

Abstract
The mechanism of action of the oncogene bcl-2, a key regulator of the apoptotic process, is still debated. We have employed organelle-targeted chimeras of the Ca2+-sensitive photoprotein, aequorin, to investigate in detail the effect of Bcl-2 overexpression on intracellular Ca2+ homeostasis. In the ER and the Golgi apparatus, Bcl-2 overexpression increases the Ca2+ leak (while leaving Ca2+ accumulation unaffected), hence reducing the steady-state [Ca2+] levels. As a direct consequence, the [Ca2+] increases caused by inositol 1,4,5 trisphosphate (IP3)-generating agonists were reduced in amplitude in both the cytosol and the mitochondria. Bcl-2 overexpression also reduced the rate of Ca2+ influx activated by Ca2+ store depletion, possibly by an adaptive downregulation of this pathway. By interfering with Ca2+-dependent events at multiple intracellular sites, these effects of Bcl-2 on intracellular Ca2+ homeostasis may contribute to the protective role of this oncogene against programed cell death.
Keywords: calcium, organelles, oncogene, aequorin, cell signaling
Introduction
In the past years, Bcl-2, the mammalian homologue of CED-9, the antiapoptotic gene initially described in Caenorhabditis elegans, has emerged as a major checkpoint controlling the escape of cells from their death fate (Kroemer 1997). In mammals, Bcl-2 is a member of a broad family of related gene products, with various degrees of sequence identity and different functions. While Bcl-2 and some members of the family (e.g., Bcl-x) protect the cells from apoptosis, others (e.g., Bax) have the opposite effect. However, while there is a general consensus on the role of Bcl-2, its mechanism of action remains elusive. On one hand, the various members of the family have been shown to dimerize and to interact with cofactors of caspases, key executors of the apoptotic machinery (Li and Yuan 1999); on the other, the specific localization of the protein on the ER and outer mitochondrial membrane (Lithgow et al. 1994) suggests that these proteins may interfere with some specific functions localized in these organelles. In particular, Bcl-2 has been shown to prevent the release of cytochrome c and AIF from mitochondria (Kluck et al. 1997; Yang et al. 1997), events that are considered essential in triggering apoptosis, whereas expression of Bax has opposite effects (Jurgensmeier et al. 1998).
Among the various functions occurring on ER and mitochondrial membranes, ion fluxes are recognized to play major roles. Thus, the intriguing possibility has emerged that members of the Bcl-2 family affect the apoptotic process by modulating ion fluxes in these organelles (Minn et al. 1997; Schendel et al. 1998). In particular, recent work in cell clones stably expressing the oncogene suggests that Bcl-2 causes an overload of Ca2+ within the ER (Lam et al. 1994; He et al. 1997, Kuo et al. 1998). However, this effect appears in contrast with the observation that Bcl-2 insertion in artificial lipid bilayers results in the formation of ionic channels of low selectivity (Minn et al. 1997). Such property should in fact cause a reduction in the ER Ca2+ content (due to an increased leak) rather than excess accumulation. In this report, we have undertaken a detailed investigation of Ca2+ homeostasis at the subcellular level using the recombinant aequorin (AEQ) approach developed in our laboratory (Rizzuto et al. 1992; De Giorgi et al. 1996). We conclude that Bcl-2 overexpression affects Ca2+ handling by reducing the state of filling of the intracellular Ca2+ stores. The implications of these results for the protective action of Bcl-2 on apoptosis are discussed.
Materials and Methods
Cell Culture and Transfection
HeLa cells were grown in DME supplemented with 10% FCS in 75-cm2 Falcon flasks. Before transfection, cells were seeded onto 13-mm glass coverslips and allowed to grow to 50% confluence. At this stage, transfection with 4 μg of DNA (3 μg Bcl-2 + 1 μg AEQ) was carried out as previously described (Rizzuto et al. 1995a) and AEQ measurements or immunocytochemistry were performed 36 h after transfection. For fura-2 measurements, the cells were seeded onto 24-mm coverslips and transfection was carried out with 8 μg plasmid DNA (6 μg Bcl-2 + 2 μg mtGFP).
Aequorin Measurements
For cytosolic AEQ (cytAEQ) and mitochondrial AEQ (mtAEQ), the coverslip with the cells was incubated with 5 μM coelenterazine for 1–2 h in DME supplemented with 1% FCS, and then transferred to the perfusion chamber. For reconstituting with high efficiency, the AEQ chimeras targeted to the Golgi apparatus and the ER (GoAEQ and erAEQ, respectively), the luminal [Ca2+] of these compartments first must be reduced. This was obtained by incubating the cells for 1 h at 4°C in KRB (Krebs-Ringer modified buffer: 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, and 20 mM Hepes, pH 7.4, at 37°C) supplemented with coelenterazine 5 μM, the Ca2+ ionophore ionomycin, and 600 μM EGTA. After this incubation, the cells were extensively washed with KRB supplemented with 2% BSA and 1 mM EGTA, and then AEQ reconstitution was carried out as described.
All AEQ measurements were carried out in KRB. Agonists and other drugs were added to the same medium. In experiments with permeabilized cells, a buffer mimicking the cytosolic ionic composition (intracellular buffer, IB: 140 mM KCl, 10 mM NaCl, 1 mM K3PO4, 5.5 mM glucose, 2 mM MgSO4, 1 mM ATP, 2 mM sodium succinate, 20 mM Hepes, pH 7.05, at 37°C) was employed. This solution was supplemented with either 100 μM EGTA or a fixed [Ca2+] (buffered with 2 mM EGTA). The experiments were terminated by lysing the cells with 100 μM digitonin in a hypotonic Ca2+-rich solution (10 mM CaCl2 in H2O), thus discharging the remaining AEQ pool. The light signal was collected and calibrated into [Ca2+] values as previously described (Brini et al. 1995; Rizzuto et al. 1995a). In brief, a 13-mm round coverslip with the transfected cells was placed in a perfused, thermostated chamber located in the close proximity of a low-noise photomultiplier, with built-in amplifier discriminator. The output of the discriminator was captured by a Thorn-EMI photon counting board and stored in an IBM-compatible computer for further analyses. The AEQ luminescence data were calibrated off-line into [Ca2+] values, using a computer algorithm based on the Ca2+ response curve of wild-type and mutant AEQs, as previously described (Brini et al. 1995; Barrero et al. 1997).
In some experiments using erAEQ, after reaching the plateau level, an artifactual slow decrease in [Ca2+] was observed. Such an artifact is due to a small fraction of missorted AEQ in a low [Ca2+] compartment and a simple practical cure for it has been described by Maechler et al. 1999.
In the experiment of Fig. 4, cells transfected with erAEQ were used to determine the steady [Ca2+]er obtained by incubating the cells in low Ca2+ medium. Two protocols were adopted that gave identical results. In the first, cells incubated in normal medium were depleted according to the standard protocol, but the Ca2+ concentration during the refilling was reduced to 100 μM. In the second protocol, the cells were preincubated in 100 μM CaCl2 for 16 h, depleted, and finally refilled with an extracellular Ca2+ concentration of 100 μM.
Figure 4.
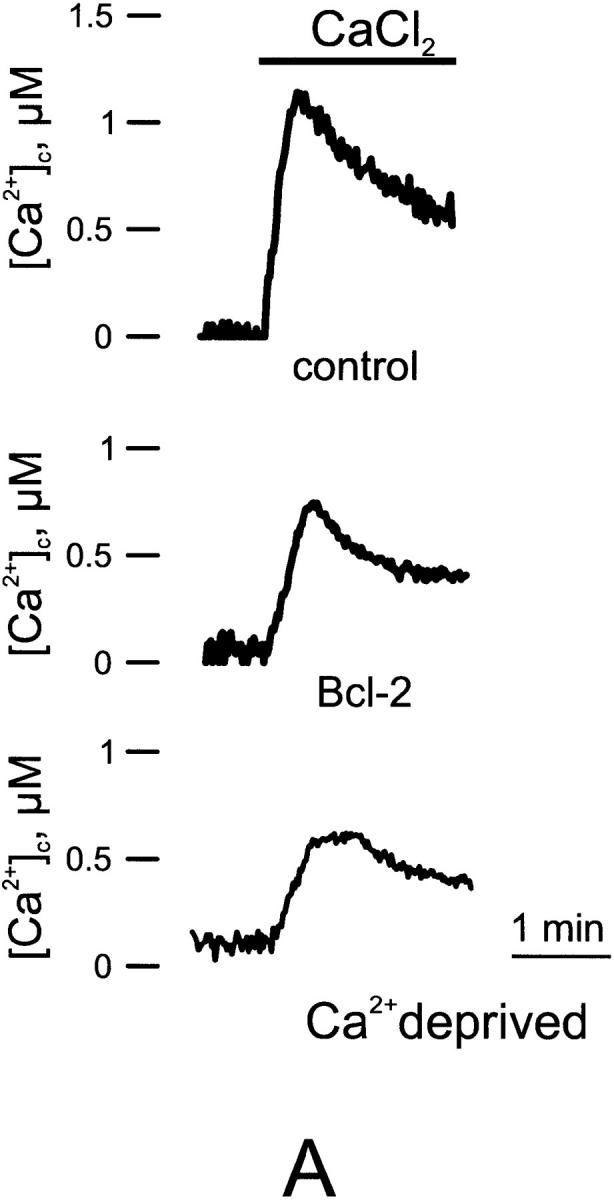

Capacitative Ca2+ influx in control, Bcl-2–transfected, and Ca2+-deprived cells. A, Representitive trace. B, Average [Ca2+] peak. Parallel batches of HeLa cells were either cotransfected with cytAEQ and Bcl-2 (Bcl-2), or transfected with cytAEQ alone (control and Ca2+-deprived). In the Ca2+-deprived cells, the cells were maintained in the 18 h preceding the experiment in KRB containing a lower (0.1 mM) CaCl2 concentration (KRB/lowCa2+). After transferring the coverslip with the cells to the luminometer chamber, the cells, perfused with KRB/EGTA, were challenged with 10 μM tBuBHQ (a specific blocker of the Ca2+ ATPase present in the ER and Golgi apparatus) added to the same medium. After 2 min, capacitative Ca2+ entry was initiated by changing the medium to KRB/Ca2+ (CaCl2). All other conditions as in Fig. 1.
Fura-2 Measurements
The coverslip with the cells was incubated with 5 μM fura-2 (added to DME + 1% FCS) at 37°C for 30 min, and after a brief washout was then transferred to the thermostated stage of a Zeiss Axiovert inverted microscope equipped with a Sutter filterwheel and 340/380 excitation filters. The fluorescence data were collected with a Princeton Instruments back-illuminated camera and expressed as emission ratios, using Metafluor software (Universal Imaging). Within the microscope field, transfected and untransfected cells were identified before carrying out the fura-2 monitoring by revealing mitochondrial-targeted green fluorescent protein (mtGFP) fluorescence (excitation 480 nm, emission 510 nm).
Results and Discussion
To directly investigate the role of Bcl-2 in Ca2+ homeostasis, we have selectively measured the [Ca2+] in different cell compartments, i.e., the cytoplasm and the organelles acting as sources (ER and Golgi apparatus) or targets (mitochondria) of the Ca2+ signal. Targeted AEQ chimeras developed in our lab (Rizzuto et al. 1992; Brini et al. 1995; Montero et al. 1995; Pinton et al. 1998) were employed. This approach has two major advantages. The first is the high subcellular specificity of the probes, which allows to clearly identify the effects of Bcl-2 expression on the signaling properties of the various compartments; the second is that the Ca2+ probe can be cotransfected with Bcl-2. We and others have previously shown that in transient cotransfection, the two recombinant proteins are expressed by the same subset of cells, thus avoiding the risks associated with the use of stable clones or single cells (Brini et al. 1995).
The effect of Bcl-2 expression on the cytosolic Ca2+ signal elicited by an agonist, ATP, acting on receptors coupled, through Gq proteins, to the production of inositol 1,4,5 trisphosphate (IP3) was first investigated. In the experiment shown in Fig. 1, HeLa cells, either coexpressing Bcl-2 and cytAEQ (Bcl-2–transfected) or expressing only cytAEQ (controls) were challenged with ATP, which acts on a Gq-coupled P2Y receptor. Both in control and Bcl-2–transfected cells, ATP stimulation causes a rapid rise in cytoplasmic Ca2+ concentration ([Ca2+]c), followed by a gradually declining sustained plateau. In Bcl-2–transfected cells, the [Ca2+] increases evoked by stimulation with ATP are significantly smaller than in controls (peak amplitude 1.53 ± 0.22 vs. 2.06 ± 0.13 μM, n = 10; Fig. 1 A).
Figure 1.

Cytoplasmic (A) and mitochondrial (B) Ca2+ homeostasis in control and Bcl-2–overexpressing HeLa cells. Parallel batches of HeLa cells were either cotransfected with the appropriate AEQ chimera (cytAEQ and mtAEQ for monitoring the cytoplasm and the mitochondria, respectively) and Bcl-2 (Bcl-2), or transfected with the AEQ chimera alone (control). 36 h after transfection, the measurement of AEQ luminescence was carried out and calibrated into [Ca2+] values, as described in Materials and Methods. Where indicated, the cells, perfused with KRB, were challenged with 100 μM ATP added to the same buffer. These and the following traces are representative of ten experiments that gave similar results.
Since Bcl-2 is localized in the mitochondria also, and thus could, at least in principle, enhance Ca2+ uptake in this organelle, a simple explanation for this result would be that cytosolic Ca2+ is more rapidly cleared by mitochondria, acting as an immobile buffer, placed in closed proximity to IP3-gated channels. Fig. 1 B, however, shows that this is not the case. Indeed, Bcl-2 overexpression does not increase, but rather reduces, the mitochondrial [Ca2+] rise induced by ATP (peak amplitude 6.56 ± 0.72 in controls vs. 4.42 ± 0.46 μM in Bcl-2–expressing cells; n = 10). Interestingly, the reduction caused by Bcl-2 overexpression appears larger in the mitochondria than in the cytosol, both in absolute terms and as percent (26 vs. 33% reduction, respectively). While not responsible for the alteration of the cytosolic Ca2+ signal, the effect of Bcl-2 on mitochondrial Ca2+ homeostasis appears of major functional relevance, given that key events occurring in the mitochondrial matrix, such as the stimulation of ATP production (Jouaville et al. 1999) and possibly the opening of the permeability transition pore, PTP (Bernardi 1999), are regulated by [Ca2+]m.
A second possible explanation for the reduction in the agonist-dependent [Ca2+]c (and [Ca2+]m) increases was next considered, i.e., a decrease in the amount of Ca2+ released by the agonist-sensitive Ca2+ stores. To investigate this possibility, the [Ca2+] in the lumen of the ER was measured in cells cotransfected with Bcl-2 and an erAEQ chimera (Montero et al. 1995). In addition, since we recently have demonstrated that the Golgi apparatus shares many of the Ca2+ homeostatic properties of the ER (Pinton et al. 1998), this latter organelle was also investigated. Fig. 2 shows the calibrated [Ca2+] values in the two compartments. To obtain reliable quantitative estimates of the [Ca2+] in the lumen of these two organelles, their [Ca2+] needs to be decreased during both the reconstitution of AEQ with coelenterazine and the subsequent initial phase of perfusion with KRB/EGTA in the luminometer (see Materials and Methods). Under those conditions, the [Ca2+] was <10 μM in both organelles. When the [Ca2+] in the perfusion medium was switched to 1 mM, the [Ca2+] in the lumen of the two compartments gradually increased, reaching in control cells plateau values of 452 ± 46 μM (n = 7) in the ER and 262 ± 47 μM (n = 7) in the Golgi apparatus, respectively. In Bcl-2 transfected cells, lower steady state values were attained in both compartments (304 ± 38 μM, n = 7 for the ER, and 186 ± 34 μM, n = 7 for the Golgi apparatus, respectively). When the cells were treated with ATP, rapid decreases in the [Ca2+] of the two compartments were observed, both in control and Bcl-2–expressing cells. In both organelles, the decrease of [Ca2+] was larger and faster in controls compared with Bcl-2–expressing cells, reflecting the higher filling state and thus the more rapid flow through the IP3-gated channels.
Figure 2.

Ca2+ homeostasis in the lumen of the ER (A) and of the Golgi apparatus (B) in control and Bcl-2–overexpressing HeLa cells. Parallel batches of HeLa cells were either cotransfected with the appropriate AEQ chimera (erAEQ and GoAEQ for monitoring the lumen of the ER and of the Golgi apparatus, respectively) and Bcl-2 (Bcl-2), or transfected with the AEQ chimera alone (control). 36 h after transfection, the organelles were depleted of Ca2+ to optimize AEQ reconstitution. After reconstitution, the cells were transferred to the luminometer chamber and AEQ luminescence was collected and calibrated into [Ca2+] values. Where indicated, the cells were stimulated with 100 μM ATP. All other conditions are as in Fig. 1.
The data reported above differ substantially from those reported in WEHI7.2 clones, where Bcl-2 overexpression results in an augmentation of the ER Ca2+ content (Lam et al. 1994; He et al. 1997). This discrepancy may be due to the use of transient vs. stable transfections, in which the results might reflect unique properties of the selected clone and/or the long-term expression of a foreign gene may result in adaptation phenomena that are only indirect consequences of the expressed protein. This interpretation might also explain the results obtained by Kuo and coworkers in clones of MCF10A cells overexpressing Bcl-2, in which an increase in SERCA2 (sarcoendoplasmic reticulum Ca2+ ATPases 2) protein leads to accelerated Ca2+ uptake and enhanced Ca2+ loading (Kuo et al. 1998). On the other hand, it is possible that, although the duration of these experiments is compatible with the time course of the apoptotic process, the events observed in transient experiments could include only some of the relevant effects of Bcl-2 on the mechanisms controlling programed cell death.
But how does Bcl-2 affect ER and Golgi Ca2+ handling? Given that the steady-state [Ca2+] in both organelles depends on the equilibrium between active accumulation and passive leak, the effect of Bcl-2 could be on either process. On one hand, Bcl-2 could reduce Ca2+ uptake by the SERCA, either by a direct effect on the pump or by reducing the resting cytosolic [Ca2+]. On the other, it could increase the passive diffusion of Ca2+ from the ER. To clarify this critical issue, we investigated these possibilities independently.
At first, the possibility that Bcl-2 affected primarily cytosolic [Ca2+] (and hence [Ca2+]er only indirectly) was verified. Given that AEQ is not accurate enough to reveal small differences of [Ca2+] ∼10−7 M, the resting [Ca2+]c was measured with the fluorescent indicator fura-2 (Grynkiewicz et al. 1985). In this experiment, to identify Bcl-2–overexpressing cells in single-cell imaging experiments, the cells were cotransfected with mitochondrially targeted GFP, mtGFP(S65T) (Rizzuto et al. 1995b). GFP-positive cells were distinguished from controls by the typical fluorescence emitted upon illumination with blue light. No difference in the resting cytoplasmic [Ca2+] between Bcl-2–transfected and control cells was observed (data not shown).
Then, we investigated whether Bcl-2 overexpression had any direct effect on the activity of the SERCA pump. For this purpose, the kinetics of Ca2+ accumulation were studied in permeabilized cells (Fig. 3 A). After reconstituting the photoprotein as in the experiment of Fig. 2, Bcl-2–transfected and control cells were transferred to the luminometer chamber and perfused with a buffer (IB, see Materials and Methods) mimicking intracellular ionic conditions, supplemented with 100 μM EGTA (IB/EGTA). After permeabilization with 100 μM digitonin added to the same medium and wash in IB/EGTA, the medium was switched to IB, containing 200 nM [Ca2+] (IB/Ca2+). Under those conditions, [Ca2+]er gradually increased, reaching, in control cells, a plateau value of 728 ± 60, n = 5. In Bcl-2–transfected cells, the maximal rate of Ca2+ accumulation was the same as in control cells (13 ± 2 vs. 12 ± 3 μM/s, n = 5), arguing that the Ca2+ pumping activity is not modified. However, the steady-state value eventually attained was distinctly lower (504 ± 52 μM, n = 5). Therefore, these data exclude that the alteration of ER Ca2+ homeostasis is due to a reduction in the activity of the SERCAs or to soluble cytosolic factors (that are released into the medium after permeabilization with digitonin). Rather, by demonstrating a difference only when ER refilling approaches the high [Ca2+]er values of intact resting cells, i.e., in conditions in which passive leak becomes significant, they suggest that Bcl-2 acts on the latter process.
Figure 3.

Kinetics of ER Ca2+ uptake and release in control and Bcl-2–transfected cells. A, ER Ca2+ refilling in permeabilized cells. Parallel batches of HeLa cells were either cotransfected with erAEQmut and Bcl-2 (Bcl-2), or transfected with erAEQmut alone (control). 36 h after transfection, depletion of Ca2+ stores and AEQ were carried out as in Fig. 2. The coverslip with the cells was then transferred to the luminometer chamber. The perfusion medium was changed to IB/EGTA, and permeabilization was carried out by perfusing 100 μM digitonin (added to the same medium) for 1 min. After a 2-min wash with IB/EGTA, Ca2+ accumulation in the ER was initiated by switching the medium to IB containing a buffered Ca2+ concentration of 0.2 μM (see text for details). AEQ calibration was carried out as described in Materials and Methods. B, Dependence on [Ca2+]er of the Ca2+ leak rate from the ER. Transfection, depletion of Ca2+ stores, and AEQ reconstitution were carried out as in A, then the coverslip with the cells was transferred to the luminometer and perfused with KRB/Ca2+ until the steady-state [Ca2+]er was reached. Ca2+ release was initiated by treating the cells with 50 μM tBuBHQ. Based on the experimental trace, the maximal rates of Ca2+ release (measured from the first derivative) at different values of [Ca2+]er were calculated and plotted for Bcl-2–transfected and control cells. The plot contains data obtained from ten independent experiments. In each experiment, duplicate samples of control and Bcl-2–overexpressing cells were measured. Due to the mixing time in the luminometer chamber, the kinetics of [Ca2+]er decrease are sigmoidal and the maximal rate is obtained 2–3 s after addition of tBuBHQ. Accordingly, we considered the maximal rates the best approximation for the initial rate of [Ca2+]er decrease. The fitting of the curve shown in B was performed using Microsoft Excel software.
We sought direct evidence for this interpretation by measuring the Ca2+ leak rate from the ER of Bcl-2–expressing and control cells. After the depletion protocol, the ER was first refilled by exposing the cells to extracellular [Ca2+] ranging from 0.2–3 mM, thus resulting in different levels of steady-state [Ca2+]er. The SERCA blocker, 2,5-di-(tert-butyl)-1,4-benzohydroquinone (tBuBHQ), was then added that initiates the release of stored Ca2+. Given that, by definition, the rates of Ca2+ uptake and leak in steady state are equal, the rate of [Ca2+]er decrease upon blockade of the SERCA must reflect the rate of Ca2+ cycling across the ER membrane and, thus, of the leak rate at any given steady-state [Ca2+]er. Fig. 3 B shows the relationship between [Ca2+]er and leak rate, which can be fitted by a power equation. In Bcl-2–overexpressing cells, the data are more scattered, but for each [Ca2+]er value, the rate of Ca2+ efflux is faster than in control cells. This result is consistent with the evidence that Bcl-2 can induce cation channels of low selectivity in artificial lipid bilayers (Minn et al. 1997).
Previously, we have demonstrated that a reduction of ∼30% in steady-state [Ca2+]er results in a substantial activation (>50%) of the so-called capacitative Ca2+ influx (Hofer et al. 1998). The reduction in steady-state [Ca2+]er caused by Bcl-2 overexpression is thus expected to cause an activation of this pathway. However, indirect evidence (namely, the observation that the resting cytosolic Ca2+ level is indistinguishable from controls) suggests that this is not the case. To further investigate the problem, we measured the capacitative Ca2+ influx in Bcl-2–transfected and control HeLa cells expressing cytAEQ (Fig. 4). The cells were treated with the SERCA blocker (10 μM tBuBHQ) while perfused with Ca2+-free medium (KRB/EGTA). This procedure first evoked a small transient increase in [Ca2+]c due to release of Ca2+ from the stores. When the release of stored Ca2+ was complete (∼3 min), Ca2+ was readded to the medium. This maneuver evoked a second, larger [Ca2+]c increase via the capacitative pathway, which was markedly lower in Bcl-2–transfected than in controls cells (1.19 ± 0.11 in controls vs. 0.68 ± 0.13 μM in Bcl-2–expressing cells, n = 6). The question then arises as to the mechanism(s) through which Bcl-2 downregulates capacitative Ca2+ influx. Two possibilities were considered: one was that the reduction of this Ca2+ influx pathway is an adaptive mechanism to the long lasting reduction in steady-state [Ca2+]er; the second was that the reduction is a direct consequence of Bcl-2 overexpression. To distinguish between these two possibilities, we verified whether a long-term reduction of [Ca2+]er, obtained independently of Bcl-2, could cause a comparable downregulation of the capacitative Ca2+ influx. Capacitative Ca2+ influx was thus compared in controls and in cells whose [Ca2+]er was reduced to values similar to those of Bcl-2–overexpressing cells by prolonged incubation with a low (100 μM) extracellular [Ca2+]. In cells that had been maintained at low external [Ca2+], and thus experienced a long-term reduction in the resting [Ca2+]er levels, the [Ca2+]c peak evoked by activation of the capacitative Ca2+ influx was markedly lower than in controls (0.59 ± 0.11 μM, n = 5). Of interest, the level of steady-state [Ca2+]er in cells incubated in low extracellular Ca2+ was very similar (269 ± 46 μM, n = 4) to that measured in Bcl-2–overexpressing cells incubated in normal (1 mM) extracellular Ca2+ concentration. The simplest explanation of these results is that the decrease in capacitative Ca2+ influx is an adaptive consequence to the prolonged reduction in steady-state [Ca2+]er, though, at the moment, it cannot be excluded that other factors (e.g., a reduction in plasma membrane potential) contribute to this effect. This reduction in capacitative Ca2+ influx, on one hand, appears critical to maintain a resting [Ca2+]c indistinguishable from that of control cells, despite a major reduction of [Ca2+] in the ER; on the other hand, it could account, at least in part, for a higher resistance of cells expressing Bcl-2 to apoptotic cell death, as the reduction of this ubiquitous Ca2+ influx pathway may prevent the overload of Ca2+, considered one of the key events in apoptosis. In agreement with this view, Ma and coworkers recently demonstrated that SERCA overexpression, by causing ER Ca2+ overload, increases spontaneous apoptosis (Ma et al. 1999). Finally, our data are not in contrast with previous results showing that the SERCA inhibitor, thapsigargin, activates programed cell death (Bian et al. 1997). In this latter case, in fact, the Ca2+ depletion is acute (1–2 min) and complete, whereas in Bcl-2–transfected cells, the drop in [Ca2+]er is modest and develops slowly. Thus, thapsigargin not only causes a massive activation of the capacitative Ca2+ influx pathway, but also such a drastic reduction in the level of [Ca2+]er to interfere with the basic activity of ER chaperonins.
We have demonstrated that overexpression of Bcl-2 causes a reduction of the steady-state [Ca2+] levels, both in the ER and in the Golgi apparatus, the two main agonist-sensitive Ca2+ stores. We also showed that this alteration affects the Ca2+ signal to which the effector systems are exposed, in particular those located in local environments highly dependent on the filling state of the intracellular stores (such as the mitochondria). The chronic reduction in [Ca2+] of the ER results in a downregulation of the capacitative Ca2+ influx. The alterations in Ca2+ handling provoked by Bcl-2 overexpression could be part of the mechanisms through which Bcl-2 exerts its role in the control of cell growth. Targets of Bcl-2 effects on Ca2+ homeostasis could be: the ER itself, where sorting and trafficking of proteins, e.g., enzymes or membrane receptors, are modulated by luminal [Ca2+] (Booth and Koch 1989); the mitochondria, where Ca2+ accumulation regulates the production of ATP (RobbGaspers et al. 1998; the concentration of which is a key determinant of the apoptosis vs. necrosis choice, Leist et al. 1997), and, possibly, the release of the apoptotic cofactor cytochrome c (Krajewski et al. 1999); or the cytoplasm, where bulk or local [Ca2+] increases activate enzymatic effectors, such as the Ca2+-dependent protease, calpain (Carafoli and Molinari 1998; Squier et al. 1999), or the phosphatase, calcineurin (Wang et al. 1999). Whether, and how, these routes interact, and how they are affected by the alteration in Ca2+ homeostasis caused by Bcl-2, awaits further investigation.
Acknowledgments
We wish to thank Valeria Tosello and Luisa Filippin for carrying out some of the experimental work and helpful discussion, and Giovanni Ronconi and Mario Santato for expert technical assistance.
This work was supported by grants from Telethon (# 845, 850), the Italian University and Health Ministries, the CNR target project “Biotechnology”, the Italian Association for Cancer Research (AIRC), and the Armenise-Harvard Foundation to R. Rizzuto and T. Pozzan.
Footnotes
Abbreviations used in this paper: AEQ, aequorin; cytAEQ, cytosolic AEQ; erAEQ, AEQ chimera targeted to the ER; GFP, green fluorescent protein; GoAEQ, AEQ chimera targeted to the Golgi apparatus; IB, intracellular buffer; IP3, inositol 1,4,5 trisphosphate; KRB, Krebs-Ringer modified buffer; mtAEQ, mitochondrial AEQ; mtGFP, mitochondrially targeted GFP; SERCA, sarcoendoplasmic reticulum Ca2+ ATPases; tBuBHQ, 2,5-di-(tert-butyl)-1,4-benzohydroquinone.
References
- Barrero M.J., Montero M., Alvarez J. Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cellsa comparative study. J. Biol. Chem. 1997;272:27694–27699. doi: 10.1074/jbc.272.44.27694. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cationschannels, exchangers, and permeability transition. Physiol. Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bian X., Hughes F.M., Jr., Huang Y., Cidlowski J.A., Putney J.W., Jr. Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. Am. J. Physiol. 1997;272:C1241–C1249. doi: 10.1152/ajpcell.1997.272.4.C1241. [DOI] [PubMed] [Google Scholar]
- Booth C., Koch G.L. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- Brini M., Marsault R., Bastianutto C., Alvarez J., Pozzan T., Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c)a critical evaluation. J. Biol. Chem. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- Carafoli E., Molinari M. Calpaina protease in search of a function? Biochem. Biophys. Res. Comm. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- De Giorgi F., Brini M., Bastianutto C., Marsault R., Montero M., Pizzo P., Rossi R., Rizzuto R. Targeting aequorin and green fluorescent protein to intracellular organelles. Gene. 1996;173:113–117. doi: 10.1016/0378-1119(95)00687-7. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G., Poenie M., Tsien R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- He H., Lam M., McCormick T.S., Distelhorst C.W. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol. 1997;138:1219–1228. doi: 10.1083/jcb.138.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer A., Fasolato C., Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ storesa study using simultaneous measurements of ICRAC and intraluminal [Ca2+] J. Cell Biol. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville L.S., Pinton P., Bastianutto C., Rutter G.A., Rizzuto R. Regulation of mitochondrial ATP synthesis by calciumevidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier J.M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Krajewski S., Krajewska M., Ellerby L.M., Welsh K., Xie Z., Deveraux Q.L., Salvesen G.S., Bredesen D.E., Rosenthal R.E., Fiskum G., Reed J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nature Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Kuo T.H., Kim H.-R.C., Zhu L., Yu Y., Lin H.-M., Tsang W. Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene. 1998;17:1903–1910. doi: 10.1038/sj.onc.1202110. [DOI] [PubMed] [Google Scholar]
- Lam M., Dubyak G., Chen L., Nunez G., Miesfeld R.L., Distelhorst C.W. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad. Sci. USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M., Single B., Castoldi A.F., Kuhnle S., Nicotera P. Intracellular adenosine triphosphate (ATP) concentrationa switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Yuan J. Deciphering the pathways of life and death. Curr. Opin. Cell Biol. 1999;11:261–266. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- Lithgow T., van Driel R., Bertram J.F., Strasser A. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ. 1994;5:411–417. [PubMed] [Google Scholar]
- Ma T.S., Mann D.L., Lee J.L., Gallinghouse G.J. SR compartment calcium and cell apoptosis in SERCA overexpression. Cell. Calcium. 1999;26:25–35. doi: 10.1054/ceca.1999.0049. [DOI] [PubMed] [Google Scholar]
- Maechler P., Kennedy E.D., Sebo E., Pozzan T., Wollheim C.B. Secretagogues modulate the calcium concentration in the endoplasmic reticulum of insulin secreting cellsstudies in aequorin-expressing intact and permeabilized INS-1 cells. J. Biol. Chem. 1999;274:12583–12592. doi: 10.1074/jbc.274.18.12583. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Velez P., Schendel S.L., Liang H., Muchmore S.W., Fesik S.W., Fill M., Thompson C.B. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Montero M., Brini M., Marsault R., Alvarez J., Sitia R., Pozzan T., Rizzuto R. Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5467–5475. doi: 10.1002/j.1460-2075.1995.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P., Pozzan T., Rizzuto R. The Golgi apparatus is an inositol 1,4,5 trisphosphate Ca2+ store, with distinct functional properties from those of the endoplasmic reticulum. EMBO (Eur. Mol. Biol. Organ.) J. 1998;18:5298–5308. doi: 10.1093/emboj/17.18.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R., Simpson A.W.M., Brini M., Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–328. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini M., Bastianutto C., Marsault R., Pozzan T. Photoprotein mediated measurement of [Ca2+] in mitochondria of living cells Meth. Enzymol. 260 1995. 417 428a [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini M., Pizzo P., Murgia M., Pozzan T. Chimeric green fluorescent proteina new tool for visualizing subcellular organelles in living cells Curr. Biol. 5 1995. 635 642b [DOI] [PubMed] [Google Scholar]
- RobbGaspers L.D., Burnett P., Rutter G.A., Denton R.M., Rizzuto R., Thomas A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendel S.L., Montal M., Reed J.C. Bcl-2 family proteins as ion-channels. Cell Growth Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- Squier M.K., Schnert A.J., Sellins K.S., Malkinson A.M., Takano E., Cohen J.J. Calpain and calpastatin regulate neutrophil apoptosis. J. Cell. Physiol. 1999;178:311–319. doi: 10.1002/(SICI)1097-4652(199903)178:3<311::AID-JCP5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Wang H.G., Pathan N., Ethell I.M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T.F., Reed J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]