

Abstract
Conventional kinesin is a highly processive molecular motor that takes several hundred steps per encounter with a microtubule. Processive motility is believed to result from the coordinated, hand-over-hand motion of the two heads of the kinesin dimer, but the specific factors that determine kinesin's run length (distance traveled per microtubule encounter) are not known. Here, we show that the neck coiled-coil, a structure adjacent to the motor domain, plays an important role in governing the run length. By adding positive charge to the neck coiled-coil, we have created ultra-processive kinesin mutants that have fourfold longer run lengths than the wild-type motor, but that have normal ATPase activity and motor velocity. Conversely, adding negative charge on the neck coiled-coil decreases the run length. The gain in processivity can be suppressed by either proteolytic cleavage of tubulin's negatively charged COOH terminus or by high salt concentrations. Therefore, modulation of processivity by the neck coiled-coil appears to involve an electrostatic tethering interaction with the COOH terminus of tubulin. The ability to readily increase kinesin processivity by mutation, taken together with the strong sequence conservation of the neck coiled-coil, suggests that evolutionary pressures may limit kinesin's run length to optimize its in vivo function.
Keywords: kinesin, tubulin, single-molecule motility, processivity, molecular motors
Introduction
Conventional kinesin is a molecular motor that transports membrane organelles (Goldstein and Philp 1999) and small vimentin particles (Prahlad et al. 1998) along microtubules. In contrast to many other members of the kinesin superfamily (Crevel et al. 1997; Stewart et al. 1998; Pierce et al. 1999), conventional kinesin is highly processive and can travel over a micron per encounter with a microtubule (Howard et al. 1989; Block et al. 1990; Hackney 1995; Vale et al. 1996). Such high processivity is likely to serve an important biological function. However, transport of large organelles may not require a highly processive motor because many motors bound to the organelle surface can function cooperatively to enable continuous transport (Howard 1997). Instead, kinesin's high processivity is likely to be particularly important for transporting cargoes whose small size precludes the attachment of many of motors (e.g., small membrane organelles and soluble mRNA or protein particles) (Carson et al. 1997; Prahlad et al. 1998; Brendza et al. 2000).
Conventional kinesin contains two identical heavy chains, each of which contains a motor domain at the NH2 terminus, two intervening coiled-coil domains, and a cargo/light chain binding domain at the COOH terminus (Vale and Fletterick 1997). A monomeric motor domain produced by truncation before the first coiled-coil domain (termed the neck coiled-coil) is sufficient to generate motion, provided that several motors are attached to the same microtubule (Yang et al. 1990; Berliner et al. 1995). However, processivity requires a dimeric motor containing at least the complete neck coiled-coil (Vale et al. 1996; Hancock and Howard 1998; Young et al. 1998). The requirement for a dimeric motor is thought to reflect an underlying coordinated hand-over-hand walking mechanism, and a number of studies have provided evidence for alternating site catalysis by the two heads of kinesin (Hackney 1994; Ma and Taylor 1997; Gilbert et al. 1998). In such a mechanism, the two heads of kinesin are thought to be bound to adjacent sites along a microtubule protofilament, but are in different nucleotide states (Vale and Milligan 2000). A nucleotide-dependent conformational change in the front head detaches the rear head from the microtubule and repositions it toward the microtubule plus end, where its rebinding to the next tubulin subunit completes an 8-nm step (Svoboda et al. 1993). This coupling between the conformational change by the forward head and the release of the rear head is believed to ensure that both heads do not detach simultaneously. This process is highly efficient since, on average, kinesin detaches from the microtubule only after taking 150 steps (99.3% chance of completing a step).
Crystal structures of kinesin (Kull et al. 1996; Kozielski et al. 1997; Sack et al. 1997) in conjunction with functional studies have begun to provide clues as to the structural basis of processive motion. A small peptide emerging from the catalytic core (termed the neck linker) appears to be the fundamental engine that drives the hand-over-hand walking mechanism (Rice et al. 1999; Case et al. 2000; Tomishige and Vale 2000). Extension and immobilization of the neck linker upon ATP binding by the forward head exerts a force that causes the rear head to release from the microtubule and be repositioned towards the plus end of the microtubule (Rice et al. 1999) (see Figure 6 in the accompanying paper, Tomishige and Vale 2000). The role of the neck coiled-coil in processive motility, on the other hand, has been more controversial. While some studies have proposed a major nucleotide-dependent unwinding of this region (Hoenger et al. 1998; Mandelkow and Johnson 1998; Mandelkow and Hoenger 1999), other studies indicate that such events, if they occur, are not essential for motility (Romberg et al. 1998; Tomishige and Vale 2000).
While a basic model for kinesin processivity is emerging (Vale and Milligan 2000), the specific factors or structural elements that determine the extent of processivity (run length) are unknown. Here we have defined a new role for the kinesin neck coiled-coil as a regulator of run length. We have engineered ultra-processive motors by increasing the positive charge of the neck coiled-coil and decreased processivity by increasing the negative charge. The gain in processivity can be abolished by treatment with high salt concentrations or by proteolytic removal of the tubulin COOH terminus. These results suggest that an electrostatic interaction between the positively charged neck coiled-coil and the negatively charged COOH terminus of tubulin plays an important role in the kinesin mechanism.
Materials and Methods
Preparation and Assays of Kinesin Proteins
K560-GFP mutants were prepared by QuikChange mutagenesis (Stratagene, Inc.) and sequenced to ensure that no unintended mutations were introduced. Proteins were prepared essentially as described (Case et al. 1997), except that a REsource Q15 column was used for the anion exchange purification step. All K560-GFP constructs were further purified by microtubule affinity (Case et al. 1997) before assaying. After the microtubule affinity purification, motors were either assayed immediately or frozen with 15% sucrose added and stored in liquid nitrogen.
Quantitation of protein concentration, ATPase assays, and microtubule gliding assays were performed as described previously (Woehlke et al. 1997). ATPase assays were performed in a microplate reader in a buffer consisting of 12 mM K+ Pipes, pH 6.8, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 3 mM NaCl, and 0.5 mg/ml casein.
Single Molecule Fluorescence Measurements
Single molecule motility measurements were performed essentially as described (Pierce and Vale 1998), except that 0.5 mg/ml casein was used as the blocking protein instead of 7.5 mg/ml bovine serum albumin. Microtubules for single molecule assays were prepared by copolymerizing Cy5-labeled tubulin with unlabeled tubulin in a 1:7 ratio. Flow cells were made using a spacer of 8-μm beads suspended in vacuum grease. Polylysine-tagged G234A K560 protein was then introduced as a microtubule glue (Hartman et al. 1998). After washing out unbound G234A, the Cy5 microtubules were introduced. After washing out unbound microtubules, the assay mixture of kinesin, ATP, oxygen scavengers, and casein was introduced. The flow cells were then flattened to minimum thickness, sealed with rubber cement, and imaged. Subtilisin-treated microtubules were prepared by incubating polymerized Cy5-labeled microtubules with 100 μg/ml subtilisin for 2 h at 37°C. The proteolytic reaction was quenched by adding PMSF (1 mM) and microtubules were separated from subtilisin and cleavage products by centrifugation through a 60% glycerol cushion or by binding to the G234A-K560 coated flow cells described above. The proteolytic cleavage was monitored by SDS-PAGE and an electrophoretic shift indicative of complete cleavage was observed, as was seen in other studies (Wang and Sheetz 2000).
Total internal reflection microscopy and run-length analysis was performed essentially as described (Shimizu et al. 2000). The laser power for total internal reflection illumination was 5 mW. Data was recorded on sVHS videotape with four frame averaging and analyzed offline using a custom set of macros in NIH-IMAGE. Segments of videotape were digitized at 10–15 fps, and the outline of the axoneme or microtubule (determined from the Cy5 image) was superimposed on the green fluorescent protein (GFP) signal. Attachment and detachment times and positions were determined manually for single fluorescent spots interacting with the axoneme. These values were then converted to run length, association time, and velocity. To determine the mean run length, we first calculated the cumulative probability distribution of the run lengths, which plots the fraction of run lengths shorter than a given run length versus the run length. The mean run length was then determined by nonlinear least squares fitting of the cumulative probability distribution from x 0 to infinity to 1 − exp[(x 0 − x)/t]. This procedure fits the data directly, without any necessity of data binning. The decay constant, t, is the only fitted parameter, and gives the mean run length of the distribution. x 0 is the lower limit for runs incorporated in the analysis and is used to exclude shorter runs that are either undersampled or not measured with the greatest accuracy by our manual tracking method. Run lengths above 1 μm can be determined with the greatest degree of accuracy in our system. Since single molecule run lengths are distributed exponentially (Block et al. 1990; Vale et al. 1996) and we had considerable data of >1 μm for wild type and the ultraprocessive mutants, we set x 0 to 1 μm for these proteins to use the most accurate data for the exponential fit. For the 4Glu mutant, which as a much shorter run length, x 0 was set to 0.2 μm, which represents the practical detection limit for unidirectional motion. Velocities were determined by fitting the measured cumulative probability distribution to the Gaussian cumulative probability distribution. All fitting was performed in MATLAB. Errors were estimated by the bootstrap technique (Press et al. 1992). Each distribution was resampled 200× and fit as described above. The standard deviation of the fitted parameter over the resampled data sets was taken as the error in the fitted quantity. Statistical significance was determined by the applying the Kolmogorov-Smirnov test to the observed run lengths (Press et al. 1992). The observed run lengths underestimate the true run length of the motor, because a moving spot disappears either when it dissociates from the axoneme or when it irreversibly photobleaches. The observed dissociation rate constant (k obs, the inverse of the association time) is given by k obs = k diss + k bleach (Pierce and Vale 1998). Both run lengths and association times were corrected for the photobleaching rate. The correction can be as large as two- or threefold for ultraprocessive motors where the observed dissociation rate is close to the photobleaching rate, and the measured are errors increased by a larger amount due to the uncertainty in the association time. The photobleaching rate was determined by measuring the rate of disappearance of GFP-kinesin nonspecifically adsorbed to the slide and was measured to be 0.086 s−1 (n = 1,769).
Optical Trapping Measurements
The optical trap used in this work has been described previously (Coppin et al. 1997). Because K560-GFP does not stick to carboxylated latex beads, we first cross-linked anti–GFP antibodies to carboxylated latex beads (Tomishige and Vale 2000). K560-GFP bound readily to these beads and exhibited motile properties comparable with full-length, wild-type kinesin attached to carboxylated beads (Svoboda and Block 1994b). Assays were performed at motor densities such that 50% or fewer beads moved when held against an axoneme for 1 min. This ensures that >98% of motility events arise from a single motor (Svoboda and Block 1994b). Assays were performed in flow cells with rhodamine-labeled axonemes bound to the glass. Beads coated with K560-GFP were then introduced in BRB80 (80 mM Pipes, 2 mM MgCl2, 1 mM EGTA) with 1 mM MgATP, 1 mg/ml casein, and an oxygen scavenger system (Coppin et al. 1997).
Bead position was determined by imaging on to a quadrant photodiode (QPD). Data was collected at 2 kHz with filtering at 4 kHz. The QPD was calibrated by rastering the QPD in steps of known size underneath a tightly held trapped bead (Coppin et al. 1997). The trap stiffness was determined by fitting of the power spectral density of a trapped bead (data collected at 4 kHz with filtering at 8 kHz) to a Lorentzian (Gittes and Schmidt 1998; Svoboda and Block 1994a). Trap stiffnesses used in these experiments ranged from 0.027 to 0.060 pN/nm, and the force was linear for the range of the experimental data shown
The data was analyzed with a custom set of programs written in Labview (National Instruments). After conversion of the QPD signal to position, motor “runs” were found by looking for rapid decreases in bead position (>25 nm between two consecutive 40-point windows; e.g., events at 4 and 4.5 s in Fig. 3 A, below), which is characteristic of motor detachment. From these detachment points, the beginning of the run was found by moving backwards in time until the bead position was within 1.75 SD of the baseline and the bead velocity was <5 nm/s averaged over a 200-point window. These runs were then baseline subtracted, and the dissociation rate as a function of load was calculated by summing the amount of time spent in 0.5 pN force bins. The force on the bead was calculated by multiplying the bead distance from the center of the trap by the trap stiffness. The number of dissociations in each force bin were also scored, and the dissociation rate at each load calculated by dividing the number of dissociations by the residence time (Coppin et al. 1997). Because the motor-bead linkage is highly compliant at low loads, the exact position at which runs start is difficult to determine, and we therefore did not analyze data below 1 pN as these results were very sensitive to the choice of analysis parameters.
Figure 3.
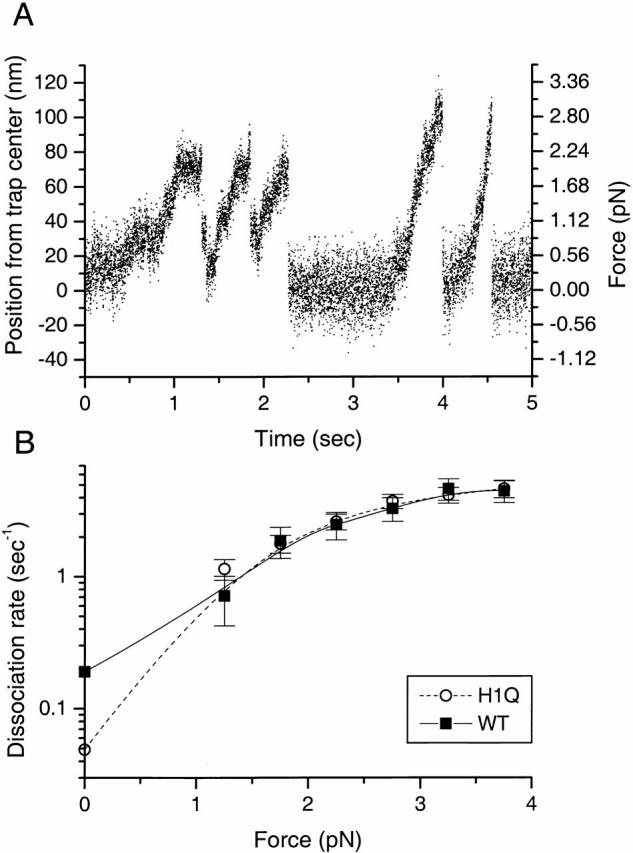
(A) Representative movements of a kinesin-coated bead along the long axis of a microtubule in an optical trap. The trap stiffness was 0.028 pN/nm, and the data was collected at 2 kHz. Five processive “runs” are shown. Dissociation of kinesin from the microtubule occurs when the bead suddenly and rapidly returns to the trap center, and the number of such events at different loads were scored. (B) Dissociation rate as a function of load for wild-type (WT) kinesin and the H1Q mutant. Dissociation rates for WT (▪) and H1Q (○) were measured by optical trapping as described in the Materials and Methods, except for the zero-load values, which are reported as the inverse of the association time in the single molecule fluorescence motility assay. 170 runs were scored for wild-type kinesin and 424 runs for H1Q in the optical trapping experiment. Error bars are calculated as the square root of the number of events in each 0.5 pN force bin divided by the residence time in that force bin. Dissociation rates could not be calculated for forces below 1 pN due to the uncertainty of precisely when the bead motion began.
Results
Quadruplication of Heptad One in the Neck Coiled-Coil Increases Processivity
In previous work, a construct containing a duplication of the first heptad of the kinesin neck coiled-coil was prepared as a control for mutagenesis experiments examining the role of the neck coiled-coil in processive motility (Romberg et al. 1998). Surprisingly, this heptad one duplication mutant (H1D) displayed an approximately threefold increase in run length, whereas all other mutations in the coiled-coil either reduced or did not change processivity. Why this mutation increased processivity was not investigated in the previous work. To test whether processivity could be further increased by including additional copies of heptad one, we prepared a mutant of K560-GFP (the first 560 amino acids of human conventional kinesin fused at its COOH terminus to the F64L/S65T variant of GFP; Romberg et al. 1998) containing a quadruplicate repeat of heptad one (termed H1Q; consists of four repeats of heptad one followed by heptads two through five; Fig. 1). To test whether any observed effects were sequence specific or instead arose from a lengthening of the coiled-coil, we also prepared a quadruplicate repeat of the fifth (terminal) heptad of the coiled-coil (termed H5Q; consists of heptads one through four followed by four copies of heptad five; Fig. 1).
Figure 1.
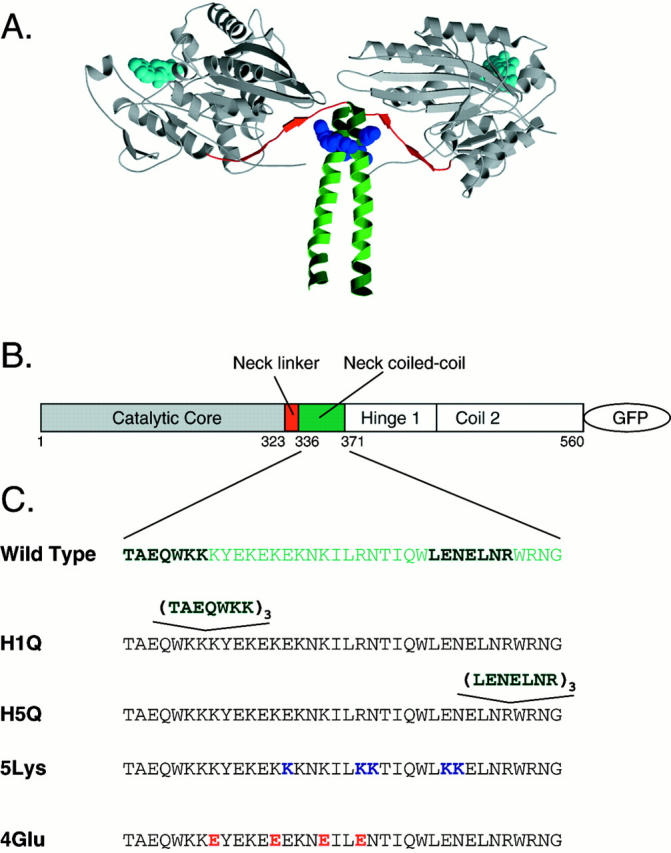
The structure of kinesin and kinesin mutations used in this study. (A) The structure of the kinesin dimer (Kozielski et al. 1997) with the catalytic core shown in gray, the neck linker in red, and the neck coiled-coil in green. The bound ADP is shown in cyan. Heptads one and five are shown in darker green, and the two terminal lysines of heptad 1 are shown as a blue space-filling model. This figure was prepared using Molscript and Raster3D (Kraulis 1991; Meritt and Murphy 1994). (B) A schematic representation of the structural domains of the first 560 amino acids of human conventional kinesin, colored as in the structure above. The catalytic core is followed by the neck linker; the core and neck linker are collectively known as the motor domain. This is followed by the neck coiled-coil, a putative unstructured hinge region (Hinge 1), and a second coiled-coil (Coil 2). The kinesin is truncated at amino acid 560 (at the end of coil 2), and GFP is fused to the COOH terminus. (C) The sequence of the mutants studied here. The wild-type coiled-coil is shown, with heptads 1 and 5 highlighted in dark green. The mutated or inserted residues for each mutant are shown in bold color.
We first investigated the basic enzymatic and motile properties of the H1Q and H5Q mutants (Table ). The microtubule gliding velocity and the ATPase k cat were within error of wild type for both mutants. The K m(MT), a parameter that reflects microtubule affinity, was similar to wild type for H1Q and slightly increased for H5Q. Thus, both H1Q and H5Q are functional motors with properties similar to wild-type kinesin.
Table 1.
ATPase and Motility Properties of Coiled-Coil Mutants
| MT-stimulated ATPase | MT gliding | ||
|---|---|---|---|
| Construct | K m(MT) | k cat | |
| μM | ATP/s per head | μm/s | |
| Wild type | 0.44 ± 0.09 | 45 ± 3 | 0.37 ± 0.11 |
| H1Q | 0.41 ± 0.10 | 35 ± 2 | 0.44 ± 0.09 |
| H5Q | 1.65 ± 0.13 | 33 ± 1 | 0.38 ± 0.12 |
| 5Lys | 1.14 ± 0.33 | 27 ± 2 | 0.35 ± 0.05 |
| 4Glu | >10 | >6 | 0.47 ± 0.05 |
K m(MT) and k cat indicate the microtubule concentration needed for half-maximal stimulation of the ATPase activity and the maximal ATPase activity at saturating microtubule concentrations, respectively. These values were obtained by fitting ATPase rates measured at eight different microtubule concentrations to a Michaelis-Menten function. Errors are given as the standard error of the fitted parameter. The ATPase activity of the 4Glu mutant did not saturate at up to 25 μM microtubule concentration, and displayed a turnover rate of 6 s−1 at that microtubule concentration. Because of their low microtubule affinity, K m(MT) and k cat could not be determined.
To measure the processivity of these mutants, we used a single molecule fluorescence assay in which individual GFP-tagged kinesins are imaged as they move along an axoneme (Vale et al. 1996; Romberg et al. 1998; Shimizu et al. 2000). Measurement of the attachment and detachment positions and times of individual movements provides a direct determination of the run length and velocity of single molecules. The run lengths are exponentially distributed (Block et al. 1990; Vale et al. 1996), and the exponential decay constant is a measure of the mean run length. H1Q displayed a run length of ∼7 μm (Table and Fig. 2). This value is more than fourfold greater than the wild-type run length and represents a larger gain in processivity than the original H1D mutant. This effect cannot be explained by a change in the association state of H1Q (such as formation of tetramers or aggregates), because the intensities of the single molecule spots were unchanged from wild-type kinesin (data not shown). The single molecule mean velocity of H1Q was the same as wild-type kinesin. In contrast, H5Q had a modest (1.7-fold) gain in processivity, which was significantly less than that obtained for H1Q. Unexpectedly, H5Q had a slightly faster velocity than wild type (18% increase). These results confirm and extend previous observations that ultraprocessive motors can be created by manipulation of the neck coiled-coil. Moreover, the mechanism does not appear to involve simply a length increase of the coiled-coil, because H5Q, which is the same length as H1Q, displayed only a small change in processivity.
Table 2.
Single Molecule Motility of Kinesin Coiled-Coil Mutants on Axonemes
| Run length | ||||
|---|---|---|---|---|
| Construct | n | Observed | Corrected | Velocity |
| μm | μm | μm/s | ||
| Wild type | 246 | 1.1 ± 0.1 | 1.5 ± 0.2 | 0.389 ± 0.005 |
| H1Q | 108 | 2.5 ± 0.3 | 6.6 ± 2.6 | 0.371 ± 0.006 |
| H5Q | 156 | 1.7 ± 0.2 | 2.5 ± 0.3 | 0.460 ± 0.007 |
| 5Lys | 128 | 1.9 ± 0.2 | 3.5 ± 0.8 | 0.392 ± 0.008 |
| 4Glu | 172 | 0.28 ± 0.03 | 0.30 ± 0.03 | 0.345 ± 0.008 |
Single molecule velocities and run lengths were measured as described in the Materials and Methods. n is the number of events that were scored for each mutant. Errors were determined by bootstrapping and are reported as 1 SD. All measurements were repeated on two separate preparations and agreed within error. The numbers reported here are derived from a single fit of all measured data. Observed run lengths were corrected for photobleaching as described in the Materials and Methods. For all mutants, the probability was <10−4 that the measured run lengths were distributed the same as the wild-type run lengths (determined using the Kolmogorov-Smirnov test).
Figure 2.
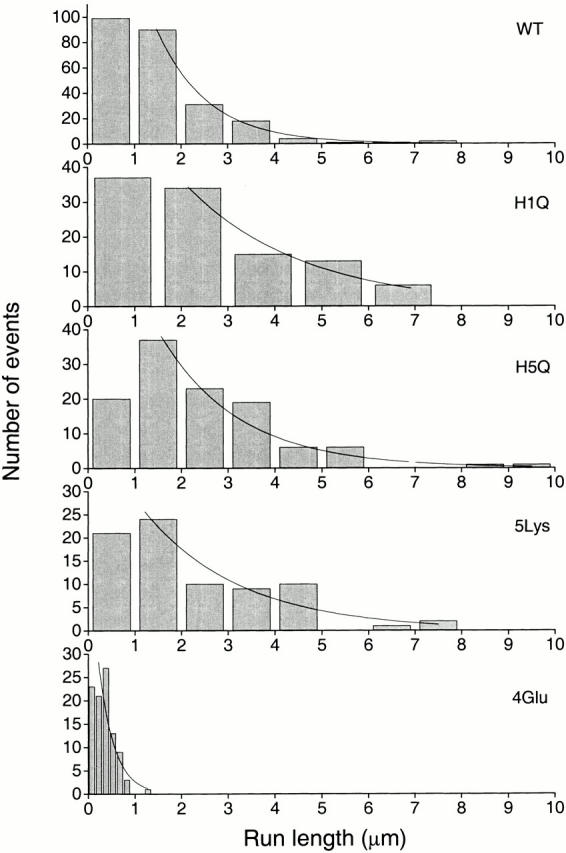
Run-length histograms of wild-type and mutant kinesins. Run lengths were measured in a single molecule fluorescence motility assay as described in the Materials and Methods. The mean run length was determined by fitting to the cumulative probability distribution of the data (see Materials and Methods) and not to the histograms shown here. The mean run lengths and their associated errors are shown in Table . Three events longer than 10 μm were scored for H1Q, but are not shown.
To further characterize the gain of processivity by the H1Q mutant, we measured processivity as a function of load using an optical trap assay. In this assay, the kinesin-coated beads are subject to a restoring force by the optical trap (Svoboda and Block 1994a), which increases the load on the motor as it moves the bead away from the center of the trap. Since the force on the kinesin is constantly changing as the motor moves outward from the trap center, we cannot simply measure the run length to determine the processivity at a given load. Instead, we measured the time bound and the number of times that the motor dissociated from the microtubule in different force regimes (0.5 pN force intervals). From this data, the motor dissociation rate could be calculated as a function of load. These measurements could be made more accurately using a force-clamping optical trap (Visscher et al. 1999), although the current setup is sufficient for comparing the relative dissociation rates as a function of load for these two mutants.
Wild-type kinesin and the H1Q mutant did not differ in their dissociation rates over the force range that could be measured accurately (1–4 pN) (Fig. 3 B). Thus, while H1Q shows a fourfold decrease in dissociation rate compared with wild type in the zero load regime of the single molecule fluorescence assay (Fig. 3 B), this difference is not apparent at higher loads. Thus, the mechanism underlying the increase in processivity is very force sensitive and does not appear to operate under loads above 1 pN. These results suggest that a weak interaction, easily disrupted by an opposing force, retains the H1Q mutant for longer times on the microtubule compared with wild type.
Electrostatic Effects Are Important for the Gain in Processivity
A notable sequence feature of heptad one (TAEQWKK) is the presence of two lysines and a net charge of +1. Thus, in the H1Q mutant, the net charge of the neck coiled-coil is increased from +4 to +7 (+8 to +14 in the dimer). These observations, together with the optical trapping data indicating that a weak interaction is responsible for the processivity gain, suggested that an electrostatic interaction might be responsible for the observed effects of the heptad one quadruplication on processivity. To test this hypothesis, we examined the effect of salt on the run length of wild-type and the H1Q mutant. Two different salt treatments were tested: 250 mM KCl, which would be expected to efficiently disrupt charge–charge interactions, and 120 mM K-acetate, a less chaotropic salt and a condition that is a closer mimic of the in vivo ionic environment (Burton 1983).
KCl reduced the processivity of wild-type kinesin by four- to fivefold, a larger effect than previously observed (Vale et al. 1996) (Table ). However, the effect of KCl on the H1Q run length was much more dramatic (12-fold decrease). K-acetate had no effect on wild-type kinesin processivity, but reduced H1Q processivity by two- to threefold. The processivity of H1Q in 120 mM K acetate is still nearly twice that of wild type. The much greater sensitivity of H1Q run length to increased salt concentration compared with wild-type kinesin strongly suggests that an electrostatic interaction is involved in the gain of processivity.
Table 3.
Effect of Salt Treatment on Kinesin Processivity
| Run length | |||||
|---|---|---|---|---|---|
| Construct | Added salt | n | Observed | Corrected | Velocity |
| μm | μm | μm/s | |||
| Wild type | 250 mM KCl | 43 | 0.30 ± 0.05 | 0.31 ± 0.05 | 0.42 ± 0.03 |
| H1Q | 250 mM KCl | 54 | 0.50 ± 0.06 | 0.55 ± 0.07 | 0.42 ± 0.01 |
| Wild type | 120 mM KAc | 75 | 1.1 ± 0.1 | 1.4 ± 0.2 | 0.416 ± 0.008 |
| H1Q | 120 mM KAc | 69 | 1.7 ± 0.2 | 2.6 ± 0.5 | 0.465 ± 0.008 |
Single molecule measurements were made as described in the Materials and Methods and in Table . n is the number of events that were scored for each mutant. The run lengths after addition of KCl differed at P < 10−16 from those without added salt. The addition of K-acetate did not produce a significant change in the wild-type run length, but did for H1Q (P = 2 × 10−3). Salts were added to a buffer of 12 mM Pipes, pH 6.8, 1 mM EGTA, and 2 mM MgCl2. The buffer pH was readjusted to 6.8 after the addition of potassium acetate (KAc).
A potential electrostatic interaction partner for the kinesin neck coiled-coil is the highly negatively charged COOH terminus, which is known to interact with a number of proteins, including kinesin (Hagiwara et al. 1994; Tucker and Goldstein 1997). Moreover, removal of the COOH terminus of tubulin by subtilisin was recently shown to reduce kinesin processivity (Wang and Sheetz 2000). To test this hypothesis, we measured the processivity of H1Q and wild-type on bovine brain microtubules before and after subtilisin cleavage (Serrano et al. 1984). Kinesin moved along microtubules with a greater velocity but slightly shorter run length compared with sea urchin sperm axonemes (Table ). The H1Q mutant showed a processivity increase on microtubules similar to that observed on axonemes. Upon treatment with subtilisin, the processivity of wild-type kinesin was only reduced by ∼30%, a smaller decrease than observed previously with a bead assay (Wang and Sheetz 2000). In contrast, subtilisin treatment reduced the processivity of the H1Q mutant by fourfold. This result clearly indicates that the COOH terminus of tubulin is the main interaction partner for kinesin's positively charged neck coiled-coil.
Table 4.
Single Molecule Motility of Kinesin Mutants on Microtubules, Before and After Subtilisin Treatment
| Run length | |||||
|---|---|---|---|---|---|
| Construct | Substrate | n | Observed | Corrected | Velocity |
| μm | μm | μm/s | |||
| Wild type | MT | 54 | 0.8 ± 0.2 | 1.0 ± 0.2 | 0.52 ± 0.02 |
| Wild type | SMT | 73 | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.40 ± 0.06 |
| H1Q | MT | 60 | 2.9 ± 0.3 | 5.8 ± 1.6 | 0.51 ± 0.02 |
| H1Q | SMT | 43 | 0.8 ± 0.1 | 1.5 ± 0.5 | 0.25 ± 0.02 |
Single molecule measurements were made as described in Materials and Methods and Table . n is the number of events that were scored for each mutant. Run lengths were significantly different on subtilisin-treated microtubules compared with untreated microtubules (P < 2 × 10−3) for both wild type and H1Q.
Engineering Kinesin Processivity by Point Mutations
Based on the previous results, we predicted that we should be able to modulate kinesin processivity by changing the charge of the neck coiled-coil without changing its length. To test this, we produced a mutant in which the terminal two residues of heptads two through four were replaced by lysine (5Lys) (Fig. 1). These residues were chosen for replacement because they are in the same positions in the heptad pattern as the added lysines in H1Q. We also produced a mutant in which four naturally occurring positively charged amino acids (three lysines and one arginine) were mutated to glutamates (4Glu). This mutant has a neck coiled-coil sequence with an overall neck coiled-coil charge of −4, which is of equal magnitude but opposite sign to the wild-type neck coiled-coil sequence (+4).
The 5Lys mutant exhibited comparable ATPase activity and microtubule gliding velocity to wild-type kinesin (Table ). The 4Glu mutant, however, showed a significant defect in ATPase activity, with a K m(MT) > 10-fold higher than wild type. Since the ATPase activity did not begin to saturate at 25 μM microtubules, we were unable to fit the ATPase data to Michaelis-Menten kinetics, and thus could not determine the K m(MT) or k cat. However, despite this reduced microtubule binding affinity, the 4Glu mutant still showed wild-type velocity in a multiple motor microtubule gliding assay. Thus, despite having substantially different charges on the neck coiled-coil than wild-type kinesin, 5Lys and 4Glu are both functional motor proteins.
In the single molecule motility assay, both mutants moved at the wild-type velocity (Table ). The 5Lys mutant, however, was approximately threefold more processive than wild type (Table and Fig. 2), while the 4Glu mutant was approximately fivefold less processive than wild type. The run lengths of these mutants are consistent with our model that the charge of the neck coiled-coil is an important determinant of kinesin processivity. These results also indicate that kinesin processivity can be engineered (either increased or decreased) with only a few point mutations.
Discussion
In conventional kinesin, processivity is generally believed to result from the hand-over-hand coordination of the two motor domains (Hackney 1994). The key element in the hand-over-hand motility cycle appears to be the neck linker, a small peptide that drives forward motion by reversibly binding to the core motor domain (Rice et al. 1999; Case et al. 2000; Tomishige and Vale 2000; Vale and Milligan 2000). Here, we have extended our understanding of the kinesin mechanism by showing that the neck coiled-coil is an important determinant of kinesin processivity. Mutations of the neck coiled-coil that make it more positively charged increase kinesin processivity, while mutations that make it more negatively charged decrease processivity. The gain in processivity is greatly diminished by high salt concentrations or by removal of the COOH terminus of tubulin, suggesting the involvement of an electrostatic interaction between kinesin's neck coiled-coil and tubulin's COOH terminus. The processivity gain of the positively charged H1Q mutant is abolished at relatively low loads, indicating that the interaction responsible for the increased processivity is weak (Bell 1978; Evans and Ritchie 1997) and distinct from the strong binding mediated by the catalytic core. Thus, we propose that the neck coiled-coil enhances processivity via an electrostatic interaction with the tubulin COOH terminus. Consistent with this hypothesis, the tubulin COOH terminus has previously been shown to interact with kinesin (Tucker and Goldstein 1997) and increase its processivity (Wang and Sheetz 2000), and deletion of the neck coiled-coil greatly reduces processivity (Romberg et al. 1998).
We propose that the neck coiled-coil functions in the kinesin mechanism by serving to tether the kinesin molecule near the microtubule surface (Fig. 4). When moving processively via a hand-over-hand mechanism, a motor must pass through a state where one head is bound to the microtubule and the other is detached and searching for the next binding site (see Figure 6 of Tomishige and Vale 2000). An electrostatic tether that keeps the kinesin molecule close to the microtubule surface would reduce the search space of the unbound head, thereby accelerating its rebinding rate. This would reduce the complete detachment of kinesin from the microtubule by dissociation from the one-head bound state and thereby enhance processivity. This mechanism is consistent with kinetic simulations of the kinesin stepping model of Rice et al. 1999 using published rate constants (Ma and Taylor 1997), which show that 80% of dissociations occur from the one-head bound state, and that a 10-fold increase in the rebinding rate can give rise to a fourfold increase in processivity (Thorn, K.S., and R.D. Vale, unpublished results). The neck coiled-coil–tubulin interaction may also enable a two-head detached kinesin to be retained near the microtubule and undergo one-dimensional diffusion along its surface (Tomishige and Vale 2000). Enhancement of binding rates for protein–protein interactions through favorable electrostatic interactions is well known (Schreiber and Fersht 1996; Janin 1997; Selzer et al. 2000). Therefore, increasing processivity via an electrostatic tether is a plausible and well understood mechanism.
Figure 4.

Model of the kinesin neck coiled-coil interaction with the tubulin COOH terminus. The kinesin neck coiled-coil is shown in blue, and the tubulin COOH-termini are shown in red. The microtubule protofilament is shown in grey, with the alpha tubulin subunit colored lighter than the beta. The state pictured is an intermediate in the hand-over-hand mechanism, where one head is bound and the second is unbound and searching for the next binding site. The coiled-coil protrudes perpendicular to the direction of motion, lying along the microtubule surface, where it can interact with the tubulin COOH termini. The interaction between the neck coiled-coil and the tubulin COOH termini reduces the volume accessible to the unbound head by diffusion and restrains it close to the microtubule surface. We propose that this tethering near the microtubule increases the second head rebinding rate, thereby enhancing kinesin processivity.
Recent structural studies are also consistent with our proposed model. Docking of the kinesin dimer structure into electron microscopic reconstructions of the kinesin-microtubule complex have predicted that the neck coiled-coil lies approximately tangential to the microtubule surface (Hoenger et al. 1998). Such a location is consistent with an interaction between the kinesin neck coiled-coil and the tubulin COOH terminus. Additionally, the tubulin COOH terminus appears to be disordered, as it is not observed in the tubulin crystal structure (Nogales et al. 1998). The flexibility and potential reach (4 nm when fully extended) of the tubulin COOH terminus (Vale 1999) may facilitate interactions with the neck coiled-coil throughout the hand-over-hand cycle.
A similar electrostatic tethering mechanism to the tubulin COOH terminus has recently been proposed to explain the processivity of a chimeric motor protein consisting of the catalytic core of KIF1A fused to the kinesin neck linker (Okada and Hirokawa 2000). In this case, the positively charged tether is a polylysine motif in loop 12 and the electrostatic interaction prevents this monomeric kinesin from diffusing away from the microtubule in its weakly bound state. This function is similar to that proposed here for the kinesin neck coiled-coil, although the mechanical properties of KIF1A and kinesin are very different. In KIF1A, a small power stroke of the motor domain may bias the one-dimensional diffusion resulting from the electrostatic tether (Tomishige and Vale 2000). In the case of conventional kinesin, the tether likely serves to keep the motor domains close to the microtubule, thereby accelerating the rebinding of the unbound motor domain and reducing dissociation events.
This neck coiled-coil electrostatic tethering mechanism may be conserved, as conventional kinesins in organisms ranging from Caenorhabditis elegans to human all have neck coiled-coils that are positively charged (ranging from +2 to +4) and well conserved in sequence, particularly in the first two heptads. Our results with various mutants also suggest that the location and distribution of charge may play some role in modulating processivity. However, fungal conventional kinesins have neutral coiled-coils, yet are processive (Crevel et al. 1999), suggesting that another region of the molecule may perform this tethering role. We also have observed significant changes of kinesin processivity due to charge reversal mutants in loops 8b and 10, which are near where the neck coiled-coil emerges from the motor domain (Thorn, K.S., and R.D. Vale, unpublished results). Thus, regions other than the neck coiled-coil also may be involved in electrostatic tethering interactions with tubulin.
The tethering interaction that we describe may be influenced by posttranslational modification of tubulin. Tubulin can be phosphorylated, acetylated, and polyglutamylated, all of which increase its negative charge (Luduena 1998). In particular, polyglutamylation can add up to seven glutamates to the COOH terminus of tubulin, greatly increasing its negative charge (Redeker et al. 1992; Luduena 1998). The biological functions of these modifications are still poorly understood. This work raises the possibility that such modifications may serve, at least in part, to modulate kinesin processivity.
Although all of our studies have been performed in vitro, we expect that the electrostatic tethering interaction described here is pertinent to in vivo function. In the presence of 120 mM potassium acetate, a reasonable mimic of the in vivo ionic conditions in the cell (Burton 1983), the H1Q mutant was still more processive than wild-type kinesin. Under the restoring force of the optical trap, however, the processivity of the two motors was the same. The forces and barriers acting upon motors in vivo, however, are not well understood (Howard 1996), and they may be quite different from the conditions that a motor experiences in an optical trap. Vesicles moving in cells undergo “saltatory” motion (Rebhun 1972; Sheetz 1999), exhibiting periods of very rapid movement separated by pauses when they are stopped. This complex motion suggests that motors are not subject to a constant load in the cytoplasm provided by viscous drag or compliant elastic elements. Instead, the sudden cessation of movement suggests that motors occasionally encounter inelastic barriers (e.g., large structures) that block its path. Ultra-high processivity may be detrimental under such circumstances since the motor would become trapped by relentlessly attempting to move along the same blocked path. On the other, if the motor(s)/organelle dissociates, it has the opportunity to find a new microtubule track that may circumvent the barrier. Thus, kinesin run length may be an evolutionarily tuned parameter that represents a compromise between long distance travel and the ability to dissociate to negotiate around barriers. The consequences of increasing or decreasing kinesin processivity for motor function in vivo would be interesting to determine, and the altered processivity mutants described here may provide useful tools for such investigations in genetically tractable organisms.
Acknowledgments
We thank Cindy Hart and Hao Zhu for preparing constructs, and Michio Tomishige, Sarah Rice, and Dan Pierce for helpful discussions, and G. Johnson for preparing 4.
K.S. Thorn is supported by a Howard Hughes Predoctoral Fellowship.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; QPD, quadrant photodiode.
References
- Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- Berliner E., Young E.C., Anderson K., Mahtani H., Gelles J. Failure of a single-headed kinesin to track parallel to microtubule protofilaments. Nature. 1995;373:718–721. doi: 10.1038/373718a0. [DOI] [PubMed] [Google Scholar]
- Block S.M., Goldstein L.S., Schnapp B.J. Bead movement by single kinesin molecules with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Brendza R.P., Serbus L.R., Duffy J.B., Saxton W.M. A function for kinesin I in the posterior transport of oskar mRNA and staufen protein. Science. 2000;289:2120–2122. doi: 10.1126/science.289.5487.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton R.F. The composition of animal cellssolutes contributing to osmotic pressure and charge balance. Comp. Biochem. Physiol. 1983;76B:663–671. doi: 10.1016/0305-0491(83)90375-9. [DOI] [PubMed] [Google Scholar]
- Carson J.H., Worboys K., Ainger K., Barbarese E. Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil. Ctyoskelet. 1997;38:318–328. doi: 10.1002/(SICI)1097-0169(1997)38:4<318::AID-CM2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Case R.B., Pierce D.W., Hom-Booher N., Hart C.L., Vale R.D. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 1997;90:959–966. doi: 10.1016/s0092-8674(00)80360-8. [DOI] [PubMed] [Google Scholar]
- Case R.B., Rice S., Hart C.L., Ly B., Vale R.D. Role of the kinesin neck linker and catalytic core in microtubule-based motility. Curr. Biol. 2000;10:157–160. doi: 10.1016/s0960-9822(00)00316-x. [DOI] [PubMed] [Google Scholar]
- Coppin C.M., Pierce D.W., Hsu L., Vale R.D. The load dependence of kinesin's mechanical cycle. Proc. Natl. Acad. Sci. USA. 1997;94:8539–8544. doi: 10.1073/pnas.94.16.8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel I., Carter N., Schliwa M., Cross R. Coupled chemical and mechanical reaction steps in a processive Neurospora kinesin. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5863–5872. doi: 10.1093/emboj/18.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel I.M., Lockhart A., Cross R.A. Kinetic evidence for low chemical processivity in ncd and Eg5. J. Mol. Biol. 1997;273:160–170. doi: 10.1006/jmbi.1997.1319. [DOI] [PubMed] [Google Scholar]
- Evans E., Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S.P., Moyer M.L., Johnson K.A. Alternating site mechanism of the kinesin ATPase. Biochemistry. 1998;37:792–799. doi: 10.1021/bi971117b. [DOI] [PubMed] [Google Scholar]
- Gittes F., Schmidt C.F. Signals and noise in micromechanical measurements. Methods Cell Biol. 1998;55:129–156. doi: 10.1016/s0091-679x(08)60406-9. [DOI] [PubMed] [Google Scholar]
- Goldstein L.S.B., Philp A.V. The road less traveledemerging principles of kinesin motor utilization. Annu. Rev. Cell Dev. Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- Hackney D.D. Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney D.D. Highly processive microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains. Nature. 1995;377:448–450. doi: 10.1038/377448a0. [DOI] [PubMed] [Google Scholar]
- Hagiwara H., Yorifuji H., Sato-Yoshitake R., Hirokawa N. Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J. Biol. Chem. 1994;269:3581–3589. [PubMed] [Google Scholar]
- Hancock W.O., Howard J. Processivity of the motor protein kinesin requires two heads. J. Cell Biol. 1998;140:1395–1405. doi: 10.1083/jcb.140.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J.J., Mahr J., McNally K., Okawa K., Iwamatsu A., Thomas S., Cheesman S., Heuser J., Vale R.D., McNally F.J. Katanin, a microtubule-severing protein, is a novel member of the AAA ATPase superfamily which contains a centrosome using a WD40-containing subunit. Cell. 1998;93:277–287. doi: 10.1016/s0092-8674(00)81578-0. [DOI] [PubMed] [Google Scholar]
- Hoenger A., Sack S., Thormahlen M., Marx A., Muller J., Gross H., Mandelkow E. Image reconstructions of microtubules decorated with monomeric and dimeric kinesinscomparison with x-ray structure and implications for motility. J. Cell Biol. 1998;141:419–430. doi: 10.1083/jcb.141.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. The movement of kinesin along microtubules. Annu. Rev. Physiol. 1996;58:703–729. doi: 10.1146/annurev.ph.58.030196.003415. [DOI] [PubMed] [Google Scholar]
- Howard J. Molecular motorsstructural adaptations to cellular functions. Nature. 1997;389:561–567. doi: 10.1038/39247. [DOI] [PubMed] [Google Scholar]
- Howard J., Hudspeth A.J., Vale R.D. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- Janin J. The kinetics of protein-protein recognition. Proteins Struct. Funct. Genet. 1997;28:153–161. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kozielski F., Sack S., Marx A., Thormahlen M., Schonbrunn E., Biou V., Thompson A., Mandelkow E.-M., Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. MOLSCRIPTa program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 1991;24:946–950. [Google Scholar]
- Kull F.J., Sablin E.P., Lau R., Fletterick R.J., Vale R.D. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luduena R.F. Multiple forms of tubulindifferent gene products and covalent modifications. Int. Rev. Cytol. 1998;178:207–275. doi: 10.1016/s0074-7696(08)62138-5. [DOI] [PubMed] [Google Scholar]
- Ma Y.-Z., Taylor E.W. Interacting head mechanism of microtubule-kinesin ATPase. J. Biol. Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- Mandelkow E., Hoenger A. Structures of kinesin and kinesin-microtubule interactions. Curr. Opin. Cell Biol. 1999;11:34–44. doi: 10.1016/s0955-0674(99)80005-2. [DOI] [PubMed] [Google Scholar]
- Mandelkow E., Johnson K.A. The structural and mechanochemical cycle of kinesin. Trends Biochem. Sci. 1998;23:429–433. doi: 10.1016/s0968-0004(98)01278-x. [DOI] [PubMed] [Google Scholar]
- Meritt E.A., Murphy M.E.P. Raster3D version 2.0a program for photorealistic molecular graphics. Acta Cryst. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- Nogales E., Wolf S.G., Downing K.H. Structure of the αβ tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Okada Y., Hirokawa N. Mechanism of the single-headed processivitydiffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc. Natl. Acad. Sci. USA. 2000;97:640–645. doi: 10.1073/pnas.97.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce D.W., Hom-Booher N., Otsuka A.J., Vale R.D. Single-molecule behavior of monomeric and heterotrimeric kinesins. Biochemistry. 1999;38:5412–5421. doi: 10.1021/bi9830009. [DOI] [PubMed] [Google Scholar]
- Pierce D.W., Vale R.D. Assaying processive movement of kinesin by fluorescence microscopy. Methods Enzymol. 1998;298:154–171. doi: 10.1016/s0076-6879(98)98016-8. [DOI] [PubMed] [Google Scholar]
- Prahlad V., Yoon M., Moir R.D., Vale R.D., Goldman R.D. Rapid movements of vimentin on microtubule trackskinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press W.H., Teukolsky S.A., Vetterling W.T., Flannery B.P. Numerical recipes in C 1992. Cambridge University Press; Cambridge, UK: pp. 623–626 [Google Scholar]
- Rebhun L.I. Polarized intracellular particle transportsaltatory movements and cytoplasmic streaming. Int. Rev. Cytol. 1972;32:93–137. doi: 10.1016/s0074-7696(08)60339-3. [DOI] [PubMed] [Google Scholar]
- Redeker V., Melki R., Prome D., J.-P. Le Caer, and J. Rossier Structure of tubulin C-terminal domain obtained by subtilisin treatment. FEBS Lett. 1992;313:185–192. doi: 10.1016/0014-5793(92)81441-n. [DOI] [PubMed] [Google Scholar]
- Rice S., Lin A.W., Safer D., Hart C.L., Naber N., Carragher B.O., Cain S.M., Pechatnikova E., Wilson-Kubalek E.M., Whittaker M. A structural change in the kinesin motor that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- Romberg L., Pierce D.W., Vale R.D. Role of the kinesin neck region in processive microtubule-based motility. J. Cell Biol. 1998;140:1407–1416. doi: 10.1083/jcb.140.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack S., Muller J., Marx A., Thormahlen M., Mandelkow E.M., Brady S.T., Mandelkow E. X-ray structure of motor and neck domains from rat brain kinesin. Biochemistry. 1997;36:16155–16165. doi: 10.1021/bi9722498. [DOI] [PubMed] [Google Scholar]
- Schreiber G., Fersht A.R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- Selzer T., Albeck S., Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat. Struct. Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- Serrano L., Avila J., Maccioni R.B. Controlled proteolysis of tubulin by subtilisinlocalization of the site for MAP2 interaction. Biochemistry. 1984;23:4675–4681. doi: 10.1021/bi00315a024. [DOI] [PubMed] [Google Scholar]
- Sheetz M.P. Motor and cargo interactions. Eur. J. Biochem. 1999;262:19–25. doi: 10.1046/j.1432-1327.1999.00340.x. [DOI] [PubMed] [Google Scholar]
- Shimizu T., Thorn K.S., Ruby A., Vale R.D. ATPase kinetic characterization and single molecule behavior of mutant human kinesin motors defective in microtubule-based motility. Biochemistry. 2000;39:5265–5273. doi: 10.1021/bi9928344. [DOI] [PubMed] [Google Scholar]
- Stewart R.J., Semerjian J., Schmidt C.F. Highly processive motility is not a general feature of the kinesins. Eur. Biophys. J. 1998;27:353–360. doi: 10.1007/s002490050142. [DOI] [PubMed] [Google Scholar]
- Svoboda K., Block S.M. Biological applications of optical forces Annu. Rev. Biophys. Biomol. Struct. 23 1994. 247 285a [DOI] [PubMed] [Google Scholar]
- Svoboda K., Block S.M. Force and velocity measured for single kinesin molecules Cell 77 1994. 773 784b [DOI] [PubMed] [Google Scholar]
- Svoboda K., Schmidt C.F., Schnapp B.J., Block S.M. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 1993;365:721–727. doi: 10.1038/365721a0. [DOI] [PubMed] [Google Scholar]
- Tomishige M., Vale R.D. Controlling kinesin by reversible disulfide cross-linkingidentifying the motility-producing conformational change. J. Cell. Biol. 2000;151:1081–1092. doi: 10.1083/jcb.151.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker C., Goldstein L.S.B. Probing the kinesin-microtubule interaction. J. Biol. Chem. 1997;272:9481–9488. doi: 10.1074/jbc.272.14.9481. [DOI] [PubMed] [Google Scholar]
- Vale R.D. Millenial musings on molecular motors. Trends Cell Biol. 1999;9:M38–M42. [PubMed] [Google Scholar]
- Vale R.D., Fletterick R.J. The design plan of kinesin motors. Annu. Rev. Cell Dev. Biol. 1997;12:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale R.D., Funatsu T., Pierce D.W., Romberg L., Harada Y., Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale R.D., Milligan R.A. The way things move looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- Visscher K., Schnitzer M.J., Block S.M. Single kinesin molecules studied with a molecular force clamp. Nature. 1999;400:184–189. doi: 10.1038/22146. [DOI] [PubMed] [Google Scholar]
- Wang Z., Sheetz M.P. The C-terminus of tubulin increases cytoplasmic dynein and kinesin processivity. Biophys. J. 2000;78:1955–1964. doi: 10.1016/S0006-3495(00)76743-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlke G., Ruby A.K., Hart C.L., Ly B., Hom-Booher N., Vale R.D. Microtubule interaction site of the kinesin motor. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]
- Yang J.T., Saxton W.M., Stewart R.J., Raff E.C., Goldstein L.S. Evidence that the head of kinesin is sufficient for force generation and motility in vitro. Science. 1990;249:42–47. doi: 10.1126/science.2142332. [DOI] [PubMed] [Google Scholar]
- Young E.C., Mahtani H.K., Gelles J. One-headed kinesin derivatives move by a nonprocessive low-duty ration mechanism unlike that of two-headed kinesin. Biochemistry. 1998;37:3467–3479. doi: 10.1021/bi972172n. [DOI] [PubMed] [Google Scholar]