

Abstract
A variable skeleto-hematopoietic phenotype was observed in collagen X null mice which mirrored the defects in transgenic (Tg) mice with dominant interference collagen X mutations (Jacenko, O., P. LuValle, and B.R. Olsen. 1993. Nature. 365:56–61). Specifically, perinatal lethality was seen in ∼10.8% of null mutants at week three after birth, and in another subset by 12 wk. In perinatal lethal mutants, growth plates were compressed, trabecular bone reduced, and hematopoietic aplasia and erythrocyte-filled vascular sinusoids were apparent in marrows. Lymphatic organs, reduced to ∼80% that of controls, displayed altered architecture and lymphocyte content. In thymuses, a paucity of cortical CD3+/CD4+/CD8+ lymphocytes was consistent with the marrow's inability to replenish maturing T cells. In spleens, an unaltered T cell distribution was coupled with diffuse staining for IgD+/B220+ B cells, whose reduction was prominent in poorly organized lymphatic nodules. Disorderly arrays of splenic macrophages surrounding periarteriolar lymphatic sheaths and a red pulp depletion further complemented the Tg perinatal lethal phenotype. Moreover, subtle growth plate compressions and hematopoietic changes were seen in all null mice. Data from Tg and null mice implicate the disruption of collagen X function in the observed skeleto-hematopoietic defects, and suggest that hypertrophic cartilage and endochondral skeletogenesis may contribute to the marrow microenvironment prerequisite for blood cell differentiation.
Keywords: endochondral ossification, hypertrophic cartilage, skeletogenesis, marrow, lymphopenia
Introduction
Hypertrophic cartilage defines each skeletal element where bone marrow formation would ensue (Chan and Jacenko 1998). Through endochondral ossification (EO), the cartilaginous blueprint of the axial and appendicular skeleton, as well as of certain cranial bones, is replaced by trabecular bone and marrow. Specifically, EO represents a substitution process where longitudinal skeletal growth involves deposition of a bone matrix upon preexisting hypertrophic cartilage, as well as replacement of the hypertrophic matrix by that of the marrow stroma. Thus, if the intricately orchestrated processes of EO do not proceed appropriately, then the marrow microenvironment, which is required for hematopoiesis, may not form adequately.
Despite a wealth of morphologic data on EO, the molecular mechanisms driving this morphogenetic process are just beginning to be defined. One distinctive feature of EO involves the short chain collagen X, a matrix molecule expressed primarily and predominantly by hypertrophic cartilage (Chan and Jacenko 1998). Due to its spatio-temporal association with EO, collagen X has been linked to processes such as mineralization, matrix stabilization or remodeling, and vascular invasion. The role of collagen X in EO has remained largely undefined partly due to the disparate phenotypes seen in three independently generated murine models with disrupted collagen X function, as well as in two human diseases resulting from collagen X mutations (Jacenko et al. 1993a; Rosati et al. 1994; McIntosh et al. 1995; Kwan et al. 1997; Chan and Jacenko 1998; Ikegawa et al. 1998; Jacenko and Chan 1998).
The first murine model represented the transgenic (Tg) mice with dominant interference collagen X mutations (Jacenko et al. 1993a,Jacenko et al. 1993b). Specifically, transgenes encoded defective chicken collagen X variants with deletions in the central, triple-helical domain. Since homotrimeric collagen X subunits assemble through associations at the COOH terminus, followed by their trimerization along the triple-helix towards the NH2 domain, the truncated molecules were proposed to compete with endogenous chains for association. Multiple independent Tg lines expressing transgenes in hypertrophic cartilage exhibited similar skeleto-hematopoietic phenotypes. Perinatal lethality ensued in >25% of mice week three after birth, and was hallmarked by acute marrow aplasia and hematopoietic defects. Survivors (∼75%) exhibited variable dwarfism and subtle hematopoietic changes (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication).
Based partly on the predominant dwarfism and skeletal phenotypes of the Tg mice, mutations in collagen X were identified in patients with Schmid metaphyseal chondrodysplasia (SMCD; Warman et al. 1993; Jacenko et al. 1994), and with spondylometaphyseal dysplasia (SMD; Ikegawa et al. 1998). To date, from >30 collagen X mutations, all but two localized to microdomains within the COOH terminus (Chan and Jacenko 1998; Chan et al. 1999; Jacenko and Chan 1998); the remaining two mutations involved the NH2 domain (Ikegawa et al. 1997). Haploinsufficiency (50% reduction; Warman et al. 1993; Chan et al. 1995, Chan et al. 1996, Chan et al. 1998) was suggested as a possible disease mechanism for SMCD, although alternate possibilities are likely for specific mutations (Chan and Jacenko 1998; McLaughlin et al. 1999). Both SMCD and SMD disease phenotypes are mild compared with the acute skeleto-hematopoietic defects in the Tg mice. SMCD manifests as dwarfism with involvement of skeletal elements that are under the greatest mechanical stress (e.g., spine, pelvis, and long bones); disease onset correlates with weight-bearing, and patients exhibit coxa vara and a waddling gait due to growth plate abnormalities (Lachman et al. 1988). SMD comprises seven disease sub-types ranging in severities from mild to lethal (Diren et al. 1992; Peeden et al. 1992), and shares multiple characteristics with SMCD. To date, no triple-helical collagen X mutations have been found.
The second and third murine models involved inactivation of collagen X by gene targeting and homologous recombination (Chan and Jacenko 1998). Rosati et al. 1994 reported that collagen X null or knockout (KO) mice developed no gross phenotypic changes. This implied that only the presence of abnormal collagen X could modify bone growth in Tg mice and humans. In contrast, Kwan et al. 1997 described a mild skeletal phenotype involving the chondro–osseous junction in their KO mice; this disease resembled SMCD, as well as certain Tg mouse features. Moreover, their data implied that collagen X may compartmentalize proteoglycans and matrix vesicles within hypertrophic cartilage during EO (Kwan et al. 1997; Chan and Jacenko 1998).
To begin resolving these phenotypic disparities in the collagen X mice, the KO mice exhibiting no obvious disease phenotype (Rosati et al. 1994) were generously provided by Drs. de Crombrugghe and Behringer (M.D. Anderson Cancer Center, University of Texas, Houston, TX). While expanding the colony, we detected an acute perinatal lethal phenotype in ∼10.8% of the null mice at week three after birth, which mirrored that seen in a subset of the Tg mice. Moreover, we observed subtle growth plate compressions primarily within the proliferative zone of the null mice, as well as hematopoietic changes. These data firstly underscore the phenotypic differences and similarities between the collagen X Tg and KO mice which may arise through different disease mechanisms, and secondly imply an intimate link between EO and marrow establishment.
Materials and Methods
Maintenance and Breeding of Mice
The collagen X null mice were bred one generation into the C57Bl/6 strain since the initial publication (Rosati et al. 1994), and generously provided along with C57Bl/6 wild-type controls (WT) by Drs. de Crombrugghe and Behringer (M.D. Anderson Cancer Center, University of Texas, Houston, TX). These mice were housed in a virus-free barrier facility in a room separate from that of the collagen X Tg mice (Jacenko et al. 1993b). Maintenance was in microisolators containing autoclaved Pine Driwood chips, and mice were fed Purina autoclaved mouse chow and water ad lib. From birth, mice were observed daily for phenotypic changes and manifestations of skeleto-hematopoietic problems, such as: decrease in size; lethargy; decrease or change in mobility; skeletal abnormalities including hunching of the back, sloping of the buttocks, and hydrocephaly; wasting; skin ulcerations; tumors; tooth overgrowth; and micropthalmia. Upon day 21, mice were weaned and genotyped (Jacenko et al. 1993a). Specifically, after DNA isolation from tail biopsy (Laird et al. 1991) and purification by phenol/chloroform, EcoRV digested DNA was analyzed by hybridizing Southern blots with a 32P random prime-labeled 1.0-kb HindIII–KpnI 5′ internal mouse collagen X probe (Rosati et al. 1994). Mice were killed with methoxyflurane (Mallinckrodt Veterinary). Weights of whole body, spleen, thymus, and right kidney were typically recorded.
Histology
For paraffin histology and growth plate histomorphometry, right tibia/femur hind limbs were dissected from WT, phenotypically normal collagen X null mice (KO), null mice exhibiting perinatal lethality (KO mutants), collagen X Tg mice with mild phenotypes (Tg), and Tg perinatal lethal mutants (Tg mutants). Hind limbs, spleens, and thymuses were fixed in 4% formaldehyde/PBS, pH 7.4, at 4°C for a 1-wk minimum. After rinses in distilled water, hind limbs were decalcified (4% formalin, 1% sodium acetate, and 10% EDTA) overnight, and all samples were dehydrated in ascending ethanol series (70, 80, 95% ethanol, 30 min; 100% ethanol 60 min × 2). Clearing with Propar (Anatech Ltd.) was followed by perfusion and embedding in paraffin. 6-μm serial longitudinal sections through the center of the tibial and femoral metaphyseal regions were heat-fixed onto Superfrost/Plus slides (Fisher Scientific). Staining for morphology involved Harris hematoxylin and eosin Y (H&E; Sigma Chemical Co.). Giemsa (Sigma Chemical Co.) was used to stain hematopoietic cells. Histomorphometric measurements were obtained with a stage micrometer and Olympus BX60 microscope; widths of overall tibial growth plate, proliferative, and hypertrophic cartilage zones were recorded.
For immunohistochemistry, spleens and thymuses were embedded in Tissue Tek OCT compound (Sakura Finetek), and 6–8-μm longitudinal cryosections were acetone-fixed (1 min), rinsed in PBS (8 min, 2 changes), and incubated in 0.3% H2O2/PBS (10 min for spleen; 5 min for thymus) to quench endogenous peroxidase. After rinsing in PBS (4 min, 2 changes) and 4% heat-inactivated newborn calf serum in PBS (NCS/PBS, 4 min, 2 changes; Sigma Chemical Co.), sections were pre-incubated in NCS/PBS (30 min) to block nonspecific binding, and reacted with anti-mouse primary antibodies (10 μg/ml) in NCS/PBS (60 min, 23°C). Primary antibodies from BD PharMingen included: thymic pan epithelium (TPE; Rat IgM, for thymic architecture); CD4/L3T4 (Rat IgG2a,κ; thymocytes and T helper cells); CD8a/Ly-2 (Rat IgG2a,κ; thymocytes and T suppresser/cytotoxic cells); CD45R/B220 (Rat IgG2a,κ; all stages of B cells); Ter 119 (Rat IgG2b,κ; erythroid lineage); and purified rat IgG2a,κ, IgG2b,κ, or IgM isotype controls. MOMA-1 antibodies (supernatant; 1:25 dilution; Rat IgG2a; macrophage subpopulations, which in spleen surround the periarteriolar lymphatic sheath) were from Serotec Ltd. After rinses with NCS/PBS (8 min, 4 changes), sections were reacted with biotinylated anti-rat IgG (5 μg/ml) secondary antibodies (Vector Laboratories, Inc.) in NCS/PBS (30 min, 23°C), then incubated in Vectastain ABC Reagent (Vector Laboratories, Inc.; 30 min, 23°C) containing avidin and biotinylated HRP, and rinsed in PBS (8 min, 4 changes). Incubation in diamino-benzidine tetrahydrochloride solution (Pierce Chemical Co.; ∼5 min) visualized the reaction. Sections were rinsed in distilled water, air dried, and mounted with Aqua-mount (Lerber Laboratories).
Sections were viewed with an Olympus BX60 light microscope with an Automated Photomicrographic System PM20.
Flow Cytometry
Spleens and thymuses were cut in 2 to 3 ∼50-mm pieces, and placed into 2 ml Tenbroeck Tissue Grinders (Wheaton) containing Ca/Mg-free HBSS (GIBCO BRL). Thymuses were homogenized by 2 rotating plunges, and spleens were homogenized by 3. After removing released cells from homogenate, tissue particles were resuspended in HBSS and subjected to a second homogenization. Next, cell suspensions were gently vortexed and settled on ice (5 min). Cells were separated from tissue fragments and pelleted by centrifugation (1,500 rpm, 10 min, 4°C) twice. A hemacytometer was used to determine cell concentration.
Cells (106/100 μl HBSS) were single-labeled with primary antibodies (10 μg/μl; listed previously) in FACS buffer (0.4% BSA/PBS; 30 min, 4°C). After washing with FACS buffer, pelleting (1,500 rpm, 5 min, 4°C), and resuspension in 100 μl FACS buffer, cells were incubated with 20 μl of FITC polyclonal anti-rat IgG secondary antibodies (5 μg/μl, 30 min, 4°C, covered; listed previously). For double-labeling, 20 μl (10 μg/μl) of directly labeled PE-CD8a (BD PharMingen) or its isotype control, PE-rat IgG2aκ (BD PharMingen) were added at this step as well. Cells were pelleted as above, and resuspended in 0.5 ml FACS buffer. Before sorting, propidium iodide (PI; 0.5 μg/μl; BD PharMingen) was added to distinguish dead cells.
The FACSCalibur was used in conjunction with CELL Quest version 3.1 software (Becton Dickinson) to acquire and analyze data. For single-labeling, fluorescence was detected on FL1 (FITC) and FL 3 (PI) without compensation. Cells were visualized on dot plots where X = FSC and Y = PI fluorescence in log. Lymphocytes were gated based on their distinct distribution (low SSC and increasing FSC), and 10,000 PI negative events per sample were acquired. Percent lymphocytes out of total live cells were recorded based on gated region statistics, and histogram plots were created to analyze the FITC positive cells in relation to both the total live cell and live lymphocyte population. Positive peaks ranged from 102–103 for CD4, CD8, and B220, and at ∼101 for B220. Erythroblasts were calculated within the live cell distribution firstly as percent lymphocytes, multiplied by percent Ter+ live lymphocytes, and divided by 100. For double-labeling, compensation between FITC, PI, and PE was required to eliminate spectral overlap. Percent double-positive cells were distinguished from single-positive cells by visualization on dot plots where X = FITC and Y = PE, and recorded in relation to total live lymphocytes.
Results
Phenotypic Variability in the Collagen X Null Mice
While expanding a homozygous collagen X KO mouse colony (Rosati et al. 1994) bred one generation into the C57Bl/6 strain, previously undetected phenotypic abnormalities were observed. Specifically, perinatal lethality ensued approximately three weeks after birth in 10.8% of the pups (103 mutants/943 mice); this manifested as dwarfism with mutant weight of ∼1/3 that of controls, lethargy, thoracolumbar kyphosis, neck lordosis, wasting, and death ensuing within 24 h. Outwardly, this acute phenotype mirrored that seen in >25% (1,220 mutants/4,428 mice; 5 lines) of the Tg mice (Fig. 1 A; Jacenko et al. 1993a; Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, submittedmanuscript for publication). Upon dissection, thymuses and spleens of perinatal lethal KO and Tg mutants were reduced, spleens were discolored (Fig. 1 B), and lymph nodes undetectable. Whereas the kidney-to-body weight ratios remained consistent in all mice, that of thymus and spleen versus kidney or body weight of perinatal lethal mutants decreased compared with controls (0.21–0.32% for thymus; 0.12–0.16% for spleen; data not shown). These ratios transcended to the number of thymic or splenic cells isolated from these organs (see below), mirrored those seen in Tg mice with perinatal lethality (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication), and underscored the disease phenotype specificity to lymphatic organs. Lastly, in perinatal lethal mutants, bright red, erythrocyte-filled marrows were readily discernible (Fig. 1 C) and highlighted this most acute phenotype.
Figure 1.
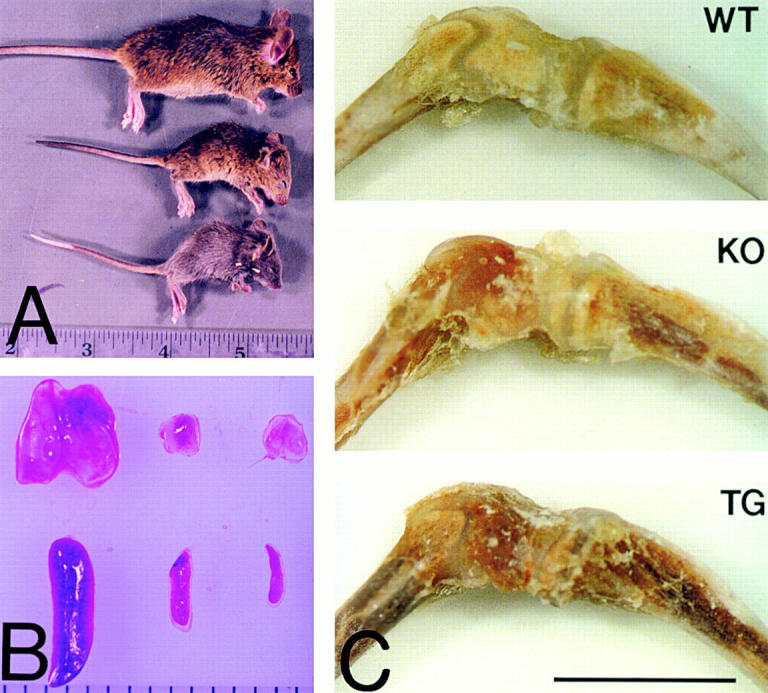
Phenotypic variability in the collagen X KO mice. A, Homozygous KO mice (top and center) with no disease phenotype (top) and exhibiting perinatal lethality (center), compared with a perinatal lethal collagen X Tg mutant (bottom). Note size reduction and hunching of backs in the mutants. B, Thymuses (top) and spleens (bottom) from KO mice with either no disease phenotype (left) or with perinatal lethality (center), compared with those from a perinatal lethal Tg mutant (right). Note size reduction of both organs, as well as a depletion of red pulp in the spleens of mutants. C, Tibia-femur hind limbs from a wild-type (WT) control, a KO perinatal lethal mutant (KO), and a Tg mutant (Tg). Note prominent red marrows in limbs from mutants. Bar, 1 mm.
Of the surviving KO mice, about two percent underwent a similar demise at two to four months, and a few at later stages. Among the survivors, a subset of mice appeared lethargic and slightly dwarfed upon weaning, however recovered; in these mice, no gross changes were detected in lymphatic organs other than a subtle decrease in spleen weight (not shown). Abnormalities in an additional approximate one to two percent of survivors included hydrocephaly and tooth overgrowth, each of which eventually resulted in death. No tumors or skin ulcerations were detected in older KO mice, as in the Tg animals. Overall, the percentage of KO mice undergoing lethality approached ∼14%.
Skeletal and Marrow Defects
Histomorphometry confirmed the subtle growth plate compressions in KO mice as reported by Kwan et al. 1997, as well as revealed differences between the Tg and KO skeletal defects. Specifically, there was an ∼14% overall decrease in growth plate width in day 21 KO mice compared with control, compared with >18% decrease in normal Tg mice (Fig. 2a and Fig. b). Furthermore, increased waviness was seen in many KO growth plates (Fig. 2 A). In KO mice with perinatal lethality, the compressions were more pronounced and somewhat variable, and approached those seen in perinatal lethal Tg mice (Fig. 2 A). Overall, when compared with controls, the proliferative zone in all KO mice was more compressed than the hypertrophic, which was opposite that seen in the Tg mice (Fig. 2 B). Moreover, in all KO mice, trabecular bony spicules were slightly decreased in size and number, with the least amount seen in the most severe mutants, as in the Tg mice (Fig. 2 A and 3 A). Likewise, most dramatic changes were seen in marrows of KO mice with perinatal lethality, where an erythrocyte predominance and leukocyte depletion were characteristic of marrow aplasia (Fig. 3). In Tg mice, the temporal onset of marrow aplasia correlated with the metaphyseal skeletal defects seen approximately two to three weeks after birth, and was proposed to underlie all the hematopoietic abnormalities (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication).
Figure 2.
Tibial growth plate histomorphometry of collagen X Tg and KO mice. A, H&E sections of day 21 tibial metaphyses of wild-type (control), and collagen X KO and Tg mice with mild (KO, TG) and acute (KO MUT, TG MUT) phenotypes. GP, Growth plate; PC, proliferating cartilage; HC, hypertrophic cartilage; T, trabecular bone; M, marrow. Note: less pronounced overall compression of growth plates in KO mice as that in TG mice; trabecular reduction in both KO and TG mice; more severe growth plate compressions and trabecular reductions in both KO MUT and TG MUT; and erythrocyte predominance and leukocyte depletion in marrows of both KO MUT and TG MUT mice. Bar, 100 μm. B, Histomorphometric analysis of growth plate zones including overall growth plate width (overall), and widths of proliferative and hypertrophic cartilages. Note greater compressions of the proliferative zone in KO mice, and greater compressions of the hypertrophic zone in Tg mice. n, Sample number; error bars = standard error.
Figure 3.
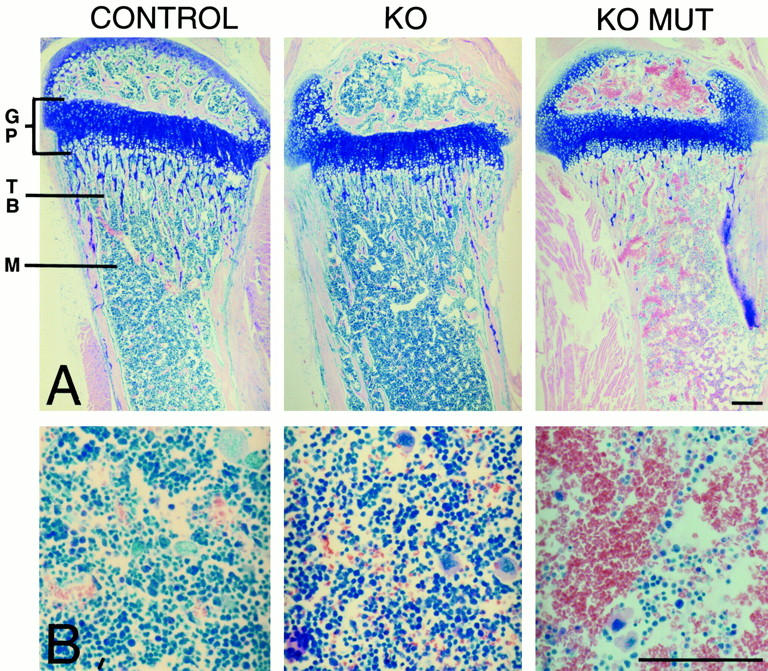
Giemsa staining of tibial longitudinal sections from control and collagen X null mice with no outward defects (KO), or with perinatal lethality (KO MUT) at week three after birth. A, Low magnification of tibiae reveals a reduction of leukocytes (blue) and a predominance of erythrocytes (red) in the KO mutant. B, Higher magnification of bone marrow from A reveals marrow aplasia in the KO mutant, manifested as a depleting hematopoietic compartment (blue). Bars, 100 μm.
Lymphatic Organ Defects
Thymus.
In KO week three perinatal lethal mutants, reduced thymuses revealed altered architecture. Specifically, the thymic cortex was diminished and virtually depleted of cells (Fig. 4H&Fig. E) when compared with the densely populated thymic cortex in control mice. This was confirmed by immunostaining for TPE, where the diminished cortex was localized as a narrow strip, compared with the wide zone comprising the majority of the thymus in controls (Fig. 4, TPE). The thymic cortex houses marrow-derived immature T cells, which after acquiring lineage identity in the marrow, migrate to the cortex, mature, and progress to the medulla (Shortman and Wu 1996). A depletion in the overall number of the immature cortical thymocytes was indicated by a paucity of double-positive CD4 and CD8 cells, whereas the medullar lymphocyte population comprised of single-positive CD4 or CD8 cells appeared virtually unaffected (Fig. 4, CD4 and CD8). In surviving KO mice, no visible differences in thymic size or architecture were detected.
Figure 4.
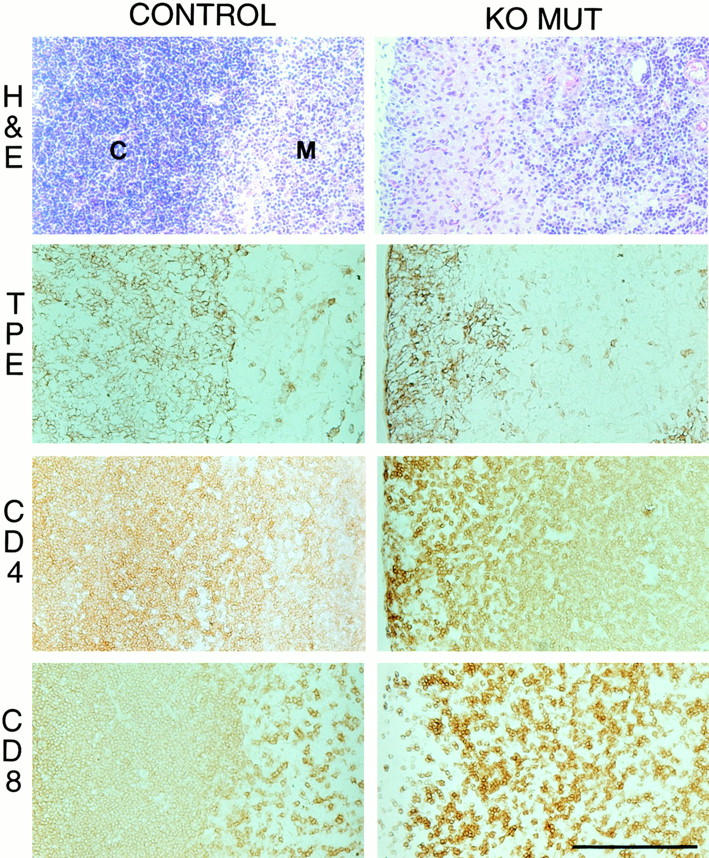
Histology and immunohistochemistry of longitudinal sections of thymus showing the cortex (C)/medulla (M) junction from week three wild-type (control) and collagen X KO mice with perinatal lethality (KO MUT). In the KO MUT, H&E staining reveals reduction of cortex and a paucity of cortical T cells, whereas TPE antibodies localize the cortex as a narrow strip compared with the extensive matrix in the control. Likewise, CD4 and CD8 T cell surface markers confirm a depletion of CD4+/CD8+ immature cortical lymphocytes in the KO MUT. Bar, 100 μm.
FACS of thymic cells confirmed a decrease in the CD4+ and CD8+ lymphocytes in week three KO mice with perinatal lethality by single- (Fig. 5 A) and double- (Fig. 5 B) labeling. Moreover, double-labeling revealed a decrease in CD4+/CD8+ cells, but an increase in single-positive T lymphocytes as well as in double-negative cells (Fig. 5 B); the latter is characteristic of autoimmune diseases. Furthermore, the lymphocyte profiles reflected those seen for Tg mice with perinatal lethality (Fig. 5). No significant differences were detected between thymic T cells from KO and Tg mice with no acute phenotype, and WT (Fig. 5), nor in KO or Tg mice at younger or older ages (data not shown). Lastly, cell isolation studies revealed a 3.5-fold reduction in cell number from thymuses of KO perinatal lethal mice when compared with controls and KO mice with no acute phenotype (data not shown). Taken together, these data suggested that, in mice exhibiting perinatal lethality, thymic cortical T cells were likely not replenished by marrow-derived immature T cells due to marrow aplasia. Alternatively, the more mature medullar T cells, in addition to those circulating in the blood, have likely escaped the effects of marrow aplasia. TUNEL assay for apoptosis and acridine orange/ethidium bromide staining for necrosis confirmed that thymic reduction in perinatal lethal mutants did not result from increased cell death (data not shown; Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication), but likely from lack of lymphocytes emigrating from the marrow.
Figure 5.
FACS analysis of thymic lymphocytes from week three wild-type (WT) and collagen X KO and Tg mice with mild (KO or Tg) or perinatal lethal (KO Mut or TG Mut) phenotypes. A, Single-label FACS reveals a reduction of CD4+/CD8+ cells in KO and Tg mice with perinatal lethality. B, Double-label FACS with FITC-conjugated antibodies labeling CD4+ lymphocytes (lower right quadrant), and PE-conjugated antibodies labeling CD8+ lymphocytes (upper left quadrant). Note depletion of cortical double-positive cells (upper right quadrant) in KO and Tg mice with perinatal lethality, with a concomitant increase in double-negative (lower left quadrant) cells, and single-positive CD4 or CD8. n, Animal number; error bars = standard error.
Spleen.
In week three KO mice with perinatal lethality, altered splenic architecture was discernible as poorly defined lymphatic nodules and a severely reduced red pulp (Fig. 6 A, H&E). MOMA-1 stained a subpopulation of splenic macrophages surrounding periarteriolar lymphatic sheaths, which revealed smaller and less-organized lymphatic nodules, with decreased internodular space (Fig. 6 A, MOMA). Ter 119 staining of the erythrocytic lineage underscored the disorganized splenic architecture that was highlighted by a diminished red pulp; the latter likely accounted for the reduced internodular distances (Fig. 6 A, TER). Furthermore, many erythrocytes appeared lysed.
Figure 6.
Histology and immunohistochemistry of longitudinal spleen sections from week three wild-type (control) and collagen X KO mice with perinatal lethality (KO MUT). A, Altered splenic architecture in the KO mutant is revealed by H&E as poorly distinguishable lymphatic nodules and red pulp reduction, by MOMA staining for splenic macrophages surrounding periarteriolar sheaths as decreased and disorderly nodules with decreased internodular distance, and by TER 119 staining of erythroid cells as a depleted red pulp. B, Immunohistochemical localization of T and B lymphocytes reveals in the KO mutant: unaltered distribution of CD4+ and CD8+ T cells, but a reduction in nodular size and internodular space, and more diffuse B220 staining for B cells. Bars, 100 μm.
The splenic T cell population comprises the mature, circulating T lymphocytes, which would likely not be affected by marrow aplasia. Thus, staining for CD4 and CD8 cell surface markers did not detect an altered T cell distribution or their deficiency in spleens of KO perinatal lethal mice (Fig. 6). Moreover, it appeared that the density of CD4+ and CD8+ cells was increased, perhaps reflecting the reduced red pulp. Alternatively, the B cell population, which is dependent on the marrow for maturation before emergence into the circulation (Takatsu 1997), was altered in the KO perinatal lethal mice. This was evidenced by more diffuse staining of lymphatic nodules than in the WT. Labeling with IgM and IgD (not shown) confirmed that, although the B cell number may be reduced, the cells appeared mature and functional, as seen in the Tg mice (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication).
Single-label FACS confirmed the histological observations by revealing an elevated splenic CD4+ T cell population in all KO mice (Fig. 7 A); this implied firstly that these cells were the least affected by marrow aplasia in mice with perinatal lethality, and second, that all KO and Tg mice had altered T lymphocyte populations. Percent CD8+ T cells remained relatively constant or slightly elevated in the KO's (Fig. 7 A), suggesting their concomitant decrease along with that of other splenic cells. These data were corroborated by double-label FACS (Fig. 7 B), which again demonstrated similar lymphocyte population changes in both the collagen X KO and Tg mice, as well as a decrease in CD4/CD8 double-negative splenic cells. Similar trends were seen in spleens from KO and Tg mice at both younger and older ages (preliminary data).
Figure 7.
FACS of splenic lymphocytes and erythroblasts from week three wild-type (WT) and collagen X KO and Tg mice with mild (KO or Tg) or perinatal lethal (KO Mut or TG Mut) phenotypes. A, Single-label FACS reveals an increase in CD4+ and CD8+ lymphocytes in all KO and Tg mice, with the largest increases in perinatal lethal mutants. B, Double-label FACS with FITC-conjugated antibodies staining CD4+ T cells (lower right quadrant), and PE-conjugated antibodies labeling CD8+ lymphocytes (upper left quadrant). Note an increase in single- and double-positive cells (upper right, left, lower right quadrants) in mutants, with a concomitant decrease in double-negative (lower left quadrant) cells. C, Single-label FACS of B220+ B cells. Note B cell reduction in perinatal lethal mutants, as well as in all Tg mice, but lack of reduction in the KO samples. D, Single-label FACS for Ter+ erythroblasts depicts an increase in KO and Tg mice with mild phenotypes, but a decrease approaching WT levels in mice with perinatal lethality. n, animal number; error bars = standard error.
B cell levels (Fig. 7 C) in KO perinatal lethal mutants were decreased by ∼45% of the control values, similar to what was seen in all Tg mice. One difference involved the subset of KO mice without an acute phenotype, where the percent B cells appeared unaltered, and even elevated in certain animals. Preliminary data indicated elevated B cell levels in KO mice at two weeks of age, followed by a slight decrease at older ages. This differed from the significant decrease in B cell levels in Tg mice, which were decreased throughout life (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication). Likewise, significant changes in Ter 119+ erythroblasts were detected only in the subset of perinatal lethal KO mutants, where erythroblasts were decreased to ∼50% that of controls (Fig. 7 D). In the Tg mice with no acute phenotype, elevated erythroblast levels implied splenic erythropoiesis; in mice, myelopoiesis persists in both spleens and marrows throughout life, whereas in humans, hematopoiesis is confined to bone marrows after their establishment. Elevated levels of Ter 119+ erythroblasts were seen in a subset of the healthy KO mice, although the overall changes were not significant. Complete blood count (CBC) analysis of marrow, spleens, and peripheral blood at different developmental stages would provide the definitive characterization of alterations in blood cell lineages. Moreover, marrow transplantations should establish whether these defects arise as a response to an altered marrow microenvironment.
Lastly, in KO perinatal lethal mutants, an ∼7.7-fold decrease was observed in splenocytes from KO perinatal lethal mutants, compared with that of splenic cells from control and KO mice with mild phenotypes (data not shown). This was comparable to the data for splenocytes from Tg perinatal lethal mutants (Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication). Likewise, the percentage of live splenic lymphocytes was reduced, which is particularly pronounced in light of red pulp reduction. Due to the latter, one would expect to see an overall increase in lymphocytes if these cells were not affected; in the KO mice, this population was significantly reduced, and supported the FACS data (Fig. 7, A–C).
Discussion
The data presented here demonstrate that mice with inactivated collagen X manifest a variable, previously undescribed phenotype that reflects certain skeleto-hematopoietic defects seen in Tg mice with dominant interference mutations for collagen X. Specifically, a subset of the KO mice develop perinatal lethality at week three after birth, which is distinguished by erythrocyte predominance and depletion of the hematopoietic compartment in marrows, and altered hematopoiesis and hypoplasia of the secondary lymphatic organs, such as the thymus, spleen, and lymph nodes. This most acute phenotype is seen in ∼10.8% of the KO mice, as opposed to >25% of the Tg mice; however, the defects in these severely affected animals appear indistinguishable. Among survivors, both KO and Tg mice continue to exhibit phenotypic variability, with milder, more subtle defects observed in the KO subset. Specifically, whereas many Tg survivors exhibit variable dwarfism during the rapid skeletal growth phase at weeks two to six, and aggressive lymphosarcomas with aging, only a small fraction of dwarfed KO mice were noticed, and no increased incidence of tumors has been detected. Alternatively, both sets of mice occasionally exhibit tooth overgrowth or hydrocephaly. However, in every mouse with altered collagen X, hematopoiesis is affected, and is manifested by altered B and T lymphocyte development (Fig. 7 A; Jacenko, O., C.J. Gress, M.R. Campbell, Z. Tao, and D.W. Roberts, manuscript submitted for publication). Although dwarfed, runted or sickly pups are occasionally observed among WT, not one of these animals has exhibited red marrows, lymphatic organ atrophy, and similar hematopoietic changes as those in the collagen X mice. In the Tg mice, the same disease phenotype was seen in multiple lines with independent transgene insertion sites, implicating the collagen X transgene as the cause of the skeleto-hematopoietic defects (Jacenko et al. 1993a). The comparable phenotypic changes reported here for the collagen X KO mice thus indicate that skeleto-hematopoietic defects in both murine sets are likely the direct and specific consequences of collagen X disruption. Moreover, these skeleto-hematopoietic defects underscore an interdependence between EO and establishment of a marrow environment required for blood cell differentiation.
Although phenotypic similarities exist between the KO and Tg mice, certain discrepancies may provide insights into the underlying pathogenic mechanisms. First, it is not unexpected for the dominant interference collagen X mutation to yield a stronger disease phenotype than one resulting from gene inactivation. The dominant interference transgene construct was designed to encode partially functional collagen X alpha chains that would compete for association with other chains at the COOH domain; however, after associations, the mutant chains would not trimerize properly due to an altered triple-helical domain. This scenario was confirmed in a cell-free translation system where the COOH domain was shown to be required for associations, and where collagen X alpha chains with triple-helical deletions were unable to trimerize with full-length chains (Chan et al. 1996; Chan and Jacenko 1998). The likely consequence of this scenario is either a depletion of hybrid trimers through a protein suicide mechanism leading to a loss-of-function, and/or persistence of these abnormal molecules leading to an impairment of collagen X structure and interactions (e.g., gain-of-function). We speculate that both loss-of-function and gain-of-function mechanisms are involved in the phenotype seen in the Tg mice. Specifically, subtle growth plate changes in the KO predominately involve the proliferating chondrocytes, whereas in the Tg mice, the most affected zone is hypertrophic cartilage. This implies that the consequences from a loss-of-function collagen X mutation may primarily affect the proliferative zone, as described by Kwan et al. 1997 in their collagen X KO mice, whereas a gain-of-function mutation that might generate a defective molecule in hypertrophic cartilage (e.g., either a mouse–chick heterotrimer and/or a chick homotrimer, as may be the case in the Tg mice), may affect the latter. Likewise, regarding hematopoiesis, B lymphocyte reduction is not observed in spleens from the surviving KO mice as it is in the surviving Tg animals. On the contrary, B cells appear to be slightly elevated in many KO mice, suggesting that these animals may be fighting infections. Again, these differences may reflect the consequences of a loss-of-function versus a dominant interference phenotype that may comprise both loss- and gain-of-function defects. Interbreeding the KO and Tg mice would ascertain the Tg contributions on a null background in contributing to the more acute Tg murine defects.
One unresolved issue involves the phenotypic variability in the KO and Tg mice, which are both homozygous at either the inactivated collagen X or at the transgene locus. Several scenarios can contribute towards such variability. The first possibility involves the presence of a strain-specific modifier, or an allelic variant at a locus other than the one being modified (Erickson 1996), whose actions may be linked to those of collagen X. In this situation, backcrossing the mice ∼12 generations into an inbred genetic background may resolve this issue, provided that these genes aren't closely linked. Both the KO and the Tg mice have been backcrossed about five generations into two strains, and continue to display phenotypic variability (our unpublished observations). A second possibility involves exposure of animals to opportunistic bacterial or fungal infections before weaning. This is supported by elevated B cells in KO mice (Fig. 7 C), as well as elevated cytokines in all mice with altered collagen X (our unpublished observations). Likewise, the significant B lymphocyte decrease in the Tg mice (Fig. 7 C) supports the more acute phenotype and a greater ratio of perinatal lethality in this murine subset. The third possibility may again reflect genetic variability due to initial crossing of two (for KO mice; Rosati et al. 1994) or three (for Tg mice; Chan and Jacenko 1998) strains while establishing these mouse lines. Variations in each mouse may involve skeletal growth, mineralization, and mobility; since mechanical stress has been linked with skeletal defects in patients with SMCD (Wasylenko et al. 1980), it is conceivable that increased stress on a weakened skeleton may yield greater perturbations of the growth plate, leading to more acute alterations in the skeleto-hematopoietic interface.
A provocative conclusion from this study is that all mice with altered collagen X, generated either through gene targeting or by transgenesis, to some extent have altered skeleto-hematopoietic differentiation. This in turn highlights a previously unforeseen yet intimate link between hypertrophic cartilage and EO, and marrow establishment. An interdependence between endochondrally derived bones and hematopoiesis has long been apparent (Taichman and Emerson 1994), however, it has not included hypertrophic cartilage as a potential contributor in hematopoietic interactions. It is conceivable that hematopoietic niches in the marrow may be compartmentalized both spatially (via trabecular bone projections or matrix components) and chemically (via bioactive molecules such as growth factors and cytokines). Defects in hypertrophic cartilage may thus contribute either alone or in concert towards skeletal defects and marrow alteration. Specifically, growth plate decompartmentalization has been reported in the KO mice (Kwan et al. 1997), and disruption of a hexagonal network in the pericellular matrix of hypertrophic chondrocytes was observed in the Tg mice (Chan and Jacenko 1998). It is speculated that collagen X may comprise the hexagonal network (Kwan et al. 1991; Chan and Jacenko 1998), and may even persist in the marrow stroma (our unpublished observations). Thus, disruption of collagen X may redistribute matrix components, such as glycosaminoglycans and proteoglycans, which have been implicated in hematopoiesis (Bruno et al. 1995; Gupta et al. 1996), or may disrupt specific hypertrophic cartilage matrix/cell interactions. Alternatively, trabecular bone reductions may directly alter both the spatial and chemical compartmentalization of the marrow; the trabecular surfaces may be essential for stem cell interactions (Taichman and Emerson 1994; Taichman et al. 1996) as well as in sequestering/synthesis of hematopoietic cytokines and growth factors involved in hematopoiesis (Bruno et al. 1995; Gupta et al. 1996).
Interestingly, skeleto-hematopoietic defects have been reported in animal models (Fletch et al. 1975; Tepper et al. 1990; Shull et al. 1992; Kulkarni and Karlsson 1993; Lewis et al. 1993; Jacenko 1995; Iotsova et al. 1997) as well as in a number of human disorders (Burke et al. 1967; Polmar and Pierce 1986; Guba et al. 1994; Turnpenny et al. 1995; Winter 1995; Young 1996), although they have not generally been viewed as being related. Our data imply that defects in hypertrophic cartilage may lead to hematopoietic disorders, including aplastic anemia, impaired immunity, and malignancies. It is noteworthy that to date >30 collagen X mutations have been implicated in causing either SMCD (Chan and Jacenko 1998) or SMD (Ikegawa et al. 1998), neither of these disorders is associated with hematopoietic abnormalities. Moreover, no mutations in the triple-helical domain of collagen X have yet been identified. It is likely that a phenotypic spectrum of disease severities may ensue from mutations in different collagen X domains, as seen in the collagen I and Osteogenesis imperfecta (Ikegawa et al. 1998). Furthermore, it is conceivable that pathogenic mechanisms involving dominant interference and gain-of-function (as is the likely mechanism of transgene action in the collagen X Tg mice) versus haplo-insufficiency (as in humans with SMCD) or complete loss-of-function (as in the collagen X KO mice), would yield the most acute skeleto-hematopoietic disease phenotypes.
Acknowledgments
We are extremely grateful to Drs. Benoit de Crombrugghe and Richard Behringer (M.D. Anderson Cancer Center, University of Texas) for generously providing the collagen X null mice and for critical review of the manuscript. We also greatly appreciate the help of Drs. Merle Elloso and Jorge Caamano (University of Pennsylvania School of Veterinary Medicine) in FACS data analysis; Dr. Chris Hunter (University of Pennsylvania School of Veterinary Medicine) for antibody-related suggestions; and Mrs. Michelle Campbell and Mr. Douglas Roberts for outstanding technical support and assistance with FACS analysis.
This work was supported by the National Institutes of Health (AR43362), an Arthritis Foundation Biomedical Research Grant, and the University of Pennsylvania Research Foundation Award to O. Jacenko.
Footnotes
Abbreviations used in this paper: EO, endochondral ossification; H&E staining, Harris hematoxylin and eosin Y staining; KO, knockout; PE, phycoerythrin; PI, propidium iodide; SMCD, Schmid metaphyseal chondrodysplasia; SMD, spondylometaphyseal dysplasia; Tg, transgenic; TPE, thymic pan epithelium; WT, wild-type controls.
References
- Bruno E., Luikart S.D., Long M.W., Hoffman R. Marrow-derived heparan sulfate proteoglycan mediates the adhesion of hematopoietic progenitor cells to cytokines. Exp. Hematol. 1995;23:1212–1217. [PubMed] [Google Scholar]
- Burke V., Colebatch J.H., Anderson C.M., Simmons M.J. Association of pancreatic insufficiency and chronic neutropenia in childhood. Arch. Dis. Child. 1967;42:147–157. doi: 10.1136/adc.42.222.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D., Cole W.G., Rogers J.G., Bateman J.F. Type X collagen multimer assembly in vitro is prevented by a Gly 618 to Val mutation in the alpha 1(X) NC1 domain resulting in Schmid metaphyseal chondrodysplasia. J. Biol. Chem. 1995;270:4558–4562. doi: 10.1074/jbc.270.9.4558. [DOI] [PubMed] [Google Scholar]
- Chan D., Jacenko O. Phenotypic and biochemical consequences of collagen X mutations in mice and humans. Matrix Biol. 1998;17:1169–1184. doi: 10.1016/s0945-053x(98)90056-7. [DOI] [PubMed] [Google Scholar]
- Chan D., Weng Y.M., Hocking A.M., Golub S., McQuillan D.J., Bateman J.F. Site-directed mutagenesis of human type X collagen. Expression of alpha 1(X) NC1, NC2, and triple-helical mutations in vitro and in transfected cells. J. Biol. Chem. 1996;271:13566–13572. doi: 10.1074/jbc.271.23.13566. [DOI] [PubMed] [Google Scholar]
- Chan D., Weng Y.M., Graham H.K., Sillence D.O., Bateman J.F. A nonsense mutation in the carboxyl-terminal domain of the type X collagen causes haploinsufficiency in schmid metaphyseal chondrodysplasia. J. Clin. Invest. 1998;101:1490–1499. doi: 10.1172/JCI1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D., Freddi S., Wang Y.M., Bateman J.F. Interaction of collagen alpha 1(x) containing engineered NCl mutations with normal alpha 1(x) in vitro. Implications for the molecular basis of schmid metaphyseal chondrodysplasia. J. Biol. Chem. 1999;274:13091–13097. doi: 10.1074/jbc.274.19.13091. [DOI] [PubMed] [Google Scholar]
- Diren H.B., Buyukgebiz A., Pirnar T. Spondylometaphyseal dysplasia, type VII. Pediatr. Radiol. 1992;22:87–89. doi: 10.1007/BF02011301. [DOI] [PubMed] [Google Scholar]
- Erickson R.P. Mouse models of human genetic diseasewhich mouse is more like a man? BioEssays. 1996;18:993–998. doi: 10.1002/bies.950181209. [DOI] [PubMed] [Google Scholar]
- Fletch S.M., Pinkerton P.H., Brueckner P.J. The Alaskan Malamute chondrodysplasia (dwarfism-anemia) syndromein review. J. Am. Anim. Hosp. Assoc. 1975;11:353–361. [Google Scholar]
- Guba S.C., Sartor C.A., Hutchinson R., Boxer L.A., Emerson S.G. Granulocyte colony-stimulating factor (G-CSF) production and G-CSF receptor structure in patients with congenital neutropenia. Blood. 1994;83:1486–1492. [PubMed] [Google Scholar]
- Gupta P., McCarthy J.B., Verafaillie C.M. Stromal fibroblasts heparan sulfate is required for cytokine-mediated ex vivo maintenance of human long-term culture-initiating cells. Blood. 1996;87:3229–3236. [PubMed] [Google Scholar]
- Ikegawa S., Nakamura K., Nagano A., Haga N., Nakamura Y. Mutations in the N-terminal globular domain of collagen X gene (Col10A1) in patients with Schmid metaphyseal chondrodysplasia. Human Mutation. 1997;9:131–135. doi: 10.1002/(SICI)1098-1004(1997)9:2<131::AID-HUMU5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Ikegawa S., Nishimura G., Nagai T., Hasegawa T., Ohashi H., Nakamura Y. Mutation in the type X collagen gene (COL10A1) causes spondylometaphyseal dysplasia. Am. J. Hum. Genet. 1998;63:1659–1662. doi: 10.1086/302158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotsova V., Caamano J., Loy J., Yang Y., Lewin A., Bravo R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nature Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- Jacenko O. c-fos and bone lossa regulator of osteoclast lineage determination. BioEssays. 1995;17:277–281. doi: 10.1002/bies.950170402. [DOI] [PubMed] [Google Scholar]
- Jacenko O., Chan D. Unraveling the consequences of collagen X mutations. J. Cell Materials. 1998;8:123–134. [Google Scholar]
- Jacenko O., LuValle P., Olsen B.R. Spondlylometaphyseal dysplasia in mice carrying a dominant negative mutation in a matrix protein specific for cartilage-to-bone transition Nature 365 1993. 56 61a [DOI] [PubMed] [Google Scholar]
- Jacenko, O., P. LuValle, K. Solum, and B.R. Olsen. 1993b. A dominant negative mutation in the α1(X) collagen gene produces spondylometaphyseal defects in mice. Progress in Clinical and Biological Research: Limb Development and Regeneration. Vol. 383B. Wiley-Liss, NY. 427–436. [PubMed]
- Jacenko O., Olsen B.R., Warman M.L. Of mice and menheritable skeletal disorders. Am. J. Hum. Genet. 1994;54:163–168. [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.B., Karlsson S. Transforming growth factor B-1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Path. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]
- Kwan A.P.L., Cummings C.E., Chapman J.A., Grant M.E. Macromolecular organization of chicken type X collagen in vitro. J. Cell Biol. 1991;114:597–604. doi: 10.1083/jcb.114.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan K.M., Pang M.K., Zhou S., Cowen S.K., Kong R.Y., Pfordte T., Olsen B.R., Sillence D.O., Tam P.P., Cheah K.S. Abnormal compartmentalization of cartilage matrix components in mice lacking collagen Ximplications for function. J. Cell Biol. 1997;136:459–471. doi: 10.1083/jcb.136.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachman R.S., Rimoin D.L., Spranger J. Metaphyseal chondrodysplasia, Schmid type. Clinical and radiographic delineation with a review of the literature. Pediat. Radiol. 1988;18:93–102. doi: 10.1007/BF02387549. [DOI] [PubMed] [Google Scholar]
- Laird P.W., Zijderveld A., Linders K., Rudnicki M.A., Jaenisch R., Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D.B., Leggitt H.D., Effman E.L., Motley S.T., Teitelbaum S.L., Jepsen K.J., Goldstein S.A., Bonadio J., Carpenter J., Perlmutter R.M. Osteoporosis induced in mice by overproduction of interleukin 4. Proc. Natl. Acad. Sci. USA. 1993;90:11618–11622. doi: 10.1073/pnas.90.24.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh I., Abbott M.H., Francomano C.A. Concentration of mutations causing Schmid metaphyseal chondrodysplasia in the C-terminal noncollagenous domain of type X collagen. Hum. Mutat. 1995;5:121–125. doi: 10.1002/humu.1380050204. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.H., Conn S.N., Bulleid N.J. Folding and assembly of type X collagen mutations that cause metaphyseal chondrodysplasia-type Schmid. Evidence for coassembly of the mutant and wild-type chains and binding to molecular chaperones. J. Biol. Chem. 1999;274:7570–7575. doi: 10.1074/jbc.274.11.7570. [DOI] [PubMed] [Google Scholar]
- Peeden J.N.J., Rimoin D.L., Lachman R.S., Dyer M.L., Gerard D., Gruber H.E. Spondylometaphyseal dysplasia, Sedaghatian. Am. J. Med. Genet. 1992;44:651–656. doi: 10.1002/ajmg.1320440525. [DOI] [PubMed] [Google Scholar]
- Polmar S.H., Pierce G.F. Cartilage hair hypoplasiaimmunological aspects and their clinical implications. Clin. Immunol. Immunopath. 1986;40:87–93. doi: 10.1016/0090-1229(86)90071-1. [DOI] [PubMed] [Google Scholar]
- Rosati R., Horan G.S., Pinero G.J., Garofalo S., Keene D.R., Horton W.A., Vuorio E., de Crombrugghe B., Behringer R.R. Normal long bone growth and development in type X collagen null mice. Nature Gen. 1994;8:129–135. doi: 10.1038/ng1094-129. [DOI] [PubMed] [Google Scholar]
- Shortman K., Wu L. Early T lymphocyte progenitors. Ann. Rev. Immunol. 1996;14:29–47. doi: 10.1146/annurev.immunol.14.1.29. [DOI] [PubMed] [Google Scholar]
- Shull M.M., Ormsby I., Kier A.B., Pawlowski S., Diebold R.J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D. Targeted disruption of mouse transforming growth factor-B1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman R.S., Emerson S.G. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J. Exp. Med. 1994;179:1677–1682. doi: 10.1084/jem.179.5.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taichman R.S., Reilly M.J., Emerson S.G. Human osteoblasts support human hematopoietic progenitor cells in in vitro bone marrow cultures. Blood. 1996;87:518–524. [PubMed] [Google Scholar]
- Takatsu K. Cytokines involved in B-cell differentiation and their sites of action. Proc. Soc. Exp. Biol. Med. 1997;215:121–133. doi: 10.3181/00379727-215-44119. [DOI] [PubMed] [Google Scholar]
- Tepper R.I., Levinson D.A., Stanger B.Z., Campos-Torres J., Abbas A.K., Leder P. IL-4 induces allergic-like inflammatory disease and alters T cell development in transgenic mice. Cell. 1990;62:457–467. doi: 10.1016/0092-8674(90)90011-3. [DOI] [PubMed] [Google Scholar]
- Turnpenny P.D., Dakwar R.A., Boulos F.N. Kyphomelic dysplasia. In: Donnai D., Winter R.M., editors. Congenital Malformation Syndromes. Chapman & Hall; London: 1995. pp. 199–205. [Google Scholar]
- Warman M.L., Abbott M.H., Apte S.S., Heffron T., McIntosh I., Cohn D.H., Hecht J.T., Olsen B.R., Francomano C.A. A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nature Genet. 1993;5:79–82. doi: 10.1038/ng0993-79. [DOI] [PubMed] [Google Scholar]
- Wasylenko M.J., Wedge J.H., Houston C.S. Metaphyseal chondrodysplasia, Schmid type. J. Bone Joint Surg. 1980;62A:660–663. [PubMed] [Google Scholar]
- Winter R.M. Dubowitz syndrome. In: Donnai D., Winter R.M., editors. Congenital Malformation Syndromes. Chapman & Hall; London: 1995. pp. 133–136. [Google Scholar]
- Young N.S. Immune pathophysiology of acquired aplastic anemia. Eur. J. Haematol. 1996;60:55–59. doi: 10.1111/j.1600-0609.1996.tb01646.x. [DOI] [PubMed] [Google Scholar]