

Summary
Software for viewing three-dimensional models and maps of viruses, ribosomes, filaments and other molecular assemblies is advancing on many fronts. New developments include molecular representations that offer better control over level of detail, lighting that improves the perception of depth, and two-dimensional projections that simplify data interpretation. Programmable graphics processors offer quality, speed and visual effects not previously possible, while 3D printers, haptic interaction devices, and auto-stereo displays show promise in more naturally engaging our senses. Visualization methods are developed by diverse groups of researchers with differing goals: experimental biologists, database developers, computer scientists, and package developers. We survey recent developments and problems faced by the developer community in bringing innovative visualization methods into widespread use.
Introduction
Viewing three-dimensional (3D) atomic models and maps of molecular assemblies is a routine aspect of determining and interpreting the architecture and operation of cellular machinery. While researchers aim to make their analyses as objective and quantitative as possible, visualization software is primarily used to make subjective assessments that guide computational modeling and conceptual understanding. Results of map filtering, automated segmentation, or model fitting are inspected by eye to judge their correctness and to choose new parameters for repeating calculations to fix errors or extract more detail from experimental data. Viewing software is also used for interactive analysis when available algorithmic methods are not adequate. Maps are segmented or models are fit by hand and eye using 3D views of data. Finally, viewing software is used to present final results in the form of images and animations.
We present some recently developed methods for displaying maps and models that have particular utility at the scale of molecular assemblies. A vast literature describes visualization methods for 3D scientific data, but we limit our review to methods that have been tried with molecular assembly data. We do not cover interactive analysis techniques. The scope of our review is still sufficiently broad that we can only highlight a few of the many recent developments.
Unlike algorithmic software, the effectiveness of visualization techniques is difficult to quantify since visualization is applied for making non-quantitative judgments. A practical measure of utility for visualization software is whether it is widely used in achieving significant biological results. Unfortunately many obstacles to widespread use have little relation to merit: research developers are principally concerned with biology or methodology rather than widespread use of their software, new visualization methods have value only in combination with old ones, and needed data standards are lacking. While a steady stream of visualization innovations are being developed as sampled below, most will not reach a large audience for lack of adequate implementation, documentation, or on-going software maintenance. The final sections of this article review obstacles to achieving widespread use of new visualization methods.
Multiple levels of detail
Molecular assemblies contain a hierarchy of structures: atoms and bonds, residues, helices and sheets, domains, macromolecules, and complexes. Insightful views highlight only a small bit of this architecture with a simplified depiction of the surroundings to provide context. For example, the binding site of an enzyme may have five conserved residues shown in atomic detail with spheres for atoms and cylinders for bonds, while nearby parts of the enzyme are shown in less detail as a ribbon. Familiar molecular depictions offer only a few levels of granularity: atoms and bonds shown using spheres, cylinders or lines, helices and strands shown as ribbons, and molecules shown using solvent-accessible molecular surfaces. Coarser granularity is needed to depict large assemblies such as virus capsids. Commonly used solvent-accessible surfaces defined by rolling an atom-size ball over the surface of the molecule have too much detail. Such a surface for a large assembly such as bluetongue virus capsid (Protein Data Bank identifier 2btv) is composed of 27 million triangles, about 30 times larger than can be moved interactively using current desktop graphics systems. The computation of the surface geometry is likewise time-consuming. While increased computing performance will improve the situation in the future, the high level of detail obtained with this approach severely diminishes the 3D appearance using current lighting methods that favor smooth, slowly varying surfaces. Lastly, these surfaces don't serve user needs where minimal detail is desired around regions of a structure shown at finer granularity.
Methods of depicting the approximate shape of domains, molecules or complexes as smooth surfaces at any desired resolution have been developed using spherical harmonics [1], density contours [2], and surface decimation [3]. Spherical harmonics produce smooth variable-resolution surfaces with few triangles, but are only easily applied to surface shapes that are topologically equivalent to a sphere, e.g., without toroidal (donut-like) holes. Density contours are simple to compute for all molecule shapes (Figure 1) but suffer from non-smooth artifacts caused by poor triangulation. Decimation methods start with a higher-resolution surface and eliminate triangles by merging vertices in a way that minimizes changes in appearance. In tests, decimation reduced the number of triangles of a solvent-accessible surface by a factor of 10, but it is not known whether they can reliably achieve a lower-resolution appearance. Decimation techniques usually preserve topology, which can prevent elimination of small holes. Decimation approaches can also be slow because of the need to calculate an initial high-resolution surface.
Figure 1. Levels of detail.

(a) Molecules depicted with low-resolution surfaces, orthoreovirus capsid (PDB 2cse). (b) Nucleic acid bases depicted as slabs [4]. (c) Carbohydrate depiction [5] with glucan ring top and bottom sides colored blue and yellow. Ball-and-stick model superimposed for reference.
Representations have been implemented at the residue level to depict bases in nucleic acid polymers [4] and sugars of carbohydrates [5] as simple geometric shapes such as rectangles and hexagons (Figures 1b and 1c) with bumps or colors indicating ring orientation. These are helpful for visualizing stacking interactions in nucleic acid and glycoprotein macromolecules and assemblies. Limiting views to comprehensible numbers of atoms is important for validating the correctness of structures. For glycoproteins, simplified oligosaccharide representations are not widely available, and it is estimated that one third of those PDB entries have errors in carbohydrate stereochemistry, atom nomenclature, glycosidic linkages, and fits with electron density [6] [7].
Enhancing depth perception
Depth perception on two-dimensional computer displays is conveyed by motion cues when objects are rotated, known as the kinetic depth effect [8], and lighting. The most common lighting technique uses one or more directional light sources. A surface appears bright in places where light bouncing off the surface would be reflected towards the viewer. This gives a strong sense of depth for smooth, slowly varying surfaces. It gives little sense of depth for surfaces with detailed features (such as molecular surfaces or space-filling sphere depictions of atomic models, or intricate contour surfaces of density maps from electron microscopy reconstructions) because the bright spots on the surface become numerous and spread over much of the surface. Recently a lighting technique called ambient occlusion was shown to be highly effective for space-filling sphere rendering [9]. This technique calculates how much light arriving from all directions can strike a patch of surface. Surface regions in recesses or concavities appear darker. This is akin to use of shadows cast by directional light sources but provides depth cues independent of how the model is oriented relative to fixed light sources (Figures 2a, 2b). Ambient occlusion is computationally demanding, and the QuteMol implementation of Tarini et al.[9] uses a computer's graphics processing unit (GPU) to effectively compute shadows from a large number of directions. QuteMol demonstrates ambient occlusion and other rendering techniques but has limited molecular analysis capabilities. Interactive ambient occlusion is not currently available in more full-featured molecular visualization software.
Figure 2. Enhancing depth perception.

(a) Standard lighting of a chaperonin model (PDB 1aon) with atoms displayed as spheres. (b) Ambient occlusion lighting of chaperonin model produced by QuteMol program. (c) Edge highlighting with thicker edges where larger jumps in depth occur. Images from [9].
Another technique to enhance 3D appearance is to add black outlines at edges where a jump in depth occurs in an image (Figure 2c). This highlights protrusions of a surface that overlay identically colored surface patches. This technique in combination with flat lighting has been used extensively by David Goodsell and is used in the Protein Data Bank molecule of the month web articles [10]. An improvement on the original algorithm employing constant-width lines is to make the line thickness depend on the magnitude of the depth change [9]. Larger depth changes are highlighted with thicker lines up to a certain maximum thickness, while small depth jumps may not be highlighted to simplify appearance.
Two-dimensional views
While methods to improve depth perception in views of molecular assemblies continue to advance, techniques that eliminate depth are also progressing. These benefit from our perceptual ease in understanding 2D illustrations and the bias of computer interfaces, mouse and screen, towards 2D representations. Goodsell [10] advocates use of cross-sectional views of cells in illustrating molecular crowding (Figure 3a), views that use flat coloring with no lighting highlights. Filaments and various molecules can be seen to overlay one another, but the views are non-immersive. The observer viewpoint is far from the collection of molecules, not within it. Electron tomograms of cells are also frequently viewed a single two-dimensional data plane at a time. For complex cellular environments, 3D representations are usually less effective because of the large number and dense packing of objects in these scenes.
Figure 3. Two-dimensional visualization methods.
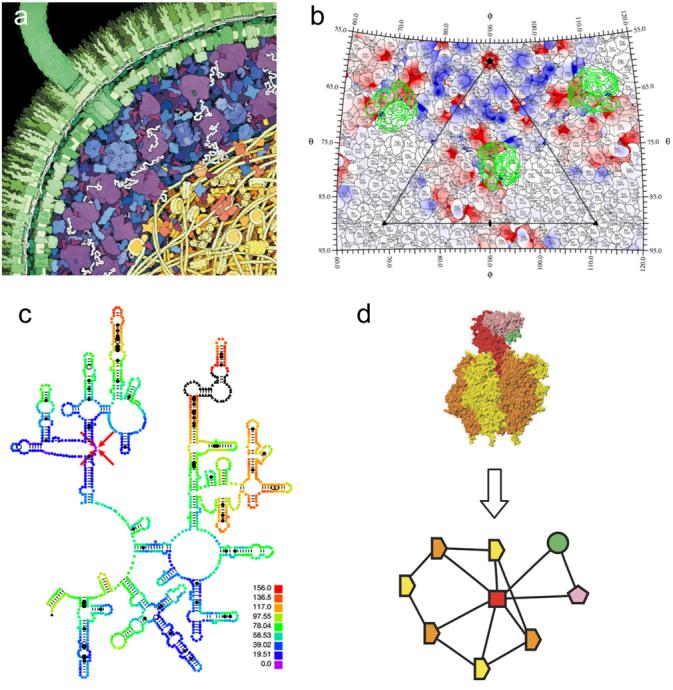
(a) E. coli cross-section [10]. The double-layered cell wall is shown in green, with a flagellum extending upward to the left. The cytoplasmic space is shown in blue, with ribosomes in purple. The nucleoid area is shown at the lower right, with DNA strands in yellow. (b) Roadmap of CVA21 virus and footprint of the ICAM-1 receptor [11]. The difference density between 145 and 160 Å radii, isolating the ICAM-1 receptor, is projected onto a stereographic diagram and contoured in green. The electrostatic potential is of CVA21 is shown in red and blue. (c) ColoRNA view of part of E. coli 23S ribosomal RNA [12] colored by distance from the residue indicated with red arrows. (d) Graph depiction of a molecular assembly from the 3D Complex database [15] shows SCOP domains [16] as symbols with lines indicating contacting domains. Homologous domains have the same symbol shape and identical domains have the same color.
Xiao et al. [11] have developed a 2D visualization of surface residues on virus capsids (Figure 3b). The residues appear like countries on a map divided by boundary lines with text labels. Stereographic projection is used as in cartography, and overlays of density contours, color-coded radial height, color-coded electrostatic potential, density contours of host cell receptors, or symmetry axes and asymmetric unit bounds can be applied. Their non-interactive implementation is specialized for virus capsids, though it seems applicable to filaments with a cylindrical projection or to non-symmetric structures, for example protein-protein interfaces, using projection onto a tangent plane.
An interactive visualization tool for ribosomal RNA structures called ColoRNA developed by LeBarron et al. [12] shows RNA secondary structure schematics. Three-dimensional information is conveyed by coloring residues to indicate distance from a chosen residue (Figure 3c), or distances between residues in two conformations, or domain structure. ColoRNA uses secondary structure layouts provided by the University of Texas RNA database [13]. A program called TopDraw for drawing 2D schematics of protein secondary structure developed by Bond [14] permits manual layout of alpha helices and beta strands. The packing of secondary structure elements can be approximated in two dimensions to make images for articles and talks. Combining TopDraw layout and ColoRNA distance-based coloring would make a more broadly applicable schematic visualization tool.
Levy et al. [15] have developed a database that classifies the patterns of contacts between molecular domains in 21037 assemblies from the Protein Data Bank. They developed a two-dimensional graphical depiction with domains shown as vertices and edges indicating contacts (Figure 3d). Identical subunits have equivalent symbol shapes and colors, while homologous subunits are distinguished by color. The symmetry of the complex is mirrored in the 2D layout. These graphs are useful for comparative studies of assemblies and could be an aid for selecting portions of assemblies in other visualization tools.
Speed optimizations
The programming interface to graphics processing units (GPUs) has changed substantially in the past few years. While older versions of the OpenGL 3D graphics library provided functions to display surfaces decomposed into triangles, shade them with lighting, and paint them with images (called textures), newer versions have added a high degree of programmability. Arbitrary computations can now be performed on the GPU on each vertex or each fragment (picture element, or pixel, of a projected surface triangle). GPU programmability allows new rendering effects such as the depth-dependent edge highlighting described earlier that would be difficult to implement and highly inefficient with the fixed functionality of older GPU interfaces. It also enables faster rendering even in cases where special effects are not employed. GPUs frequently have an order of magnitude more floating-point computation speed than the main computer processing unit; this has spurred the development of GPU programs for non-graphical purposes, such as fast molecular dynamics calculations for protein folding by the Folding@Home project (URL: http://folding.stanford.edu/FAQ-highperformance.html) or for computing thermal conductivities [17]. Programming languages for GPUs such as Cg [18] and OpenGL Shading Language [19] are still being standardized.
Rendering atomic models can be sped up by a factor of 10 or more using GPU programmability. Standard rendering methods represent atoms as spheres that are approximated by surfaces composed of tens (low quality) or hundreds (high quality) of triangles that are processed by the GPU. The program TexMol [20] eliminates the triangles, instead using 2D raster images called textures in OpenGL to represent the sphere. Different textures encode the depth and surface normal vector as a function of the position on the sphere, and the GPU interpolates values in the textures to render lit spheres of any size and position in the scene. The QuteMol program [9] forgoes the triangles and the textures and instead computes using the GPU the depth and normal vectors across the sphere from equations as needed. The TexMol and QuteMol techniques are called textural and procedural impostors and increase rendering speed by more than a factor of 10. Current GPUs are able to render molecular assemblies of 100,000 atoms (e.g. a ribosome model) as spheres using the conventional triangle approach at interactive rates, generally considered to be anything faster than 10 frames per second. For larger systems, showing the entire assembly as individual atoms provides more detail than is useful. Still, the speed improvements are valuable, for example enabling fast ambient occlusion calculations that involve rendering the scene from hundreds of directions to determine the degree of visibility of each part of a surface.
Tactile models and stereoscopic viewing
Exotic computer hardware having applications to molecular assemblies is becoming more capable and less expensive: printers that create three-dimensional objects, displays that produce stereoscopic depth without special glasses, and force feedback (haptic) devices that provide a sense of touch. Prototype applications have demonstrated ways to use these devices in molecular visualization, but will require further reductions in cost to enable widespread use.
Gillet et al. [21] use 3D printers to create physical models of molecules and assemblies (Figure 4a). They developed software that can add virtual overlays such as electrostatic potential and ligands to the hand-held models. The software composites a view of the physical model seen by a video camera with computer-generated overlays and projects the results on the computer screen. Other researchers focusing on teaching methodologies at the Center for BioMolecular Modeling (URL: http://www.rpc.msoe.edu/cbm/) have created a lending library of physical molecular models [22] and an affiliated company, 3D Molecular Designs (Wauwatosa, WI, USA), sells the models.
Figure 4. Tactile models.

(a) Physical molecular models created with 3D printers from Z Corporation (Burlington, MA, USA) and Stratasys (Eden Prairie, MN, USA) using Python Molecular Viewer software [21]. (b) Force feedback device from SensAble Technologies (Woburn, MA, USA) guides atomic models to maximum correlation locations in manual fitting with Sculptor [23] software.
Force feedback devices allow interactive position and orientation input and can provide forces and sometimes torques to the user's hand (Figure 4b). Birmanns et al. [23] have developed software, part of their Sculptor program, to manually fit atomic models into density maps from electron microscopy with force feedback to increase the correlation between model and map. A principle challenge is that force update rates of approximately 1000 Hz, much higher than typical graphics update rates of 30 Hz, are required for fine control and stability. Advances in fast correlation measures [24] make improved feedback possible while the costs of the devices have dropped approximately 5-fold in the past few years. Inexpensive devices still cost roughly as much as a desktop computer, a limiting factor in wider application of force feedback to molecular visualization.
More than half of all computer displays are now LCD flat panels, which have insufficient refresh rates to support field sequential, or time multiplexed, stereoscopic display. This has led to less frequent use of stereoscopic molecular visualization using shutter-style glasses than when CRTs were the predominant display technology. Auto-stereoscopic computer displays use an alternative technology that allows three-dimensional viewing without special glasses by presenting different views to each eye based on the viewing direction [25]. While many vendors now offer these displays, the resolutions for modestly priced devices are typically half of conventional display resolutions in either horizontal or vertical dimensions, limiting their utility.
Developer community
We now consider why few visualization innovations achieve wide use within the community of molecular assembly researchers. Some innovations are highly specialized, while others offer minimal advantages over existing techniques. We consider other factors that limit use: the goals and means of the developers, the need to combine new methods with established ones, and the need for data file format standards.
Methods for viewing molecular assemblies are developed by three groups: biologists, computer scientists, and application developers. Biologists are the immediate users of the software and have the best view of what is needed to understand molecular assemblies. They frequently develop software for their own labs and collaborating labs but it is not their primary interest to polish and distribute the software more widely, an effort that typically consumes at least three-fold more time than implementations of in-house software [26]. Computer visualization researchers develop methods and illustrate them using molecular systems and data from other scientific disciplines. Their implementations demonstrate the value of a technique in isolation, and the complexity of their techniques can become unmanageable when combined with the array of visualization capabilities needed to make a complete tool. Application developers are primarily concerned with combining methods created by others and themselves to make an effective and widely distributed program.
Productive use of new visualization techniques is achieved by combining these with other capabilities to solve a problem. The ambient occlusion method described earlier would be valuable in many contexts, for example, when screening docked ligands, examining sequence conservation at protein-protein interfaces, or fitting atomic models into density maps. Packages such as Chimera [27], PyMol [28], VISION [3] and VMD [29] incorporate many capabilities that achieve a synergistic effect, increasing the utility of the separate tools. Each of these packages can be extended with new capabilities using the Python programming language. Sanner [3] advocates use of Python to implement reusable modules that can be shared among molecular visualization applications. Pettersen et al. [27] advocates Python for its simplicity, which permits coding by biologists who are not primarily programmers. While many application developers have adopted Python to enable others to extend their molecular visualization packages, all of those packages continue to be primarily developed by an individual or central group rather than by decentralized contributors. Most new visualization methods continue to be prototyped as stand-alone applications of limited utility rather than incorporated into existing packages.
The file formats and visualization software currently used for molecular assemblies were developed when few structures of large assemblies were known. The traditional Protein Data Bank (PDB) [30] format for atomic models is limited to 62 macromolecule chains (named with single letters and digits a-z, A-Z, 0-9) and 100,000 atoms, inadequate for handling current ribosome models, which must be split into two files. Dutta et al. [31] review the PDB large assembly models and discuss the mmCIF [32] format, which was introduced 9 years ago to lift some of the restrictions. However, the need to maintain compatibility with the older PDB file format has limited the use of the extended capabilities of mmCIF. The Electron Microscopy (EM) Database [33] and the Virus Particle Explorer database [34] make use of the CCP4 density map format from crystallography for archiving data from single-particle cryoEM reconstructions. The original format is not able to specify an arbitrary physical origin, nor can it record the symmetries imposed in single-particle reconstruction. As a format for EM tomography, it lacks unsigned 8-bit integer data values and the ability to define rotations of extracted subregions that do not align with the data axes. Also, it cannot accommodate the subsampled arrays needed to handle large data sets. These many limitations impair its utility for representing EM maps of assemblies. Heymann et al. [35] recommend conventions for coordinate systems and symmetries for EM maps, but do not recommend specific file formats. Tang et al. [36] have adopted the HDF5 file format (URL: http://hdf.ncsa.uiuc.edu/HDF5/) for maps in the single-particle reconstruction package EMAN2 to address the limitations of existing formats.
Archiving symmetry
Particularly critical for archiving molecular assembly data are standards for recording symmetries of models and maps. These provide bookkeeping for the placements of identical molecular subunits in atomic models, for the symmetry imposed on a map in single-particle reconstruction, and for locations of atomic models fit into density maps. For multimeric atomic models the PDB provides Biological Unit Coordinate files [31], which contain coordinates of identical copies of molecules forming the presumed native oligomerization state. The Protein Quaternary Structure server [37] also uses this approach. The files can contain millions of atom coordinates for virus capsid structures. This representation is inefficient and the symmetry cannot easily be determined and used by visualization software. The PDB file format has an alternate method that provides rotation matrices and translation vectors for placing molecules using “remark 350 BIOMT” biological unit matrices. This is a more useful representation of symmetry but the remarks are not rigidly formatted and are sometimes not computer-readable. The mmCIF file format puts these matrices into machine-readable form, but software to utilize symmetry from mmCIF format is not yet widely available.
The symmetry imposed in single-particle reconstructions is not currently included in CCP4 [38] maps in the EM Database. While the CCP4 format supports crystallographic point and space group symmetries using conventional nomenclature, EM reconstructions exhibit helical symmetries as well and are generally not positioned in a standard coordinate frame. A common use of such symmetry is to fit a monomeric model into the map and then replicate it using the map symmetry. Yu et al. [39] use an automated method to determine symmetry directly from the map for use in segmentation. In cases where the symmetry was imposed in the reconstruction, it is preferable to use that symmetry instead of trying to re-derive it from the map.
Representations for symmetry using rotation matrices, quaternions, or Euler angles combined with a translation vector are straightforward. Missing are file format specifications and visualization and computational software that can use such specifications. The databases for atomic models and EM maps should play a prominent role in establishing these conventions and promoting their use by software developers. Macias et al. [40] have developed standards and the viewing software PeppeR as part of a distributed annotation system that overlays map, model and sequence data from many independent databases. They specify adapter formats to allow databases using alternate conventions to plug into their meta-database viewer software. The success of visualization tools that integrate content from multiple databases requires unambiguous standards for alignments in those databases.
Conclusion
A diverse group of experimentalists, computer scientists, application developers and public database curators are addressing visualization challenges posed by molecular assemblies. Innovative methods are frequently proposed and implemented by all of these groups. A central problem is how to turn innovations into useful products: well documented, widely distributed, high-quality visualization software for the research community. Because the contributors to the effort have divergent goals and no one group leads the effort, coordination and development of standards for data interchange are major problems. Current visualization of large molecular assemblies is based on modifications to software and data formats that are a decade or more old and ill-suited for the purpose. The pace of progress toward a new generation of software and data formats that easily accommodate molecular assemblies will be determined by the degree of community cooperation.
Acknowledgements
We thank Elaine C. Meng for her helpful comments in preparing this manuscript. Research reported in this review from the author's laboratory was supported by National Institutes of Health grant P41RR01081.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Macke TJ, Duncan BS, Goodsell DS, Olson AJ. Interactive modeling of supramolecular assemblies. J Mol Graph Model. 1998;16:115–120. doi: 10.1016/s1093-3263(98)00016-3. 162-113. [DOI] [PubMed] [Google Scholar]
- 2 •.Goddard TD, Huang CC, Ferrin TE. Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure. 2005;13:473–482. doi: 10.1016/j.str.2005.01.006. Describes software for viewing atomic models of viruses and other molecular assemblies composed of up to thousands of macromolecules. [DOI] [PubMed] [Google Scholar]
- 3.Sanner MF. A component-based software environment for visualizing large macromolecular assemblies. Structure. 2005;13:447–462. doi: 10.1016/j.str.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Couch GS, Hendrix DK, Ferrin TE. Nucleic acid visualization with UCSF Chimera. Nucleic Acids Res. 2006;34:e29. doi: 10.1093/nar/gnj031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuttel M, Gain J, Burger A, Eborn I. Techniques for visualization of carbohydrate molecules. J Mol Graph Model. 2006;25:380–388. doi: 10.1016/j.jmgm.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Crispin M, Stuart DI, Jones EY. Building meaningful models of glycoproteins. Nat Struct Mol Biol. 2007;14:354. doi: 10.1038/nsmb0507-354a. [DOI] [PubMed] [Google Scholar]
- 7.Berman HM, Henrick K, Nakamura H, Markley J. Reply to: Building meaningful models of glycoproteins. Nat Struct Mol Biol. 2007;14:354–355. doi: 10.1038/nsmb0507-354a. [DOI] [PubMed] [Google Scholar]
- 8.Wallach H, O'Connell DN. The kinetic depth effect. J Exp Psychol. 1953;45:205–217. doi: 10.1037/h0056880. [DOI] [PubMed] [Google Scholar]
- 9 ••.Tarini M, Cignoni P, Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Trans Vis Comput Graph. 2006;12:1237–1244. doi: 10.1109/TVCG.2006.115. The authors describe highly effective methods to enhance the perception of depth in images of molecular structures using shading and edge highlighting. [DOI] [PubMed] [Google Scholar]
- 10.Goodsell DS. Visual methods from atoms to cells. Structure. 2005;13:347–354. doi: 10.1016/j.str.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 11 •.Xiao C, Rossmann MG. Interpretation of electron density with stereographic roadmap projections. J Struct Biol. 2007;158:182–187. doi: 10.1016/j.jsb.2006.10.013. Implements a 2D schematic depiction of surface residues on virus capsids together with optional overlayed properties such as electrostatic potential and ligand binding sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeBarron J, Mitra K, Frank J. Displaying 3D data on RNA secondary structures: coloRNA. J Struct Biol. 2007;157:262–270. doi: 10.1016/j.jsb.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bond CS. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- 15 •.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. Introduces the 3D Complex database that classifies the quaternary structure of complexes found in the Protein Data Bank by symmetry and contacts of constituent SCOP domains. Establishes a basis for comparative studies of molecular assembly architecture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Wang Y, Chen Y. GPU accelerated molecular dynamics simulation of thermal conductivities. Journal of Computational Physics. 2007;221:799–804. [Google Scholar]
- 18.Mark W, Glanville R, Akeley K, Kilgard M. Cg: A system for programming graphics hardware in a C-like language. ACM Transactions on Graphics. 2003;22:896–907. [Google Scholar]
- 19.Rost RJ. OpenGL Shading Language. edn 1st Pearson Education, Inc; 2004. [Google Scholar]
- 20.Bajaj C, Djeu P, Siddavanahalli V, Thane A. IEEE Visualization 2004 Oct 10-15, 2004. Austin, Texas: 2004. TexMol: interactive visual exploration of large flexible multi-component molecular complexes; pp. 243–250. [Google Scholar]
- 21 •.Gillet A, Sanner M, Stoffler D, Olson A. Tangible interfaces for structural molecular biology. Structure. 2005;13:483–491. doi: 10.1016/j.str.2005.01.009. Uses 3D printer technology to create physical models of molecular assemblies and develops methods to display computer-generated overlays on such models. [DOI] [PubMed] [Google Scholar]
- 22.Herman T, Morris J, Colton S, Batiza A, Patrick M, Franzen M, Goodsell DS. Tactile teaching: Exploring protein structure/function using physical models. Biochemistry and Molecular Biology Education. 2006;34:247–254. doi: 10.1002/bmb.2006.494034042649. [DOI] [PubMed] [Google Scholar]
- 23.Birmanns S, Wriggers W. Interactive fitting augmented by force-feedback and virtual reality. J Struct Biol. 2003;144:123–131. doi: 10.1016/j.jsb.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Birmanns S, Wriggers W. Multi-resolution anchor-point registration of biomolecular assemblies and their components. J Struct Biol. 2007;157:271–280. doi: 10.1016/j.jsb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Dodgson N. Autostereoscopic 3D displays. Computer. 2005;38:31–36. [Google Scholar]
- 26.Brooks F. The Mythical Man-Month: Essays on Software Engineering. Addison-Wesley; 1995. [Google Scholar]
- 27.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 28.DeLano W. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto: 2002. [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27-38. [DOI] [PubMed] [Google Scholar]
- 30.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutta S, Berman HM. Large macromolecular complexes in the Protein Data Bank: a status report. Structure. 2005;13:381–388. doi: 10.1016/j.str.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Bourne P, Berman H, Watenpaugh K, Westbrook J, Fitzgerald P. The macromolecular Crystallographic Information File (mmCIF) Meth. Enzymol. 1997;277:571–590. doi: 10.1016/s0076-6879(97)77032-0. [DOI] [PubMed] [Google Scholar]
- 33.Tagari M, Newman R, Chagoyen M, Carazo JM, Henrick K. New electron microscopy database and deposition system. Trends Biochem Sci. 2002;27:589. doi: 10.1016/s0968-0004(02)02176-x. [DOI] [PubMed] [Google Scholar]
- 34.Shepherd CM, Borelli IA, Lander G, Natarajan P, Siddavanahalli V, Bajaj C, Johnson JE, Brooks CL, 3rd, Reddy VS. VIPERdb: a relational database for structural virology. Nucleic Acids Res. 2006;34:D386–389. doi: 10.1093/nar/gkj032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heymann JB, Chagoyen M, Belnap DM. Common conventions for interchange and archiving of three-dimensional electron microscopy information in structural biology. J Struct Biol. 2005;151:196–207. doi: 10.1016/j.jsb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 38.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Yu Z, Bajaj C. Automatic ultrastructure segmentation of reconstructed cryoEM maps of icosahedral viruses. IEEE Trans Image Process. 2005;14:1324–1337. doi: 10.1109/tip.2005.852770. [DOI] [PubMed] [Google Scholar]
- 40 •.Macias JR, Jimenez-Lozano N, Carazo JM. Integrating electron microscopy information into existing Distributed Annotation Systems. J Struct Biol. 2007;158:205–213. doi: 10.1016/j.jsb.2007.02.004. Develops standards and viewer software for simultaneous visualization of EM density maps, atomic models, fits of models within maps, and genomic sequence data accessed in realtime from hundreds of databases. [DOI] [PubMed] [Google Scholar]