

Abstract
Although TGF-β isoforms (TGF-β1-3) display very similar biochemical characteristics in vitro, it has been determined that they demonstrate different or even opposing effects in vivo. During embryogenesis, TGF-βs play important roles in several developmental processes. Tgfb3 is strongly expressed in the prefusion palatal epithelium, and mice lacking Tgfb3 display a cleft of the secondary palate. To test whether the effect of TGF-β3 in palatogenesis is isoform-specific in vivo, we generated a knockin mouse by replacing the coding region of exon1 in the Tgfb3 gene with the full length Tgfb1 cDNA, which resulted in the expression of Tgfb1 in the Tgfb3 expressing domain. The homozygote knockin mice display a complete fusion at the mid-portion of the secondary palate, while the most anterior and posterior regions fail to fuse appropriately indicating that in vivo replacement of TGF-β3 with TGF-β1 can only partially correct the epithelial fusion defect of Tgfb3 knockout embryos. Palatal shelves of Tgfb1 knockin homozygote mice adhere, intercalate, and form characteristic epithelial triangles. However, decreased apoptosis in the midline epithelium, slower breakdown of the basement membrane and a general delay in epithelial fusion were observed when compared to control littermates. These results demonstrate an isoform-specific role for TGF-β3 in the palatal epithelium during palate formation, which cannot be fully substituted with TGF-β1.
Keywords: TGF-β, palatogenesis, cleft palate, Transforming growth factor, development, mouse
Introduction
Cleft palate and cleft lip are among the most common congenital birth defects in humans occurring once in every 500-700 births (Murray, 2002;Rice, 2005). While our understanding of molecular control of palate formation (=palatogenesis) has greatly improved during the last decade, many questions still remain unanswered (Chai and Maxson, Jr., 2006;Schutte and Murray, 1999). The mammalian secondary palate develops from bilateral outgrowth of palatal shelves from the maxillary processes. Two key cell types underlying palatogenesis are epithelial cells derived from the pharyngeal ectoderm and mesenchymal cells derived mostly from the neural crest (Dudas and Kaartinen, 2005;Gritli-Linde, 2007;Schutte and Murray, 1999). The interactions between the ectoderm and the underlying mesenchyme promote the vertical growth of palatal shelves beside the tongue. Accompanied by the enlargement of the lower jaw and forward displacement of the tongue, palatal shelves elevate to a horizontal position above the tongue. Subsequently, they make contact, become adherent, and fuse (Ferguson, 1988).
Many growth factors, signaling pathways and nuclear factors have been implicated in palatogenesis (Dudas and Kaartinen, 2005;Gritli-Linde, 2007;Hilliard et al., 2005). Among them, TGF-βs have been shown to play important roles in regulating epithelial-mesenchymal interactions leading to appropriate growth and fusion of palatal shelves (Nawshad et al., 2005); Tgfb2 is expressed in the palatal mesenchyme, whereas Tgfb1 and Tgfb3 expression was proposed to be limited to the pre-fusion palatal midline epithelium (Fitzpatrick et al., 1990;Pelton et al., 1990). Both Tgfb2 and Tgfb3 knockout mice display cleft palate phenotypes, which are caused by different pathogenetic mechanisms (Kaartinen et al., 1995;Martinez-Alvarez et al., 2000;Proetzel et al., 1995;Sanford et al., 1997;Taya et al., 1999). Palatal shelves of Tgfb2 knockout mice fail to grow and elevate, suggesting a role for TGF-β2 in the growth of the palatal mesenchyme. Interestingly, palatal shelves of Tgfb3 knockout mice elevate and become apposed, but fail to fuse leading to cleft palate. Although TGF-β1 is also expressed in the epithelial tips of prefusion palatal shelves, Tgfb1 null mutants do not display a cleft palate phenotype (Kulkarni et al., 1993;Shull et al., 1992).
TGF-β isoforms share a high degree of homology in the mature domain of the growth factor and are qualitatively indistinguishable in in vitro cell culture assays (Roberts and Sporn, 1992). During embryogenesis, TGFβ isoforms display overlapping but distinct expression patterns, and they are often expressed at sites where commitment of a specific cell fate occurs (Akhurst et al., 1990;Pelton et al., 1990). Non-overlapping phenotypes associated with the null mutation of different TGF-β isoforms suggest that biological activities of TGF-β isoforms may be different in vivo. Indeed, functional analyses of the TGF-β isoforms ex vivo have revealed that they have different, or even opposing, biological activities in certain model systems (Foitzik et al., 1999;Li et al., 1999;Shah et al., 1995). In a skin organ culture model, TGF-β1 treatment resulted in the inhibition of hair follicle development and keratinocyte proliferation, whereas TGF-β2 treatment potently stimulated hair follicle formation and concomitantly produced epidermal hyperplasia (Foitzik et al., 1999). In skin grafts, TGF-β3, but not TGF-β1, was shown to protect keratinocytes against TPA-induced cell death (Li et al., 1999). Moreover, in a rat cutaneous wound healing model, TGF-β1 and TGF-β2 were shown to be involved in cutaneous scarring, while TGF-β3 acted as an anti-scarring agent (Shah et al., 1995).
In this study, we tested whether the role of TGF-β3 in vivo during palatal fusion is isoform specific by replacing the coding region of exon1 in the Tgfb3 gene with Tgfb1 cDNA using gene-targeting technology. The resulting Tgfb1-knockin (Tgfb1-KI) mice, which express Tgfb1 in the Tgfb3 locus and are null for Tgfb3, displayed a remarkable improvement in the cleft palate phenotype when compared to Tgfb3 null mutants. A mid-portion of the secondary palate demonstrated a complete fusion while the anterior secondary palate still failed to fuse to the primary palate, and the posterior palate displayed a submucous cleft. The rate of apoptosis was attenuated and breakdown of the basement membrane was reduced in Tgfb1-KI samples when compared to wild-type littermates. These findings demonstrate a partial isoform-specific role for TGF-β3 in the palatal epithelium during the development, which cannot be fully compensated by TGF-β
Materials and Methods
Gene targeting of Cre cDNA into Tgfb3 locus (Tgfb3-Cre mice)
A targeting vector was generated by combining a 9-kb clone isolated from the B6xCBA (F1) mouse genomic library (right arm), a 1.6- kb PCR fragment amplified from the R1-ES cell DNA (left arm), and a cassette containing a promoterless Cre gene and a neomycin-resistant gene driven by the phosphoglycerate kinase promoter (PGK-Neo) to replace the coding region of the ATG containing exon 1 of the Tgfb3 gene. Briefly, a 1.6-kb PCR-amplified 5' XbaI-BamHI genomic fragment, the Cre-pgk-Neo, and a 10-kb 3' NotI-EcoRI genomic fragment (generated by fusing a 2.6-kb Not1-Kpn1 PCR fragment with a 7.6-kb KpnI-EcoRI fragment from the library clone) containing exons 2 and 3 were subcloned into the pKOdt plasmid (Stratagene). R1-ES cells were cultured, electroporated and screened as previously described (Kaartinen et al., 1995;Kaartinen et al., 2004). Targeted colonies were initially identified by PCR amplification of a 2-kb fragment using a 5'-arm outside sense primer1 (GCATGCTCCAGACTGCCTTGGGA) and a Cre anti-sense primer 2 (CCTCATTCACTCGTTGCATCGACCGG). Southern blot analyses of genomic DNAs, digested with both ClaI and KpnI, were used to confirm homologous recombination of the targeted allele using the Cre and Neo probes. A total of 7 positive homologous recombinant ES cells clones were obtained from 260 G418-resistant clones. ES cells from two independent targeted clones were microinjected into C57/BL6J blastocysts by the transgenic core facility at the University of Southern California. Three male chimeras (male/female ratio was 9:3) were used to produce heterozygote Tgfb3-Cre mice.
Gene targeting of Tgfb1 cDNA into Tgfb3 locus (Tgfb1-KI mice)
A 1.6-kb PCR-amplified XbaI-BamHI Tgfb3 genomic fragment, a Tgfb1 cDNA with phosphoglycerate kinase (pgk) polyA sequence, a loxP-floxed neomycin-resistant gene driven by the pgk promoter, and a 10-kb PCR-amplified NotI-EcoRI genomic fragment containing exons 2 and 3 of Tgfb3 gene were subcloned into a pKOdt vector (Stratagene). Targeted colonies were initially identified by PCR amplification of a 2-kb fragment using a 5'-arm outside sense primer 1 (GCATGCTCCAGACTGCCTTGGGA) and a Tgfb1 anti-sense primer 3 (GGAGTGGGAGCAGAAGCGGCAGTAGCC). Southern blot analysis of genomic DNAs digested with NheI was used to confirm homologous recombination of the targeted allele using a Neo probe. One positive homologous recombinant ES cell clone was obtained from 192 G418-resistant clones. This cell line produced two highly chimeric males, both of whom were germ line transmitters.
Mouse breeding, embryo isolation, and genotype assays
ROSA26 Cre reporter mice were obtained from the Jackson laboratory (Bar Harbor, ME, USA). Tgfb3 −/− mice were previous made in the laboratory (Kaartinen et al., 1995). Tgfb3-Cre and Tgfb1-K mice were generated as described above. All mice were bred to have a mixed genetic background. Mice were mated during the dark period of the controlled light cycle and female mice acquiring vaginal plugs were designated as day 0. At the time interval indicated in respective figures (E14 to E17), females were euthanized by CO2 and embryos were collected in PBS (Invitrogen) followed by further analyses. All studies and procedures performed on mice were carried out at the Animal Care Facility of the Saban Research Institute, and were approved by the CHLA Animal Care and Use Committee (IACUC). Mice tail biopsies were collected and genotyped by PCR. For the Tgfb1-KI allele the following oligos were used; sense: GAGTCAGAGCCCGGCAGAACCTGTT, antisense: GCCGGTTACCAAGGTAACGCCAGGA. Oligos for genotyping Cre and Tgfb3-null alleles have been described elsewhere (Kaartinen et al., 2004;Shi et al., 1999).
Histological analyses, X-gal staining, scanning electron microscopy and In situ Hybridization
Mouse embryos or tissues were fixed in 4% formaldehyde for 24 h, and paraffin sections were stained with hematoxylin-eosin using standard procedures. Frozen sections of embryos were stained for lacZ activity as described (Hogan et al., 1994). For scanning electron microscopy analysis, samples were fixed in 4% formaldehyde plus 0.5% glutaraldehyde for 48 hours and processed at the Tissue Imaging Core in DEI/USC Norris Cancer Center. Pictures were recorded using a scanning electron microscope with computerized digital capture (Hitachi S-570). RNA in situ hybridization was performed as previous described (Moorman et al., 2001). A 455-bp fragment of the mouse Tgfb1 cDNA (nts 206-661) was subcloned into pBSks, digested with KpnI, and transcribed with T3 RNA polymerase to prepare the anti-sense probe, according to the manufacturers' protocol (Boehringer Mannheim).
Organ culture, immunostainings, and TUNEL assays
Palatal shelves were cultured for 48 hours in BGJb medium (Invitrogen) (Kaartinen et al., 1997) and analyzed with serial sectioning. Paraffin sections were stained for both laminin and K14 using an anti-laminin antibody (1:250, Sigma) and an anti-K14 antibody (1:250, Lab Vision) (Harlow E and Lane D, 1988). Images from the anti-laminin staining (red color) and the anti-K14 staining (green color) were superimposed using Adobe Photoshop software. Immunostaining for phospho-Smad2/3 was done in paraffin sections using Phospho-Smad2 Ser465/467 antibody (1:200, Chemicon), after antigen retrieval by boiling in 10 mM Tris buffer (pH 10). Apoptotic cells were detected in paraffin sections using DeadEnd Fluorometric TUNEL system (Promega).
Western Blotting
Tips of palatal shelves were dissected out from E14 embryos (n=3 for each genotype) and frozen in an ethanol-dry ice bath. Samples were lysed at 90°C for 10 minutes in 40 μl 1%SDS buffer (50 mM Tris pH 7.5, 1% SDS, 1 mM EDTA, 10% Glycerol) plus protease inhibitors (Aprotinin 2μg/ml, Leupeptin 20μg/ml, Pepstatin A 1μM, PMSF 1 mM) and phosphatase inhibitors (NaF 10 mM, Sodium Vanadate 1mM). Five μg of protein per lane was resolved by 9% SDS PAGE and electroblotted onto PVDF membrane (Hybond-P, Amersham). The membrane was blocked and incubated either overnight at 4°C with diluted anti-phospho Smad2 antibody (Cell signaling, 1:1000 dilution in blocking solution) or 1 hour with diluted anti-Smad2 antibody (Zymed, 1:500 dilution in blocking buffer). The membrane was then probed with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody and developed with HRP substrate (Immobilon Western, Millipore). The chemiluminescent signal was exposed to X-ray films and the signal intensity was quantified by densitometric analysis using Un-Scan it Gel software (Silk Scientific, Inc.). Signal intensities of phospho-Smad2/3 bands from wild-type and mutant samples were normalized to signal intensities of Smad2, using the total pixel value obtained by the ‘Un-Scan it’ gel software.
Total RNA preparation, reverse transcription, and gene expression analyses
Total RNA was isolated from tips of E14 palatal shelves using the RNeasy mini kit (Qiagen), and cDNAs were synthesized using the Omniscript reverse transcription kit (Qiagen) according to the manufacturers' protocols. Real-time PCR was carried out using a LightCycler 1.5 (Roche) with the LightCycler Taqman Master mix and universal probe/primer sets for the following genes: Tgfb1 (mouse universal probe #72, Left primer TGGAGCAACATGTGGAACTC, and right primer GTCAGCAGCCGGTTACCA), Tgfb3 (mouse universal probe #25, left primer CCCTGGACACCAATTACTGC, and right primer TCAATATAAAGGGGGCGTACA), Mmp-13 (mouse universal probe # 62, left primer CAGTCTCCGAGGAGAAACTATGA, and right primer GGACTTTGTCAAAAAGAGCTCAG), and β-actin (universal probe #106, left primer TGACAGGATGCAGAAGGAGA, and right primer CGCTCAGGAGGAGCAATG). Relative quantification of gene expression between samples, and absolute quantification of gene expression within a sample were done using the LightCycler software 4.0. The amplification efficiencies, which were calculated from standard curves generated by dilutions of cDNA samples at known concentrations; for Tgfb1, Tgfb3, and β-actin, were 1.803, 1.911, and 1.742, respectively.
CHO cell transfection and luciferase reporter assay
The mouse TGF-β1 cDNA was purchased from ATCC (#3586216), PCR-amplified, and cloned into the pGK plasmid. Three independent bacterial clones were chosen to transfect the CHO cells using lipofectamine 2000 (Invitrogen). The supernatant from transfected cells was diluted with an equal volume of DMEM and applied to MLE cells stably integrated with plasminogen activator inhibitor-1/luciferase reporter plasmid. Luiciferase activity was measured by using the Luciferase Reporter Assay System (Promega) with a Luminometer.
Results
Generation of Tgfb1-KI and control Tgfb3-Cre mice
To test whether TGF-β1 is able to substitute for TGF-β3 when expressed in vivo in the Tgfb3-expressing domain, we used gene targeting to replace the coding region in exon 1 of the Tgfb3 gene with a full-length cDNA encoding the mouse Tgfb1 gene followed by the loxP-Pgk-Neo-loxP cassette (Fig. 1A). Since the Tgfb1-KI allele lacks the sequences encoded by Tgfb3 exon 1, and since all the promoter and regulatory elements of the Tgfb3 gene were essentially preserved, we predicted that i) Tgfb1 expression would faithfully recapitulate the endogenous expression pattern of the Tgfb3 gene and ii) the Tgfb1-KI allele would be a true null allele for Tgfb3. First, we verified that our PCR-generated mouse Tgfb1 cDNA clones produced a biologically active ligand. Culture media collected from CHO cell cultures transfected with the Tgfb1 expression vector were analyzed by the plasminogen activator inhibitor-1/luciferase reporter assay (Abe et al., 1994). The positive clone #1 was used to construct the targeting vector, which in turn was used to generate chimeric mice and subsequently mice heterozygous for the Tgfb1 knockin allele (Tgfb1-KI) (Fig. 1C).
Fig. 1. Generation of Tgfb1 knockin and Cre knockin mice.
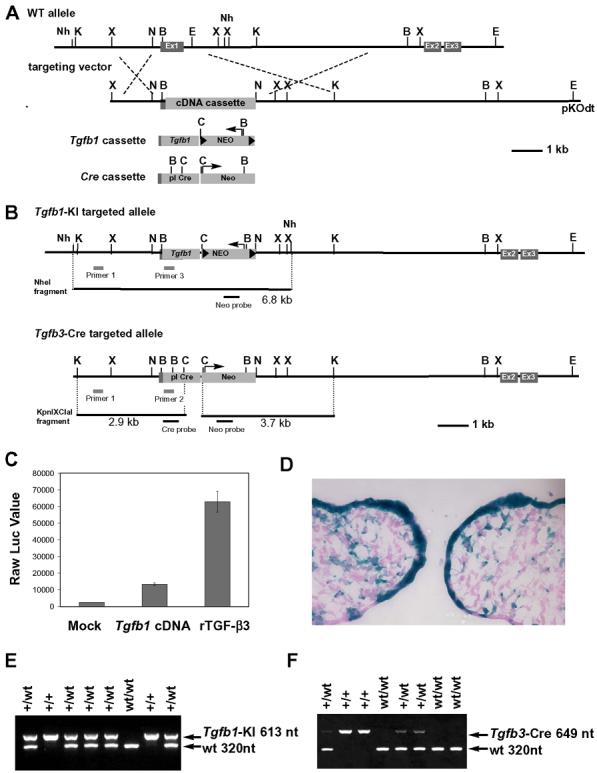
(A) A schematic diagram of Tgfb3 allele and targeting vectors for Tgfb1 knockin (Tgfb1-KI) and Cre knockin (Tgfb3-Cre). Ex1, Ex2, Ex3: Exon1, Exon2, and Exon3; Tgfb1 cassette: Tgfb1 cDNA with phosphoglycerate kinase polyA sequence and the pgk promoter driven neomycin-resistant gene flanked by loxP sites; Cre cassette: the promotorless Cre gene with the neomycin-resistant gene driven by the pgk promoter; Nh: NheI, K: KpnI, X: XhoI, B: BamHI, E: EcoRI, C: ClaI, N: NotI. (B) A schematic diagram for the targeted alleles. PCR and Southern blot screening strategies are illustrated. (C) Luciferase reporter assay for PCR-amplified Tgfb1 cDNA. MLE cells stably integrated with plasminogen activator inhibitor-1/luciferase reporter plasmid were cultured with supernatants from mock transfected cells (Mock) or Tgfb1 cDNA expression plasmid-transfected cells (Tgfb1 cDNA). The supernatant containing recombinant TGF-β3 at 10 ng/ml served as a positive control (rTGF-β3). Data are presented as raw luciferase values against each supernatant, n=2. (D) A lineage tracing assay for Tgfb3 expressing cells in the prefusion palate at E14: X-gal staining of the frontal frozen section from Tgfbr3-Cre+/−;R26R+/− embryos. (E-F) Genotyping of Tgfb1-KI and Tgfb3-Cre mice using PCR analysis.
To prove that the generated Tgfb1-KI allele is a true null allele for Tgfb3, and that a resultant phenotype is due to an in vivo replacement of TGF-β3 with TGF-β1, we generated another mouse line by inserting the promoterless Cre recombinase-Pgk-Neo cassette into Tgfb3 exon1 by homologous recombination essentially as described above for the Tgfb1 knockin allele (Fig. 1A). Concordant with the published expression pattern for Tgfb3 (Fitzpatrick et al., 1990;Pelton et al., 1990), Cre-induced recombination was strikingly strong in the prefusion palatal epithelium (Fig. 1D). Moreover, the homozygote Tgfb3-Cre mice displayed a cleft palate phenotype identical to that of the previously described Tgfb3 knockout mice (Kaartinen et al., 1995;Proetzel et al., 1995) confirming that homozygous Tgfb3-Cre mice were true null mutants for the Tgfb3 gene, and suggesting that the analogous Tgfb1-KI allele is a true null allele (Fig. 2).
Fig. 2. Scanning electron microscopic and histological analyses of palates from wildtype, Tgfb3-Cre homozygote, and Tgfb1 knockin homozygote embryos.
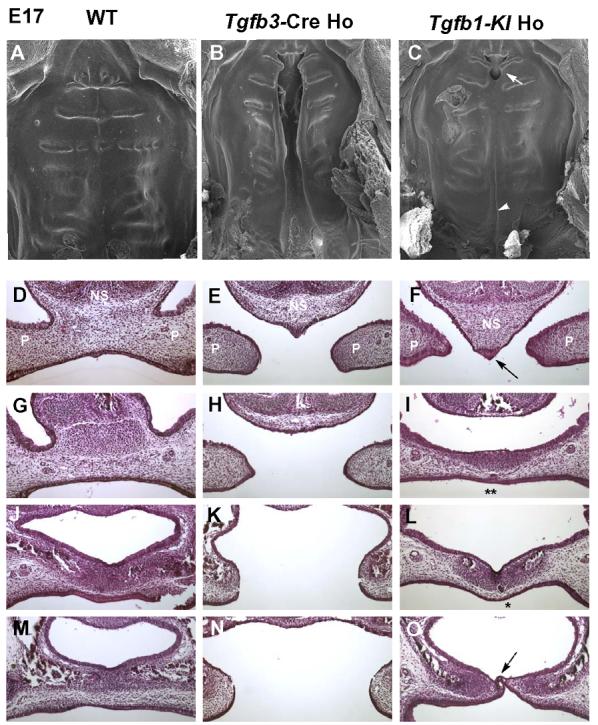
(A-C) Scanning electron microscopic images of palate from wildtype (WT), Tgfb3-Cre homozygote (Tgfb3-Cre Ho), and Tgfb1 knockin homozygote (Tgfb1KI Ho) mice at E17. A wildtype specimen displays a fully fused palate. Tgfb3-Cre homozygote mouse exhibits a complete bilatateral clefting of the secondary palate, whereas the Tgfb1-KI homozygote mouse only has a cleft on the junction of primary and secondary palate (arrow) and a submucous cleft in the posterior region (arrowhead). (D-O) Samples from E17 embryos were sectioned serially in the frontal orientation and stained by hematoxylin-eosin. Sections at four different levels are shown for wildtype (D, G, J, M), Tgfb3-Cre Ho (E, H, K, N), and Tgfb1-KI Ho (F, I, L, O) embryos. In the Tgfb1-KI homozygote sample, the nasal septum fails to fuse with palatal shelves in the anterior region of the palate (F, arrow); palatal shelves fused in the middle region of the palate with the epithelial islands (I, L; single and double stars), indicating an ongoing process of palatal fusion; a submucous cleft is present in the posterior region of the palate (O, dotted arrow). In the wildtype sample, the palatal shelves demonstrate a complete fusion. In the Tgfb3 knockout sample, the palatal shelves display a failure to fuse along the anterior-posterior axis. NS: nasal septum; P: palatal shelf.
In vivo replacement of TGF-β3 with TGF-β1 leads to a partial rescue of the Tgfb3- null cleft palate phenotype
Crossing the Tgfb1-KI heterozygote male mice with heterozygote female mice produced an expected Mendelian ratio of wild-types, heterozygotes and homozygotes at E14 (n=103) and at E17 (n=36). However, the homozygote knockin mice failed to suckle as demonstrated by a complete lack of milk in the stomach (data not shown). At two weeks of age we could not detect any surviving homozygote Tgfb1-KI mice (n=45). While a superficial macroscopic examination of the homozygote Tgfb1-KI newborn mice failed to demonstrate any obvious cleft palate phenotypes, detailed scanning electron microscopy and histological analyses exposed a submucous posterior cleft and a failure of the primary palate to fuse to the secondary palate (Fig 2). It is noteworthy that while the nasal septum failed to fuse with the anterior palate in homozygote Tgfb1 knockin embryos, a large segment of the mid-palate displayed complete epithelial fusion with continuous palatal mesenchyme; a phenotype seen in neither conventional Tgfb3 knockouts (Kaartinen et al., 1995;Proetzel et al., 1995) nor epithelial specific Tgfb receptor knockouts (Dudas et al., 2006;Xu et al., 2006).
Tgfb1 is strongly expressed in the tips of palatal shelves in Tgfb1 knockin mice
The partial rescue seen in the Tgfb1 knockin mice could result from a failure to achieve an appropriate expression level of Tgfb1 in the knockin mice. Therefore, we first carefully quantified Tgfb1 and Tgfb3 mRNAs in developing palates of embryos derived from the crossing of Tgfb1-KI heterozygote mice. The tissues composed of both the palatal epithelium and mesenchyme were dissected from distal tips of prefusion palatal shelves of wild-type, Tgfb1-KI heterozygote and homozygote embryos at E14 (Fig. 3A). Total RNA was isolated and reverse-transcribed, and the obtained cDNA was then quantified using the TaqMan real-time PCR approach (Fig. 3B). In embryos heterozygous for both the Tgfb1 knockin and the endogenous Tgfb3 alleles, the level of Tgfb1 mRNA in the distal edges of palatal shelves was approximately 50% higher, and the level of Tgfb3 mRNA was approximately 2-fold lower, than the level in corresponding wild-type samples. In contrast, in homozygous Tgfb1-KI mice, there was no detectable Tgfb3 expression, while the level of Tgfb1 mRNA was approximately 2-fold higher than that in the corresponding wild-type sample. These results indicated that mice with the Tgfb1 knockin allele can express Tgfb1 mRNA under the control of the endogenous Tgfb3 promoter.
Fig. 3. Gene expression analyses of the Tgfb1 knock-in mice.
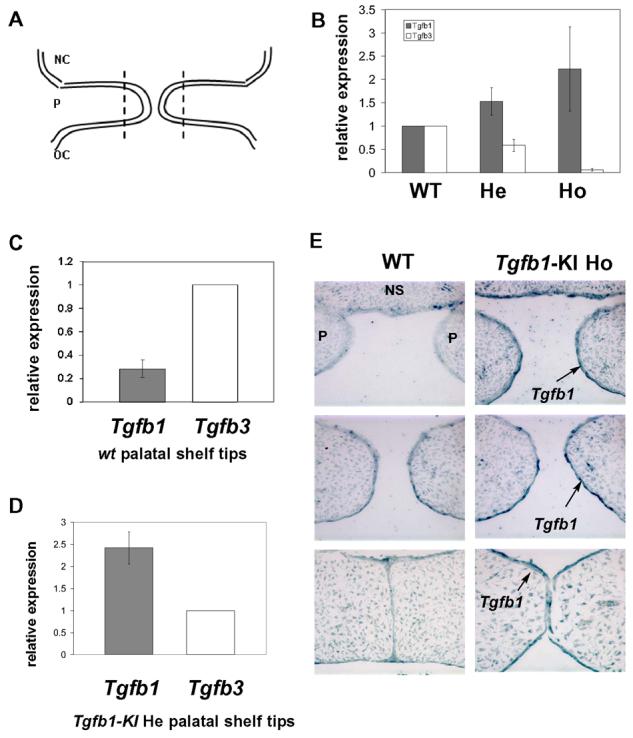
(A) A schematic presentation of prefusion palatal shelves. Distal tips of prefusion palatal shelves to be dissected for real-time PCR analysis are indicated by a dotted line. NC: nasal cavity, P: palatal shelf, OC: oral cavity. (B) Real-time quantitative PCR analysis for the expression of Tgfb1 and Tgfb3 in tips of palatal shelves dissected from wildtype (WT), Tgfb1-KI heterozygous (He), and Tgfb1KI homozygous (Ho) embryos at E14. In embryos heterozygous for the Tgfb1-KI allele, there is a 50% decrease in Tgfb3 expression and a concomitant 50% increase in Tgfb1 expression when compared to the wild-type sample. In embryos homozygous for the Tgfb1-KI allele, Tgfb3 expression is barely detectable, while expression of Tgfb1 has increased 2-fold when compared to the wild-type sample. Bar graph: mean +/− s.d., n=3; The expression level of Tgfb1 and Tgfb3 in the wildtype specimen has been arbitrarily set at 1; results shown are from three independent litters. (C) Real-time absolute quantitative PCR analysis for gene expression of Tgfb1 and Tgfb3 in the tips of wildtype (WT) prefusion palatal shelves at E14. Bar graph: mean +/-s.d., n=4; Tgfb3 expression has been arbitrarily set at 1. (D) Real-Time absolute quantitative PCR analysis for the expression level of Tgfb1 and Tgfb3 in tips of palatal shelves dissected from Tgfb1KI heterozygotes at E14. Bar graph: mean +/− s.d., n=5; Tgfb3 expression has been arbitrarily set at 1. (E) In situ hybridization for Tgfb1 in the prefusion palatal shelves of the wild-type and Tgfb1-KI homozygote embryos (n=3). E14 embryos were processed for serial sectioning in a transverse orientation. Sections from the anterior, the middle, and the posterior region of the palate were hybridized with the Tgfb1 anti-sense probe. The palatal epithelium shows a strong Tgfb1 hybridization signal along the entire anterior-posterior axis (arrow).
Next, we dissected tips of prefusion palatal shelves at E14.0 as outlined above and analyzed them using qPCR with an absolute quantification method to compare Tgfb1 and Tgfb3 expression levels in wild-types and Tgfb1-KI heterozygotes. Based on standard curves, absolute quantification revealed that the total level of Tgfb3 expression in wild-type palatal shelves was about 3-fold higher than that of Tgfb1 (Fig. 3C). An absolute quantification assay for Tgfb1 and Tgfb3 mRNA expression was also applied to the Tgfb1-KI heterozygote samples and the result indicated that Tgfb1 mRNA was expressed at a level about 2.5-fold higher than Tgfb3 mRNA (Fig. 3D). In summary, these analyses revealed that the failure of palatal shelves to fuse appropriately in the homozygote samples was not caused by defective global expression of Tgfb1 at the edges of the prefusion palatal shelves.
To verify that the fusion failure in the anterior and posterior palate was not a result of regional defects in Tgfb1 expression, we performed in situ hybridization analyses on serial sections of the control and Tgfb1-KI homozygote embryos at three different levels (Fig. 3E). The hybridization signal detected in the palatal epithelium was stronger in the homozygote knockin samples than in control samples, and demonstrated comparable intensity along the entire anterior-posterior axis.
Tgfb1-KI homozygotes and wild-type controls display a comparable level of canonical TGF-β signaling activity
Upon TGF-β binding, TGF-β type II receptors, which are constitutively active, are brought into a complex with type I receptors and subsequently activate type I receptors, resulting in the phosphorylation and activation of intracellular effectors Smad2 and Smad3. To confirm that the Tgfb1-KI allele generated a functional TGF-β1 protein, we analyzed the phosphorylation level of Smads2 and -3 (pSmad2/3) in the midline epithelium of E14 palatal shelves from wild-type, Tgfb3-Cre homozygote, and Tgfb1-KI homozygote embryos by immunostaining with an anti-pSmad2/3 antibody (Fig. 4). The pSmad2/3-positive staining was seen in the periderm and midline epithelium in wild-type and Tgfb1-KI homozygote embryos, whereas staining in the midline epithelium of Tgfb3-Cre homozygote embryos was barely detectable. Importantly, we did not observe any significant differences in the phosphorylation level of Smad2/3 in the midline epithelium between wild-type and Tgfb1-KI homozygote embryos in the anterior, mid- or posterior palate (Fig. 4A). In addition, we dissected tips of prefusion palatal shelves from wild-type, Tgfb1-KI and Tgfb3-Cre homozygote embryos and analyzed their Smad2/3 phophorylation levels using a quantitative western blot analysis (Fig. 4B and C). While phosphorylation between wild-type and Tgfb1-KI samples was comparable, the homozygote Tgfb3-Cre samples consistently displayed about 10% reduction in phosphorylation, when compared to the wild-type controls or Tgfb1-KI samples. The relatively strong, albeit reduced Smad2/3 phosphorylation in Tgfb3 null mutant samples is likely due to the fact that most of the cells in the harvested tissues were of mesenchymal origin, which also exhibit phosphorylated Smad2/3, as can be seen in Fig. 4A. To conclude, these results suggest that the Tgfb1-KI allele can express a functional protein, and that in vivo replacement of TGF-β3 with TGF-β1 results in a comparable level of canonical TGF-β signaling activity.
Fig. 4. Comparison of the phospho-Smad2/3 state in the midline palatal epithelium.
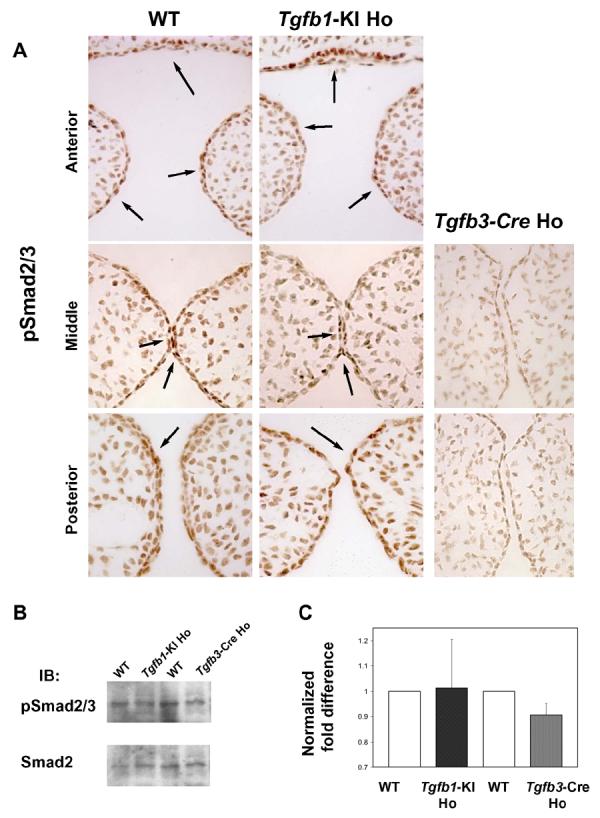
A, Paraffin sections from wild-type (left panel), Tgfb3-Cre homozygote (Tgfb3-Cre Ho, right panel), and Tgfb1-KI homozygote (Tgfb1-KI Ho, mid panel) were immunostained with a phospho-specific antibody against phosphorylated Smad2 and Smad3 (anterior to posterior from top to bottom). In wild-type and Tgfb1-KI Ho samples, cells in the periderm, palatal epithelium and in the nasal septum (epithelium) display a strong positive signal (arrows left and mid panels), whereas in the Tgfb3-Cre sample (right panel) the pSmad2/3 staining is undetectable (n=3).
B, Tissues harvested from tips of prefusion palatal shelves were analyzed for Smad2/3 phosphorylation using western blotting with anti-phopho-Smad2/3 antibodies. C, The histogram represents the relative quantitation of the scanned images. The wild-type (control) phospho-Smad2/3 to Smad2/3 ratio was arbitrarily set at 1 (n=3).
Palatal shelves of Tgfb1-KI homozygote mice display a delayed fusion
Palatal shelves isolated from mice lacking TGF-β type I or type II receptors in the palatal epithelium, as well as palatal shelves from Tgfb3 −/− mice, display an persistent intact midline seam under organ culture conditions (Dudas et al., 2006;Kaartinen et al., 1997;Taya et al., 1999;Xu et al., 2006). To more closely compare the fusion of palatal shelves between Tgfb1-KI and control mice, we dissected prefusion palatal shelves from control and mutant embryos and cultured them under chemically defined conditions in the air-medium interface for 48 hours. The control samples demonstrated an almost complete fusion characterized by a loss of epithelial cells in the midline region (Fig. 5A, C, E, G). In contrast, the Tgfb1-KI samples displayed a midline seam that was only partially degraded displaying a patchy loss of epithelial cells (Fig. 5B, D, F, H). Interestingly, the basement membrane was still clearly visible even in locations where epithelial cells were lost, as demonstrated by immunostaining for the basement membrane marker (laminin) and for the epithelial cell marker (cytokeratin-14) (Fig. 5I-K). To conclude, our organ culture analyses reveal that the fusion process is significantly delayed in knockin samples when compared to controls despite the fact that Tgfb1-KI samples demonstrate a seemingly normal successful fusion in the mid-palate in vivo.
Fig. 5. Comparison of the palatal fusion between Tgfb1-KI homozygote and control embryos in vitro.
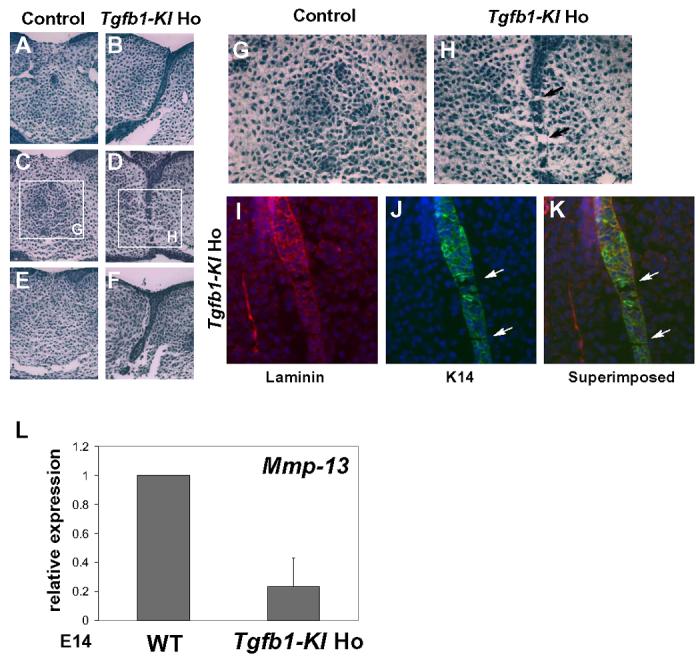
Prefusion palatal shelves were dissected from E14 embryos, cultured under chemically defined conditions for 48 hours and analyzed using serial sectioning. Wildtype (A, C, E) and Tgfb1-KI homozygote (B, D, F) samples were sectioned at three different levels and stained with hematoxylin & eosin. (G, H), higher magnification of the rectangular regions indicated in the (C, D). In control samples, the midline seam disappears with only a few epithelial islands left, indicating an almost complete fusion (G). In Tgfβ1-KI homozygous samples, the midline seam is in the process of breaking down, displaying a patchy loss of epithelial cells (H). The basement membrane is visible even at sites where the epithelial seam is breaking down (arrows in H). (I-K) Paraffin-sections from the palatal shelf organ culture of Tgfb1-KI homozygote were immunostained for laminin (M) and K14 (N). The superimposed image is shown in (K). Arrows in (J) and (K) point out the breakdown of the midline epithelial seam. (L) Real-time quantitative PCR analysis of Mmp-13 expression in tips of palatal shelves dissected from wild-type (WT) and Tgfb1-KI homozygote (Tgfb1-KI Ho) embryos at E14. Mmp-13 expression in the Tgfb1KI homozygote is about 30% of that expressed in the wild-type. Bar graph: mean +/−s.d., n=3; WT level has been arbitrarily set at 1.
It has been previously suggested that the disappearance of midline epithelium requires intensive remodeling of the extracellular matrix, and that one of the key enzymes in this process is matrix metalloproteinase-13 (MMP-13), the expression of which is upregulated in the midline epithelium seam and the surrounding mesenchyme during palatal fusion (Blavier et al., 2001). In Tgfb3 knockout mice, Mmp-13 expression domain is greatly reduced during palatogenesis, suggesting that its expression is controlled by TGF-β3 (Blavier et al., 2001). Therefore, we compared Mmp-13 expression in the tips of palatal shelves between wild-type and Tgfb1-KI samples (Fig. 5L). Our quantitative real-time PCR analysis demonstrated that misexpression of Tgfb1 in the Tgfb3 locus failed to restore Mmp-13 expression in Tgfb1-KI homozygous samples to the level seen in wild-type littermates.
Palatal shelves of Tgfb1-KI homozygotes become adherent but display reduced apoptosis in the midline palatal epithelium during palatal fusion
Previous studies have emphasized the importance of intercalation of the apposing epithelia as a necessary initial step during palatal fusion (Martinez-Alvarez et al., 2000), and apoptosis as a main mechanism for the removal of the midline epithelium during subsequent stages of palatogenesis (Cuervo et al., 2002). Therefore, we examined both the histology and apoptosis of the midline epithelium seam during the fusion in Tgfb1-KI homozygote, Tgfb3-Cre homozygote, and control embryos (Fig. 6). Histological analysis of frontal sections showed that apposing palatal shelves of control and Tgfb1-KI homozygote samples became adherent, and that the midline epithelia were able to intercalate and form comparable oral and nasal epithelial triangles. In contrast, palatal shelves of Tgfb3-Cre homozygote samples made contact, but failed to intercalate and form epithelial triangles. TUNEL assays showed that control samples exhibited a large number of apoptotic cells, particularly in the nasal and oral epithelial triangles along the anterior-posterior axis, while a number of apoptotic nuclei were clearly detectable, but notably reduced in Tgfb1-KI samples. As a negative control, Tgfb3-Cre homozygote samples did not show any TUNEL-positive cells. Therefore, attenuated apoptosis rather than reduced adherence likely accounts for the delayed fusion of Tgfb1-KI palatal shelves.
Fig. 6. Histological analysis and TUNEL assays on palatal shelves from control, Tgfb3-Cre homozygote, and Tgfb1-KI homozygote mice.
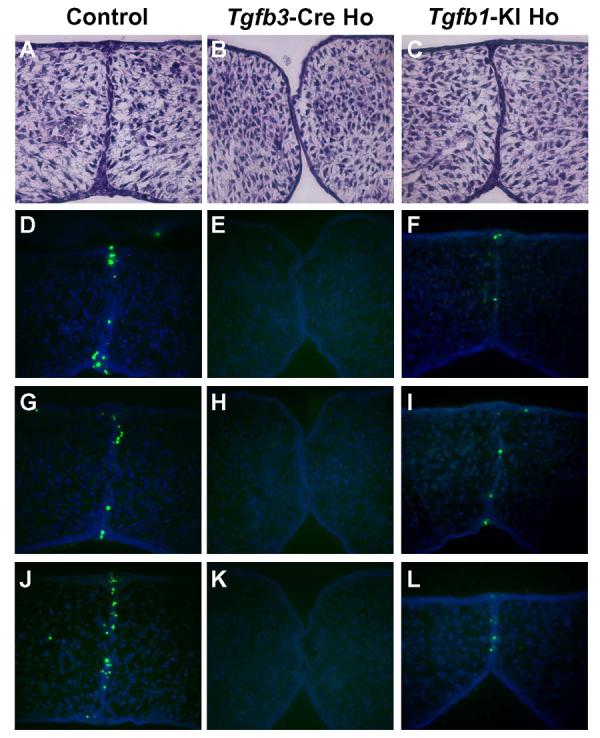
Embryos of each genotype indicated were harvested at E14 and sectioned serially (frontal orientation). (A-C) Histological analysis of frontal sections at comparable levels, staining with hematoxylin & eosin. Palatal shelves of Tgfb3-Cre homozygote made contact but failed to adhere and form epithelial triangles (B). In contrast, palatal shelves of the Tgfb1-KI homozygote (C) were adherent and formed epithelial triangles, which were similar to those from the control sample (A). (D-L) TUNEL assays on frontal sections at three different levels from each genotype (n=3). The Tgfb1-KI homozygote sample (F, I, L) exhibits a reduced number of apoptotic cells when compared to a control (D, G, J). Apoptotic cells were undetectable in the midline epithelium of the Tgfb3-Cre homozygote sample (E, H, K).
Discussion
During palatogenesis, TGFβ signaling is involved in the appropriate growth of palatal shelves and disappearance of the midline epithelial seam (Hay, 1995;Nawshad et al., 2004;Shuler, 1995;Young et al., 2000). Despite specific expression of both the Tgfb1 and Tgfb3 genes in distal edges of the prefusion palatal shelves, particularly in the medial edge epithelium, endogenously expressed Tgfb1 is not able to rescue the cleft palate phenotype caused by the lack of Tgfb3. Yet, mice heterozygous for the Tgfb3-null allele have normal palate formation. Whether this failure to rescue the Tgfb3 null phenotype was due to in vivo isoform-specific effects or simply reflected a significant difference in gene expression was not previously known. Given that three specific isoforms of TGF-β exhibit distinct and also overlapping expression patterns during development of different organs, we believe that use of the endogenous Tgfb3 locus to express Tgfb1 is the most reliable strategy to address the question of functional interchangeability.
Mice homozygous for Tgfb3-Cre allele displayed a cleft palate phenotype identical to that of the previously described Tgfb3 knockout mice. Since an identical strategy was used to generate the Tgfb1-KI and Tgfb3-Cre alleles, it can be deduced that the Tgfb1-KI allele is also a true null allele for Tgfb3, and that the partially fused palatal phenotype in Tgfb1-KI embryos is due to in vivo replacement of TGF-β3 with TGF-β1. Moreover, we demonstrated that Tgfb1 knockin allele can express Tgfb1 mRNA under the control of the endogenous Tgfb3 promoter by analyzing the level of Tgfb1 and Tgfb3 mRNA expression in distal tips palatal shelves isolated from wild-type, Tgfb1-KI heterozygote and homozygote embryos. In addition, our real-time absolute quantitative PCR data showed that Tgfb1 was expressed at an approximately 2.5-fold higher level than Tgfb3 in heterozygote Tgfb1-KI mice. Given that a total level of Tgfb3 expression in wild-type palatal shelves was about 3-fold higher than that of Tgfb1, quantitative PCR data of Tgfb1-KI heterozygote samples reveal that Tgfb1 mRNA expressed in distal edges of prefusion palatal shelves in homozygote Tgfb1-KI embryos is at a comparable level to that of Tgfb3 in wild-type embryos. Furthermore, we detected similar phospho-Smad2/3 levels in midline epithelium between wild-type and Tgfb1-KI homozygote embryos. Taken together, our experiments indicate that the failure of TGF-β1 to fully substitute for the function of TGF-β3 in palatogenesis does not result from an inadequate level of gene expression and subsequent protein synthesis.
Our in vivo and in vitro experiments suggest that the fusion process is delayed in Tgfb1-KI homozygotes, leading to a partially fused palatal phenotype. During palatogenesis, the mid-anterior portion of palatal shelves make contact first, and the anterior and posterior parts come in contact later, displaying slightly rounded contact surface between the two opposing palatal shelves (Dudas and Kaartinen, 2005). As the fusion progresses, the midline epithelial seam gradually disappears along the sites where contact was first established, leading to a tighter contact between the opposing shelves and enabling closer contact between other parts of the palate. Delayed epithelial fusion in Tgfb1-KI samples, characterized by attenuated apoptosis and slower breakdown of the basement membrane, may provide a scenario for an in vivo situation, where the anterior and the posterior parts of the midline seam are insufficiently degraded, such that palatal shelves become stretched by a lateral growth of the head, resulting in the anterior cleft and the posterior submucous cleft. However, our present do not rule out the possibility that TGF-β3 plays an isoform-specific role in the most anterior and posterior regions of the palate. Interestingly, it has been reported that addition of TGF-β3 induced the formation of filopodia-like structures in the palatal epithelium in vitro, while TGF-β1 and TGF-β2 were relatively inefficient in inducing filopodia, but rather induced lamellipodia in organ cultures (Taya et al., 1999). Perhaps, filopodia-like structures, which are more efficiently produced by TGF-β3 play a particularly significant role in the fusion of these regions, which fail to fuse in Tgfb1-KI embryos.
Mice deficient in TGF-β3, including both the traditional Tgfb3 knockout mice (Kaartinen et al., 1995;Proetzel et al., 1995) and the new Tgfb3-Cre knockin mice described in this study, display either complete clefting of the secondary palate, or posterior clefting and superficial anterior adherence. A spectrum of cleft palate phenotypes can also be seen in humans; these vary from a total cleft to a submucous posterior cleft, and even to bifid uvula, which can be viewed as the mildest form of oral fusion defects (Dudas and Kaartinen, 2005). The etiology of these conditions is currently not well understood. We demonstrated that in vivo replacement of TGF-β3 with TGF-β1 results in a consistent mild cleft phenotype, characterized by an anterior small cleft and a posterior submucous cleft. At the molecular level, the knock-in TGF-β1 can restore the Smad-dependent TGF-β signaling activity in the Tgfb3−/− background, while it cannot resore the Mmp13 level to that of the wild-type littermate. Despite the fact that TGF-β1 exerted an opposing effect to TGF-β3 in a rat cutaneous model (Shah et al., 1995), the partially rescued cleft palate phenotype seen in Tgfb1-KI homozygotes demonstrates that TGF-β1 and -β3 have partially overlapping functions in the palatal epithelium. This is consistent with earlier studies, which have demonstrated that addition of TGF-β1 or TGF-β2 can induce an almost complete fusion of Tgfb3−/− palatal shelves in organ cultures in vitro (Taya et al., 1999). Taken together data from our present study suggest that mildly perturbed developmental signals may lead to less severe cleft palate phenotypes. Since mice lacking both alleles of Tgfb1 do not exhibit a cleft palate phenotype, it would be of interest to know, what is the role of TGF-β1 in the prefusion palatal epithelium? TGF-β1 may play an auxiliary role secondary to TGF-β3 during palatogenesis by fine-tuning the signaling field setup by TGF-β3, or it may compensate for the haploid deficiency of Tgfb3 since Tgfb3 heterozygotes do not display cleft palate. However, there may be cell type-specific differences in the processing and activation of TGF-β precursors, which may, in part, explain the observed differences between the effects of TGF-β1 and -β3 in the palatal epithelium.
To conclude, our experiments demonstrate that TGF-β isoforms display cell type specific differences during palatogenesis in vivo. We propose that in some cell types overlapping expression patterns may modulate the strength of signaling rather than create a field for signal specificity, while in other cell types, e.g., the palatal epithelium, there can be notable differences between the action of different isoforms.
Acknowledgements
We thank A. Nagy for technical assistance. LTY was supported by a grant from the CIRM (T2-00005) and VK by grants from the NIH (HL074862 and DE013085).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ, Fitzpatrick DR, Gatherer D, Lehnert SA, Millan FA. Transforming growth factor betas in mammalian embryogenesis. Prog. Growth Factor Res. 1990;2:153–168. doi: 10.1016/0955-2235(90)90002-2. [DOI] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol. Biol. Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Cuervo R, Valencia C, Chandraratna RA, Covarrubias L. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev. Biol. 2002;245:145–156. doi: 10.1006/dbio.2002.0620. [DOI] [PubMed] [Google Scholar]
- Dudas M, Kaartinen V. Tgf-beta superfamily and mouse craniofacial development: interplay of morphogenetic proteins and receptor signaling controls normal formation of the face. Curr. Top. Dev. Biol. 2005;66:65–133. doi: 10.1016/S0070-2153(05)66003-6. 65-133. [DOI] [PubMed] [Google Scholar]
- Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006;296:298–314. doi: 10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103(Suppl):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Denhez F, Kondaiah P, Akhurst RJ. Differential expression of TGF beta isoforms in murine palatogenesis. Development. 1990;109:585–595. doi: 10.1242/dev.109.3.585. [DOI] [PubMed] [Google Scholar]
- Foitzik K, Paus R, Doetschman T, Dotto GP. The TGF-beta2 isoform is both a required and sufficient inducer of murine hair follicle morphogenesis. Dev. Biol. 1999;212:278–289. doi: 10.1006/dbio.1999.9325. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. Molecular control of secondary palate development. Dev. Biol. 2007;301:309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. A Laboratory Manual. CSH Press; New York: 1988. Antibodies. [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J. Anat. 2005;207:655–667. doi: 10.1111/j.1469-7580.2005.00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. A laboratory manual. Cold Spring Harbor Laboratory Press; New York: 1994. Manipulating the mouse embryo. [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U. S. A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Foitzik K, Calautti E, Baden H, Doetschman T, Dotto GP. TGF-beta3, but not TGF-beta1, protects keratinocytes against 12-O- tetradecanoylphorbol-13-acetate-induced cell death in vitro and in vivo. J. Biol. Chem. 1999;274:4213–4219. doi: 10.1074/jbc.274.7.4213. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Bonelli R, Tudela C, Gato A, Mena J, O'Kane S, Ferguson MW. Bulging medial edge epithelial cells and palatal fusion. Int. J. Dev. Biol. 2000;44:331–335. [PubMed] [Google Scholar]
- Moorman AF, Houweling AC, de Boer PA, Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J. Histochem. Cytochem. 2001;49:1–8. doi: 10.1177/002215540104900101. [DOI] [PubMed] [Google Scholar]
- Murray JC. Gene/environment causes of cleft lip and/or palate. Clin. Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- Nawshad A, Lagamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch. Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues. Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Dickinson ME, Moses HL, Hogan BL. In situ hybridization analysis of TGF beta 3 RNA expression during mouse development: comparative studies with TGF beta 1 and beta 2. Development. 1990;110:609–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP. Craniofacial anomalies: from development to molecular pathogenesis. Curr. Mol. Med. 2005;5:699–722. doi: 10.2174/156652405774641043. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. Differential expression of the TGF-beta isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol. Reprod. Dev. 1992;32:91–98. doi: 10.1002/mrd.1080320203. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non- overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte BC, Murray JC. The many faces and factors of orofacial clefts. Hum. Mol. Genet. 1999;8:1853–1859. doi: 10.1093/hmg/8.10.1853. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- Shi W, Heisterkamp N, Groffen J, Zhao J, Warburton D, Kaartinen V. TGF-beta3-null mutation does not abrogate fetal lung maturation in vivo by glucocorticoids. Am. J. Physiol. 1999;277:L1205–L1213. doi: 10.1152/ajplung.1999.277.6.L1205. [DOI] [PubMed] [Google Scholar]
- Shuler CF. Programmed cell death and cell transformation in craniofacial development. Crit Rev. Oral Biol. Med. 1995;6:202–217. doi: 10.1177/10454411950060030301. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya Y, O'Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr., Urata MM, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Young DL, Schneider RA, Hu D, Helms JA. Genetic and teratogenic approaches to craniofacial development. Crit Rev. Oral Biol. Med. 2000;11:304–317. doi: 10.1177/10454411000110030201. [DOI] [PubMed] [Google Scholar]