

Abstract
Syndapin I (SdpI) interacts with proteins involved in endocytosis and actin dynamics and was therefore proposed to be a molecular link between the machineries for synaptic vesicle recycling and cytoskeletal organization. We here report the identification and characterization of SdpII, a ubiquitously expressed isoform of the brain-specific SdpI. Certain splice variants of rat SdpII in other species were named FAP52 and PACSIN 2. SdpII binds dynamin I, synaptojanin, synapsin I, and the neural Wiskott-Aldrich syndrome protein (N-WASP), a stimulator of Arp2/3 induced actin filament nucleation. In neuroendocrine cells, SdpII colocalizes with dynamin, consistent with a role for syndapin in dynamin-mediated endocytic processes. The src homology 3 (SH3) domain of SdpI and -II inhibited receptor-mediated internalization of transferrin, demonstrating syndapin involvement in endocytosis in vivo. Overexpression of full-length syndapins, but not the NH2-terminal part or the SH3 domains alone, had a strong effect on cortical actin organization and induced filopodia. This syndapin overexpression phenotype appears to be mediated by the Arp2/3 complex at the cell periphery because it was completely suppressed by coexpression of a cytosolic COOH-terminal fragment of N-WASP. Consistent with a role in actin dynamics, syndapins localized to sites of high actin turnover, such as filopodia tips and lamellipodia. Our results strongly suggest that syndapins link endocytosis and actin dynamics.
Keywords: pheochromocytoma, Arp2/3 complex, dynamin, neural Wiskott-Aldrich syndrome protein, filopodia
Introduction
Endocytosis allows eukaryotic cells to internalize parts of their plasma membrane and constituents of the extracellular medium. The extracellular molecules are engulfed by a part of the lipid bilayer and sequestered through the formation of a sealed endocytic vesicle that subsequently detaches from the plasma membrane. The formation of vesicles for receptor-mediated endocytosis in higher organisms is driven by the assembly of clathrin and the adaptor protein 2 complex (reviewed in Schmid 1997), and involves a large array of accessory proteins (reviewed in Marsh and McMahon 1999). One is the GTPase dynamin, which is targeted to the necks of endocytic coated pits where it functions to regulate the fission reaction leading to clathrin-coated vesicle formation (Schmid et al. 1998; Sever et al. 1999). Dynamin interacts via its proline-rich domain (PRD) with several src homology 3 (SH3) domain-containing proteins implicated in clathrin-mediated endocytosis (Shupliakov et al. 1997; Wigge et al. 1997; Owens et al. 1998; Wigge and McMahon 1998; Sengar et al. 1999; Simpson et al. 1999).
In yeast, endocytosis is critically dependent on actin filament organization (Riezman et al. 1996) and genetic analyses of endocytosis have identified gene products that are required both for the internalization step at the plasma membrane and proper actin organization (reviewed in Geli and Riezman 1998; Wendland et al. 1998). They include actin itself and proteins that affect actin dynamics, such as Arp2, a component of the Arp2/3 complex which enhances actin filament polymerization (reviewed in Machesky and Insall 1999). The ability of the Arp2/3 complex to nucleate actin filaments in vitro is greatly enhanced by members of the Wiskott-Aldrich syndrome protein (WASP) family (Machesky et al. 1999; Rohatgi et al. 1999; Winter et al. 1999; Yarar et al. 1999). Deletion of the WASP homologue, Las17p, resulted in both the loss of cortically attached actin patches (Li 1997) and a block of fluid-phase uptake in yeast (Naqvi et al. 1998). Similarly, lymphocytes from WASP knockout mice exhibited both a reduction in actin polymerization and defects in T cell receptor endocytosis (Zhang et al. 1999). However, the association between the endocytic machinery and the actin cytoskeleton in mammalian cells is not well understood. Evidence for the actin requirement for clathrin-dependent and -independent internalization in mammalian cells, based on the use of drugs modulating the degree of actin polymerization (reviewed in Geli and Riezman 1998) and on mutant forms of the Rho family of small GTPases (Schmalzing et al. 1995; Lamaze et al. 1996), appears to be conflicting and seems to depend on the cell type and the assay used. If endocytosis and the actin cytoskeleton are also functionally connected in mammalian cells, it should be possible to identify molecular links. Candidate molecules are the G-actin binding proteins profilin I and II, which bind to molecules of the endocytic pathway (Witke et al. 1998) and HIP1R, a new component of clathrin-coated pits and vesicles (Engqvist-Goldstein et al. 1999). Another attractive possible linker is SdpI. The dynamin-associated protein SdpI also interacts with the neural WASP (N-WASP; Qualmann et al. 1999), a highly brain-enriched multidomain protein (Miki et al. 1996). The COOH terminus of N-WASP, which greatly activates the ability of the Arp2/3 complex to nucleate actin filaments (Rohatgi et al. 1999), is usually masked and inactivated through an intramolecular interaction (Miki et al. 1998); however, N-WASP can be activated by binding of the small GTPase Cdc42 and phosphatidylinositol (4,5) bisphosphate (Rohatgi et al. 1999) most likely by exposing the COOH terminus of the protein. These observations suggest an exciting link between endocytosis and the regulation of actin polymerization.
Protein associations measured in vitro can be misleading. Although the SH3 domains of Grb2, spectrin, and phospholipase Cγ, for example, bind to dynamin they failed to block receptor-mediated endocytosis (Wang and Moran 1996; Wigge et al. 1997). Here, we confirm the involvement of syndapin in endocytosis by inhibiting internalization of transferrin with syndapin fragments. In addition, overexpression of full-length syndapin modulates the cortical actin cytoskeleton in vivo to induce filopodia formation. The ability to modify endocytosis and actin polymerization is shared by SdpII, a ubiquitously expressed protein highly homologous to the brain-specific SdpI. Thus, the role of syndapin in connecting endocytosis and actin organization is not specific for synaptic vesicle recycling, but represents a general, fundamental mechanism in mammalian cells.
Materials and Methods
Cloning of Syndapin II
During PCR amplification of the SdpI gene from a rat brain cDNA library, an additional, slightly larger, insert was generated (Qualmann et al. 1999). Sequencing of the insert revealed a gene product different from the SdpI gene, but related. The nucleotide sequence was extended to the 5′ and 3′ ends by nested PCR, using 10 ng cDNA prepared from a rat brain MATCHMAKER cDNA library in pGAD10 (Clontech Laboratories, Inc.) per reaction as template. The primers for the nested PCR were synthesized to hybridize with the gene sequence: first round at the 5′ end, 5′-TCAAGCTCCTTCAGGGC-3′ and at the 3′ end, 5′-ACACGGGCAGCAGCGTCA-3′; second round at the 5′ end, 5′-GAGGGATGGATCTGCTT-3′ and at the 3′ end, 5′-GAAGTGCGAGTTCGAGCC-3′; and with vector sequences flanking the cDNA inserts. PCR products from the second round were ligated into the pT-Adv vector (Clontech Laboratories, Inc.).
To generate specifically SdpII full-length clones, a gene product containing noncoding sequence at the 5′ and 3′ end was amplified by PCR reaction and used as a template for a second round of PCR with primers BQ056 (5′-CGCGGATCCATGTCTGTCACCTACGATGA-3′) and BQ057 (5′-CGGAATTCTCACTGGATAGCCTCAGCATAG-3′). The PCR products of the second round (sizes ∼1,400–1,600 bp) were digested with BamH1 and EcoRI and subcloned into the pGEX-2T vector (Pharmacia Biotech). The nucleotide sequences have been deposited in the Genbank/EMBL/DDBJ database with accession numbers AF139492, AF139493, AF139494, and AF139495.
All PCRs were carried out with the EXPAND High Fidelity PCR System (Boehringer Mannheim Corp.). DNA of all PCR products was sequenced in the UCSF/BRC sequencing facility.
DNA Constructs and Recombinant Proteins
Glutathione S-transferase (GST) fusion proteins containing full-length SdpI or domains thereof were generated as described (Qualmann et al. 1999). The following GST–SdpII constructs were produced by performing PCR on the appropriate full-length constructs: SdpII–SH3 (amino acid residues 419–488 of SdpII-l) with primer BQ058 (5′-CGCGGATCCAACCCGTTTGACGAGGACAC-3′) and primer BQ057; SdpII-l–N (residues 1–426) and SdpII-s–N (residues 1–387) with primer BQ056 and with primer BQ059 (5′-CGGAATTCTCATCCTGAGGTAGTGTCCTCG-3′). The resulting DNA fragments were cloned into the BamH1–EcoRI sites of the pGEX-2T vector. A construct encompassing residues 305–387 of SdpII-l for antibody generation (SdpII-l–Ab) was generated with primers BQ076 (5′-CGGAATTCGAGTGGTCTGCAGATCTGA-3′) and BQ077 (5′-CGCGTCGACTAGCTGACATTTTTGGCCTTA-3′). The PCR product was digested with Sal1 and EcoRI and cloned into the Sal1–EcoRI sites of pGEX-5X. GST fusion proteins were expressed in Escherichia coli BL21 cells according to standard methods and purified from cell lysates on glutathione agarose (Sigma Chemical Co.) columns as described before (Qualmann et al. 1999). GST for control experiments was expressed from the plasmid pGEX-2T.
A construct to express a maltose binding protein (MBP) fusion protein of SdpII for affinity purification of anti-SdpII antibodies was obtained by subcloning SdpII-l–Ab into the Sal1–EcoRI sites of the pMAL-c2 vector (New England Biolabs). MBP fusion proteins were expressed and purified over an amylose column following the recommendations of the manufacturer.
For expression in mammalian cells, constructs encoding the full-length proteins or fragments thereof were subcloned into the pcDNA3.1/His vector (Invitrogen). Since expression of the SH3 domains was very low, new plasmids containing slightly larger COOH-terminal fragments were generated by PCR using the appropriate plasmids as template. SdpI–SH3, wild-type and mutant (residues 339–441), were generated with forward primer BQ070 (5′-CGCGGATCCGGGGACCGTGGCAGTGTCA-3′) and reverse primer BQ026 (Qualmann et al. 1999), SdpII–SH3 (residues 383–488 of SdpII-l) with primer BQ068 (5′-CGCGGATCCAAGGCCAAAAATGTCAGCAG-3′) and primer BQ057. The PCR products were subsequently cloned into the BamH1–EcoRI sites of pcDNA3.1/His.
A construct for expression of the COOH-terminal part of rat N-WASP containing the verpolin homology, cofilin, and acidic domains (VCA; amino acids 391–501, N-WASP–VCA) in mammalian cells was generated by PCR with primers BQ092 (5′-CCGCTCGAGGGTGACCATCAAGTTCCAG-3′) and BQ093 (5′-CGGAATTCAGTCTTCCCACTCATCATC-3′) using rat N-WASP cDNA as a template. The PCR product was cloned into the XhoI–EcoRI sites of a derivative of the pEGFP-C1 vector (Clontech), in which GFP was replaced by the HA peptide.
Antibodies
Polyclonal anti-SdpII antibodies were raised in rabbit (3685) and guinea pig (P339; Alpha Diagnostic International., Inc.) against a purified GST fusion protein of amino acid residues 305–387 of the long SdpII splice variant (SdpII-l–Ab). Antibodies were affinity-purified on an analogous MBP fusion protein of SdpII-l–Ab blotted to nitrocellulose membranes (Qualmann et al. 1999).
SdpI-specific antibodies (antiserum 2703) were raised and affinity-purified as described previously (Qualmann et al. 1999). Rabbit antisera 2521, 2703, and 2704 also served as the source for affinity-purified anti-GST antibodies. Antisynaptojanin antibodies, anti–N-WASP antibodies, and anti-Arp3 antibodies were kindly provided by Dr. P. McPherson (McGill University, Montreal, Canada), Dr. H. Miki (University of Tokyo, Japan), and Dr. M.D. Welch (University of California, Berkeley, CA), respectively. Antibodies against dynamin-1 (hudy-1) and synapsin I were purchased from Upstate Biotechnology and Biogenesis, respectively. Mouse ascites fluid containing mAbs against human transferrin receptor (H68.4) was generated by Berkeley Antibody Co. from cells kindly provided by Dr. I.S. Trowbridge (Salk Institute, La Jolla, CA).
Tissue Homogenates and Cell Extracts
Postnuclear supernatants and subcellular fractions from different rat tissues (brain, liver, kidney, spleen, lung, skeletal muscle, heart, and testis) were prepared and processed for Western blots as described (Qualmann et al. 1999). To generate cellular extracts, cells grown to 80–90% confluency were rinsed with PBS and lysed in 0.1% Triton X-100 in buffer A (10 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM EGTA, 0.1 mM MgCl2) supplemented with protease inhibitors (10 μg/ml aprotinin, 5 μg/ml leupeptin, 2 μg/ml antipain, 10 μg/ml benzamidine, 1 μg/ml chymostatin, 5 μg/ml pepstatin, 1 mM PMSF) for 30 min at 4°C. The lysates were cleared by centrifugation for 10 min at 16,000 g at 4°C.
Blot Overlay
Blot overlays with recombinant fusion proteins were performed as described previously (Roos and Kelly 1998). In brief, samples were resolved on SDS-PAGE gels and transferred to nitrocellulose. Membranes were blocked and incubated overnight at 4°C with the GST fusion protein in 5% nonfat dry milk powder in PBS/0.05% Tween 20. Fusion protein binding was detected by subsequently incubating the overlays with affinity-purified antibodies directed against GST, goat anti–rabbit HRP-conjugated secondary antibody (Cappel/ICN), and the ECL detection system (Amersham).
Coprecipitation Assays
For the generation of solubilized brain extracts, frozen rat brains (Pel-Freeze) were diced and homogenized at 4°C in buffer A (1:3, wt/vol) supplemented with protease inhibitors via 20 passes in a glass Teflon Dounce homogenizer. The homogenate was incubated with Triton X-100 at a final concentration of 1% (vol/vol) for 1 h at 4°C and insoluble material was removed by centrifugation for 1 h at 40,000 g. Recombinant GST fusion proteins (25 μg each) were immobilized on glutathione Sepharose 4B beads (Pharmacia Biotech) in PBS. 25 μl of beads were incubated with rat brain extracts (1 mg protein) overnight at 4°C. Beads were extensively washed with buffer A and eluted with 30 μl of 20 mM glutathione in 50 mM Tris, pH 8.0, 120 mM NaCl for 30 min at room temperature. Proteins were then separated on 4–15% SDS-PAGE and assayed by Coomassie staining and by Western blotting with various antibodies.
Immunoprecipitation
Pheochromocytoma cells (PC12) grown to 80–90% confluency were rinsed with PBS and collected by centrifugation (1,000 g, 5 min). The cell pellet was resuspended in IP buffer (10 mM Hepes, pH 7.4, 10 mM NaCl, 1 mM EGTA, 0.1 mM MgCl2, 1% Triton X-100) supplemented with protease inhibitors (see above), and lysed for 30 min at 4°C. Cell lysates were cleared by centrifugation at 14,000 g for 20 min at 4°C. Immunoprecipitations from PC12 cell extracts were performed with affinity-purified SdpII-specific antibodies (from serum 3685) or unrelated rabbit IgG prebound to protein G–Sepharose as described previously (Qualmann et al. 1999).
Immunofluorescence
PC12 cells for immunocytochemistry were grown in DME H-21 supplemented with 0.5% FCS and 1% horse serum at 37°C in 10% CO2 on Lab-Tek Permanox slides (Nunc) that were coated with 50 μg/ml poly-l-lysine (Sigma Chemical Co.) and 75 μg/ml rat tail collagen (Collaborative Biomedical Products). Cells were differentiated with 50 ng/ml NGF (Boehringer Mannheim Corp.) for 2 d; the medium was replaced every 2 d. For immunofluorescence staining, cells were washed twice in 50 mM MES, pH 6.3, 5 mM MgCl2, 3 mM EGTA, and fixed in 4% freshly depolymerized paraformaldehyde (Sigma Chemical Co.) and 0.1% glutaraldehyde (Fluka) in PBS for 30 min. After quenching free aldehyde groups with 25 mM glycine in PBS twice for 10 min and an additional wash in PBS, the cells were permeabilized and blocked for 1 h in 2% BSA, 1% fish skin gelatin, and 0.02% saponin in PBS (block solution). The slides were then incubated with primary antibodies for 90 min at room temperature and subsequently washed three times with block solution. Fluorophore-conjugated secondary antibodies and, when indicated, phalloidin-Texas red (1:667 dilution; Molecular Probes) were applied for 1 h at room temperature in block solution. After several washes, cells were mounted in Moviol (Calbiochem-Novabiochem Corp.).
As primary antibodies, affinity-purified antibodies from serum 2703 and 3685/P339, which specifically recognize SdpI and -II, respectively, were used at 1:100. The expression of constructs in the pcDNA3.1/His vector was detected with anti-Xpress antibodies (1:500; Invitrogen), the expression of HA-tagged N-WASP fragments with the monoclonal anti-HA antibody HA.11 (1:1,000; Berkeley Antibody Co.). Secondary antibodies used in this study were fluorescein-conjugated goat anti–rabbit IgG (Cappel), fluorescein-conjugated goat anti–mouse IgG (Cappel), fluorescein-conjugated goat anti–guinea pig IgG (Cappel), Texas red-conjugated goat anti–mouse IgG (Jackson ImmunoResearch Laboratories), Texas red-conjugated rabbit anti–mouse IgG (Cappel), rabbit anti–mouse FITC (Jackson ImmunoResearch Laboratories, Inc.), and Alexa Fluor™ 350 goat anti–mouse IgG (Molecular Probes).
Incubations were viewed under a Leica TCS NT laser confocal microscope using the Leica TCS software package or with an inverted Nikon Eclipse TE300 fluorescence microscope and recorded digitally (ImageProPlus; Phase3 Imaging Systems). Images were processed using Adobe Photoshop software.
Endocytosis Assay and Actin Cytoskeleton Analysis
To analyze the effect of overexpression of the full-length syndapin proteins or fragments thereof, HeLa cells were transiently transfected using the FuGENE™ 6 transfection reagent (Boehringer Mannheim Corp.) with the appropriate SdpI or -II cDNAs subcloned into the pcDNA3.1/His vector and/or cDNA encoding N-WASP fragments. HeLa cells were maintained in DME H-21 supplemented with 10% FCS at 37°C in 10% CO2 and replated onto glass coverslips (18-mm diam). Transfection was carried out in serum-containing medium following the recommendations of the manufacturer. For cotransfection experiments, equal amounts of the appropriate plasmids were used. Cotransfection efficiency was >95%, as judged by double immunofluorescence staining. 24 h after transfection, N-butyric acid (Sigma Chemical Co.) was added at a final concentration of 6 mM to increase protein expression levels. After a further 24 h, cells were processed to study receptor-mediated endocytosis and cytoskeletal architecture as follows.
HeLa cells used for the transferrin internalization assays were serum-starved in DME H-21 containing 1 mg/ml BSA for 45 min at 37°C and subsequently incubated with 25 μg/ml FITC-transferrin (Molecular Probes) in DME H-21/BSA. After three washes with ice-cold PBS (containing 0.3 mM CaCl2 and 0.3 mM MgCl2), cell surface labeling was reduced by incubating the cells twice with 50 μM of the iron chelator, deferoxamine mesylate (Sigma Chemical Co.), in 150 mM NaCl, 2 mM CaCl2, 25 mM sodium acetate/acetic acid, pH 4.5, for 10 min on ice according to Stoorvogel et al. 1991. The cultures were fixed in 4% freshly depolymerized paraformaldehyde in PBS for 30 min and processed for immunofluorescence as described. The percentage of transferrin uptake-positive cells was determined by counting cells from at least three independent experiments. Cells were only scored to be uptake-negative when no FITC signal was observed.
To assess cytoskeletal changes, cells were washed twice in 50 mM MES, pH 6.3, 5 mM MgCl2, 3 mM EGTA, and fixed in 4% freshly depolymerized paraformaldehyde and 0.1% glutaraldehyde in PBS for 30 min and processed for immunofluorescence staining. Filamentous actin was visualized with Texas red-phalloidin (Molecular Probes), microtubuli were stained with mouse anti–α-tubulin (Nycomed Amersham Inc.). Quantitation of the percentage of cells with filopodia induction was performed as follows. According to the cell morphology and cortical actin organization observed on the red fluorescence channel, cells were classified into those exhibiting numerous filopodia or those with almost no filopodia (see Fig. 7 k) at the cell surface. Thereafter, cells in both groups were analyzed for expression of full-length syndapin or fragments thereof as determined by labeling with syndapin- or tag-specific primary antibodies and FITC- or fluorescein-conjugated secondary antibodies. For each construct and for untransfected cells, the phenotype induction was analyzed in at least three independent experiments. Each bar corresponds to >250 cells counted.
Figure 7.
Only full-length proteins are sufficient to induce cytoskeletal rearrangements upon overexpression. HeLa cells transiently transfected with SdpI full-length (a and b), but not the SdpI SH3 domain alone (c and d), the SdpI full-length P434L mutant (e and f), the NH2 terminus of SdpII-l (g and h), or SdpII-s (i and j) exhibit filopodia. 48 h after transfection, cells were fixed and stained for expression of the syndapin constructs (anti-Xpress labeling; left) and filamentous actin (phalloidin–Texas red staining; right). Bar, 25 μm. k, Overexpression of syndapin full-length proteins dramatically increased the number of filopodia observed at the cell surface of HeLa cells from 2.9 ± 2.3 filopodia/100 μm in untransfected to 52.4 ± 9.9, as observed by confocal microscopy (n = 15 cells each; Σ surface length analyzed = 2,361 μm and 1,630 μm for SdpI-overexpressing and untransfected cells, respectively. l, HeLa cells transfected with plasmids encoding wild-type or mutant syndapin proteins, or fragments thereof, were classified into those exhibiting numerous filopodia or those with almost no filopodia at their cell. Thereafter, these cells were analyzed for expression of the protein encoded by the transfected construct. Bars represent the mean ± standard deviation of the percentage of cells with filopodia induction from at least three independent experiments (n > 250 for each construct). Only wild-type full-length syndapin proteins potently triggered filopodia formation. Massive filopodia induction was observed in 97.7 ± 0.8% of cells overexpressing SdpI and 86.5 ± 2.4% and 70.0 ± 2.5% of cells overexpressing the short and long splice variants of SdpII (SdpII-s and SdpII-l), respectively. Percentages of filopodia-positive in cells overexpressing truncated syndapin constructs (5.2 ± 2.7% and 6.9 ± 1.4% for the SH3 domain of SdpI and -II, respectively, and 6.4 ± 1.6%, 9.7 ± 1.5%, and 6.8 ± 4.5% for the NH2-terminal parts of SdpI, -II-s, and -II-l, respectively) were quite similar to levels observed in untransfected cells (4.5 ± 2.2%). Even a single point mutation in the SH3 domain, P434L, abolished the ability of SdpI to trigger filopodia formation at the cell cortex (4.0 ± 1.7%; SdpIm).
Syndapin expression was visualized either with anti-Syndapin or anti-Xpress (Invitrogen) antibodies.
Results
Identification and Cloning of SdpII
Dynamin and endophilin have brain-specific forms and forms with wider tissue distribution (Ringstad et al. 1997; Urrutia et al. 1997), implying that they are universally used in endocytosis. This also appears to be true for syndapin. During cloning of the gene for the synaptic dynamin-associated protein, SdpI (Qualmann et al. 1999), we identified additional 1,400–1,600 bp messages in the rat cDNA library that all encoded different splice variants of the same putative protein, which we named SdpII because it has 67% sequence identity with SdpI (Fig. 1). The open reading frame of the longest gene identified encodes a putative protein of 488 amino acids with a predicted molecular weight of 56.1 kD. We identified additional messages encoding splice variants of SdpII lacking amino acids 303–304 and/or amino acids 346–386 (Fig. 1 b). The polymorphism at the first splice site has no detectable effects on the properties of SdpII (data not shown). The long form of SdpII containing the second insert will be referred to as SdpII-l, while for the shorter form lacking the insert we will use the term SdpII-s. The first sequence reported for a syndapin family member was FAP52, a focal adhesion protein found in chicken (Meriläinen et al. 1997). This protein is most similar to SdpII-s (Fig. 1, a and c). Recently, PACSIN2, the mouse homologue of SdpII-l, has been described (Ritter et al. 1999).
Figure 1.
Sequence analysis of SdpII. a, The amino acid sequence of the shortest and the longest of the four splice variants of the novel rat proteins SdpII, SdpII-s, and SdpII-l, respectively, is shown aligned to SdpI (GenBank/EMBL/DDBJ accession number AF104402), the chicken protein FAP52 (GenBank/EMBL/DDBJ accession number Z50798), and murine PACSIN 2 (GenBank/EMBL/DDBJ accession number AAD41780). Boxes indicate residues of ≥80% identity between all five sequences. b, Schematic cartoon of the domain structure of SdpII. Members of the syndapin protein family share a similar domain structure with a highly conserved SH3 domain at the COOH terminus, two predicted coiled coil domains, and several NPF motifs. The multidomain protein is alternatively spliced at at least two different sites (amino acids 303–304 and/or amino acids 346–386 of SdpII-l). c, Comparison of sequence identities between SdpII and related proteins. The COOH-terminal SH3 domain, corresponding to amino acids 430–488 in SdpII-l, represents the part of the protein that is most highly conserved between members of the syndapin protein family. These sequence data for SdpII are available from GenBank/EMBL/DDBJ under accession numbers AF139492, AF139493, AF139494, and AF139495.
Members of the syndapin protein family share a similar domain structure with a highly conserved SH3 domain at the COOH terminus, two predicted coiled coil domains, and several arginine–proline–phenylalanine (NPF) motifs (Fig. 1 b). The COOH-terminal SH3 domain represents the part of the protein that is most highly conserved between members of the syndapin protein family (Fig. 1 c). The region containing the two splice sites in SdpII shows the lowest degree of sequence similarity between syndapin-like proteins and might, therefore, correspond to a flexible linker region. However, this region contains multiple copies of the tripeptide motif NPF, two copies in SdpI and SdpII-s, and an additional copy in the insert in SdpII-l. NPF motifs mediate protein–protein interactions to Eps15 homology domains, modules that appear to have an important role in endocytic function.
Syndapin II Splice Variants Show a Distinct Expression Pattern in a Variety of Different Tissues and Cell Lines
Polyclonal antisera against SdpII were produced in rabbits and guinea pigs by injecting a GST fusion protein encoding the region with the lowest degree of sequence similarity between SdpI and -II, amino acids 305–387 of SdpII-l. After affinity purification, antibodies were tested for specificity on extracts of cells transiently transfected with the full-length proteins SdpI, SdpII-s, SdpII-l, or vector alone transferred to nitrocellulose membranes. Affinity-purified guinea pig antibodies (P339) specifically recognized both forms of SdpII; the rabbit antibodies (3685), however, showed a preference for SdpII-l (Fig. 2 a). Antibodies raised against the NH2 terminus of SdpI (Qualmann et al. 1999) did not cross-react with SdpII (Fig. 2 a). An additional weak band at 65 kD could be detected in all cell lysates, including the vector-only control after long exposures of the blots incubated with antibodies from P339 and 3685 (data not shown). This signal most likely corresponds to endogenous SdpII-l present in the HeLa cells.
Figure 2.

SdpII-specific antibodies identify different splice variants of the protein in a number of different cell lines and tissues. a, Lysates from HeLa cells transfected with the full-length proteins SdpI, SdpII-s, SdpII-l, or vector alone were separated on SDS-PAGE, transferred to nitrocellulose, and probed with either tag-specific antibodies (anti-Xpress) or affinity-purified antibodies raised against SdpI (antiserum 2703) and SdpII (antisera P339 and 3685). b, Postnuclear supernatants from various rat tissues and cellular extracts were resolved by SDS-PAGE, transferred to nitrocellulose, and subjected to Western blot analysis. SdpII-specific antibodies recognized a 65-kD band predominantly in the PC12 cell line and heart, and, less prominent, in testis and additional cellular extracts. In addition, a protein of an electrophoretic mobility of 52 kD was detected in all tissues analyzed, which appears to be enriched in brain, kidney, testis, and skeletal muscle.
Western blot analysis of cellular extracts and postnuclear supernatants from a variety of rat tissues demonstrate that SdpII is ubiquitously expressed with a different tissue distribution for the short and long splice variants. SdpII-l is preferentially expressed in the PC12 cell line and heart, whereas SdpII-s was detected in most tissues examined (Fig. 2 b) consistent with the expression pattern described for the chicken focal adhesion protein, FAP52 (Meriläinen et al. 1997). Using antibody dilutions on Western blots that were calibrated against material expressed in HeLa cells, SdpII is far less abundant than SdpI (data not shown).
Investigation of SdpII-binding Partners
SdpI has been shown to recognize a subset of highly brain-enriched proteins via its SH3 domain. By an overlay technique, the protein bands recognized by full-length SdpII (Fig. 3 a) were almost identical to the binding pattern previously shown for SdpI full-length. Thus, the high degree of similarity in the SH3 domain of SdpI and -II is reflected in similar binding patterns. The 41-amino acid insert in SdpII-l did not appear to significantly change the binding pattern of the affinities displayed. Binding partners of SdpII were only observed in nonneural tissues after longer exposure. Nonneural tissues may have a low abundance of binding partners consistent with the similarly low expression levels of SdpII. A major protein detected with SdpII in all tissues analyzed comigrated with N-WASP.
Figure 3.

Characterization of SdpII protein–protein interactions. a, Overlay analysis of postnuclear supernatants from various rat tissues with GST fusion proteins of SdpII full-length molecules. 50 μg of each fraction was resolved on 4–15% SDS-PAGE gels, transferred to nitrocellulose, and overlaid with GST–SdpII-l and GST–SdpII-s. Binding of fusion proteins was detected with affinity-purified anti-GST antibodies. Both SdpII splice variants bind a similar subset of brain-enriched or brain-specific proteins. After a long exposure, the interaction of SdpII with a protein of 65 kD was also detectable in nonneural tissues (arrow). b, Triton X-100 soluble proteins from rat brain were affinity-purified onto GST–SdpII SH3 domain, GST–SdpI SH3 domain wild-type, or the P434L point mutant immobilized on glutathione Sepharose. Material specifically bound was eluted with glutathione containing buffer and analyzed per SDS-PAGE and Western blotting. The wild-type SH3 domains from both SdpI and -II, but not the mutant form, specifically coprecipitate synaptojanin, dynamin I, synapsin I, and N-WASP. c, Coimmunoprecipitation of dynamin with SdpII. Immunoprecipitates from PC12 extracts were generated with affinity-purified antibodies against SdpII (from rabbit serum 3685) or with unrelated rabbit IgG immobilized onto protein G–Sepharose. Precipitated material was analyzed by Western blotting using antidynamin antibodies (hudy-1) and anti-SdpII antibodies (from guinea pig serum P339).
To identify some of the major binding partners of SdpII, GST fusion proteins comprising either the wild-type SdpII SH3 domain, the wild-type SdpI SH3 domain, or the P434L mutant SdpI SH3 domain, immobilized on glutathione Sepharose beads, were incubated with Triton X-100 extracts from rat brains homogenized in buffer A. Precipitated material was eluted with glutathione, separated by SDS-PAGE, and assayed by immunoblotting. The SH3 domain of SdpII coprecipitated the PRD-containing proteins synaptojanin, dynamin I, synapsin I, and the N-WASP as shown in Fig. 3 b. The amount of precipitated material was comparable for the SH3 domains of SdpII and SdpI; however, the SdpI SH3 domain exhibited a greater affinity for synapsin I. A GST fusion protein of the P434L mutated form of the SdpI SH3 domain was included as a control and did not show any binding to synaptojanin, dynamin, synapsin, or N-WASP (Fig. 3 b).
Coimmunoprecipitation analyses from PC12 cell extracts furthermore demonstrate that SdpII is associated with dynamin. Affinity-purified anti-SdpII antibodies, but not unrelated rabbit IgG, coimmunoprecipitated dynamin (Fig. 3 c).
Our results indicate that syndapins are not restricted to nerve cells, and that the neuronal and ubiquitous forms have similar properties, particularly the ability to bind dynamin and N-WASP.
Syndapin II Colocalizes with Dynamin in NGF-differentiated PC12 Cells
Antibodies specific to SdpII allowed us to investigate the distribution of endogenous SdpII in a differentiating neuroendocrine cell line. Rat PC12 cells, which resemble chromaffin cells under normal growth conditions, respond to NGF by differentiating into nonreplicating, neuron-like cells. Within 24–48 h, NGF-treated cells flatten and begin to send out short neurite-like processes, which extend with time (Fig. 4). The endogenous SdpII colocalized with dynamin at the edges of the cell bodies (Fig. 4, a–c) and, to an even higher degree, at the tips and in varicosities along the processes (Fig. 4, d–i), as observed by confocal microscopy. Striking was the concentration of both markers in punctae of ∼0.7 μm in diameter. These are reminiscent of hot spots of endocytosis seen in Drosophila melanogaster nerve terminals (Estes et al. 1996; González-Gaitán and Jäckle 1997; Roos and Kelly 1998). Colocalization of the two markers in the punctae suggested that SdpII is involved in endocytosis.
Figure 4.

SdpII colocalizes with dynamin in NGF-stimulated PC12 cells. PC12 cells, differentiated with 50 μg/ml NGF for 2 d, were fixed and double-labeled with antibodies directed against SdpII (a, d, e, and j), dynamin (b, f, and g), or transferrin receptor (k). SdpII localizes to discrete areas in cortical cell regions (a), in growth cones (e), and in the varicosities of processes (d) that are marked by the endocytic machinery of the cells, as indicated by staining for dynamin (b, f, and g). Colocalization of SdpII with transferrin receptor is partial and most prominent at the cell surface (l). Bars: (a–c, j–l) 5 μm; (d–i) 3 μm.
The transferrin receptor could also be recovered in small punctae (Fig. 4 k). The subcellular distribution of SdpII in PC12 cells overlapped only partially with transferrin receptor-positive compartments. Colocalization was at its maximum, but still not complete when the focal plane was close to the plasma membrane (Fig. 4, j–l). However, SdpII does not codistribute with the transferrin receptor at perinuclear compartments, since overlap was minimal in confocal sections through the center of the cells (data not shown). Thus, it seems that the patches of dynamin, SdpII, and transferrin receptor only show overlap when they are on or near the cell surface. Intracellular endosomes do not appear to contain SdpII.
Syndapin SH3 Domains Inhibit Receptor-mediated Endocytosis In Vivo
SdpI and -II are associated via a PRD/SH3 domain interaction with the large GTPase dynamin (this study; Qualmann et al. 1999) which has been demonstrated to be an essential component in the formation of clathrin-coated vesicles in the receptor-mediated endocytosis pathway (Damke et al. 1994). To assess the relevance of syndapin interactions on receptor-mediated endocytosis in vivo, we transiently overexpressed different domains of syndapin in HeLa cells and assayed the endocytic accumulation of FITC-labeled transferrin. In untransfected cells, the transferrin internalized for 30 min at 37°C accumulated in vesicular structures spread throughout the cytoplasm. Overexpression of the wild-type SH3 domains of SdpI and -II in HeLa cells inhibited endocytosis as evidenced by a block in accumulation of intracellular FITC–transferrin (green signal) in transfected cells (red fluorescence), compared with neighboring untransfected cells (Fig. 5 a). In contrast, transfection of cells with the NH2 terminus or the mutant SH3 domain (P434L) of SdpI had no effect on transferrin accumulation (Fig. 5 a). Quantitation of the results by assessing the percentage of cells with detectable transferrin uptake demonstrates that the SH3 domains of syndapin can interfere with receptor-mediated endocytosis in vivo. Whereas almost all cells overexpressing the NH2 terminus of Sdp (97.4 ± 0.3%, n = 956) or the P434L mutant of the SdpI SH3 domain (97.0 ± 0.3%, n = 366) were capable of clathrin-mediated endocytosis, the fraction of cells that internalized transferrin was significantly reduced among those overexpressing the wild-type SdpI or -II SH3 domain (Fig. 5 b). Only 44.4 ± 3.4% (n = 519) of the cells overexpressing the wild-type SdpI SH3 domain and 47.6 ± 4.4% (n = 450) of cells overexpressing the SdpII SH3 domain contained some internalized transferrin. The fraction of cells that could still take up transferrin was also reduced by overexpression of the SH3 domains of amphiphysin I and II (24.0 ± 3.1%, n = 641, and 73.1 ± 6.5%, n = 433 uptake-positive cells, respectively) as has been reported previously (Wigge et al. 1997; Owens et al. 1998).
Figure 5.
A surplus of SdpI and -II SH3 domain inhibits transferrin internalization in vivo. a, HeLa cells transiently transfected with SdpI SH3 domain, SdpII SH3 domain, the P434L mutant form of the SdpI SH3 domain, or the SdpI NH2 terminus were incubated at 37°C with FITC-transferrin for 30 min, fixed, and processed for immunofluorescence microscopy. Expression of syndapin constructs was visualized with anti-Xpress antibodies, followed by a Texas red-conjugated secondary antibody. Panels show superimposition of FITC–transferrin with the staining of the epitope-tagged protein fragments. SdpI and -II SH3 domains inhibited endocytosis as evidenced by a block in accumulation of intracellular transferrin (green signal) in transfected cells (red fluorescence) compared with neighboring untransfected cells. In contrast, transfection of cells with the NH2 terminus or the mutant SH3 domain had no effect on transferrin accumulation. b, Quantitation of the results by assessing the percentage of transferrin uptake-positive cells demonstrates that a surplus of the SH3 domain of syndapin interferes with receptor-mediated endocytosis in vivo. Whereas 97.4 ± 0.3% (n = 956) of the HeLa cells overexpressing the NH2 terminus of SdpI and 97.0 ± 0.3% (n = 366) of cells overexpressing the P434L mutant of the SdpI SH3 domain were capable of clathrin-mediated endocytosis, only 44.4 ± 3.4% (n = 519) of the cells overexpressing the wild-type SdpI SH3 domain and 47.6 ± 4.4% (n = 450) of cells overexpressing the SdpII SH3 domain contained some internalized transferrin.
The results might underestimate the amount of inhibition since only cells exhibiting virtually no FITC–transferrin signal were counted as uptake-negative, whereas partial inhibition was neglected.
Syndapin Overexpression Induces Rearrangements of the Cortical Actin Cytoskeleton
SdpI and -II interact with the N-WASP. N-WASP in turn interacts with the Arp2/3 complex, leading to a stimulation of actin filament nucleation and polymerization. This prompted us to investigate the consequences of syndapin overexpression on the actin cytoskeleton in HeLa cells.
Syndapin overexpression induced a striking cortical actin phenotype. Cells transiently transfected with full-length SdpI exhibited actin-rich filopodia (Fig. 6, a–d) all over the cell surfaces. Similar filopodia formation was triggered by overexpression of both SdpII-l (Fig. 6e and Fig. f) and SdpII-s (Fig. 6g and Fig. h); however, the phenotype was less striking for SdpII than SdpI. Massive filopodia induction (Fig. 7 k) was observed in almost all SdpI-transfected cells (97.7 ± 0.8%) and in 86.5 ± 2.4% and 70.0 ± 2.5% of HeLa cells overexpressing the short and the long splice variant of SdpII, respectively (Fig. 7 l). This overexpression phenotype was only observed in transfected cells; in contrast, untransfected cells displayed only a few short filopodia (Fig. 6, a–h). SdpI-positive cells exhibited 52.4 ± 9.9 filopodia over a surface length of 100 μm, compared with 2.9 ± 2.3 in untransfected cells (Fig. 7 k). Filopodia at the surface of syndapin-overexpressing cells were in average two to three times longer and thicker than those at the surface of untransfected cells. In addition to peripheral actin microspikes, SdpI overexpression also frequently resulted in the induction of lamellipodial-like protrusions (Fig. 6c and Fig. d, and see Fig. 9, a, b, e, and f). Syndapin localized to the periphery of these lamellipodia (Fig. 6 c, and see Fig. 9, a and e). In general, syndapin did not colocalize with stable F-actin stress fibers, but instead localized to areas characterized as sites of actin polymerization and turnover. The observed cytoskeletal changes were not specific to HeLa cells, but were induced as well in NIH 3T3 fibroblasts (data not shown). Western blot analysis confirmed the expression of the transfected genes, revealing appropriately sized proteins (Fig. 2 a, and data not shown).
Figure 6.
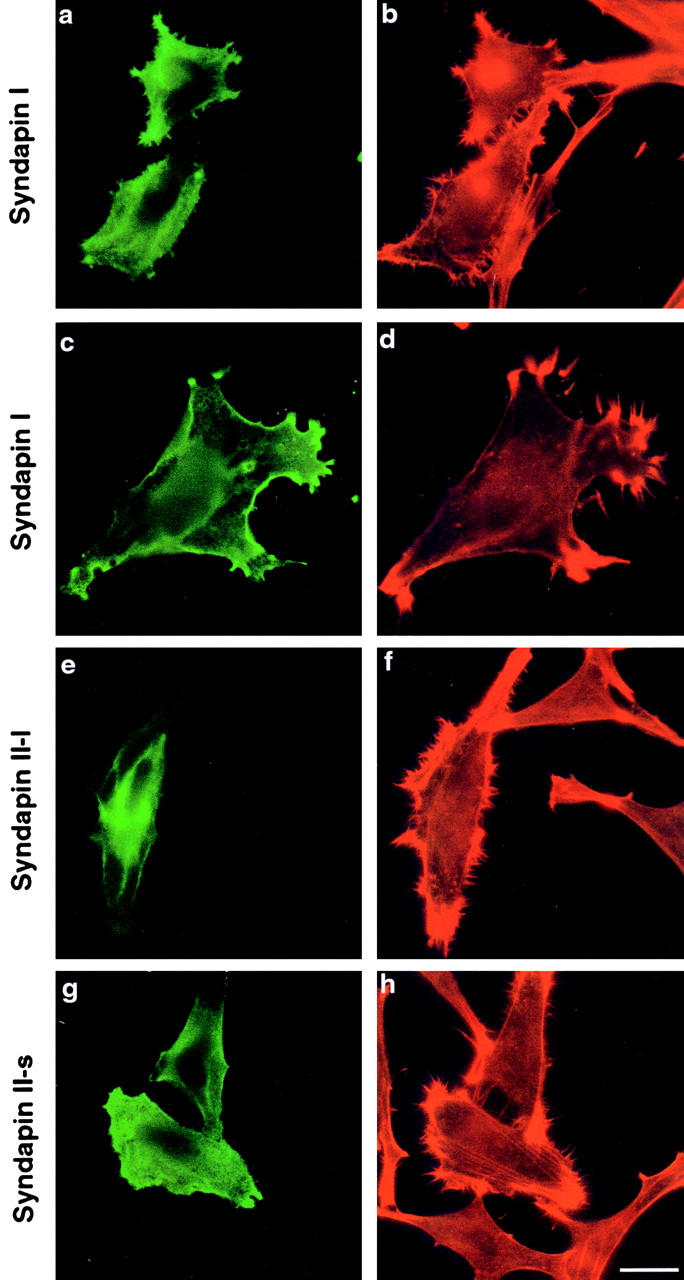
SdpI and -II induce a striking cortical actin phenotype upon overexpression. HeLa cells transiently transfected with SdpI full-length (a, b, c, and d), SdpII-l (e and f), and SdpII-s (g and h) (green fluorescence) exhibit actin microspikes (filopodia) all over the cell surfaces in addition to protrusion-like structures (lamellipodia). This overexpression phenotype was only observed in transfected cells, untransfected cells were unaffected and displayed smooth cell surfaces. Right panels show phalloidin–Texas red staining of F-actin to visualize actin rearrangements. Antitag labeling to identify syndapin-overexpressing cells is shown in the left panels. Bar: (a, b, e–h) 25 μm; (c and d) 15.75 μm.
Figure 9.
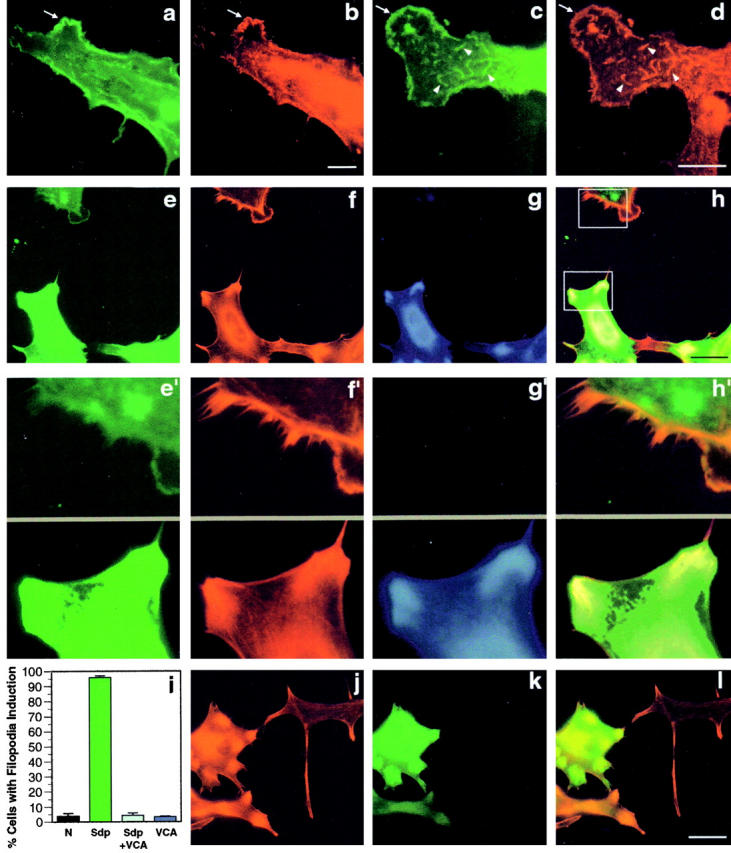
Syndapin-induced cortical actin rearrangements require the Arp2/3 complex. The Arp2/3 complex, visualized with an antibody directed against Arp3 (b and d) colocalizes with SdpI (a and c) at lamellipodial (arrows) and other structures (arrowheads) induced upon overexpression of SdpI in transiently transfected HeLa cells. Expression of syndapin was detected with antibodies against the Xpress-tag (a and c). Coexpression of the COOH-terminal VCA fragment of N-WASP (amino acids 391–501) suppressed the syndapin-induced cortical actin phenotype completely (e–i). Cells expressing only SdpI, as identified by staining with anti-SdpI antibodies and FITC-conjugated secondary antibodies (e and e′), exhibit lamellipodia and numerous filopodia detected by phalloidin (red fluorescence; f and f′). Other cells, coexpression of N-WASP–VCA, as identified by staining with anti-HA–antibodies and Alexa Fluor™ 350-conjugated secondary antibodies (g), impaired the customary induction of lamellipodia and filopodia (f and f′). Anti-Sdp, phalloidin, and anti-HA staining are overlaid in h. e′–h′ show 3.5-fold enlargements of selected surface areas from SdpI only (top) or SdpI and N-WASP–VCA coexpressing cells (bottom), as indicated by the boxes in h. Quantitation of the percentage of cells with filopodia induction (i) demonstrates that coexpression of the COOH-terminal VCA fragment of N-WASP reduced the phenotype induction from 95.9 ± 1.2% in HeLa cells overexpressing syndapin alone to 4.4 ± 1.5%. This value equals those in untransfected cells (3.7 ± 2.0%) and cells expressing N-WASP–VCA alone (3.5 ± 0.4%). Transfection of HeLa cells with N-WASP–VCA (k, green fluorescence) did not cause cortical actin rearrangements, but resulted in an increase in phalloidin-stainable actin, especially in the perinuclear region (j) even at low expression levels of N-WASP–VCA (k and l). Bars: (a–d) 10 μm; (e–h and j–l) 30 μm.
To determine structural domains of syndapin involved in these peripheral actin rearrangements, various constructs were transiently expressed in HeLa cells. The morphological changes were not observed with the SH3 domain of SdpI (Fig. 7c and Fig. d) or SdpII alone, with only 5.2 ± 2.7% and 6.9 ± 1.4% of filopodia-positive cells detected (Fig. 7 l), indicating that binding of syndapin via its SH3 domain alone to the actin-modulating protein N-WASP is not sufficient to induce these cytoskeletal rearrangements. Furthermore, the NH2 terminus of SdpI, SdpII-l (Fig. 7g and Fig. h), or SdpII-s (Fig. 7i and Fig. j), as well as the full-length SdpI protein harboring the P434L mutation in the SH3 domain (SdpIm; Fig. 7e and Fig. f) were incapable of stimulating filopodial outgrowth. The percentages of filopodia-positive cells (Fig. 7 l), 6.4 ± 1.6%, 9.7 ± 1.5%, and 6.8 ± 4.5% of cells overexpressing the NH2-terminal parts of SdpI, II-s, and II-l, respectively, and 4.0 ± 1.7% of cells overexpressing SdpIm, were very similar to levels observed in untransfected cells (4.5 ± 2.2%). These results indicate that syndapin-induced filopodia formation depends on an intact NH2-terminal and an intact SH3 domain.
Strikingly, SdpI frequently localized to the very tips of the actin-rich filopodia induced upon overexpression (Fig. 8). These structures were observed to be very fragile and were easily ripped off during incubations. This may be the reason why they are not seen on every cell exhibiting filopodia.
Figure 8.

SdpI localizes to the very tips of actin-rich filopodia induced upon overexpression. HeLa cells transiently transfected with SdpI full-length were fixed and stained for filamentous actin (phalloidin–Texas red) and SdpI (anti-Xpress/FITC–anti-mouse). Bar, 15 μm.
Syndapin-induced Cortical Actin Rearrangements Are Mediated by the Arp2/3 Complex
The Arp2/3 complex can enhance the nucleation of new actin filaments in vitro and is required for actin-based motility in vivo (reviewed in Machesky and Insall 1999). In SdpI-transfected HeLa cells, the Arp2/3 complex, visualized with an antibody directed against Arp3, localized to lamellipodia (arrows) and other structures, which appeared to be cortical (arrowheads; Fig. 9b and Fig. d), induced upon overexpression of syndapin and colocalized at these sites with SdpI (Fig. 9, a and c). These results suggested that syndapin-induced cortical actin rearrangements involved the targeting of the Arp2/3 complex to the sites of dynamic actin remodelling at the cell cortex. To test this hypothesis, we coexpressed the COOH-terminal VCA-region of N-WASP with SdpI in HeLa cells. This COOH-terminal segment binds to actin (Miki et al. 1996) and to the Arp2/3 complex, and greatly enhances its ability to nucleate actin polymerization in vitro (Rohatgi et al. 1999). In Swiss 3T3 fibroblasts, a similar COOH-terminal fragment of Scar1 or WASP disrupted lamellipodia formation and impaired the localization of the Arp2/3 complex to the cell periphery (Machesky and Insall 1998).
Coexpression of the VCA fragment of N-WASP abolished the syndapin-induced cortical actin phenotype completely. Whereas HeLa cells overexpressing only syndapin showed the customary formation of lamellipodia and filopodia (Fig. 9e and Fig. f, and Fig. e′ and f′), cells expressing both SdpI and N-WASP–VCA did neither. Their cell surfaces appeared smooth (Fig. 9, e–h and e′–h′). This effect is not caused by reduced SdpI expression levels in double-transfected cells because severe cortical actin rearrangements were already triggered by moderate expression levels of SdpI (Fig. 9e and Fig. f, and Fig. e′ and f′). In contrast, in HeLa cells coexpressing N-WASP–VCA, SdpI overexpressed at much higher levels failed to induce filopodia formation as measured by phalloidin-Texas red staining (Fig. 9, e–h and e′–h′). N-WASP–VCA protein-positive cells were detected by antibodies to the HA-epitope and Alexa Fluor™ 350-conjugated secondary antibodies (blue fluorescence; Fig. 9g and Fig. g′). The effects were quantified by assessing the percentage of cells with filopodia induction (Fig. 9 i). Whereas the cortical actin phenotype was observed in almost every cell overexpressing SdpI alone (95.9 ± 1.2%), coexpression of N-WASP–VCA reduced the percentage to 4.4 ± 1.5%, which equals values observed in untransfected cells (3.7 ± 2.0%) and HeLa cells single-transfected with N-WASP–VCA (3.5 ± 0.4%). HeLa cells expressing N-WASP–VCA (green fluorescence; Fig. 9, j–l) exhibited a striking increase in diffuse phalloidin-stainable actin within the cytosol, especially in the perinuclear region (Fig. 9 j). This was also true when N-WASP–VCA was coexpressed with SdpI (Fig. 9, e–h and e′–h′).
These data suggest that syndapin-induced cortical actin rearrangements require the Arp2/3 complex and its translocation to the cell periphery.
Discussion
Based on protein–protein interactions, the synaptic dynamin-associated protein, SdpI, has been proposed to link actin dynamics and endocytic processes in the nerve terminal (Qualmann et al. 1999). Whereas SdpI is brain-specific, SdpII, described here, exhibits a wide tissue distribution. The colocalization of endogenous SdpII with dynamin in PC12 cells is consistent with a role in endocytosis. The behavior of cells transfected with either of the two syndapins or their fragments, the distribution of exogenous syndapin, and the interaction of SdpI and -II with synaptojanin, dynamin I, synapsin I, and N-WASP strongly suggest a center role for the syndapins in linking endocytosis to the actin cytoskeleton. The broad tissue distribution of SdpII and the existence of syndapin-related proteins in a wide array of multicellular organisms (Echinococcus, Caenorhabditis elegans, rat, mouse, and human) furthermore suggest that a connection of endocytosis and actin organization via syndapin is based on a conserved machinery in many different organisms and cell types.
To establish a function of syndapin in endocytosis, we tested the physiological significance of the determined syndapin protein interactions by assaying the effect of overexpression of both SdpI and -II SH3 domains on transferrin uptake. We observed a potent block in receptor-mediated endocytosis in vivo. This is in striking contrast to the SH3 domains of Grb2, spectrin, and phospholipase Cγ that bind to dynamin in vitro, but do not block receptor-mediated endocytosis (Wang and Moran 1996; Wigge et al. 1997). Therefore, both syndapin isoforms appear to play a role in clathrin-mediated endocytosis, although SdpI, which is expressed at much higher levels in brain, might be specialized for synaptic vesicle endocytosis. The transfection studies are consistent with earlier in vitro reconstitution assays in which the SdpI SH3 domain interfered with endocytic coated vesicle formation (Simpson et al. 1999). The reconstitution studies also suggested that the interaction of the syndapin SH3 domain with one of its partners is required for late events in endocytosis, leading to vesicle fission. Late events in coated vesicle formation were also selectively affected by a dynamin mutant defective in GTP binding and the actin monomer sequestering drugs, latrunculin B and thymosin beta4 (Damke et al. 1994; Lamaze et al. 1997).
Syndapins interact with synaptojanin, synapsin I, and N-WASP, all proteins implicated in cytoskeletal reorganization (Bähler and Greengard 1987; Miki et al. 1996; Sakisaka et al. 1997). In this study, we have provided in vivo evidence that syndapin modulates the actin cytoskeleton. Syndapin overexpression triggered the formation of filopodia, protrusions from the cell surface containing bundled actin filaments that are found particularly in motile cells and at the ends of growth cones in neurons (O'Connor and Bentley 1993). Full-length syndapin proteins, but not the SH3 domain or the NH2 terminus alone, induced filopodia upon overexpression. Strikingly, syndapin localizes to the very tips of filopodia induced upon overexpression, which represent the actual sites of actin polymerization (Okabe and Hirokawa 1991) and to the edges of lamellipodia. Neither location is a known site of endocytosis.
Our data suggest that syndapin-induced cortical actin rearrangements are mediated by the Arp2/3 complex. First, syndapin and Arp3 colocalize at lamellipodial structures induced upon syndapin overexpression. Likewise, GFP–dynamin 2, but not other parts of the endocytosis machinery, like clathrin, has been reported to localize to cortical membrane ruffles and lamellipodia (Cao et al. 1998). Second, coexpression of a COOH-terminal fragment of N-WASP completely suppressed syndapin-triggered filopodia formation (Fig. 9). An analogous fragment of Scar or WASP was shown by Machesky and Insall 1998 to impair cortical localization of the Arp2/3 complex and lamellipodia formation. Thus, syndapin-induced filopodia formation appears to require the Arp2/3 complex and its translocation to the cell cortex. In contrast, filopodia formation triggered by constitutively active Cdc42 mutants was not inhibited by the coexpression of the Scar COOH terminus (Machesky and Insall 1998). This is consistent with the observation that filopodia induced by membrane recruitment of Cdc42 differed from those induced by WASP in morphology and protein composition (Castellano et al. 1999), suggesting that there may be two types of filopodia formation and syndapins may only induce one class.
Overexpression of the dynamin K44A mutant (Damke et al. 1994) and K+-depletion or cytosolic acidification (Altankov and Grinnell 1993) were shown to block receptor-mediated endocytosis, but also to alter cell shape and redistribute actin stress fibers. In these cases, the defects in cell morphology and cytoskeletal organization might be secondary to an endocytosis block. We have shown here that different domains of syndapin affect membrane trafficking and cytoskeletal architecture in vivo. While the SH3 domain alone was sufficient to block endocytosis, it did not induce filopodia. To trigger these actin rearrangements, overexpression of the full-length protein was necessary, showing that the syndapin-induced cytoskeletal rearrangements, at least, are not an indirect consequence of the inhibition of receptor-mediated endocytosis.
Several potential mechanisms may explain the syndapin-induced rearrangement of the cortical actin cytoskeleton. First, syndapin may mimic N-WASP activators, such as Cdc42 (Rohatgi et al. 1999), causing a conformational change of N-WASP, which allows its COOH terminus to interact with and stimulate the Arp2/3 complex actin polymerization machinery. Second, syndapin may recruit N-WASP to the plasma membrane and this recruitment by itself may be sufficient for filopodia formation. In support of this, overexpression of N-WASP alone was insufficient to alter cell morphology or cytoskeletal architecture (Miki et al. 1998), whereas the local recruitment of WASP to a membrane receptor resulted in the formation of membrane protrusions (Castellano et al. 1999). A third potential mechanism is syndapin could act upstream of Cdc42 and activate the GTPase. And fourth, syndapin itself may have actin-modulating properties in conjunction with the Arp2/3 complex.
In general, the actin cytoskeleton could be involved in endocytic processes in several different ways, implicating different potential functional roles for syndapin. Cytoskeletal structures may help to organize the endocytic machinery at the plasma membrane. Specific sites, or “hot spots”, for synaptic vesicle recycling have been observed in Drosophila nerve terminals (Estes et al. 1996; González-Gaitán and Jäckle 1997; Roos and Kelly 1998). Since syndapin protein family members represent multidomain proteins, they could provide the linkage between the endocytic machinery and the cytoskeleton. A rigid cortical actin cytoskeleton has an inhibitory effect on membrane traffic (Trifaró and Vitale 1993). Syndapin might thus, via its interaction with N-WASP, promote local actin treadmilling, removing physical barriers to endocytosis. A third potential function of the dynamin-associated syndapin involves the membrane fission event. By in vitro reconstitution studies, syndapin was implicated in the late, coated-pit scission event (Simpson et al. 1999), which is also selectively affected by a dynamin mutant (Damke et al. 1994) and by actin monomer sequestering drugs in mammalian A431 cells (Lamaze et al. 1997). Perhaps the actin cytoskeleton provides the force to drive membrane fission. Actin polymerization may also promote the movement of newly formed endocytic vesicles into the cytoplasm. Both pinosomes and clathrin-coated vesicles have recently been described associated with actin comet tails in the cytoplasm (Frischknecht et al. 1999; Merrifield et al. 1999). Finally, it is possible that syndapin regulation allows either endocytosis to take place at a patch of membrane or actin rearrangement, but not both simultaneously.
Our results strongly suggest that SdpI links endocytosis and cytoskeletal dynamics in mature nerve terminals, whereas SdpII performs a similar role in other cell types.
Acknowledgments
We are grateful to Dr. H. Miki for rat N-WASP cDNA and anti–N-WASP antibody, to Dr. P. McPherson for antisynaptojanin antibodies, and to Dr. M.D. Welch for anti-Arp3 antibodies. We thank Dr. M.M. Kessels for advice and stimulating discussion. We thank Drs. Y. Lichtenstein, H. Palokangas, and J. Roos for critically reading the manuscript.
This work was supported by National Institutes of Health grants (NS15927, NS09878, and DA10154) to R.B. Kelly and by fellowships from the Deutsche Forschungsgemeinschaft (Qu 116/1-1 and Qu 116/2-1) and the Max-Planck-Gesellschaft to B. Qualmann.
Footnotes
Britta Qualmann's present address is Leibnitz Institute for Neurobiology, D-39118 Magdeburg, Germany.
Abbreviations used in this paper: GST, glutathione S-transferase; MBP, maltose binding protein; N-WASP, neural Wiskott-Aldrich syndrome protein; PC12, pheochromocytoma cells; PRD, proline-rich domain; Sdp, syndapin; SH3, src homology 3; VCA, verpolin, homology, cofilin and acidic domains at the COOH terminus of N-WASP; WASP, Wiskott-Aldrich syndrome protein.
References
- Altankov G., Grinnell F. Depletion of intracellular potassium disrupts coated pits and reversibly inhibits cell polarization during fibroblast spreading. J. Cell Biol. 1993;120:1449–1459. doi: 10.1083/jcb.120.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler M., Greengard P. Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature. 1987;326:704–707. doi: 10.1038/326704a0. [DOI] [PubMed] [Google Scholar]
- Cao H., Garcia F., McNiven M.A. Differential distribution of dynamin isoforms in mammalian cells. Mol. Biol. Cell. 1998;9:2595–2609. doi: 10.1091/mbc.9.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano F., Montcourrier P., Guillemot J.-C., Gouin E., Machesky L., Cossart P., Chavrier P. Inducible recruitment of Cdc42 or WASP to a cell-surface receptor triggers actin polymerization and filopodium formation. Curr. Biol. 1999;9:351–360. doi: 10.1016/s0960-9822(99)80161-4. [DOI] [PubMed] [Google Scholar]
- Damke H., Baba T., Warnock D.E., Schmid S.L. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein A., Kessels M.M., Chopra V.S., Hayden M.R., Drubin D.G. An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J. Cell Biol. 1999;147:1503–1518. doi: 10.1083/jcb.147.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes P.S., Roos J., van der Bliek A., Kelly R.B., Krishnan K.S., Ramaswami M. Traffic of dynamin within individual Drosophila synaptic boutons relative to compartment-specific markers. J. Neurosci. 1996;16:5443–5456. doi: 10.1523/JNEUROSCI.16-17-05443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F., Cudmore S., Moreau V., Reckmann I., Röttger S., Way M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella . Curr. Biol. 1999;9:89–92. doi: 10.1016/s0960-9822(99)80020-7. [DOI] [PubMed] [Google Scholar]
- Geli M.I., Riezman H. Endocytic internalization in yeast and animal cellssimilar and different. J. Cell Sci. 1998;111:1031–1037. doi: 10.1242/jcs.111.8.1031. [DOI] [PubMed] [Google Scholar]
- González-Gaitán M., Jäckle H. Role of Drosophila α-adaptin in presynaptic vesicle recycling. Cell. 1997;88:767–776. doi: 10.1016/s0092-8674(00)81923-6. [DOI] [PubMed] [Google Scholar]
- Lamaze C., Chuang T.-H., Terlecky L.J., Bokoch G.M., Schmid S.L. Regulation of receptor-mediated endocytosis by Rho and Rac. Nature. 1996;382:177–179. doi: 10.1038/382177a0. [DOI] [PubMed] [Google Scholar]
- Lamaze C., Fujimoto L.M., Yin H.L., Schmid S.L. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- Li R. Bee1, a yeast protein with homology to Wiscott-Aldrich syndrome protein, is critical for the assembly of the cortical actin cytoskeleton. J. Cell Biol. 1997;136:649–658. doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Machesky L.M., Insall R.H. Signaling to actin dynamics. J. Cell Biol. 1999;146:267–272. doi: 10.1083/jcb.146.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L.M., Mullins R.D., Higgs H.N., Kaiser D.A., Blanchoin L., May R.C., Hall M.E., Pollard T.D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M., McMahon H.T. The structural era of endocytosis. Science. 1999;285:215–220. doi: 10.1126/science.285.5425.215. [DOI] [PubMed] [Google Scholar]
- Meriläinen J., Lehto V.-P., Wasenius V.-M. FAP52, a novel, SH3 domain-containing focal adhesion protein. J. Biol. Chem. 1997;272:23278–23284. doi: 10.1074/jbc.272.37.23278. [DOI] [PubMed] [Google Scholar]
- Merrifield C.J., Moss S.E., Ballestrem C., Imhof B.A., Giese G., Wunderlich I., Almers W. Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nature Cell Biol. 1999;1:72–74. doi: 10.1038/9048. [DOI] [PubMed] [Google Scholar]
- Miki H., Miura K., Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- Miki H., Sasaki T., Takai Y., Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- Naqvi S.N., Zahn R., Mitchell D.A., Stevenson B.J., Munn A.L. The WASp homologue Las17p functions with the WIP homologue End5p/verprolin and is essential for endocytosis in yeast. Curr. Biol. 1998;8:959–962. doi: 10.1016/s0960-9822(98)70396-3. [DOI] [PubMed] [Google Scholar]
- O'Connor T.P., Bentley D. Accumulation of actin in subsets of pioneer growth cone filopodia in response to neural and epithelial guidance cues in situ. J. Cell Biol. 1993;123:935–948. doi: 10.1083/jcb.123.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S., Hirokawa N. Actin dynamics in growth cones. J. Neurosci. 1991;11:1918–1929. doi: 10.1523/JNEUROSCI.11-07-01918.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens D.J., Wigge P., Vallis Y., Moore J.D.A., Evans P.R., McMahon H.T. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5273–5285. doi: 10.1093/emboj/17.18.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B., Roos J., DiGregorio P.J., Kelly R.B. Syndapin I, a synaptic dynamin-binding protein that associates with the neural Wiskott-Aldrich syndrome protein. Mol. Biol. Cell. 1999;10:501–513. doi: 10.1091/mbc.10.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riezman H., Munn A., Geli M.I., Hicke L. Actin-, myosin- and ubiquitin-dependent endocytosis. Experientia. 1996;52:1033–1041. doi: 10.1007/BF01952099. [DOI] [PubMed] [Google Scholar]
- Ringstad N., Nemoto Y., De Camilli P. The SH3p4/Sh3p8/SH3p13 protein familybinding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc. Natl. Acad. Sci. USA. 1997;94:8569–8574. doi: 10.1073/pnas.94.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter B., Modregger J., Paulsson M., Plomann M. PACSIN 2, a novel member of the PACSIN family of cytoplasmic adapter proteins. FEBS Lett. 1999;454:356–362. doi: 10.1016/s0014-5793(99)00830-3. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Roos J., Kelly R.B. Dap160, a neural-specific Eps15 homology and multiple SH3 domain-containing protein that interacts with Drosophila dynamin. J. Biol. Chem. 1998;273:19108–19119. doi: 10.1074/jbc.273.30.19108. [DOI] [PubMed] [Google Scholar]
- Sakisaka T., Itoh T., Miura K., Takenawa T. Phosphatidylinositol 4,5-bisphosphate phosphatase regulates the rearrangement of actin filaments. Mol. Cell. Biol. 1997;17:3841–3849. doi: 10.1128/mcb.17.7.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalzing G., Richter H.-P., Hansen A., Schwarz W., Just I., Aktories K. Involvement of the GTP binding protein Rho in constitutive endocytosis in Xenopus laevis oocytes. J. Cell Biol. 1995;130:1319–1332. doi: 10.1083/jcb.130.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S.L. Clathrin-coated vesicle formation and protein sortingan integrated process. Annu. Rev. Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Schmid S.L., McNiven M.A., De Camilli P. Dynamin and its partnersa progress report. Curr. Opin. Cell Biol. 1998;10:504–512. doi: 10.1016/s0955-0674(98)80066-5. [DOI] [PubMed] [Google Scholar]
- Sengar A.S., Wang W., Bishay J., Cohen S., Egan S.E. The EH and SH3 domain Ese proteins regulate endocytosis by linking dynamin and Eps15. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1159–1171. doi: 10.1093/emboj/18.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever S., Muhlberg A.B., Schmid S.L. Impairment of dynamin's GAP domain stimulates receptor-mediated endocytosis. Nature. 1999;398:481–486. doi: 10.1038/19024. [DOI] [PubMed] [Google Scholar]
- Shupliakov O., Löw P., Grabs D., Gad H., Chen H., David C., Takei K., De Camilli P., Brodin L. Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- Simpson F., Hussain N.K., Qualmann B., Kelly R.B., Kay B.K., McPherson P.S., Schmid S.L. SH3-domain–containing proteins function at distinct steps in clathrin-coated vesicle formation. Nature Cell Bio. 1999;1:119–124. doi: 10.1038/10091. [DOI] [PubMed] [Google Scholar]
- Stoorvogel W., Strous G.J., Geuze H.J., Oorschot V., Schwartz A.L. Late endosomes derive from early endosomes by maturation. Cell. 1991;65:417–427. doi: 10.1016/0092-8674(91)90459-c. [DOI] [PubMed] [Google Scholar]
- Trifaró J.-M., Vitale M.L. Cytoskeleton dynamics during neurotransmitter release. Trends Neurosci. 1993;16:466–472. doi: 10.1016/0166-2236(93)90079-2. [DOI] [PubMed] [Google Scholar]
- Urrutia R., Henley J.R., Cook T., McNiven M.A. The dynaminsredundant or distinct functions for an expanding family of related GTPases? Proc. Natl. Acad. Sci. USA. 1997;94:377–384. doi: 10.1073/pnas.94.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Moran M.F. Requirement for the adaptor protein GRB2 in EGF receptor endocytosis. Science. 1996;272:1935–1939. doi: 10.1126/science.272.5270.1935. [DOI] [PubMed] [Google Scholar]
- Wendland B., Emr S.D., Riezman H. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr. Opin. Cell Biol. 1998;10:513–522. doi: 10.1016/s0955-0674(98)80067-7. [DOI] [PubMed] [Google Scholar]
- Wigge P., McMahon H.T. The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci. 1998;21:339–344. doi: 10.1016/s0166-2236(98)01264-8. [DOI] [PubMed] [Google Scholar]
- Wigge P., Vallis Y., McMahon H.T. Inhibition of receptor-mediated endocytosis by the amphiphysin SH3 domain. Curr. Biol. 1997;7:554–560. doi: 10.1016/s0960-9822(06)00254-5. [DOI] [PubMed] [Google Scholar]
- Winter D., Lechler T., Li R. Activation of the yeast Arp2/3 complex by Bee1p, a WASP-family protein. Curr. Biol. 1999;9:501–504. doi: 10.1016/s0960-9822(99)80218-8. [DOI] [PubMed] [Google Scholar]
- Witke W., Podtelejnikov A.V., Di Nardo A., Sutherland J.D., Gurniak C.B., Dotti C., Mann M. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:967–976. doi: 10.1093/emboj/17.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D., To W., Abo A., Welch M.D. The Wiskott-Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr. Biol. 1999;9:555–558. doi: 10.1016/s0960-9822(99)80243-7. [DOI] [PubMed] [Google Scholar]
- Zhang J., Shehabeldin A., da Cruz L.A.G., Butler J., Somani A.-K., McGavin M., Kozieradzki I., dos Santos A.O., Nagy A., Grinstein S. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 1999;9:1329–1341. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]