

Abstract
Death receptors can trigger cell demise dependent or independent of caspases. In WEHI-S fibrosarcoma cells, tumor necrosis factor (TNF) induced an increase in cytosolic cathepsin B activity followed by death with apoptotic features. Surprisingly, this process was enhanced by low, but effectively inhibiting, concentrations of pan-caspase inhibitors. Contrary to caspase inhibitors, a panel of pharmacological cathepsin B inhibitors, the endogenous cathepsin inhibitor cystatin A as well as antisense-mediated depletion of cathepsin B rescued WEHI-S cells from apoptosis triggered by TNF or TNF-related apoptosis-inducing ligand. Thus, cathepsin B can take over the role of the dominant execution protease in death receptor-induced apoptosis. The conservation of this alternative execution pathway was further examined in other tumor cell lines. Here, cathepsin B acted as an essential downstream mediator of TNF-triggered and caspase-initiated apoptosis cascade, whereas apoptosis of primary cells was only minimally dependent on cathepsin B. These data imply that cathepsin B, which is commonly overexpressed in human primary tumors, may have two opposing roles in malignancy, reducing it by its proapoptotic features and enhancing it by its known facilitation of invasion.
Keywords: apoptosis, cancer, caspase independent, cathepsins, tumor necrosis factor
Introduction
Tumor necrosis factor (TNF) is a multifunctional cytokine capable of inducing several biological responses like apoptosis, inflammation, and stress response (Ashkenazi and Dixit 1999; Wallach et al. 1999). The numerous biological effects of TNF are signaled via two distinct cell surface receptors, TNF receptor (TNF-R) 1 and TNF-R2, the former being the major signaling receptor in most cells. The intracellular part of the TNF-R1 contains a sequence called a death domain, which is highly conserved among various death-inducing receptors of TNF-R superfamily and is required for the induction of apoptosis by them. Binding of TNF to TNF-R1 leads to the trimerization of the death domains and their subsequent binding to cytosolic death domain–containing proteins, TNF-R1-associated death domain protein, and Fas-associated death domain protein. The latter can then recruit a cysteine protease, caspase-8, to the receptor complex, where it is activated possibly by proteolytic cleavage mediated by itself. Such autoactivation of caspase-8 is believed to initiate an amplifying caspase cascade leading to the activation of so-called effector caspases, caspase-3 and caspase-7, that cleave a limited set of cellular proteins resulting in apoptotic morphology and death (Thornberry and Lazebnik 1998).
Several studies with caspase inhibitors and mice deficient for various caspases have supported the involvement of caspases in various forms of mammalian apoptosis, including that induced by TNF and related death-inducing ligands, TNF-related apoptosis-inducing ligand (TRAIL) and CD95 ligand (Thornberry and Lazebnik 1998; Ashkenazi and Dixit 1999; Wallach et al. 1999). However, noncaspase proteases such as cathepsins D (Deiss et al. 1996) and B (Guicciardi et al. 2000), calpains (Vanags et al. 1996), and various serine proteases (Ruggiero et al. 1987; Wright et al. 1994) have been reported as essential downstream effectors of caspases, for instance, in TNF-mediated apoptosis. It has only lately become evident that apoptosis can also occur in the complete absence of caspase activation (Lavoie et al. 1998; Mathiasen et al. 1999; Nylandsted et al. 2000). In parallel, it was found that death receptors, which were originally believed to induce cell demise only via the direct activation of the caspase cascade, may simultaneously activate mechanistically different death pathways, leading either to necrosis or apoptosis. The first demonstration of the existence of such overlapping death pathways took advantage of the requirement for ATP for the apoptotic process. The predepletion of Jurkat or HeLa cells of ATP prevented caspase activation, and the stimulation of CD95 in such ATP-depleted cells resulted in necrosis (Eguchi et al. 1997; Leist et al. 1997). A similar switch from TNF-induced apoptosis to necrosis was then observed in L929 and U937 cells in the presence of a potent peptide caspase inhibitor z-Val-Ala-DL-Asp-CH2F (zVAD-fmk) (Vercammen et al. 1998; Khwaja and Tatton 1999). Interestingly, in NIH3T3 cells, the inhibition of caspase activation did not change the mode of cell death, but enhanced apoptosis-like process induced by TNF, TRAIL, or agonistic antibody against CD95 (anti-CD95; Luschen et al. 2000). The mediators of this caspase-independent/receptor-induced death have remained obscure.
This study was initiated by our surprising observation that low but effective concentrations of pan-caspase inhibitors sensitized WEHI-S fibrosarcoma cells to TNF-induced cytotoxicity. Characterization of this death process clearly demonstrated that TNF-treated WEHI-S cells died in an identical apoptosis-like manner both in the presence and absence of caspase inhibitors. These findings prompted us to search for alternative noncaspase mediators of apoptosis and resulted in the identification of cathepsin B as an essential mediator of TNF-induced tumor cell apoptosis.
Materials and Methods
Cells and Treatments
WEHI-S, ME-180as (ME-ashsp2), and MCF-7S1 cells were propagated as described (Jäättelä et al. 1998). Murine embryonic fibroblasts (MEFs) from wild-type, cathepsin B–deficient (Deussing et al. 1998), or cathepsin S–deficient (Shi et al. 1999) mice were used for experiments at passages 3–5. Primary murine hepatocytes were prepared as described (Leist et al. 1994).
The recombinant human TNF (rhTNF) was provided by A. Cerami (Kenneth Warren Laboratories, Tarrytown, NY), and Flag-TRAIL, by J. Tschopp (University of Lausanne, Epalinges, Switzerland). Recombinant mTNF was from R&D Systems, and anti-CD95 (Jo-2) from was from BD PharMingen. Protease inhibitors used included z-Phe-Lys-2,4,6-trimethylbenzoyloxymethylketone (zFK-mbmk), zVAD-fmk from Bachem, z-Val-Asp(OMe)-Val-Ala-Asp(OMe)-CH2F (zVDVAD-fmk), N-Acetyl-leu-Leu-Nle-CHO (ALLN), PD 150606, clasto-lactacystin-β-lactone (lactacystine) from Calbiochem-Novabiochem, CA-074-Me, Boc-Asp-CH2F (Boc-D-fmk) from Peptides International, z-Phe-Ala-CH2F (zFA-fmk) from Enzyme System Products, pepstatin A, N-a-Tosyl-l-Lys-chloromethyl ketone (TLCK), tosyl-l-Phe-chloromethyl ketone (TPCK) from Roche Biochemicals, acetyl-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO), and acetyl-Ile-Glu-Thr-Asp-aldehyde (IETD-CHO) from Biomol.
Survival Assays and Detection of Apoptotic Markers
The 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction and lactate dehydrogenase (LDH) release assays were used to analyze the survival of the cells as described (Jäättelä et al. 1998; Latta et al. 2000). The clonogenic survival of ME-180as cells was determined by replating cells treated as indicated at serial dilutions on cloning plates (Greiner) and counting viable colonies 7 d later.
Apoptotic nuclear changes were identified by staining of cells with a mixture of 0.5 μg/ml H-33342 and 1 μM SYTOX (Molecular Probes) as described (Leist et al. 1997).
The externalization of phosphatidylserine (PS) was visualized by staining with FITC-annexin V (Molecular Probes) of propidium iodide-negative cells. Confocal microscopy, equipped with a TCS-4D system including UV laser (DMB-IRB; Leica), was performed as described (Leist et al. 1997). The electron microscopy (Philips 201) was performed as described (Nylandsted et al. 2000), except that cells were fixed in situ for 10 min in 50% Karnovsky solution before the scraping.
Time-Lapse Microscopy
Cells were grown in four-well dishes (Greiner) to 40% confluence. For experiments, serum-free L15 medium (CO2-independent pH buffering) was used, and the culture dishes were equipped with a needle thermosensor and mounted on an electronically controlled heating block fixed on a microscope stage (35 ± 0.3°C). Images were recorded by a 40× Varell contrast microscopy (Axiovert-25; ZEISS) at 10-s intervals for 750 min at a resolution of 768 × 576 pixels. The images in Fig. 2 C correspond to 120 × 120 pixels of the original video frame, and the processed video sequences correspond to the pictures shown in the figure. The number of video frames was reduced to a total of 600 for all sequences during the transformation into Quicktime format, and replay speed is 15 frames/s, with every second corresponding to 6 min in real time.
Figure 2.
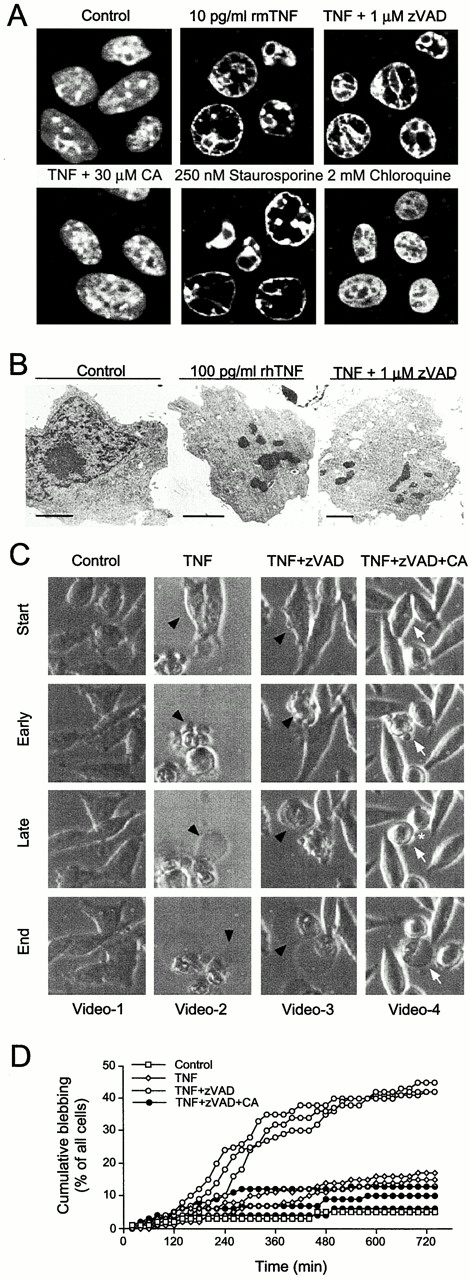
TNF-treated WEHI-S cells display apoptotic morphology in the absence of caspase activity. (A) Chromatin of WEHI-S cells was imaged by confocal microscopy. Cells treated with rmTNF or TNF plus zVAD-fmk are shown at the initiation of chromatin condensation. Cotreatment with 30 μM CA-074-Me inhibited chromatin condensation. For comparison, chromatin of untreated control cells is shown. Chloroquine- and staurosporine-exposed cells are shown as control for necrosis and apoptosis, respectively. (B) WEHI-S cells were treated for 6 h as indicated, fixed in situ, and prepared for transmission electron microscopy. Condensed and fragmented chromatin lumps, nuclear fragmentation, and loss of cell surface microvilli were induced by TNF in the presence or absence of zVAD-fmk. Bars, 2 μm. (C) WEHI-S cells were exposed to 5 pg/ml rmTNF or indicated combinations of 0.2 pg/ml rmTNF, 1 μM zVAD-fmk, and CA-074-Me (30 μM) and recorded by time-lapse microscopy (also see supplementary videos available at http://www.jcb.org/cgi/content/full/153/5/999/DC1). Note the continuous membrane movements in control cells. Two different types of blebbing in TNF–treated cells are marked as early (highly dynamic, multifocal, apoptotic blebbing) and late (formation of a single large bleb on one side of the cell) events. In necrotic death, only the latter occurs (data not shown). Blebbing characteristics were not significantly modified by zVAD-fmk, and quantitative analysis showed an average blebbing time of 21 ± 2 min (n = 43) for TNF and 18 ± 2 min (n = 37) for TNF plus zVAD-fmk. The very few cells that died in the presence of CA-074-Me showed a distinct blebbing behavior with average blebbing time of 50 ± 9 min (white arrows). During that time cells recovered from blebbing and looked normal (*) for 13 ± 5 min (n = 20), before the final lethal bleb formed. (D) WEHI-S cells were exposed to indicated combinations of rmTNF (0.2 pg/ml), zVAD-fmk (1 μM), and CA-074-Me (30 μM) and recorded by time-lapse microscopy. For quantitative analysis of bleb formation, every cell (40–50) in the field was included, and the time between the addition of TNF and the start of the blebbing was measured. Data for each experiment is presented as cumulative frequency of cells that have initiated blebbing at a given time point.
Analysis of Protease Inhibitors
For the determination of apparent IC 50 values for irreversible inhibitors and IC 50 values for reversible inhibitors, inhibitors were incubated at serial dilutions with the cell lysate (4 × 105 cells/ml) or 10 pg/ml of caspase-2, caspase-3, or caspase-8 (R&D Systems) in lysis buffer (25 mM Hepes, 5 mM MgCl2, 1 mM EGTA, 0.5% Triton-X-100, 5 mM DTT, 1 mM pefablock, pH 7.5) or 1.5 U/ml rabbit skeletal muscle calpain (Sigma) in calpain assay buffer (50 mM Tris, pH 7.5, 8 mM DTT) for 20 min at 25°C before the measurement. The enzyme activities were estimated by adding one volume of 20 μM zVDVAD-7-amino-trifluoromethylcoumarin (AFC) (Calbiochem-Novabiochem) for caspase-2, Ac-DEVD-AFC (Biomol) for caspase-3, zRR-7-amino-4-methylcoumarin (AMC) (Calbiochem-Novabiochem) or zFR-AFC (Enzyme System Products) for cathepsins B and L, and 100 μM succinyl-LLVY-AMC (Bachem) plus 2 mM Ca2+ for calpain measurements in appropriate reaction buffers; i.e., caspase reaction buffer (100 mM Hepes, 20% glycerol, 0.5 mM EDTA, 0.1% CHAPS, 5 mM DTT, 1 mM pefablock, pH 7.5), cathepsin reaction buffer (50 mM sodium acetate, 4 mM EDTA, 8 mM DTT, 1 mM pefablock, pH 6.0), cathepsin L reaction buffer (4 M urea, 20 mM sodium acetate, 4 mM EDTA, 8 mM DTT, 1 mM pefablock, pH 5.0), and calpain assay buffer. The V max of the liberation of AFC (excitation 400 nM, emission 489 nM) or AMC (380 nM, 442 nM) was measured over 20 min at 30°C with a Spectramax Gemini fluorometer (Molecular Devices). The specific cathepsin B activity was calculated from the difference of the V max values in the absence and presence of 0.1 μM zFK-mbmk. IC 50 values were calculated by a four-parameter fit procedure using Graph Pad-Prism software.
Measurement of Total Cellular Cysteine Protease Activities
Cells were treated as indicated and washed twice with PBS before the lysis and analysis of intracellular protease activities was performed as described above. The caspase-2 activity was measured in the presence of 50 nM DEVD-CHO.
Measurement of Cytosolic Cathepsin Activities
To measure cysteine cathepsin activity in the cytosol, experiments were performed under serum-free conditions. After the removal of the medium, extraction buffer (25 μg/ml digitonin, 250 mM sucrose, 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM pefablock, pH 7.5) was added, and cells were incubated for 5 min (ME-180as) or 15 min (WEHI-S) on ice. The digitonin concentration and treatment times were optimized to result in the total release of cytosolic LDH activity without disruption of lysosomes. Cathepsin and LDH activities in the resulting supernatant were determined as described above, and the cytosolic cathepsin activity is expressed as the ratio of the cathepsin activity and the LDH activity within one sample.
Transfections
Transient transfections were performed using lipofectamine reagents (Life Technologies). The plasmids pBK-CystA (Jones et al. 1998) and pEGFP-hCathB (Roberts et al. 1997) encoding for rat cystatin A and GFP–cathepsin B, respectively, were provided by G. Gores (Mayo Clinic, Rochester, MN). The antisense cathepsin B plasmids were created by cloning PCR products complementary to bases 116–482 (pcDNA-as-mCathB-1) or 642–843 (pcDNA-as-mCathB-2) of the murine cathespsin B (EMBL/GenBank/DDBJ under accession no. NM_007798) and bases 175–551 (pcDNA-as-hCathB-1) or 561–911 (pcDNA-as-hCathB-2) of the human sequence (EMBL/GenBank/DDBJ under accession no. M14221) into the cloning site of the pcDNA3-neo (Invitrogen). Similar constructs containing antisense cathepsin D were used as controls. The transfected cells were visualized by co-transfection (1:10) of pEGFP-N1 (CLONTECH Laboratories, Inc.).
Analysis of Nuclear Factor κB Activation
Transient transfections were performed by electroporation using 5 μg of p3K-INF-LUC containing three copies of a consensus nuclear factor κB (NF-κB) binding sequence in front of human minimal interferon promoter and Photinius pyralis luciferase cDNA and 3 μg of pEBS-β-Gal per 106 cells (Jäättelä et al. 1995). 2 d after the transfection, cells were treated as indicated, harvested, and analyzed for luciferase and β-galactosidase activities as described previously (Jäättelä et al. 1996).
Analysis of Receptor Binding, Internalization, and Degradation
Subconfluent cells growing on six-well plates (Nunc) were treated with protease inhibitors as indicated for 1 h before a 90-min incubation with 1 nM 125I-labeled TNF (>30 μCi/μg; NEN Life Science Products) on ice, surface bound, and internalized. Degraded 125I-labeled TNF were analyzed as described previously (Tsujimoto et al. 1985).
Immunocytochemistry, Lysosomal Staining, and Immunoblotting
Cells were fixed in methanol, treated with 1% H2O2, and stained with cathepsin B antibody (1:200; Oncogene Research Products), biotinylated anti–mouse-IgG (Dako), peroxidase-conjugated streptavidin–biotin complex, and diaminobenzidine/H2O2 as substrate according to the manufacturer's instructions (StreptABC, Vector Laboratories). Images were recorded with an Olympus digital camera mounted on an Olympus BX60 microscope.
The lysosomal–cytoplasmic pH gradient was visualized by a Leica TCS-4D confocal microscope equipped with a 40× long-distance lens in cells incubated for 1 min with 10 nM LysoTracker red (Molecular Probes). The specificity was demonstrated by complete inhibition of staining after pre-treatment with 40 mM NH4Cl. The cytosol was counterstained by a 2-min preincubation with calcein-AM (0.5 μM; Molecular Probes).
Immunodetection of proteins separated by SDS-PAGE and transferred to nitrocellulose was performed with enhanced chemiluminesence Western blotting reagents (Amersham Pharmacia Biotech). Rabbit polyclonal antibodies to caspase-3 (BD PharMingen), murine caspase-7 (P. Vandenabeele, University of Gent, Gent, Belgium), Bid (S. Korsmeyer, Harvard Medical School, Boston, MA), or cPLA2 (Wissing et al. 1997) and mouse monoclonal anti-Hsc70 (B. Margulis, Russian Academy of Sciences, St. Petersburg, Russia) were used as primary antibodies. Peroxidase-conjugated secondary antibodies were from Dako.
Online Supplementary Material
Four video sequences of dying WEHI-S cells demonstrate that TNF induces morphologically indistinguishable apoptosis in the presence and absence of 1 μM zVAD-fmk, and that CA-074-Me inhibits all apoptotic changes observed. Videos are available at http://www.jcb.org/cgi/content/full/153/5/999/DC1.
Results
Caspase Inhibitors Sensitize WEHI-S Cells to Death Receptor–induced Apoptosis
TNF induces both caspase activation and apoptosis in WEHI-S murine fibrosarcoma cells (Jäättelä et al. 1996; Faraco et al. 1999). To further characterize this death pathway, we evaluated the necessity of caspase activity for apoptosis induction in this model system. First, we compared the concentration dependence of zVAD-fmk for the inhibition of effector caspase (DEVDase) activity and apoptosis. Although 0.8 μM zVAD-fmk completely inhibited TNF-induced DEVDase activity in WEHI-S cells, concentrations of over 30 μM were required to confer protection against TNF-induced apoptosis (Fig. 1, A–C). As the initiator caspase-8 is even more sensitive to inhibition by zVAD-fmk than effector caspases (Table ) (Garcia-Calvo et al. 1998), the classical death receptor–mediated apoptosis pathway is likely to be completely blocked by 1 μM zVAD-fmk. Since caspase-2 is less susceptible to the inhibition by zVAD-fmk than other caspase family members (Garcia-Calvo et al. 1998), the requirement of high zVAD-fmk concentrations for the protection of WEHI-S cells against TNF could be due to the involvement of this isozyme in the death signaling. However, neither had the potent caspase-2 inhibitor zVDVAD-fmk any effect on TNF-induced death of WEHI-S cells (Fig. 1 D), nor was any significant caspase-2–like activity detectable in WEHI-S cells treated with TNF (not shown).
Figure 1.
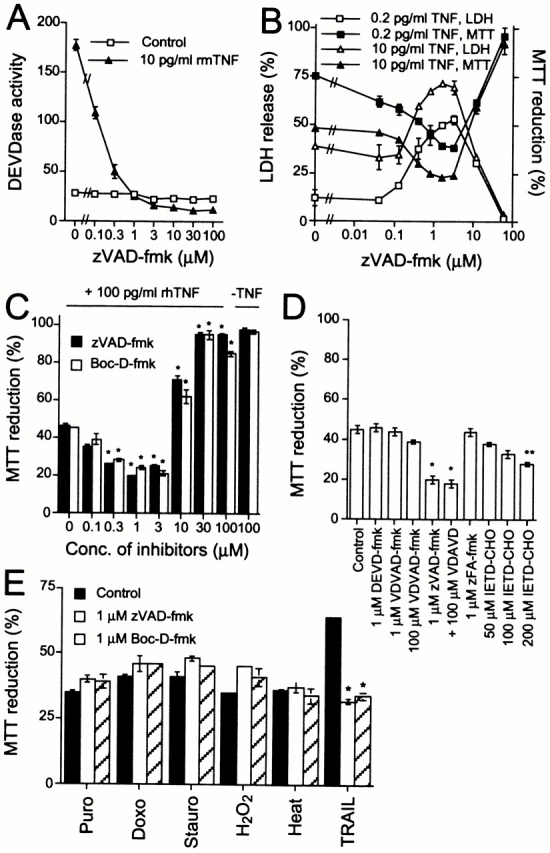
Inhibition of caspase activation sensitizes WEHI-S cells to death receptor–mediated apoptosis. (A) The DEVDase activity in lysates of cells treated as indicated for 4 h was quantitated by measurement of the cleavage of DEVD-AFC. The enzyme activity is expressed as arbitrary units. (B) Cells were incubated with the indicated concentrations of zVAD-fmk for 1 h before a 10-h treatment with rmTNF. The viability of the cells was measured by LDH-release and MTT assays. (C) Cells were treated for 1 h with indicated concentrations of pan-caspase inhibitors before an 11-h treatment with or without rhTNF. The survival of the cells was measured by the MTT assay. (D) WEHI-S cells were pretreated with indicated protease inhibitors for 1 h before the addition of 100 pg/ml of rhTNF. The survival of the cells was analyzed 12 h later by the MTT assay. (E) Cells were treated with caspase inhibitors for 1 h before a 22-h treatment with 4 μg/ml of puromycin (Puro) or doxorubicin (Doxo), 100 ng/ml of staurosporine (Stauro), or 50 μM H2O2, a 16-h treatment with 75 ng/ml of Flag-TRAIL plus 2 μg/ml of anti-Flag antibody (TRAIL), or a 45-min treatment at 43.5°C followed by 18 h at 37°C (Heat). The survival of the cells was measured by the MTT assay. *p-value < 0.001, **p-value < 0.01; as compared with the control. All values represent means of triplicate determinations ± SD.
Table 1.
Specificity Profile of Protease Inhibitors Used to Define Cathepsin B–mediated Apoptosis
| Apparent IC50 | Supposed specificity | Rec. casp3 | Rec. casp2 | Rec. casp8 | DEVDase casp3/7 | zFRase (cathB) | zFRase (cathL) | Muscle calpain |
|---|---|---|---|---|---|---|---|---|
| μM | ||||||||
| zVAD-fmk | pan-casp | 0.15 | 0.3 | <0.1 | <0.1 | 8 | >100 | >100 |
| Boc-D-fmk | pan-casp | 0.5 | 100 | 1 | 3 | 20 | >100 | >100 |
| DEVD-CHO | casp3 | <0.1 | 2.3 | <0.1 | <0.1 | 50 | ND | >100 |
| IETD-CHO | casp8 | 0.1 | 8 | <0.1 | 0.1 | 90 | ND | 95 |
| CA-074-Me | cathB | >100 | >100 | >100 | >100 | 0.5 | 55 | >100 |
| zFA-fmk | cathB/L | >100 | 60 | >100 | 100 | <0.1 | <0.1 | ND |
| zFK-mbmk | cathB | 80 | 80 | 90 | 50 | <0.1 | 10 | 3 |
| pepstatin | cathD | >100 | >100 | >100 | >100 | >100 | ND | >100 |
| TPCK | ser. prot. | >100 | >100 | >100 | >100 | 50 | ND | >100 |
| ALLN | calpain | >100 | >100 | >100 | >100 | <0.1 | <0.1 | 1.2 |
casp, caspase; cath, cathepsin; Rec., recombinant; ser. prot., serine protease.
Low zVAD-fmk concentrations (0.2–3 μM) correlating with the progressive inhibition of the DEVDase activity not only failed to rescue WEHI-S cells from apoptosis, but sensitized them up to 50-fold to recombinant murine TNF (rmTNF) (Fig. 1 B). Caspase inhibition appeared to have sensitized WEHI-S cells to TNF-R1–mediated apoptosis, because 1 μM zVAD-fmk enhanced also apoptosis induced by rhTNF, which binds selectively to the murine TNF-R1 (Tartaglia et al. 1991). Also, another pan-caspase inhibitor (Boc-D-fmk) sensitized WEHI-S cells to rhTNF at low concentrations (0.3–3 μM) and conferred full protection only at concentrations >30 μM (Fig. 1 C). The sensitizing effect was, however, specific with respect to the stimulus and the sensitizer: 1 μM zVAD-fmk or Boc-D-fmk had no effect on death induced by puromycin, doxorubicin, staurosporine, H2O2, or heat, whereas apoptosis induced by TRAIL was greatly enhanced (Fig. 1 E). Moreover, micromolar concentrations of DEVD-fmk, zFA-fmk, or VDVAD-fmk did not interfere with TNF signaling by a potentially unspecific effect of the fmk group, whereas a structurally different inhibitor of caspase-8 (IETD-CHO) sensitized WEHI-S cells to TNF (Fig. 1 D).
As judged by chromatin condensation, the mode of TNF-triggered death was clearly apoptotic, independently of whether caspases were inhibited or not (Fig. 2 A). These observations were confirmed by electron microscopy (Fig. 2 B), and also the analysis of time-lapse video microscopy sequences showed that low zVAD-fmk concentrations accelerated the onset of death but did not change its morphological features (Fig. 2C and Fig. D; supplementary videos available at http://www.jcb.org/cgi/content/full/153/5/999/DC1). Thus, caspase activation may only be an epiphenomenom of apoptosis in this model. Accordingly, TNF concentrations used in this study failed to induce caspase-8 activity (IETDase) as well as processing of caspase-7, caspase-8, and several known caspase substrates in WEHI-S cells (Fig. 3; data not shown). A significant percentage of pro-caspases or caspase substrate proteins was cleaved only when over 100-fold higher concentration of TNF was used (Fig. 3). Together with the fact that the inhibition of DEVDase activity accelerated the kinetics and extent of TNF-induced WEHI-S cell apoptosis, it is likely that the death pathway triggered by low TNF concentrations is caspase-independent also in the absence of zVAD-fmk.
Figure 3.

Absence of significant caspase-mediated intracellular proteolysis in WEHI-S cells exposed to low but lethal concentration of TNF. Equal amounts of protein from WEHI-S cells treated for indicated times with 0.1 or 50 ng/ml of rhTNF were analyzed for cleavage of caspase-7 or caspase substrates cPLA2 and Bid by immunoblot analysis. The arrowhead indicates a caspase cleavage product of cPLA2 (Wissing et al. 1997). Hsc70 serves as a loading control.
Since full sensitization by zVAD-fmk was observed even when it was added 2 h after TNF, receptor availability and the potential for activation of receptor-driven signaling in general appeared to have been unaffected (Fig. 4 A). In line with this, pretreatment of cells with 1 μM zVAD-fmk had no effect on the TNF-induced activation of NF-κB (Fig. 4 B) or on the binding, internalization, or degradation of TNF (Fig. 5 C).
Figure 4.
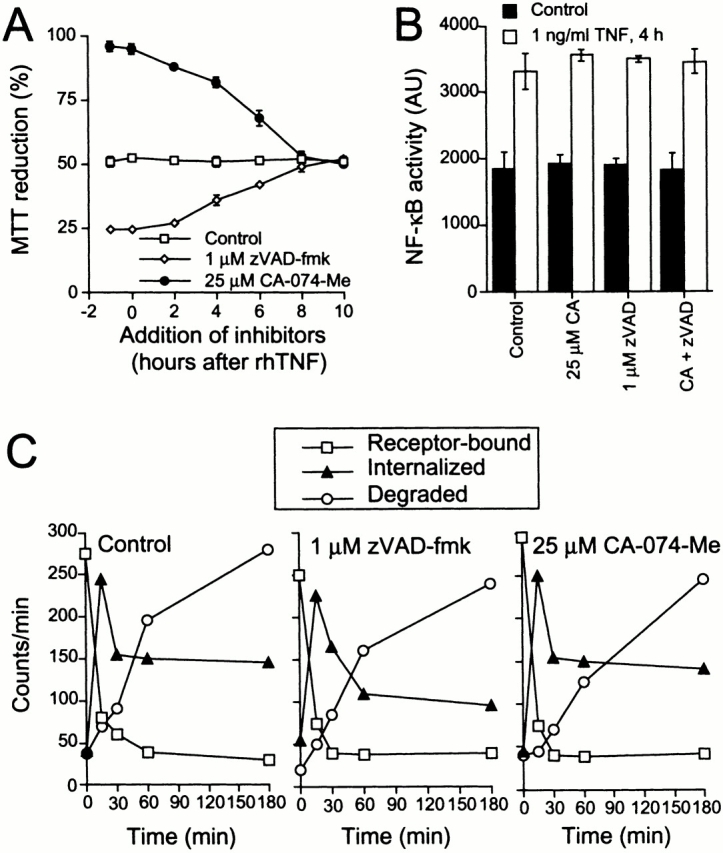
Inhibition of caspase or cathepsin B activities does not affect TNF-induced NF-κB activation or binding, internalization, or degradation of TNF. (A) WEHI-S cells were treated with 100 pg/ml rhTNF for 11 h and protease inhibitors as indicated. The survival of the cells was measured by the MTT assay. (B) WEHI-S cells transfected with p3K-INF-LUC and pEBS7-β-Gal were left untreated (control) or treated with 1 ng/ml of rhTNF for 4 h. 1 μM zVAD-fmk, 25 μM CA-074-Me, or their combination was added to the cells 1 h before the addition of TNF. NF-κB activity is expressed as arbitrary units of luciferase activity relative to β-galactosidase activity. (A and B) The values represent means of triplicate determinations ± SD, and the experiments were repeated twice with similar results. (C) Subconfluent WEHI-S cells were treated with indicated protease inhibitors for 1 h before a 90-min incubation with 1 nM 125I-labeled TNF on ice. After careful washing, prewarmed complete medium containing the indicated protease inhibitors was added, and the cells were placed at 37°C (0 min). Receptor-bound, internalized, and degraded TNF at indicated time points are shown. The values represent means of triplicate determinations. The experiment was repeated once with similar results.
Figure 5.

Inhibition of cathepsin B activity or expression protects WEHI-S cells from death receptor–induced apoptosis. (A) WEHI-S cells were pretreated with indicated protease inhibitors for 1 h before the addition of 100 pg/ml rhTNF. The survival of the cells was analyzed 12 h later by the MTT assay. (B) WEHI-S cells were treated for 11 h with indicated concentrations of CA-074-Me and rhTNF in the presence or absence of 1 μM zVAD-fmk or 10 μM cycloheximide (CHX). The cytotoxicity was measured by the LDH release assay. (C) The survival of WEHI-S cells pretreated with CA-074-Me for 1 h before the treatment with cytotoxic agents, as indicated in the legend for Fig. 1 E (except for TRAIL which was used at 150 ng/ml) was measured by the MTT assay. (D) WEHI-S cells were transfected with indicated plasmids plus pEGFP-N1 and treated 22 h later with rmTNF alone or with 1 μM zVAD-fmk or 20 μM CA-074-Me. The survival of green cells was determined 12 h later. The values represent means of triplicate determinations (A–C) or means of 10 (D) randomly chosen fields of ≥100 cells ± SD. *p-value < 0.01, as compared with control cells (A and C) or to similarly treated vector-transfected cells (D).
Cathepsin B Is a Mediator of Death Receptor–triggered Apoptosis of WEHI-S Cells
Next, we examined whether the protective activities of high concentrations of pan-caspase inhibitors could be due to the inhibition of proteases other than caspases. For instance, zVAD-fmk has been shown to inhibit lysosomal cysteine proteases (Schotte et al. 1999), which have recently been reported to contribute to hepatocyte apoptosis (Roberts et al. 1997; Guicciardi et al. 2000). Thus, we studied the effect of a panel of protease inhibitors on TNF-induced cell death. Compounds reported to inhibit lysosomal cysteine proteases (ALLN and zFA-fmk) as well as those more specific for cathepsin B (CA-074-Me and zFK-mbmk) conferred significant protection against TNF-induced apoptosis in WEHI-S cells, whereas inhibitors of calpains (PD 150606), the proteasome (lactacystin), serine proteases (TLCK and TPCK) or aspartic proteases including cathepsin D (pepstatin A), had no significant effect when applied at serial dilutions up to maximal tolerated concentrations (Fig. 5 A). Thorough in vitro analysis of the efficacy profiles of the protease inhibitors revealed that the protective effect agreed best with the inhibition of the cathepsin B–like activity (Table ). Therefore, we chose CA-074-Me for the further studies. According to all viability parameters, WEHI-S cells treated with 30 μM CA-074-Me were protected against TNF, even in the presence of sensitizing concentrations of zVAD-fmk or the protein synthesis inhibitor cycloheximide (Fig. 2 and Fig. 5 B). CA-074-Me also blocked TRAIL-induced apoptosis of WEHI-S cells but did not affect the cytotoxicity induced by puromycin, doxorubicin, staurosporine, H2O2, or heat (Fig. 5 C). To further challenge the specific requirement of cathepsin B in apoptosis induced by death receptors, we followed two strategies independent of chemical inhibitors. First, forced ectopic expression of cystatin A, an endogenous inhibitor of cysteine cathepsins, attenuated intracellular cathepsin activity by 59 ± 1.5% and TNF-induced apoptosis of WEHI-S cells almost completely (Fig. 5 D). Second, expression of two independent and nonoverlapping antisense cDNAs complementary to the murine cathepsin B sequence in WEHI-S inhibited cathepsin activity in transfected cells by 53 ± 1.9% or 44 ± 3.8%, correlating well with their ability to protect cells from TNF (Fig. 5 D). The expression of a similar antisense construct directed against murine cathepsin D had no effect on WEHI-S cell sensitivity to TNF (not shown).
Cathepsin B Is Required for All TNF-induced Apoptosis-related Events in WEHI-S Cells
In individual WEHI-S cells appearing perfectly normal, the blebbing was very abruptly initiated after variable times (2–8 h) of TNF treatment (Fig. 2C and Fig. D; supplementary videos available at http://www.jcb.org/cgi/content/full/153/5/999/DC1). The time from the initiation of the blebbing to the lysis of the cell was, however, remarkably constant (∼20 min). The release of cytochrome c into the cytosol, PS exposure, and chromatin condensation were only detectable in blebbing cells (not shown). All these events were completely absent in WEHI-S cells rescued by CA-074-Me, suggesting that the entire death process is cathepsin B dependent. Processing of TNF as well as TNF-induced NF-κB activation were, however, unaffected by pretreatment with CA-074-Me, indicating that TNF-R1 remained functional (Fig. 4B and Fig. C). In line with this, CA-074-Me conferred significant protection against TNF even when added 2–4 h after TNF (Fig. 4 A).
Cathepsin B Is an Essential Downstream Mediator of Caspase-initiated Tumor Cell Apoptosis
To test whether the requirement of cathepsin B–like enzymes was a particularity of WEHI-S cells, we tested the effect of cathepsin B inhibition on ME-180as cervix carcinoma cells. First, we demonstrated that these cells display classical apoptotic morphology and die in a caspase-dependent manner upon TNF treatment (Fig. 6A and Fig. B). Interestingly, CA-074-Me as well as the expression of two independent antisense cDNAs complementary to the human cathepsin B sequence potently inhibited also this caspase-dependent cell death (Fig. 6B and Fig. C). The functionality of the two antisense constructs was demonstrated by their ability to inhibit the expression of GFP–cathepsin B fusion protein by 81 ± 4.4% and 68 ± 2.8%, respectively. The expression of GFP–actin was unaffected by antisense constructs in a similar assay. Importantly, the effect of cathepsin B inhibition was not merely a delay of the death process, as the protected cells retained the ability of clonal growth after the TNF challenge (Fig. 6 D). Also MCF-7 breast carcinoma cells dying in a caspase-dependent manner were rescued from TNF-induced apoptosis by inhibitors of cysteine but not aspartic cathepsins (Fig. 6 E). Inhibition of TNF- or anti-CD95–induced apoptosis of primary murine hepatocytes (Fig. 7 A) by CA-074-Me was relatively minor compared with the effects observed in tumor cells, even when very high (>100 μM) inhibitor concentrations were used. zFA-fmk (50–150 μM) failed to confer any protection (data not shown). When primary fibroblasts (early passages of MEFs) were exposed to TNF, the protective effect due to pretreatment with cathepsin inhibitors or genetic deletion of cathepsin B was also clearly less pronounced than that observed in tumor cells (Fig. 7 B).
Figure 6.

Cathepsin B is a key mediator in TNF-induced caspase-dependent apoptosis of tumor cells. (A) ME-180as cells were treated as indicated for 24 h and analyzed by transmission electron microscopy. Nuclear and chromatin condensation as well as formation of apoptotic bodies was induced by TNF treatment. Bar, 2 μm. (B) The survival of ME-180as cells pretreated with 200 μM DEVD-CHO or IETD-CHO, 1 μM zVAD-fmk, or 5 μM CA-074-Me for 1 h before a 48-h treatment with 20 ng/ml of rhTNF was analyzed by the MTT assay. (C) ME-180as cells were transfected with indicated plasmids plus pEGFP-N1 and treated 48 h later as indicated. The survival of the green cells was analyzed after 48-h treatment. (D) After 3 d of treatment with indicated combinations of 100 ng/ml rhTNF, 5 μM zVAD-fmk, and 5 μM CA-074-Me in serum-free medium, ME-180as cells were replated on cloning plates in medium containing 6% FCS, and the clonogenic potential was determined 7 d later by counting viable colonies. (E) The survival of MCF-7 breast cancer cells pretreated with 1 μM zVAD-fmk, 100 μM zFA-fmk, or 12.5 μM ALLN for 1 h before a 32-h treatment with 10 ng/ml of rhTNF was analyzed by the MTT assay. The values represent means of triplicate determinations (B and E) or means of 10 randomly chosen fields of ≥100 cells (C and D) ± SD. *p-value < 0.01, as compared with control cells (B, D, and E) or similarly treated vector-transfected cells (C).
Figure 7.

Cathepsin B plays a minor role in death receptor–induced apoptosis of primary cells. (A) The survival of hepatocytes pretreated with the indicated concentrations of CA-074-Me for 1 h before the addition of 200 nM actinomycin D plus 28 ng/ml of rmTNF or 100 ng/ml of anti-CD95 antibody (Jo-2) was analyzed after 22-h treatment by the LDH release assay. (B) MEF (passage 4) originating from wild-type, cathepsin B–, or cathepsin S–deficient mice were left untreated or treated with 50 μM CA-074-Me or 100 μM zFA-fmk for 1 h before an 18-h treatment with 100 ng/ml of rhTNF plus 10 μM cycloheximide. The survival was analyzed by the MTT assay. The values represent means of triplicate determinations ± SD. *p-value < 0.01, as compared with control cells.
TNF Induces a Translocation of Cathepsin B from Lysosomes to Cytosol
Next, we studied whether and how TNF-induced apoptosis involves a modulation of the constitutive activity of cathepsin B. Neither a significant increase in the protein levels of pro-cathepsin B or mature cathepsin B nor in the total cysteine cathepsin activity was observed. However, TNF treatment resulted in cellular redistribution of the protein. Cathepsin B disappeared from perinuclear granules (colocalizing with lysosomal markers) and distributed diffusely throughout the cell (Fig. 8 A). To analyze the cytosolic cathepsin B activities in intact cells, we developed a rapid digitonin extraction method not requiring mechanical disruption of cells. Time course analysis revealed that the appearance of cathepsin B activity in the cytosol was a late event that could not be temporally separated from the LDH release in asynchronously dying cell populations (Fig. 8 B). However, on the single-cell level, lysosomal cathepsin release clearly preceded plasma membrane lysis since cytosolic cathepsin activity was measured in cells with intact plasma membranes. Moreover, our finding of cathepsin B in the cytosol of cells protected by CA-074-Me (Fig. 8 A) suggests that the release of the protease from lysosomes occurs independently of the subsequent lysis of the plasma membrane. Interestingly, lysosomes of blebbing TNF-treated cells with a clear apoptotic morphology retained the ability to accumulate acidic organelle-selective probes, indicating that the cytosolic–lysosomal pH gradient was not dissipated, and lysosomes were, at least in part, intact (Fig. 8 C).
Figure 8.
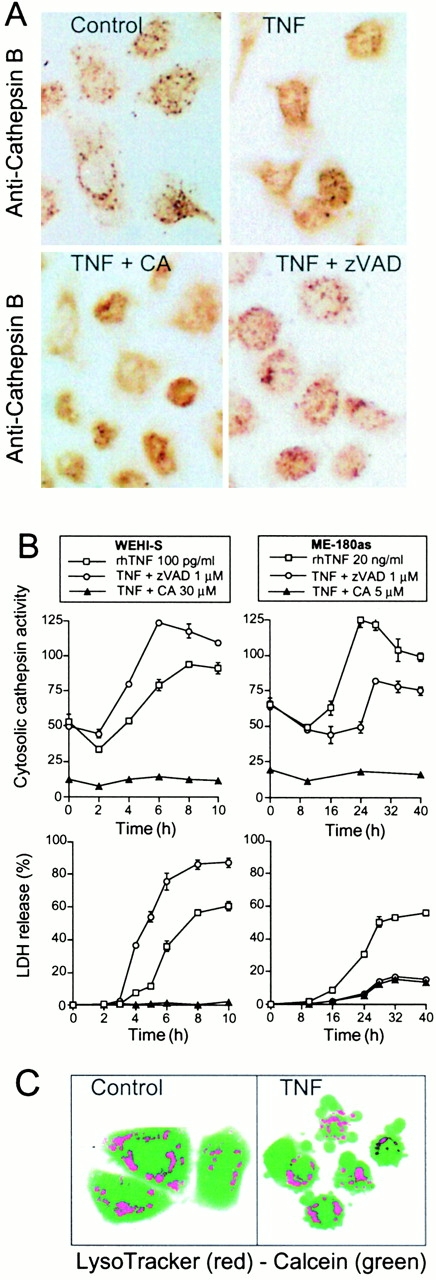
TNF induces the appearance of active cysteine cathepsins in the cytosol. (A) ME-180as cells were left untreated (control) or treated with 20 ng/ml rhTNF, TNF plus 5 μM CA-074-Me, or TNF plus 1 μM zVAD-fmk and stained with anti–cathepsin B after 16-h treatment. (B) WEHI-S or ME-180as cells were treated as indicated and analyzed for cytosolic cysteine cathepsin activity within intact cells and for released LDH activity. The values represent means of a triplicate determination ± SD. (C) ME-180as cells were left untreated (control) or treated with 20 ng/ml rhTNF for 30 h and stained with LysoTracker and calcein-AM.
Cathepsin B Acts Downstream of Caspases in ME-180as Cells
The inhibition of ME-180as cell apoptosis by 1 μM zVAD-fmk was accompanied by the inhibition of the appearance of cathepsin B protein and activity in the cytosol (Fig. 8A and Fig. B). Conversely, the inhibition of cathepsin B activity had no effect on TNF-induced caspase activation as analyzed by either DEVDase enzyme assay or immunoblotting using antibodies against caspase-3, -7, and -8 or caspase substrates (cPLA2 and PARP) (Fig. 9A and Fig. B; data not shown). The initial rounding, NF-κB activation, and cathepsin B translocation were also unaffected by protective concentrations of CA-074-Me, whereas PS exposure, formation of apoptotic bodies, and chromatin condensation were completely inhibited even in the rounded cells (Fig. 8 and Fig. 9 C; data not shown). Thus, in TNF-treated ME-180as cells, caspase activation appears to be a reversible event leading to the cathepsin B translocation, whereas cathepsin B activity appears indispensable for the final phases of the execution as well as the display of phagocytosis markers.
Figure 9.
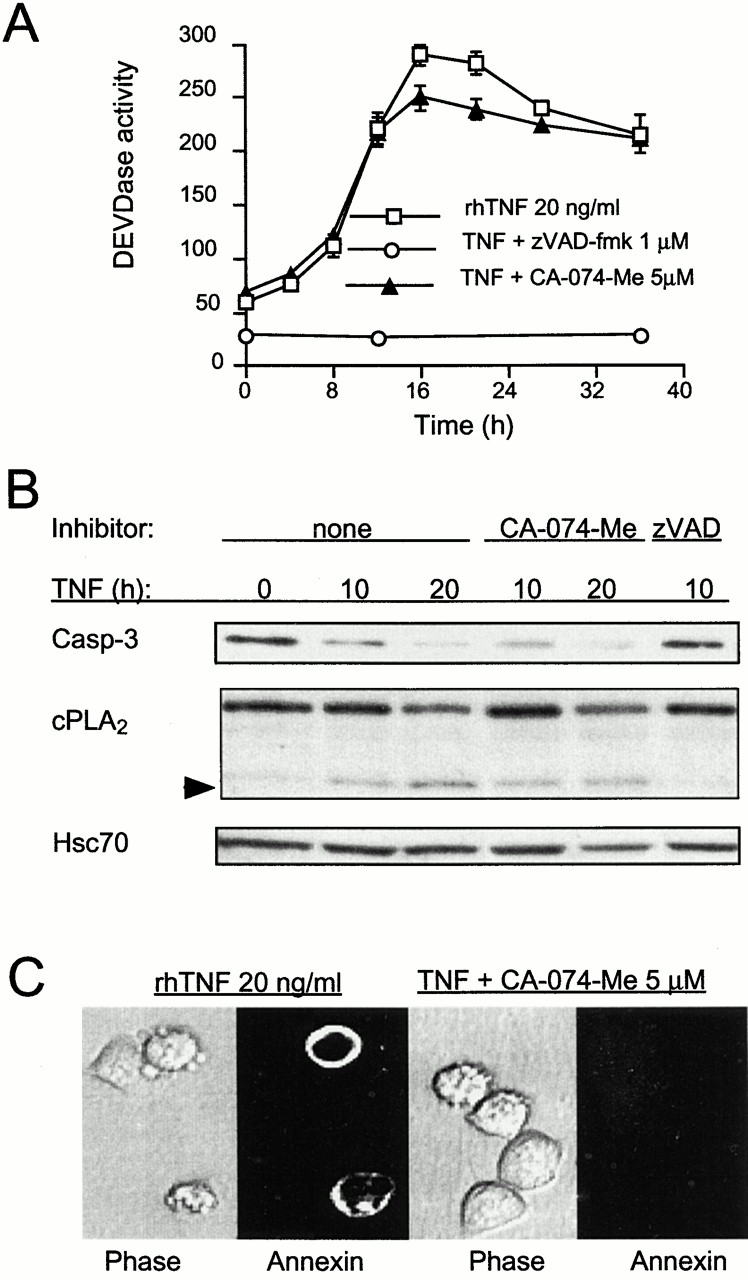
TNF induces cathepsin B–independent activation of caspases and cathepsin B–dependent translocation of PS in ME-180as cells. (A and B) Equal amount of protein from cells treated for indicated times with 20 ng/ml of rhTNF in the presence or absence of 1 μM zVAD-fmk (zVAD) or 5 μM CA-074-Me (CA) was analyzed for caspase-3–like activity by DEVDase assay (DEVDase activity is expressed as arbitrary units and values represent means of triplicate determinations ± SD) or immunoblot analysis of caspase-3 or its substrate cPLA2. The arrowhead indicates a cleavage product of cPLA2. Hsc70 serves as a loading control. (C) ME-180as cells treated as indicated for 30 h were analyzed for PS translocation by staining with FITC–annexin V. Note that in the presence of CA-074-Me, TNF induces cellular shrinkage and rounding in the absence of PS translocation.
Discussion
Since the demonstration that the developmentally programmed death of male linker cells in Caenorhabditis elegans does not require the ced-3 gene product (Ellis and Horvitz 1986), there has been several further reports providing evidence for caspase-independent death programs in vitro (Lavoie et al. 1998; Mathiasen et al. 1999; Nylandsted et al. 2000) and in vivo (Chautan et al. 1999; Doerfler et al. 2000). Some of these death routines become only evident when the caspase-dependent pathway is inhibited, e.g., by pan-caspase inhibitors (Vercammen et al. 1998; Khwaja and Tatton 1999; Luschen et al. 2000), mutations in caspases (Kawahara et al. 1998), or ATP depletion (Eguchi et al. 1997; Leist et al. 1997). However, in other models, cell death proceeds in the absence of any sign of caspase activation (Lavoie et al. 1998; Mathiasen et al. 1999; Nylandsted et al. 2000). Also, in such cases, death retains the character of a program in the sense that selective biochemical inhibitors can block death without interfering with the initial triggering event. In this report, we identified and characterized death receptor–mediated tumor cell death that occurs independently of effector caspase activation. This conclusion is based on three lines of argumentation: (i) caspase substrates do not appear to be cleaved, even though some DEVDase activity is triggered in the cells; (ii) cell death proceeds equally well (or rather faster) when caspase inhibitors are added; and (iii) cell death has an apoptosis-like morphology that is not changed by addition of caspase inhibitors.
It is still a matter of dispute whether the morphological appearance of cell death reflects a single defined mechanism. Although there is evidence that also caspase-independent death can lead to some of the typically apoptotic changes such as chromatin condensation, PS exposure, and early phagocytic removal (Lavoie et al. 1998; Hirt et al. 2000; Nylandsted et al. 2000), the classical apoptotic morphology has been most closely linked to caspase activation. We examined the mode of death in the WEHI-S model established here with special attention on the effect of caspase inhibitors. Electron microscopy, light microscopic video imaging, as well as fluorescent microscopy focussing on various apoptosis-related end points suggested an apoptosis-like death process that was not affected by the inhibition of the cellular DEVDase activity. This set of data supports our suggestion that death of WEHI-S cells induced by low TNF concentrations proceeds independently of known caspases, and the DEVDase activity that can be detected in these cells is an epiphenomenom, either not linked to cell death at all or possibly involved in its counter regulation (Luschen et al. 2000).
Although strong evidence is accumulating for programmed cell death occurring independent of caspases (Wyllie and Golstein 2001), the molecular identity of the mediators remains to be elucidated in most cases. The knowledge is particularly sparse for the well-characterized model of death receptor–mediated alternative cellular demise. There is vast evidence suggesting that many noncaspase proteases like calpains (Vanags et al. 1996), the proteasome (Grimm and Osborne 1999), cathepsins (Deiss et al. 1996; Roberts et al. 1999), and serine proteases (Wright et al. 1997) can act in concert with caspases in standard apoptotic processes. For instance, cathepsin B can be involved in the upstream death signals of TNF in hepatocytes by linking the death receptor to the mitochondrial enhancement loop of caspase activation (Guicciardi et al. 2000). One molecular mechanism by which cathepsins might affect mitochondria has been identified recently to be a selective proteolysis of Bid (Stoka et al. 2001). Our study now gives complementary evidence that cathepsin B can also be a downstream mediator of caspases in triggering TNF-signaled death in many human tumor cell lines. Putting together these pieces of evidence, it appears possible that cathepsin B might, in certain situations, be sufficient for all stages of apoptosis without requiring caspases. This appears indeed to be the case in the TNF-induced death of WEHI-S cells.
It is possible that different proteases may take a similar role as cathepsin B in other circumstances. For example there is good evidence for a dominant role of cathepsin D in other systems (Deiss et al. 1996; Roberg and Ollinger 1998). This might be due to the ratios of different proteases and their inhibitors/activators in different cell types. To get further evidence on the exact molecular identity of the relevant protease in the model of WEHI-S cells exposed to low concentrations of TNF, we first performed an extensive screen of possible proteases using careful titrations with a large panel of chemical protease inhibitors. This seemed to exclude cathepsin D and pointed instead to a cathepsin B–like protease. Further evidence was then obtained by overexpression of cystatin A, which blocks cysteine cathepsins like cathepsin B, but not calpains or cathepsin D (Sloane et al. 1990). Cathepsin D was finally excluded on the basis of antisense experiments, although these experiments provided final definite evidence for cathepsin B as the key protease involved in the WEHI-S cell death model.
How is it possible that a specific cell death program can be triggered by the rather unspecific digestive power of a lysosomal protease? It appears as if a specific translocation process could be a key to the understanding of this phenomenon. For instance the selective translocation of cathepsin B from lysosomes to cytosol and nucleus is well documented for bile salt–induced and TNF-triggered hepatic apoptosis (Roberts et al. 1999; Guicciardi et al. 2000). Similarly, there is clear evidence of early cathepsin D translocation from secondary lysosomes to the cytosol under conditions of oxidative stress–induced apoptosis (Roberg and Ollinger 1998). A second possibility is a quantitative relationship between the amount of lysosomal rupture and the mode of cell death. According to this model, low stress intensities would trigger a limited release of lysosomal enzymes to the cytoplasm followed by apoptotic death, whereas high intensity stresses would lead to a generalized lysosomal rupture and rapid cellular necrosis (Brunk et al. 1997). A causal association between a limited lysosomal rupture and apoptosis has been supported by experiments showing cytoprotection by membrane stabilizing agents (Roberg and Ollinger 1998) as well as triggering of cell death by selective lysosomal disrupters (Li et al. 2000). Our experiments showing clonal survival of cells protected by the cathepsin inhibitor CA-074-Me, although cathepsin B was released to the cytosol, suggest that translocation of lysosomal contents to the cytosol need not necessarily be fatal for a cell. However, the mechanisms and extent of cathepsin release awaits further elucidation.
The reasons why a single death receptor can trigger cellular demise by several mechanisms remain a subject of speculation at the current stage of knowledge. The caspase-independent mechanisms could represent a backup program especially important in pathological situations. Tumor cells have frequently impaired standard apoptosis routines due to, for example, mutations (Lowe and Lin 2000), gene silencing (Soengas et al. 2001), or overexpression of survival proteins (Jäättelä 1999). On the other hand, lysosomal proteolytic efficacy has to be increased in many transformed cells due to the higher protein turnover, and also for enhanced invasiveness (Mai et al. 2000). This would agree with our findings that the death receptor–triggered cathepsin B–dependent death pathway appears to take a more dominant role in tumor cells than in primary cells. This selectivity could prove useful in cancer therapy and it may be due to the fact that tumor cells have frequently increased the expression of cysteine cathepsins (Kos and Lah 1998). Conversely, tumors that manage to suppress the alternative death programs might have a selective growth advantage. For instance, cathepsin-inactivating cystatins may reduce the apoptosis susceptibility of tumor cells. This latter antiapoptotic effect may contribute to the recently reported aggressive phenotype of cystatin A expressing tumors (Kuopio et al. 1998).
In conclusion, our data define two potent cathepsin B–dependent apoptosis pathways in tumor cells that can be triggered by TNF: one in WEHI-S cells, where cathepsin B functions as the dominant execution protease even in the presence of pan-caspase inhibitors, and the other in ME-180as cells with cathepsin B, as a downstream effector of receptor-activated caspases. Our results further imply that cathepsin B–like proteases and their endogenous inhibitors may have a dual role in tumor progression: the proteases reducing malignancy by their pro-apoptotic features but enhancing it by promoting invasion, and the opposite applying to the inhibitors.
Supplemental Material
Acknowledgments
We thank H. Hentze, M. Latta, E. Gawlitta-Gorka, D. Lützhøft, H. Naumann, B. Poulsen, and T. Rignes for excellent technical assistance.
This work was supported by the Danish Cancer Society, the Danish Medical Research Council, the Novo Foundation, the Danish Cancer Research Foundation, the German Research Council (program on endogenous tissue destruction), and the Leo Pharmaceuticals Foundation.
Footnotes
The online version of this article contains supplemental material.
M. Leist and M. Jäättelä share senior authorship.
Abbreviations used in this paper: AFC, 7-amino-trifluoromethylcoumarin; ALLN, N-Acetyl-leu-Leu-Nle-CHO; anti-CD95, agonistic antibody against CD95; Boc-D-fmk, Boc-Asp-CH2F; DEVD-CHO, acetyl-Asp-Glu-Val-Asp-aldehyde; IETD-CHO, acetyl-Ile-Glu-Thr-Asp-aldehyde; lactacystine, clasto-lactacystin-β-lactone; LDH, lactate dehydrogenase; MEF, murine embryonic fibroblast; MTT, 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide; NF-κB, nuclear factor κB; PS, phosphatidylserine; rhTNF, recombinant human TNF; rmTNF, recombinant murine TNF; TLCK, N-α-Tosyl-l-Lys-chloromethyl ketone; TNF, tumor necrosis factor; TNF-R, TNF receptor; TPCK, tosyl-l-Phe-chloromethyl ketone; TRAIL, TNF-related apoptosis-inducing ligand; zFA-fmk, z-Phe-Ala-CH2F; zFK-mbmk, z-Phe-Lys-2,4,6-trimethylbenzoyloxymethylketone; zVAD-fmk, z-Val-Ala-DL-Asp-CH2F; zVDVAD-fmk, z-Val-Asp(OMe)-Val-Ala-Asp(OMe)-CH2F.
References
- Ashkenazi A., Dixit V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell Biol. 1999;11:255–260. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- Brunk U.T., Dalen H., Roberg K., Hellquist H.B. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic. Biol. Med. 1997;23:616–626. doi: 10.1016/s0891-5849(97)00007-5. [DOI] [PubMed] [Google Scholar]
- Chautan M., Chazal G., Cecconi F., Gruss P., Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr. Biol. 1999;9:967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- Deiss L.P., Galinka H., Berissi H., Cohen O., Kimchi A. Catepsin D protease mediates programmed cell death induced by interferon-γ, Fas/APO-1, and TNF-α. EMBO J. 1996;15:3861–3870. [PMC free article] [PubMed] [Google Scholar]
- Deussing J., Roth W., Saftig P., Peters C., Ploegh H.L., Villadangos J.A. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc. Natl. Acad. Sci. USA. 1998;95:4516–4521. doi: 10.1073/pnas.95.8.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler P., Forbush K.A., Perlmutter R.M. Caspase Enzyme activity is not essential for apoptosis during thymocyte development. J. Immunol. 2000;164:4071–4079. doi: 10.4049/jimmunol.164.8.4071. [DOI] [PubMed] [Google Scholar]
- Eguchi Y., Shimizu S., Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- Ellis H.M., Horvitz H.R. Genetic control of programmed cell death in nematode C. elegans . Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Faraco P.R., Ledgerwood E.C., Vandenabeele P., Prins J.B., Bradley J.R. Tumor necrosis factor induces distinct patterns of caspase activation in WEHI-164 cells associated with apoptosis or necrosis depending on cell cycle stage. Biochem. Biophys. Res. Commun. 1999;261:385–392. doi: 10.1006/bbrc.1999.1042. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Grimm L.M., Osborne B.A. Apoptosis and the proteasome. Results Probl. Cell Differ. 1999;23:209–228. doi: 10.1007/978-3-540-69184-6_10. [DOI] [PubMed] [Google Scholar]
- Guicciardi M.E., Deussing J., Miyoshi H., Bronk S.F., Svingen P.A., Peters C., Kaufmann S.H., Gores G.J. Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt U.A., Gantner F., Leist M. Phagocytosis of nonapoptotic cells dying by caspase-independent mechanisms. J. Immunol. 2000;164:6520–6529. doi: 10.4049/jimmunol.164.12.6520. [DOI] [PubMed] [Google Scholar]
- Jäättelä M. Escaping cell deathsurvival proteins in cancer. Exp. Cell Res. 1999;248:30–43. doi: 10.1006/excr.1999.4455. [DOI] [PubMed] [Google Scholar]
- Jäättelä M., Benedict M., Tewari M., Shayman J.A., Dixit V.M. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene. 1995;10:2297–2305. [PubMed] [Google Scholar]
- Jäättelä M., Mourizen H., Elling F., Bastholm L. A20 zinc finger protein inhibits TNF and IL-1 signaling. J. Immunol. 1996;156:1166–1173. [PubMed] [Google Scholar]
- Jäättelä M., Wissing D., Kokholm K., Kallunki T., Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–6134. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B., Roberts P.J., Faubion W.A., Kominami E., Gores G.J. Cystatin A expression reduces bile salt-induced apoptosis in a rat hepatoma cell line. Am. J. Physiol. 1998;275:G723–G730. doi: 10.1152/ajpgi.1998.275.4.G723. [DOI] [PubMed] [Google Scholar]
- Kawahara A., Ohsawa Y., Matsumura H., Uchiyama Y., Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A., Tatton L. Resistance to the cytotoxic effects of tumor necrosis factor α can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J. Biol. Chem. 1999;274:36817–36823. doi: 10.1074/jbc.274.51.36817. [DOI] [PubMed] [Google Scholar]
- Kos J., Lah T.T. Cysteine proteinases and their endogenous inhibitorstarget proteins for prognosis, diagnosis and therapy in cancer. Oncol. Rep. 1998;5:1349–1361. doi: 10.3892/or.5.6.1349. [DOI] [PubMed] [Google Scholar]
- Kuopio T., Kankaanranta A., Jalava P., Kronqvist P., Kotkansalo T., Weber E., Collan Y. Cysteine proteinase inhibitor cystatin A in breast cancer. Cancer Res. 1998;58:432–436. [PubMed] [Google Scholar]
- Latta M., Kunstle G., Leist M., Wendel A. Metabolic depletion of ATP by fructose inversely controls CD95- and tumor necrosis factor receptor 1–mediated hepatic apoptosis. J. Exp. Med. 2000;191:1975–1986. doi: 10.1084/jem.191.11.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie J.N., Nguyen M., Marcellus R.C., Branton P.E., Shore G.C. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol. 1998;140:637–645. doi: 10.1083/jcb.140.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M., Gantner F., Bohlinger I., Germann P.G., Tiegs G., Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-α requires transcriptional arrest. J. Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- Leist M., Single B., Castoldi A.F., Kuhnle S., Nicotera P. Intracellular adenosine triphosphate (ATP) concentrationa switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Yuan X., Nordgren G., Dalen H., Dubowchik G.M., Firestone R.A., Brunk U.T. Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett. 2000;470:35–39. doi: 10.1016/s0014-5793(00)01286-2. [DOI] [PubMed] [Google Scholar]
- Lowe S.W., Lin A.W. Apoptosis in cancer. Carcinogenesis. 2000;21:485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- Luschen S., Ussat S., Scherer G., Kabelitz D., Adam-Klages S. Sensitization to death receptor cytotoxicity by inhibition of FADD/Caspase signalingrequirement of cell cycle progression. J. Biol. Chem. 2000;275:24670–24678. doi: 10.1074/jbc.M003280200. [DOI] [PubMed] [Google Scholar]
- Mai J., Waisman D.M., Sloane B.F. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim. Biophys. Acta. 2000;1477:215–230. doi: 10.1016/s0167-4838(99)00274-5. [DOI] [PubMed] [Google Scholar]
- Mathiasen I.S., Lademann U., Jäättelä M. Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res. 1999;59:4848–4856. [PubMed] [Google Scholar]
- Nylandsted J., Rohde M., Brand K., Bastholm L., Elling F., Jäättelä M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA. 2000;97:7871–7876. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg K., Ollinger K. Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am. J. Pathol. 1998;152:1151–1156. [PMC free article] [PubMed] [Google Scholar]
- Roberts L.R., Kurosawa H., Bronk S.F., Fesmier P.J., Agellon L.B., Leung W.-Y., Mao F., Gores G.J. Cathepsin B contributes to bile salt-induced apoptosis of rat hepatocytes. Gastroenterology. 1997;113:1714–1726. doi: 10.1053/gast.1997.v113.pm9352877. [DOI] [PubMed] [Google Scholar]
- Roberts L.R., Adjei P.N., Gores G.J. Cathepsins as effector proteases in hepatocyte apoptosis. Cell Biochem. Biophys. 1999;30:71–88. doi: 10.1007/BF02737885. [DOI] [PubMed] [Google Scholar]
- Ruggiero V., Johnson S.E., Baglioni C. Protection from tumor necrosis factor cytotoxicity by protease inhibitors. Cell. Immunol. 1987;107:317–325. doi: 10.1016/0008-8749(87)90240-1. [DOI] [PubMed] [Google Scholar]
- Schotte P., Declercq W., Van Huffel S., Vandenabeele P., Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- Shi G.P., Villadangos J.A., Dranoff G., Small C., Gu L., Haley K.J., Riese R., Ploegh H.L., Chapman H.A. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10:197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- Sloane B.F., Moin K., Krepela E., Rozhin J. Cathepsin B and its endogenous inhibitorsthe role in tumor malignancy. Cancer Metastasis Rev. 1990;9:333–352. doi: 10.1007/BF00049523. [DOI] [PubMed] [Google Scholar]
- Soengas M.S., Capodieci P., Polsky D., Mora J., Esteller M., Opitz-Araya S., McCombie R., Herman J.G., Gerald W., Lazebnik Y.A. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- Stoka V., Turk B., Schendel S.L., Kim T.H., Cirman T., Snipas S.J., Ellerby L.M., Bredesen D., Freeze H., Abrahamson M. Lysosomal protease pathways to apoptosis. Cleavage of Bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- Tartaglia L.A., Weber R.F., Figari I.S., Reynolds C., Palladino M.A., Goeddel D.V. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. USA. 1991;88:9292–9296. doi: 10.1073/pnas.88.20.9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspasesenemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Tsujimoto M., Yip Y.K., Vilcek J. Tumor necrosis factorspecific binding and internalization in sensitive and resistant cells. Proc. Natl. Acad. Sci. USA. 1985;82:7626–7630. doi: 10.1073/pnas.82.22.7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanags D.M., Porn-Ares M.I., Coppola S., Burgess D.H., Orrenius S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 1996;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D., Varfolomeev E.E., Malinin N.L., Goltsev Y.V., Kovalenko A.V., Boldin M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- Wissing D., Mouritzen H., Egeblad M., Poirier G.G., Jäättelä M. Involvement of caspase-dependent activation of cytosolic phospholipase A2 in tumor necrosis factor-induced apoptosis. Proc. Natl. Acad. Sci. USA. 1997;94:5073–5077. doi: 10.1073/pnas.94.10.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.C., Wei Q.S., Zhong J., Zheng H., Kinder D.H., Larrick J.W. Purification of a 24-kD protease from apoptotic tumor cells that activates DNA fragmentation. J. Exp. Med. 1994;180:2113–2123. doi: 10.1084/jem.180.6.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.C., Schellenberger U., Wang H., Kinder D.H., Talhouk J.W., Larrick J.W. Activation of CPP32-like proteases is not sufficient to trigger apoptosisinhibition of apoptosis by agents that suppress activation of AP24, but not CPP32-like activity. J. Exp. Med. 1997;186:1107–1117. doi: 10.1084/jem.186.7.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie A.H., Golstein P. More than one way to go. Proc. Natl. Acad. Sci. USA. 2001;98:11–13. doi: 10.1073/pnas.98.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.