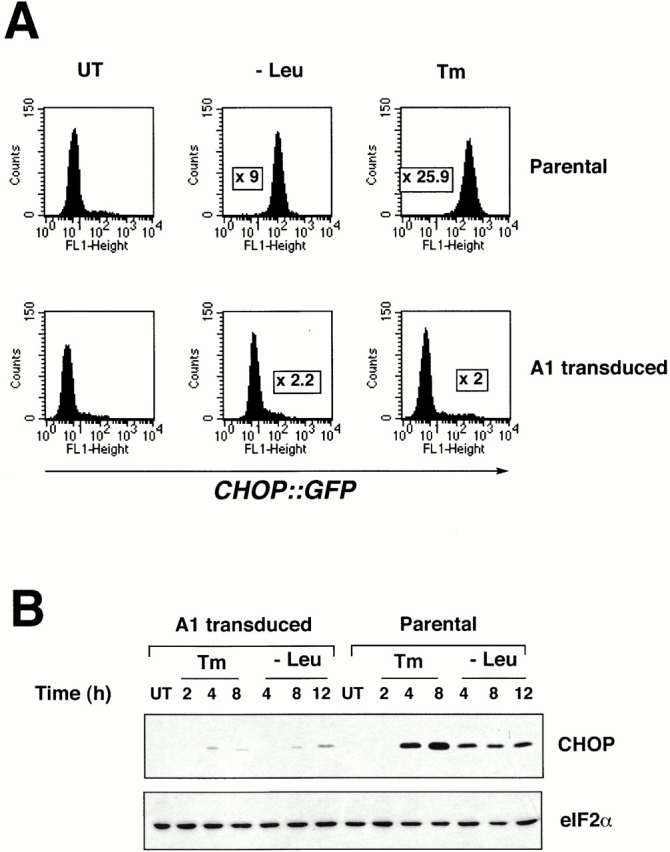
A1 is a GSE attenuating CHOP expression by ER stress and amino acid deprivation. (A) GFP expression levels analyzed by FACS® in parental CHOP::GFP-containing CHO cells and in cells transduced by the A1 retroviral clone. Cells were untreated and cultured in complete media (UT), cultured for 16 h in media lacking leucine (−Leu), or exposed to tunicamycin (2 μg/ml) for 16 h (Tm). The fold increase in GFP compared with the untreated cells is indicated. (B) Immunoblot of endogenous CHOP in the parental and A1-transduced cells after exposure to tunicamycin (2 μg/ml) or leucine-free media (upper panel). Anti-eIF2α immunoblot of the same membrane serves as a control of protein loading (lower panel).
Pools of cells transduced with the retroviral library were exposed to tunicamycin, an agent that causes ER stress by inhibiting protein glycosylation, and cells that failed to activate the resident CHOP::GFP transgene were selected by FACS®. The GFP-dull population was enriched by successive cycles of sorting. Clones of GFP-dull cells were isolated by limit dilution, expanded, and the cDNA insert was recovered by PCR of the integrated viral genome. The insert containing the putative GSE was used to construct a new recombinant retrovirus to recapitulate the inhibition of CHOP::GFP expression. Fig. 1 A shows the effect on CHOP::GFP expression of an inhibitory retrovirus containing one such GSE isolated by this screen that we refer to as A1. Amino acid deprivation and ER stress, manipulations that activate the eIF2α kinases GCN2 and PERK, respectively (Harding et al. 2000a, and see below), induce the CHOP::GFP reporter in the parental cells, but not in the A1 GSE–transduced cells. The block in reporter gene activity correlated with a marked impairment in the activation of the endogenous CHOP gene, reflected by the absence of CHOP protein in stressed cells (Fig. 1 B). These results indicate that the A1 GSE inhibits the essential component(s) of the pathway leading to CHOP activation by ER stress and amino acid deprivation.
The A1 GSE Interferes with eIF2α Phosphorylation and Blocks the Integrated Stress Response
ATF4 is a transcription factor that activates CHOP in the integrated stress response (Fawcett et al. 1999). ATF4 is translationally upregulated by the UPR (Harding et al. 2000a). We found that the A1 GSE profoundly inhibited ATF4 protein accumulation in tunicamycin-treated CHO cells (Fig. 2 A). This result suggests that the A1 GSE interfered with CHOP activation at some proximal step in the integrated stress response. Mutations in upstream components of the integrated stress response also interfere with activation of the BiP gene, encoding an important ER chaperone (Harding et al. 2000a). Northern blot analysis of tunicamycin-treated cells showed that the activation of BiP was also impaired in tunicamycin-treated, A1-transduced CHO cells (Fig. 2 B). This result indicates that the defect induced by the A1 GSE is at a step (or steps) common to the activation of both CHOP and BiP.
Figure 2.
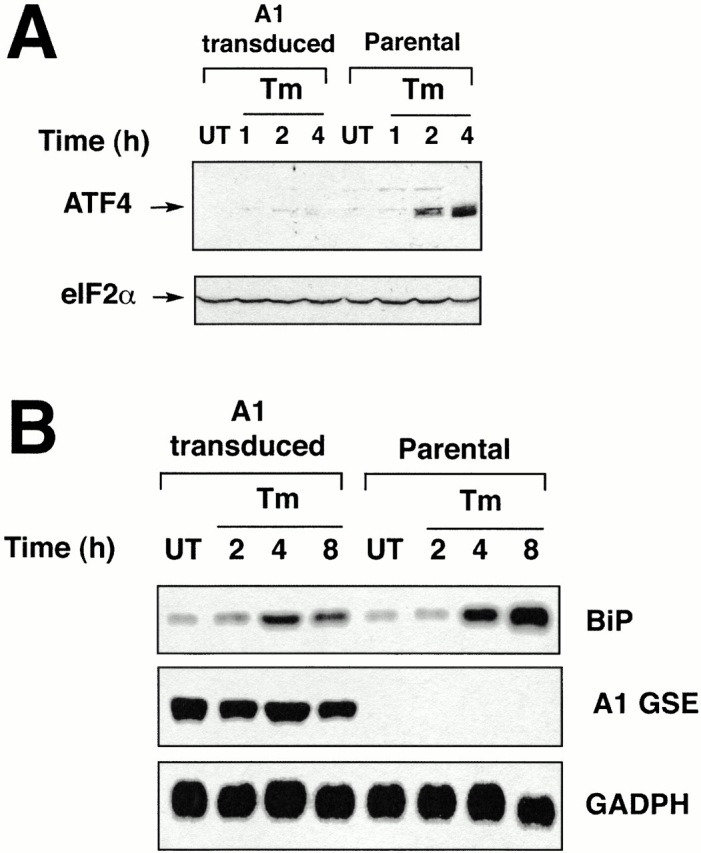
The A1 GSE interferes with a step common to the activation of BiP and ATF4 in the UPR. (A) Immunoblot of ATF4 in tunicamycin-treated parental and A1-transduced CHO cells (upper panel). Anti-eIF2α immunoblot of the same membrane serves as a control of protein loading (lower panel). (B) Northern blot of RNA from tunicamycin-treated parental and A1-transduced CHO cells. The blot was hybridized sequentially with BiP, the insert of the A1 retrovirus, and GAPDH radiolabeled probes.
Phosphorylation of eIF2α on serine 51 activates ATF4 translation, and is a key step in signaling cell stress. Therefore, we compared the phosphorylation of eIF2α in parental CHO cells with that of the A1-transduced cells. Immunoblotting of lysates from stressed cells with an antiserum that specifically detects eIF2α phosphorylated on serine 51 revealed a profound defect in eIF2α phosphorylation in response to both ER stress and amino acid starvation in A1-transduced cells (Fig. 3A and Fig. B). The protein kinases PERK and GCN2 are responsible for eIF2α phosphorylation by ER stress and amino acid deprivation, respectively (Harding et al. 2000a,Harding et al. 2000b). Activity of these kinases correlates with their phosphorylation status, and is easily detected as a shift in mobility on SDS-PAGE (in the case of PERK; Harding et al. 1999), or by an epitope-specific antiserum that detects the active, phosphorylated form (in the case of GCN2; Harding et al. 2000a). PERK activation in response to tunicamycin and thapsigargin (the latter activates the UPR by depleting ER calcium stores) was indistinguishable in parental and A1-transduced cells, and GCN2 activation by amino acid starvation was similarly unimpaired (Fig. 3A and Fig. B). These results indicate that the A1 GSE exerts its effect on a common step required for eIF2α phosphorylation, acting downstream of the eIF2α kinases PERK and GCN2.
Figure 3.

The A1 GSE interferes with eIF2α phosphorylation and downstream signaling but does not block stress-activation of the eIF2α kinases PERK and GCN2. (A) Immunoblot of PERK immunoprecipitated from parental and A1-transduced cells after treatment with tunicamycin (2 μg/ml, Tm) or thapsigargin (100 nM, Tg) (upper panel). The positions of the higher mobility, inactive form of the protein (PERK0) and the lower mobility, phosphorylated active form of the protein (P-PERK) are indicated. Immunoblot of the same lysate with antisera that detects eIF2α phosphorylated on serine 51 (P-eIF2α), or total eIF2α. (B) Immunoblot of GCN2 immunoprecipitated from parental and A1-transduced cells cultured in leucine-deficient media where indicated (−Leu). GCN2 is revealed by immunoblot with an antiserum that detects the phosphorylated, active form of the kinase (P-GCN2), or an antiserum that reacts with all forms of the kinase (GCN2) (upper panels). Immunoblot of the same lysates with antisera to eIF2α as in A (lower panels). (C) Autoradiography of a SDS-PAGE gel of whole cell lysates from parental and A1-transduced CHO cells that had been treated with DTT (2 mM) for the indicated period of time to rapidly induce ER stress, and that had been pulsed with [35S]methionine. The lower panel shows a Coomassie blue stain of the same gel to control for equal loading of the lanes. (D) FACS® analysis of GFP activity in A1-transduced or parental CHOP::GFP CHO cells mock-transfected or transfected with expression plasmids that encode the CD2 cell surface marker alone, CD2 and wild-type eIF2α (eIF2αWT), or CD2 and S51D mutant eIF2α (eIF2αS51D). Note the population of double positive (CD2+ and GFP+) cells present only in the pool transfected with the mutant eIF2α (rectangle).
ER stress and the attendant PERK-induced eIF2α phosphorylation inhibits protein synthesis (Harding et al. 1999). To determine the functional significance of the defect in eIF2α phosphorylation induced by the A1 GSE, we compared new protein synthesis rates in ER-stressed parental CHO cells with A1-transduced cells. Cells were treated with DTT, an agent that strongly activates PERK and inhibits protein synthesis in a strictly PERK-dependent manner (Harding et al. 2000b). Protein synthesis measured by pulsing cells with radiolabeled amino acids showed the expected reduction in ER-stressed parental cells, but the A1-transduced cells exhibited no decrease in incorporation of tracer into newly synthesized polypeptides (Fig. 3 C). This result indicates that the defect in eIF2α phosphorylation induced by the A1 GSE leads to loss of translational regulation.
Some of the effects of phosphorylation of eIF2α can be mimicked by an activating substitution (serine 51 to aspartic acid, S51D; Srivastava et al. 1998). To determine if the constitutively active S51D eIF2α could rescue the defect in CHOP::GFP expression in A1-transduced cells, we transfected cells with a plasmid expressing the mutant eIF2α. In addition to the effector protein, the plasmid expressed the cell surface marker CD2 that can be detected with a specific antibody identifying the transfected cells. The effect of eIF2α S51D on the activity of the integrated CHOP::GFP reporter was measured by dual channel FACS® analysis of transfected cells. Plasmids expressing CD2 alone or CD2 and wild-type eIF2α had no effect on CHOP::GFP activity, whereas the S51D mutant eIF2α reproducibly activated CHOP::GFP in a small subset of transfected cells. The relatively small number of transfected cells expressing CHOP::GFP may be due to the toxicity of the activated eIF2α protein. This was equally apparent in parental and A1 GSE–transduced cells (Fig. 3 D). Rescue by the activated eIF2α S51D suggests that the A1 GSE exerts its effect on CHOP expression at the level of eIF2α phosphorylation.
The A1 GSE Encodes the COOH Terminus of GADD34 and Its Function Is Dependent on Interaction with the Catalytic Subunit of PP1
The insert of retroviral clone A1 was sequenced and found to encode the COOH-terminal 299 amino acids (aa 292–590) of the hamster GADD34 protein (also known as MYD116) (Lord et al. 1990; Zhan et al. 1994). To determine if the full-length GADD34 is also capable of attenuating signaling in the integrated stress response, we constructed an expression plasmid encoding the mouse GADD34 (for which a full-length EST clone was available) and expressed it transiently in CHOP::GFP cells. Two deletion mutants of the mouse protein were constructed and the hamster-derived A1 GSE was also expressed from the same plasmid. To identify the transfected cells, we used the expression plasmid encoding the cell surface marker CD2 (described above), and measured the effect of GADD34 and its derivatives on the activity of the integrated CHOP::GFP reporter by dual channel FACS® analysis of stressed cells. The plasmid encoding CD2 alone had no effect on CHOP::GFP activity in tunicamycin-treated cells, whereas the A1 GSE and the comparable COOH-terminal fragment of the murine GADD34 (aa 241–657, ΔN) profoundly inhibited CHOP::GFP expression in this transient transfection assay (Fig. 4 A). The normal induction of CHOP::GFP in the CD2-negative, untransfected portion of the cells provided a built-in positive control for the induction of ER stress in each experiment.
Figure 4.
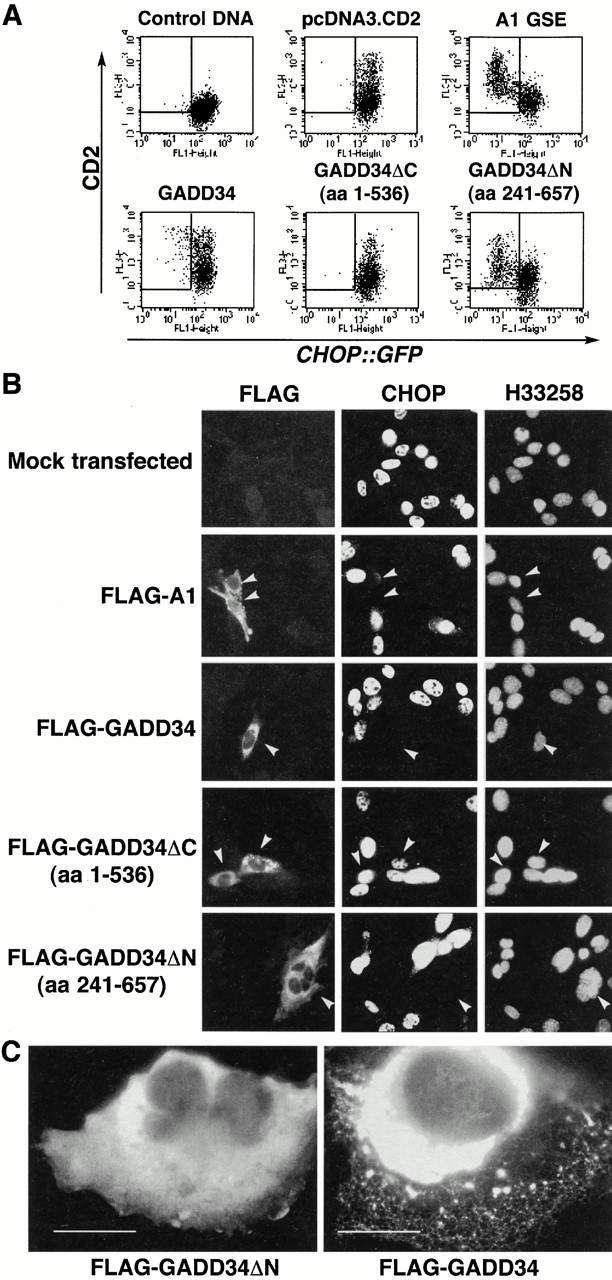
GADD34 inhibits CHOP activation in the UPR. (A) FACS® analysis of CHO cells expressing the CHOP::GFP reporter and transfected with CD2-expressing plasmids that coexpress the indicated derivatives of mouse GADD34, the A1 GSE, or no additional protein. The cells were treated with tunicamycin (2 μg/ml, 16 h) to activate CHOP::GFP. Note the presence of CD2-positive and GFP-negative cells (rectangle) in the pools transfected with A1, wild-type GADD34, and GADD34ΔN. (B) Immunostaining of mouse embryonic fibroblast cells transfected with expression vectors encoding FLAG-tagged A1 or GADD34, and the indicated deletion mutants of GADD34. The cells were treated with tunicamycin (2 μg/ml, 8 h) and immunostained with an antibody to FLAG that detects the recombinant proteins and an antiserum to CHOP that detects endogenous CHOP. The karyophilic dye H33258 stains the nuclei of all the cells in the field. The arrows mark the cells expressing the recombinant GADD34 proteins. (C) High magnification (60×) view of transfected cells expressing the full-length mouse GADD34 or the ΔN deletion. The recombinant proteins are revealed by immunostaining of the FLAG epitope tag.
Full-length murine GADD34 reproducibly inhibited CHOP::GFP, but the level of inhibition was significantly less than that induced by the A1 GSE and the comparable murine COOH-terminal fragment (Fig. 4 A). Deletion of the 121 COOH-terminal residues (GADD34ΔC, aa 1–536) abolished the inhibitory effect, attesting to the importance of COOH-terminal region of GADD34 in the inhibition of the integrated stress response (Fig. 4 A).
To determine if GADD34 and its derivatives were also able to inhibit endogenous CHOP activation by stress, we treated transfected mouse embryonic fibroblast cells with tunicamycin and stained them for endogenous CHOP and the recombinant GADD34 proteins (the latter were detected by an NH2-terminal FLAG epitope tag). Endogenous CHOP expression was significantly attenuated by the A1 GSE, the homologous murine fragment (aa 241–657), and by the full-length GADD34, but not by the COOH-terminal deletion mutant that also failed to attenuate the CHOP::GFP reporter (aa 1–536; Fig. 4 B). The fraction of CHOP-positive cells among the cells expressing the recombinant proteins was reduced from 69.7% positive cells among those transfected with the inactive GADD34ΔC fragment (aa 1–536) to 3.5% in GADD34 transfectants, 4.7% in the A1 GSE transfectants, and 4% in GADD34 fragment 241–657 transfectants. In this assay we could not detect differences between the activity of the A1 GSE–like COOH-terminal fragments of GADD34 and the full-length protein. This may be due to the nonquantitative nature of the immunocytochemical assay compared with the highly quantitative FACS® analysis.
In the course of these experiments we noticed that the staining pattern of the A1-like COOH-terminal fragment of mouse GADD34 was very different from that of the full-length GADD34: the former exhibited a diffuse cytosolic pattern, whereas the latter was distributed in a reticular pattern (Fig. 4 C). The possible implications of this finding are discussed below.
The COOH terminus of GADD34 is related in sequence to the COOH terminus of the herpes simplex virus (HSV)-encoded protein γ134.5 (Chou and Roizman 1994, Figure 6A therein). γ134.5 plays an important role in evading the consequences of PKR-mediated shutdown of host protein synthesis in virally infected cells, and the COOH-terminal fragment of GADD34 can substitute for this activity of γ134.5 (He et al. 1996). This activity of γ134.5 is dependent on its ability to associate with the catalytic subunit of PP1 (PP1c), and correlates with an increase in a cellular phosphatase activity that dephosphorylates eIF2α (He et al. 1997, He et al. 1998). To determine if expression of the A1 GSE and GADD34 affected eIF2α phosphorylation levels by promoting dephosphorylation, we compared the phosphatase activity directed against eIF2α in lysates of cells expressing the A1 GSE or GADD34 with that of parental cells. The eIF2α in reticulocyte lysate was phosphorylated in vitro on serine 51 using bacterially expressed GST-PERK (Harding et al. 1999), and incubated with aliquots of detergent lysates prepared from parental CHO cells and cells expressing the A1 GSE or the COOH-terminal fragment of mouse GADD34. eIF2α was efficiently dephosphorylated in lysates from cells expressing the GADD34 derivatives, whereas the dephosphorylation activity in lysates from parental cells was considerably lower (Fig. 5 A). Expression of the GADD34 fragments specifically promoted the dephosphorylation of eIF2α, as the dephosphorylation of phospho–GST-PERK and an unidentified phosphoprotein of 110 kD that are both present in the radiolabeled reticulocyte lysate progressed to similar degree in lysates of parental CHO cells and cells expressing GADD34 (Fig. 5 A).
Figure 5.
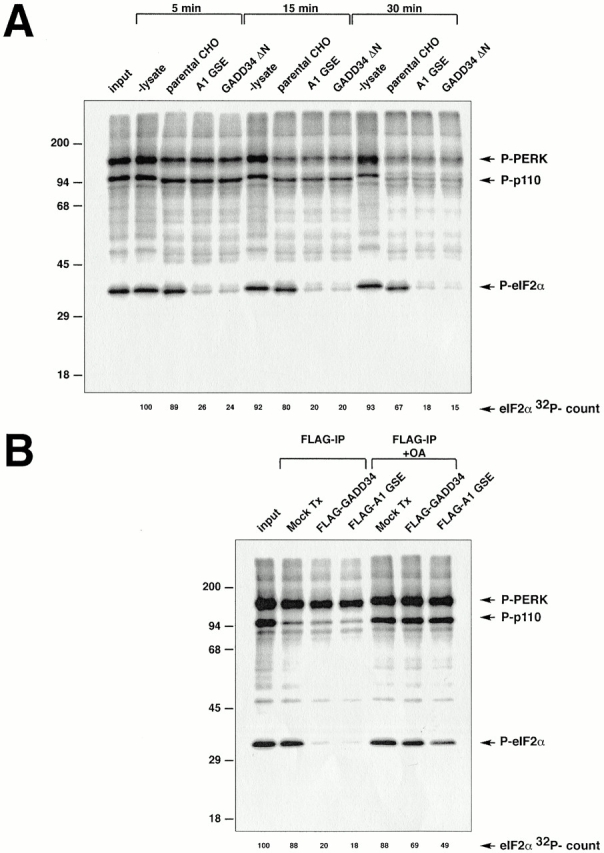
Cells expressing GADD34 have increased phosphatase activity directed towards eIF2α. (A) Autoradiogram revealing the in vitro dephosphorylation of radiolabeled eIF2α by lysates of cells expressing the A1 GSE or the mouse GADD34 COOH-terminal fragment (GADD34 ΔN). eIF2α in rabbit reticulocyte lysate was radiolabeled in vitro on serine 51 with GST-PERK, and incubated for the specified time with lysates of CHO cells expressing the indicated GADD34 derivatives. The position on the SDS-PAGE of radiolabeled eIF2α (P-eIF2α), GST-PERK (P-PERK), and an unidentified phosphoprotein of 110 kD present in the reticulocyte lysate (P-p110) are indicated. The latter two serve as controls for the specificity of the activity that dephosphorylates eIF2α. The radioactive signal associated with eIF2α is quantified in arbitrary units (eIF2α 32P-count). (B) Autoradiogram revealing the in vitro dephosphorylation of radiolabeled eIF2α by complexes immunoprecipitated from 293T cells transfected with epitope-tagged, full-length mouse GADD34 (FLAG-GADD34) or the A1 GSE (FLAG-A1 GSE), or mock-transfected cells (Mock Tx). The in vitro dephosphorylation assays were carried out in the absence or presence of phosphatase inhibitor (2 μM okadaic acid, +OA).
To determine if the eIF2α phosphatase activity induced by GADD34 is physically associated with the GADD34 proteins, we performed the same in vitro eIF2α dephosphorylation assay using purified immune complexes recovered from transfected cells. Immunopurified complexes containing FLAG epitope–tagged A1 GSE or full-length mouse GADD34 were both able to efficiently dephosphorylate eIF2α. The phosphatase activity of the immune complex was reduced by okadaic acid, an inhibitor of PP1 and PP2A (Fig. 5 B). Okadaic acid also inhibited eIF2α dephosphorylation by crude lysates of cells expressing GADD34 (data not shown). Proteins immunoprecipitated from mock-transfected cells had only background eIF2α phosphatase activity. Phosphatase activity directed towards the 110-kD phosphoprotein and phospho–GST-PERK was similar in all three immunecomplexes, providing a built-in control for the activity of the eIF2α phosphatase. These results implicate GADD34 in dephosphorylation of eIF2α through its physical association with a PP1- (or PP2A-) like activity.
To determine if the inhibitory effect of GADD34 on the integrated stress response correlated with its binding to PP1c, we constructed mutant derivatives of mouse GADD34 that would be predicted not to bind PP1c (Fig. 6 A); the predictions were based on the crystal structure of PP1c (Egloff et al. 1997). GADD34 mutants that failed to associate with the PP1c in an in vivo coimmunoprecipitation assay (Fig. 6 C; GADD34, aa 241–562, aa 241–536) also failed to inhibit CHOP::GFP (Fig. 6 B). Most striking in this regard is the single residue substitution, V549E, in the R/K-V/I-X-F PP1c binding motif common to many regulatory subunits of the phosphatase (Egloff et al. 1997). This mutation eliminated all interaction of GADD34 with PP1c, and abolished all inhibitory activity on CHOP::GFP (Fig. 6B and Fig. C). These experiments establish a strong correlation between PP1c binding by GADD34 and inactivation of the integrated stress response. Mutants of GADD34 that were incapable of inhibiting CHOP::GFP were also inactive in promoting eIF2α phosphatase activity in vitro (Fig. 6 D), supporting a role for eIF2α dephosphorylation in the inhibition of the integrated stress response.
Figure 6.
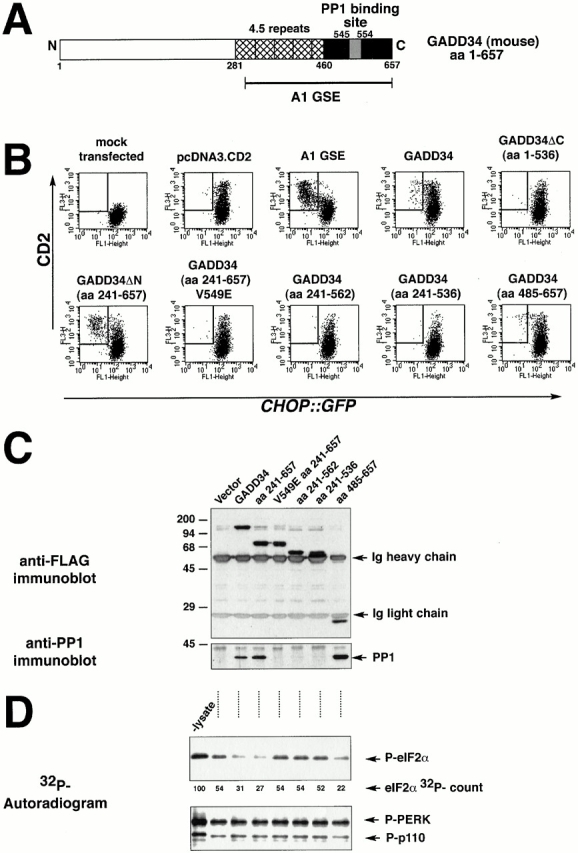
GADD34 inhibition of CHOP expression requires interaction with PP1. (A) Structure of mouse GADD34. The conserved repeat motifs are hatched, the COOH-terminal region with sequence similarity to HSV γ134.5 is colored black, and the peptide motif presumed to interact with PP1c is colored gray. (B) FACS® analysis of CHO cells expressing the CHOP::GFP reporter and transfected with CD2-expressing plasmids that coexpress the indicated derivatives of mouse GADD34, the A1 GSE, or no additional protein. The cells were treated with tunicamycin (2 μg/ml, 16 h) to activate CHOP::GFP. Note the presence of CD2-positive, GFP-negative cells (rectangle) in the pools transfected with A1, GADD34, GADD34ΔN, and GADD34 (aa 485–657), but not in the COOH-terminal deletions of GADD34 (aa 1–536, 241–562, 241–536) or in the V549E mutant form. (C) Anti-FLAG immunoprecipitation followed by immunoblot of proteins from lysates of cells transfected with FLAG-tagged GADD34 and the indicated derivatives as in B. The recombinant proteins in the immunoprecipitates were detected by immunoblotting with an antibody to the FLAG epitope (upper panel), and coimmunoprecipitating endogenous PP1c was detected by a specific polyclonal serum (bottom panel). The position of the precipitating immunoglobulin heavy and light chains and the PP1c band are indicated by arrows. (D) Autoradiogram of eIF2α that had been phosphorylated in vitro with 32P on serine 51 (P-eIF2α) after incubation for 15 min with lysates of cells transfected as in C (upper panel). The lane marked “-lysate” reports on the 32P–eIF2α signal from sample incubated under the same conditions in the absence of lysate. The radioactivity signal of the 32PeIF2α is quantified in arbitrary units below (eIF2α 32P-count). Autoradiogram of the signal from 32PGST-PERK and an unidentified radiolabeled phosphoprotein of 110 kD that were present in the preparation of 32PeIF2α and serve as control substrates for the phosphatase activity (lower panel).
Immediately NH2 terminal to the PP1c-binding site, GADD34 has a series of conserved repetitive elements (hamster, 3.5 repeats; humans, 4 repeats; mouse, 4.5 repeats; Fig. 6 A). A 172-residue fragment of GADD34 (aa 485–657) that lacks the repeats still bound to PP1c and also inhibited CHOP::GFP activity, albeit less strongly than the A1 fragment that contains some of the repeats (Fig. 6 B). This result indicates that the repeats may play a role in inactivating the integrated stress response, but are not absolutely required.
Expression of GADD34 Is Mediated by the Integrated Stress Response
GADD34 is a stress-inducible gene whose expression profile parallels that of CHOP (Fornace et al. 1989; Zhan et al. 1994). We sought to determine if GADD34 itself was regulated by the integrated stress response. Northern blot analysis showed that GADD34 induction in the UPR was dependent on the activity of the ER stress–inducible eIF2α kinase PERK, whereas GADD34 induction by amino acid starvation was dependent on GCN2 (Fig. 7). These results indicate that the integrated stress response plays an important role in GADD34 activation. There are also PERK- and GCN2-independent pathways to activate GADD34, as the induction of the gene by the alkylating agent methyl methanesulfonate (MMS) was preserved in PERK−/− and partially preserved in GCN2−/− cells. We do not know if this stress-causing agent induced GADD34 by promoting eIF2α phosphorylation through an alternative kinase or by other activating signaling pathways that are independent of eIF2α phosphorylation.
Figure 7.
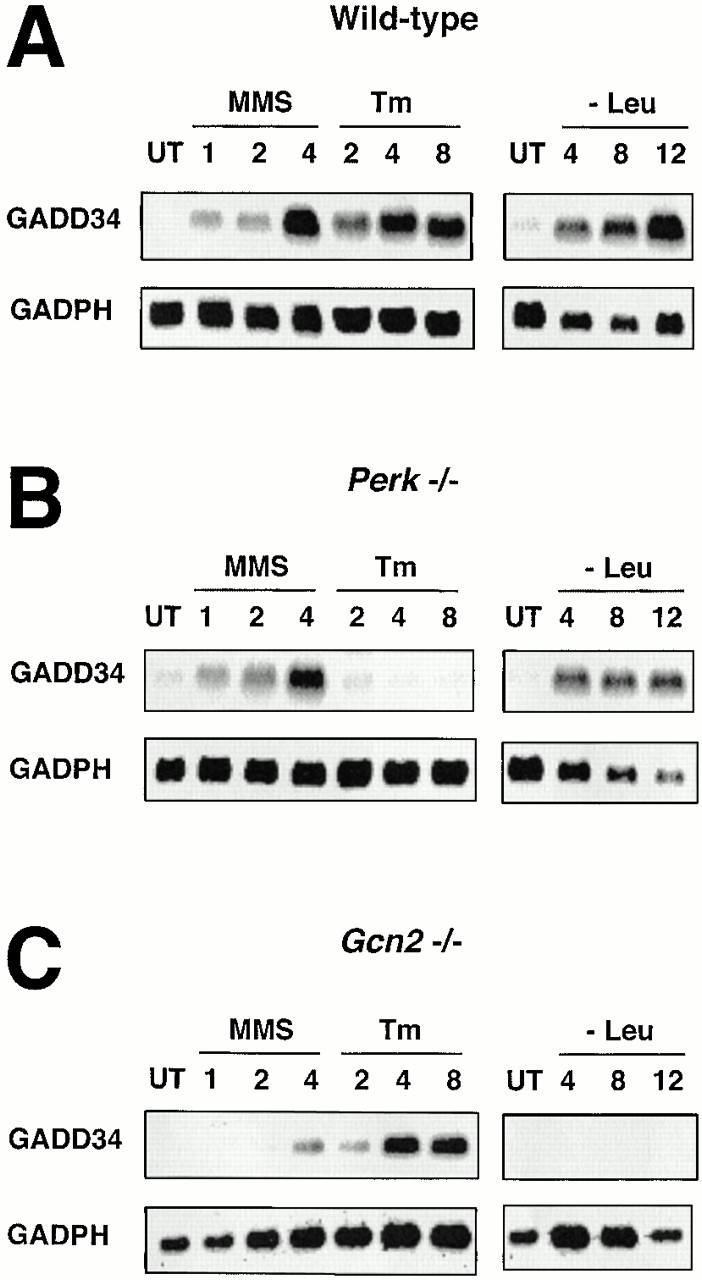
GADD34 expression depends on stress-induced eIF2α kinases. Northern blot analysis of GADD34 and GAPDH mRNA in (A) wild-type cells, (B) PERK−/− cells, and (C) GCN2−/− cells. Cells were exposed to tunicamycin (Tm, 2 μg/ml), the alkylating agent MMS (100 μg/ml), or cultured in the absence of leucine (−Leu) for the indicated period of time.
Discussion
Signaling in the general control response (the yeast counterpart of the mammalian integrated stress response) is dependent on phosphorylation of eIF2α, as substitution of the residue corresponding to serine 51 in yeast eIF2α prevents activation of genes by amino acid starvation (Williams et al. 1989; Dever et al. 1992). We have found previously that the integrated stress response requires the activity of the eIF2α kinases PERK and GCN2, and is mediated by translational regulation of ATF4 (Harding et al. 2000a). Here, we extend these observations to show that a GSE that promotes the dephosphorylation of eIF2α is able to block the integrated stress response. Activation of the stress-induced eIF2α kinases is unaffected by this GSE, indicating that the latter does not interfere with the development of the proximal stress signal. Our findings further support the notion that the activation of gene expression by the integrated stress response is mediated by eIF2α phosphorylation in mammalian cells.
The GSE we identified is a product of the GADD34 gene. Its ability to interfere with signaling in the integrated stress response correlates with its ability to bind the catalytic subunit of PP1 and to dephosphorylate eIF2α. Binding to PP1c is shared by the structurally related HSV protein γ134.5, and lysates of cells infected with γ134.5-expressing virus also have markedly increased activity of an eIF2α phosphatase (He et al. 1997, He et al. 1998). Together, these findings suggest that GADD34 exerts its activity on the integrated stress response by promoting the dephosphorylation of eIF2α. It seems likely that HSV has coopted this cellular mechanism to prevent host protein synthesis shutdown when the eIF2α kinase PKR is activated by viral infection, as suggested originally by Roizman and colleagues (He et al. 1998). Although γ134.5's essential role in viral infectivity is well established, it is not known if inhibition of host gene activation is part of this essential role. In this regard, it is interesting to note that a dominant negative mutation in the yeast PP1c homologue, GLC7, is able to derepress a transcriptional program that is dependent on signaling by the eIF2α kinase, Gcn2p (Wek et al. 1992).
Suppression of the integrated stress response by the COOH-terminal fragment of GADD34 is significantly greater than that by the full-length protein. This difference does not appear to be due to the higher level of expression of the smaller fragment, or to significantly reduced ability to associate with the catalytic subunit of the phosphatase (Fig. 6 C). Furthermore, the full-length GADD34 was able to impart high levels of phosphatase activity to lysates of transfected cells or to immune complexes obtained from transfected cells (Fig. 5 B and Fig. 6 D). This suggests that its ability to promote the dephosphorylation of eIF2α was unmasked by cell lysis and detergent extraction. We noted that the COOH-terminal active fragment of GADD34 is distributed throughout the cytoplasm, whereas the full-length protein is localized to a reticular structure costained with antiserum to ER markers (Fig. 4 C and data not shown). It is tempting to speculate that the reduced activity of the full-length protein may be due to its sequestration at an inactive site. If correct, this suggests a means to regulate GADD34 activity posttranslationally as well as at the level of the gene's expression.
What might be the physiological significance of GADD34's potential activity as an inhibitor of the integrated stress response? It had previously been noted that eIF2α phosphorylation and protein synthesis inhibition are transient in stressed cells (Prostko et al. 1992, Prostko et al. 1993). GADD34 is activated under physiologically stressful conditions (Fornace et al. 1989; Doutheil et al. 1999), and its profile of induction is very similar to other targets of the integrated stress response such as CHOP and BiP. Indeed, we find that GADD34 induction by ER stress is dependent on the activity of the eIF2α kinases PERK and GCN2 (Fig. 7B and Fig. C). Therefore, it seems likely that transcriptional induction of GADD34 is part of a negative feedback loop that attenuates signaling in the integrated stress response. Other stress-responsive signaling pathways have elaborated components that serve similar negative feedback functions. For example, the mitogen-activated protein kinase pathway is inhibited by dual-specificity phosphatases that are transcriptionally induced by mitogen-activated protein kinase signaling (English et al. 1999; Camps et al. 2000), and cytokine signaling is inhibited by products of suppressor of cytokine signaling (SOCS) genes that are themselves positively regulated at the transcriptional level by binding of cytokines to their receptors (Krebs and Hilton 2000). The possible application of this principle to the integrated stress response is depicted in cartoon form in Fig. 8.
Figure 8.

Model depicting GADD34's role in inhibiting signaling through the integrated stress response pathway. GADD34 is induced transcriptionally by the eIF2α kinases PERK and GCN2. It associates with PP1c to reduce levels of eIF2α phosphorylation and feed-back negatively on the pathway. ATF4 is a transcription factor that is activated translationally by eIF2α phosphorylation and plays a role in the transcriptional activation of CHOP in the integrated stress response (Harding et al. 2000a). We do not know if it plays a similar role in GADD34 activation and for this reason it is displayed by a shadowed font.
There are several theoretical reasons why the phosphorylation of eIF2α may have evolved as a transient signal in stressed cells. Sustained eIF2α phosphorylation is lethal to cells in culture (Srivastava et al. 1998), and in ischemic neurons, sustained eIF2α phosphorylation correlates with death in vivo (DeGracia et al. 1997; Paschen and Doutheil 1999). Early in the UPR, high levels of eIF2α phosphorylation inhibit translation of most proteins, including some like BiP that are induced later in the course of the response (Harding et al. 2000a, Figure 5C therein). We speculate that in addition to inhibiting further signaling, the activation of GADD34 and the attendant delayed dephosphorylation of eIF2α provide a means for cells to translate mRNAs like BiP, whose induction occurred earlier, during the active phase of the integrated stress response.
Acknowledgments
We thank J. Hirst (New York University) for expert assistance in fluorescent activated cell sorting, D. Littman (New York University) for plasmids used in retroviral production and for use of the FACScan™, and I. Mohr, D. Levy, and E.Y. Skolnik for useful discussions.
This study was supported by grants ES08681 and DK47119 from the National Institutes of Health. I. Novoa was supported by a Basque Government Fellowship. D. Ron is a Scholar of the Leukemia and Lymphoma Society of America.
Footnotes
Abbreviations used in this paper: ATF, activating transcription factor; BiP, immunoglobulin binding protein of B cells; CHOP, C/EBP homologous protein; eIF2α, eukaryotic translation initiation factor 2 α; GADD, growth arrest and DNA damage gene; GAPDH, glutaraldehyde 3-phosphate dehydrogenase; GCN, general control nonrepressed; GFP, green fluorescent protein; GSE, genetic suppressor element; GST, glutathione S-transferase; HSV, herpes simplex virus; MMS, methyl methanesulfonate; PERK, PKR-like ER kinase; PKR, double-stranded RNA–activated-kinase; PP1, protein phosphatase 1; UPR, unfolded protein response.
References
- Berlanga J.J., Santoyo J., De Haro C. Characterization of a mammalian homologue of the GCN2 eukaryotic initiation factor 2alpha kinase. Eur. J. Biochem. 1999;265:754–762. doi: 10.1046/j.1432-1327.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- Bertolotti A., Zhang Y., Hendershot L., Harding H., Ron D. Dynamic interaction of BiP and the ER stress transducers in the unfolded protein response. Nat. Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Brostrom C.O., Brostrom M.A. Regulation of translational initiation during cellular responses to stress. Prog. Nucleic Acid Res. Mol. Biol. 1998;58:79–125. doi: 10.1016/s0079-6603(08)60034-3. [DOI] [PubMed] [Google Scholar]
- Camps M., Nichols A., Arkinstall S. Dual specificity phosphatasesa gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- Chou J., Roizman B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA. 1994;91:5247–5251. doi: 10.1073/pnas.91.12.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens M. Protein kinases that phosphorylate eIF2 and eIF2B, their role in eukaryotic cell translational control. In: Hershey J., Mathews M., Sonenberg N., editors. Translational Control. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1996. pp. 139–172. [Google Scholar]
- Clemens M.J., Elia A. The double-stranded RNA-dependent protein kinase PKRstructure and function. J. Interferon Cytokine Res. 1997;17:503–524. doi: 10.1089/jir.1997.17.503. [DOI] [PubMed] [Google Scholar]
- DeGracia D.J., Sullivan J.M., Neumar R.W., Alousi S.S., Hikade K.R., Pittman J.E., White B.C., Rafols J.A., Krause G.S. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J. Cereb. Blood Flow Metab. 1997;17:1291–1302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Dever T.E., Feng L., Wek R.C., Cigan A.M., Donahue T.F., Hinnebusch A.G. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- Doutheil J., Althausen S., Gissel C., Paschen W. Activation of MYD116 (gadd34) expression following transient forebrain ischemia of ratimplications for a role of disturbances of endoplasmic reticulum calcium homeostasis. Brain Res. Mol. Brain Res. 1999;63:225–232. doi: 10.1016/s0169-328x(98)00276-9. [DOI] [PubMed] [Google Scholar]
- Egloff M.P., Johnson D.F., Moorhead G., Cohen P.T., Cohen P., Barford D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997;16:1876–1887. doi: 10.1093/emboj/16.8.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J., Pearson G., Wilsbacher J., Swantek J., Karandikar M., Xu S., Cobb M.H. New insights into the control of MAP kinase pathways. Exp. Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- Fawcett T.W., Martindale J.L., Guyton K.Z., Hai T., Holbrook N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Fornace A.J., Neibert D.W., Hollander M.C., Luethy J.D., Papathanasiou M., Fragoli J., Holbrook N.J. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov A.V., Roninson I.B. Isolation of genetic suppressor elements (GSEs) from random fragment cDNA libraries in retroviral vectors. Methods Mol. Biol. 1997;69:221–240. doi: 10.1385/0-89603-383-x:221. [DOI] [PubMed] [Google Scholar]
- Harding H., Zhang Y., Ron D. Translation and protein folding are coupled by an endoplasmic reticulum resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding H., Novoa I., Zhang Y., Zeng H., Schapira M., Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells Mol. Cell. 6 2000. 1099 1108a [DOI] [PubMed] [Google Scholar]
- Harding H., Zhang Y., Bertolotti A., Zeng H., Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response Mol. Cell 5 2000. 897 904b [DOI] [PubMed] [Google Scholar]
- He B., Chou J., Liebermann D.A., Hoffman B., Roizman B. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J. Virol. 1996;70:84–90. doi: 10.1128/jvi.70.1.84-90.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Gross M., Roizman B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 1998;273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- Krebs D.L., Hilton D.J. SOCSphysiological suppressors of cytokine signaling. J. Cell Sci. 2000;113:2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- Landau N., Littman D. Packaging system for rapid production of murine leukemia virus vectors with variable tropism. J. Virol. 1992;66:5110–5113. doi: 10.1128/jvi.66.8.5110-5113.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord K.A., Hoffman-Liebermann B., Liebermann D.A. Sequence of MyD116 cDNAa novel myeloid differentiation primary response gene induced by IL6. Nucleic Acids Res. 1990;18:2823. doi: 10.1093/nar/18.9.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern J.P., Land H. Advanced mammalian gene transferhigh titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- Paschen W., Doutheil J. Disturbances of the functioning of endoplasmic reticuluma key mechanism underlying neuronal cell injury? J. Cereb. Blood Flow Metab. 1999;19:1–18. doi: 10.1097/00004647-199901000-00001. [DOI] [PubMed] [Google Scholar]
- Prostko C.R., Brostrom M.A., Malara E.M., Brostrom C.O. Phosphorylation of eukaryotic initiation factor (eIF) 2 alpha and inhibition of eIF-2B in GH3 pituitary cells by perturbants of early protein processing that induce GRP78. J. Biol. Chem. 1992;267:16751–16754. [PubMed] [Google Scholar]
- Prostko C.R., Brostrom M.A., Brostrom C.O. Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol. Cell. Biochem. 1993;127–128:255–265. doi: 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- Samuel C.E., Kuhen K.L., George C.X., Ortega L.G., Rende-Fournier R., Tanaka H. The PKR protein kinase—an interferon-inducible regulator of cell growth and differentiation. Int. J. Hematol. 1997;65:227–237. doi: 10.1016/s0925-5710(96)00544-0. [DOI] [PubMed] [Google Scholar]
- Scorsone K.A., Panniers R., Rowlands A.G., Henshaw E.C. Phosphorylation of eukaryotic initiation factor 2 during physiological stresses which affect protein synthesis. J. Biol. Chem. 1987;262:14538–14543. [PubMed] [Google Scholar]
- Sood R., Porter A.C., Ma K., Quilliam L.A., Wek R.C. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress Biochem. J. 346Pt. 22000. 281 293a [PMC free article] [PubMed] [Google Scholar]
- Sood R., Porter A.C., Olsen D., Cavener D.R., Wek R.C. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha Genetics. 154 2000. 787 801b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S.P., Kumar K.U., Kaufman R.J. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J. Biol. Chem. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- Vallejo M., Ron D., Miller C.P., Habener J.F. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc. Natl. Acad. Sci. USA. 1993;90:4679–4683. doi: 10.1073/pnas.90.10.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Z., Lawson B., Brewer J., Zinszner H., Sanjay A., Mi L., Boorstein R., Kreibich G., Hendershot L., Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP homologous protein (CHOP/GADD153) Mol. Cell. Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Z., Kuroda M., Sok J., Batchvarova N., Kimmel R., Chung P., Zinszner H., Ron D. Identification of novel stress-induced genes downstream of chop EMBO J. 17 1998. 3619 3630a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-Z., Harding H.P., Zhang Y., Jolicoeur E.M., Kuroda M., Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses EMBO J. 17 1998. 5708 5717b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek R.C., Cannon J.F., Dever T.E., Hinnebusch A.G. Truncated protein phosphatase GLC7 restores translational activation of GCN4 expression in yeast mutants defective for the eIF-2 alpha kinase GCN2. Mol. Cell. Biol. 1992;12:5700–5710. doi: 10.1128/mcb.12.12.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N.P., Hinnebusch A.G., Donahue T.F. Mutations in the structural genes for eukaryotic initiation factors 2 alpha and 2 beta of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. USA. 1989;86:7515–7519. doi: 10.1073/pnas.86.19.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q., Lord K.A., Alamo I., Jr., Hollander M.C., Carrier F., Ron D., Kohn K.W., Hoffman B., Liebermann D.A., Fornace A.J., Jr. The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth. Mol. Cell. Biol. 1994;14:2361–2371. doi: 10.1128/mcb.14.4.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]