

Abstract
Cancer vaccines can induce the in vivo generation of tumor Ag-specific T cells in patients with metastatic melanoma yet seldom elicit objective clinical responses. Alternatively, adoptive transfer of autologous tumor-infiltrating lymphocytes (TIL) can mediate tumor regression in 50% of lymphodepleted patients, but are logistically and technically difficult to generate. In this study, we evaluated the capability of vaccine-induced PBMC to mediate tumor regression after transfer to patients receiving the same chemotherapy-induced lymphodepletion used for TIL transfer therapy. Autologous PBMC from nine gp100-vaccinated patients with metastatic melanoma were stimulated ex vivo with the gp100:209–217(210M) peptide and transferred in combination with high-dose IL-2 and cancer vaccine. Transferred PBMC contained highly avid, gp100:209–217 peptide-reactive CD8+T cells. One week after transfer, lymphocyte counts peaked (median of 14.3 × 103 cells/μl; range of 0.9–59.7 × 103 cells/μl), with 56% of patients experiencing a lymphocytosis. gp100:209–217 peptide-specific CD8+T cells persisted at high levels in the blood of all patients and demonstrated significant tumor-specific IFN-γ secretion in vitro. Melanocyte-directed autoimmunity was noted in two patients; however, no patient experienced an objective clinical response. These studies demonstrate the feasibility and safety of using vaccine-induced PBMC for cell transfer, but suggests that they are not as effective as TIL in adoptive immunotherapy even when transferred into lymphodepleted hosts.
Successful cancer immunotherapy requires the generation and persistence, in vivo, of sufficient numbers of antitumor T cells with appropriate phenotypic and functional characteristics to home to tumor and mediate tumor destruction. The identification of multiple MHC-restricted epitopes from human melanomas has facilitated the application of peptide-based vaccination as an attractive immunotherapeutic approach for the treatment of patients with cancer (1). The melanocyte differentiation Ag gp100 has been identified as a frequent target of tumor-infiltrating lymphocytes (TIL),3 and adoptive cell transfer (ACT) of TIL containing gp100-reactive T cells has been shown to mediate tumor regression in patients with metastatic melanoma (2). The gp100:209–217 epitope (referred to as g209) is naturally processed and presented by HLA-A*0201 molecules on human melanoma cells with an intermediate MHC binding affinity (3, 4). Substitution of methionine for threonine in position 2 of the g209 peptide increased peptide-HLA-A*0201 binding affinity and enhanced the capacity for eliciting gp100-reactive T cells through in vitro stimulation of PBMC from melanoma patients (5). Vaccination with the modified g209 peptide (referred to as g209-2M) was more efficient than the g209 peptide in successfully immunizing HLA-A*0201 patients with metastatic melanoma (6, 7). Multiple courses of g209-2M peptide vaccination of patients at risk of recurrence induced high frequencies of g209-specific CD8+T cell responses in some patients (8, 9). Despite the reproducible generation, in vivo, of tumor Ag-specific T cell responses in these and other vaccination trials, recurrence can occur in patients successfully immunized in the adjuvant setting, and objective clinical responses are seldom observed in immunized patients with solid tumor (9, 10).
In contrast to the general lack of clinical effectiveness of current cancer vaccines, adoptive transfer of tumor-reactive lymphocytes, derived from autologous tumor, to patients with metastatic melanoma following nonmyeloablative but lymphodepleting conditioning resulted in objective clinical responses in ~50% of treated patients (11, 12). The capacity of adoptively transferred T cells to persist in the peripheral blood correlated well with in vivo tumor regression (13). In murine studies of ACT, effector cell persistence, proliferation, and antitumor efficacy was substantially improved when hosts were lymphoablated and subsequently administered a tripartite regimen of tumor Ag-specific T cells, IL-2, and cancer vaccine, showing that, in addition to lymphodepleting preconditioning, vaccination and cytokine administration can augment the antitumor effectiveness of transferred T cells (14). Further studies suggest that CD8+T cells that possess properties of less differentiated effector cells are best suited to proliferate, persist, and mediate tumor regression after transfer in vivo (15–17).
Adoptive immunotherapy circumvents many of the barriers confronting cancer vaccines because large numbers of tumor Ag-reactive T cells can be selected and activated ex vivo for transfer into a lymphoablated host, devoid of suppressive and competing cells (14, 18, 19). Because tumor Ag-specific T cells generated by cancer vaccines seldom elicited objective clinical responses in the vaccinated host, we hypothesized that, by analogy to the experience with TIL, harvest of these immune cells from the circulation, ex vivo stimulation with specific Ags and subsequent autologous transfer to lymphodepleted patients might augment their function and increase their therapeutic potency. In this study, we thus assessed whether autologous ex vivo expanded vaccine-specific T cells from the peripheral blood of cancer vaccine recipients can mediate objective clinical responses in patients with metastatic melanoma when transferred under favorable conditions, which include nonmyeloablative but lymphodepleting preconditioning, exogenous cytokine administration, and cancer vaccination.
Materials and Methods
Treatment regimen
All patients in this study had metastatic melanoma and were entered on institutional review board-approved protocols in the Surgery Branch of the National Cancer Institute. Informed consent was obtained from all subjects. Enrolled patients were previously vaccinated with either gp100:209–217(210M) peptide emulsified in Montanide ISA 51 (IFA) or recombinant fowlpox virus (rF-gp100P209; Therion Biologics) vaccine, as described elsewhere (Table I) (9, 20). Nine patients with metastatic melanoma, all refractory to treatment with IL-2 and with progressive disease, underwent lymphodepleting conditioning with 2 days of cyclophosphamide (60 mg/kg) followed by 5 days of fludarabine (25 mg/m2). On the day following the final dose of fludarabine (day 0), all patients received cell infusion with autologous gp100 peptide-reactive, in vitro-stimulated PBMC cultures, high-dose IL-2 therapy, and cancer vaccination. Cancer vaccination consisted of either i.v. injection of 6 × 109 PFU of rF-gp100P209 fowlpox virus delivered on day 0 and day 28 with a second course of IL-2, or s.c. injection of 1 mg of gp100:209–217(210M) peptide emulsified in IFA for five consecutive days beginning on day 0, and weekly thereafter for 1 mo.
Table I.
Patient demographics, administered cells, and vaccinations
| Infused g209-2M Peptide-Stimulated PBMCa |
Post-ACT Vaccination
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Age/Gender | Prior Therapiesb | Sites of Disease | Number (× 10−10) | No. of stimulations | Age of culture (days) | %CD8, %CD4c | % g209+ CD8c | Vaccined | Number of IL-2 Doses |
| 1 | 57/F | g209-2M/Tyr
Salmonella NY-ESO F2M F2M + HD IL-2 |
s.c. | 4.5 | 1 + 2 | 18, 18, 18 | 29, 51 | 29.9 | Peptide | 4 |
| 2 | 22/M | IFN + IL-2
g209-2M + CTLA-4 |
Shoulder, lung | 3.4 | 1 | 9, 10, 11 | 61, 21 | 33.2 | Peptide | 4 |
| 3 | 54/M | F2M
F2M + HD IL-2 |
s.c. | 12.0 | 1 | 8, 9, 10 | 91, 6 | 45.1 | Peptide | 6 |
| 4 | 30/M | TRP2 + HD IL-2
g209-2M + CTLA-4 |
s.c. adrenal | 10.4 | 1 + 2 | 15, 16, 17, 17, 17 | 35, 40 | 56.9 | Virus | 5 |
| 5 | 54/M | g209-2M/Tyr
HD IL-2 |
Shoulder, lung | 10.2 | 1 | 11, 12, 13 | 84, 10 | 63.3 | Virus | 5 |
| 6 | 54/M | Radiation, HD IL-2
g209-2M + CTLA-4 |
Neck, lung | 1.3 | 1 | 9, 10, 11 | 25, 45 | 5.8 | Virus | 5 |
| 7 | 30/F | Allogenic TC vaccine
IFN |
Axilla, spleen, liver, lung, hilum, brain | 7.0 | 1 | 10, 11, 12 | 35, 43 | 5.8 | Virus | 5 |
| 8 | 55/F | IFN + IL-2
HD IL-2 g209-2M + CTLA-4 |
Periaortic nodes, lung | 4.0 | 1 | 10, 11, 12 | 29, 55 | 35.2 | Virus | 9 |
| 9 | 38/M | g209-2M/Tyr
HD IL-2 |
Intra-abdominal, s.c. | 7.0 | 1 | 10, 11, 12 | 89, 10 | 20.4 | Virus | 11 |
PBMC from previously vaccinated patients were in vitro stimulated once or twice with 1 μM g209-2M peptide and grown in IL-2 (300 IU/ml) for the indicated number of days.
Before treatment, all patients had been vaccinated with either g209-2M peptide in IFA in combination with tyrosinase peptide (g209-2M/Tyr) or CTLA4-blocking Ab (g209-2M + CTLA-4), or F2M virus with or without high-dose IL-2.
Cell composition was determined by flow cytomtry using CD8- and CD4-specific Abs. Tetramer analysis of CD8+T cells was performed using g209 peptide-containing HLA-A2 tetramers (g209+).
Beginning on the day of cell transfer, patients received g209-2M peptide in IFA vaccine administered for 5 days followed by three weekly vaccinations or F2M virus vaccine administered on the day of cell transfer and at 1 mo postinfusion along with IL-2. All patients received high-dose (HD; 720,000 IU/kg) exogenous IL-2 administration every 8 h to tolerance subsequent to cell transfer. gp100:209–217(210M), peptide (g209-2M); gp100:209–217 peptide (g209); recombinant fowlpox virus encoding the g209-2M epitope (F2M); tyrosinase peptide (Tyr); anti-hCTLA-4 Ab (CTLA-4); tyrosinase-related protein 2 (TRP2); tumor cell TC.
Patient cell samples
Peptide-stimulated PBMC cultures for transfer were established as follows. Briefly, PBMC obtained by cytopheresis were enriched for lymphocytes by Ficoll-Paque (Amersham Pharmacia Biotech) gradient separation and stimulated with 1 μM g209-2M peptide and 300 IU/ml IL-2. Fresh IL-2 was added every 2–3 days. After ~12 days, PBMC cultures were screened to confirm tumor Ag-reactivity, harvested from culture bags, prepared for patient treatment, and aliquots were cryopreserved for future experimental analysis. Posttreatment PBMC were purified on Ficoll-Hypaque step gradients and cryopreserved.
Cell culture medium
Complete medium (CM) consisted of RPMI 1640 (Invitrogen Life Technologies) supplemented with 2 mM glutamine (Biofluids), 25 mM HEPES buffer (Biofluids), 100 U/ml penicillin (Biofluids), 100 μg/ml streptomycin (Biofluids), 50 μM 2-ME (Invitrogen Life Technologies), and 10% heat-inactivated human AB sera (Gemini Bioproducts).
Peptides and recombinant fowlpox virus
The nonamer native gp100:209–217 peptide (g209, ITDQVPFSV), the modified gp100:209–217(210M) peptide (g209-2M, IMDQVPFSV), MART-1:26–35(27L) peptide (ELAGIGILTV), tyrosinase:368–376 (370D) peptide (YMDGTMSQV), and the gp100:280–288 peptide (g280, YLEPGPVTA) were produced to Good Manufacturing Practice grade by solid phase synthesis by Multiple Peptide Systems. The g209 and the g280 peptide were provided by the National Cancer Institute Cancer Therapy Evaluation Program. Recombinant fowlpox viruses for vaccination were constructed and manufactured by Therion Biologics as described previously (20, 21).
Tetramers, mAbs, and flow cytometric immunofluorescence analysis
Allophycocyanin-labeled gp100:209–217 (ITDQVPFSV) peptide/HLA-A*0201 tetramer complexes were obtained from Immunotech, Beckman Coulter. gp100:209–217 (ITDQVPFSV) peptide-loaded HLA-A2 (wtA2) and D227K/T228A HLA-A2(CD8-nullA2) monomers were obtained from the National Institutes of Health Tetramer Facility. Tetrameric complexes were produced by mixing PE-conjugated streptavidin (Molecular Probes) and biotinylated peptide/HLA-A2 monomers at a 1:4 ratio. FITC-conjugated anti-CD25, CD27, CD45RO, CD69, and perforin, and PE-labeled anti-CD4, CD62L (L-selectin), CD28, CD45RA, CD38, and 41BB Abs were obtained from BD Biosciences. FITC-labeled anti-CD3, CD57, HLA-DR, and PerCP-labeled CD8 Abs were obtained from BD Pharmingen. PE-labeled IL-7Rα (CD127) and FITC-labeled CCR7 Abs were purchased from R&D Systems. Biotinylated FOXP3 Ab (eBioscience) was used in combination with streptavidin-PE (BD Biosciences). PE-conjugated granzyme B Ab was obtained from Caltag Laboratories. PE-labeled CD25 (Miltenyi Biotec) was used for detection of regulatory T (Treg) cells. Pro-pidium iodide was used for nonviable cell exclusion and analysis performed using a FACSCalibur (BD Biosciences). For intracellular perforin and granzyme B staining, cells were stained with tetramer and anti-CD8 Ab and then permeabilized and fixed using Cytofix/Cytoperm buffers (BD Pharmingen). Cells were then stained with FITC-conjugated perforin or granzyme B Abs, washed twice, and analyzed. Intracellular FOXP3 protein staining was performed according to the manufacturer’s instructions (eBioscience).
Ex vivo proliferation analysis
Cells used for infusion were thawed and immediately cultured in CM containing 10 μM BrdU for 5–7.5 h. For patients 8 and 9, 5 h BrdU labeling was performed using fresh lymphocytes. Posttransfer peripheral blood lymphocytes were directly ex vivo cultured in CM containing 10 μM BrdU for 5 h. All BrdU-treated cells were washed and cryopreserved for subsequent measurement of incorporation. For evaluation of incorporation, cells were thawed, stained with extracellular Abs, and fixed with 1% formaldehyde. Fixed cells were permeabilized with Cytofix/Cytoperm buffers and Cytoperm Plus buffers (BD Pharmingen), exposed to DNase (Sigma-Aldrich), stained with BrdU Ab (BD Pharmingen), and subsequently analyzed in a FACSCalibur (BD Biosciences). Alternatively, freshly collected PBMC were washed and resuspended in CM at 5 × 106 cells/ml. A total of 200 μl of cell suspension (106) was then added per well to a 96-well U-bottom plate, in quadruplicate. Cell cultures were then incubated at 37°C for 18 h in the presence of 1 μCi [3H]thymidine per well. Plates were harvested onto nylon filters using the beta plate system, and radioactivity was quantified using a beta plate counter. Results are expressed in cpm as the mean of quadruplicate cultures.
Cytokine release and chromium release assays
Cryopreserved PBMC samples were thawed and cultured overnight in CM with or without recombinant human IL-2 (300 IU/ml). Cytokine release and chromium release assays were performed as described previously (8).
TCRβchain V region (TRBV) analysis
The 5′ RACE analysis has been described previously (22). Briefly, a primer that was complementary to the TCRβ-chain C region, 5′-CTCTT GACCATGGCCATC-3′, was used with the SMART RACE cDNA Amplification kit (BD Biosciences) to amplify the TRBV region sequences expressed by polyclonal TIL samples. Germline genes that encoded the expressed TRBV products were identified by aligning the cloned sequences with the known TRBV gene sequences using the VectorNTI AlignX protocol (Invitrogen Life Technologies), and the highly variable sequences that resulted from joining the TRBV genes to the D and J regions were then compared to identify T cell clonotypes.
RNA isolation, cDNA synthesis, and real-time PCR
Real-time PCR was performed as described previously (23). Breifly, total RNA was isolated using RNeasy columns (Qiagen) according to the manufacturer’s instructions. RNA was eluted in 50 μl of RNase-free water and used as template for one round of reverse transcription for cDNA synthesis. The ABI PRISM 7700 Sequence Detector System (Applied Biosystems) was used for quantitative mRNA expression analysis. Primers and probes for β-actin (24) (forward 5′-GCGAGAAGATGACCCAGGATC-3′, reverse 5′-CCAGTGGTACGGCCAGAGG-3′; TaqMan probe 5′ (FAM)-CCAGCCATGTACGTTGCTATCCAGGC-(TAMRA)3′) were synthesized by Applied Biosystems. For analysis of FOXP3, Assay on Demand (Applied Biosystems) primers and probes were used with TaqMan Universal Master Mix (Applied Biosystems). The conditions used for PCR were 50°C for 2 min, 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. Each cDNA sample was tested in duplicate, and the mean values were calculated.
Results
Patient selection and treatment
Nine HLA-A*0201+ patients with progressive metastatic melanoma refractory to treatment with high-dose IL-2 received the current treatment regimen (Table I). All patients had received gp100-specific vaccination, either with g209-2M peptide or recombinant fowlpox virus encoding the g209-2M epitope. PBMC were cultured with 1 μM g209-2M peptide and 300 IU/ml IL-2 for 8–18 days (mean of 12 days). PBMC from two patients were stimulated a second time in vitro with peptide-pulsed autologous, irradiated PBMC. Autologous PBMC cultures were screened for peptide reactivity and administered to the patient after a 7-day nonmyeloablative but lymphodepleting chemotherapy regimen of fludarabine and cyclophosphamide as described previously (11, 12). Transferred cell numbers ranged between 1.3 and 12.0 × 1010 cells and were comprised of both CD8+ and CD4+T cells (Table I). On the day of cell transfer, treated patients also received high-dose IL-2 and either gp100-specific vaccination with g209-2M peptide in Montanide ISA for five sequential days followed by single weekly injections for 3 wk (patients 1–3) or recombinant fowlpox virus encoding the g209-2M epitope at the time of transfer and 1 mo later with high-dose IL-2 (patients 4–9). The number of administered IL-2 doses ranged from 4 to 11 doses. Autoimmunity was observed in patient 1, who developed pronounced vitiligo after therapy, and patient 2, who experienced a transient induction of uveitis, which was controlled with steroid eyedrops. However, no patient experienced an objective clinical response. To understand the limiting factors for this therapeutic approach, we evaluated the properties and fate of the cells used for therapy.
Characteristics of T cells used for adoptive immunotherapy
Before treatment, low frequencies of g209-specific T cells were detected in the peripheral blood of 6 of 9 patients by tetramer staining analysis using commercially available tetramers with a single mutation in the CD8 binding region (0–4% of CD8+T cells; data not shown). Ex vivo stimulation of patient PBMC with g209-2M peptide and IL-2 resulted in the expansion of native g209-specific CD8+T cells with frequencies ranging from 5.8 to 63.3% of CD8+T cells (Table I).
To evaluate whether g209-reactive CD8+T cells expressed high-affinity TCR, PBMC for transfer were stained with “CD8-null” HLA-A2/peptide tetramer complexes, with two mutations in the CD8 binding region (D227K/T228A substitutions in the α3 domain of HLA-A2 that completely abrogate HLA/CD8 interaction (25)), as a measure of TCR binding affinity (Fig. 1A). The frequency of transferred CD8+ lymphocytes that bound to the native g209 peptide containing CD8-null tetramer (g209/nullA2) ranged between 2.6 and 43.7%, whereas higher frequencies bound to g209 peptide containing tetramer with an unaltered HLA-A2 domain (g209/wtA2; range of 23.6 to 94.6% of CD8+T cells). The staining intensity of g209/nullA2 tetramer bright cells in PBMC was similar to that for a g209-specific CD8+T cell clone, JR209, isolated from the blood of a peptide vaccine recipient. Transferred PBMC exhibited no to very low binding to the control MART-1: 26–35(27L) peptide containing tetramer (MART-1/A2). Because no TIL from patients responding to ACT were solely gp100-reactive, direct comparisons could not be made.
FIGURE 1.

Characteristics of in vitro-stimulated, g209 peptide-specific CD8+T cells used for adoptive immunotherapy. A, Detection of CD8+T cells expressing high-affinity g209-specific TCR. In vitro-stimulated PBMC used for therapy, a gp100-reactive clone (JR209) isolated from the peripheral blood of a gp100 vaccine recipient, and a MART-1-reactive TIL subculture (DMF4) were stained with unaltered HLA-A2 molecules bearing tetramer containing gp100:209–217 (g209/wtA2) or MART-1:26–35(27L) peptide (MART-1/wtA2) or with a CD8 null tetramer containing the gp100:209–217 peptide (g209/nullA2). Histograms were gated on viable CD8+ lymphocytes, and the frequency of tetramer-positive events is shown. B, Phenotypic analysis of g209-specific CD8+T cells used for the treatment of patients 2, 5, 8, and 9. Tumor Ag-specific T cells were identified using commercially available gp100:209–217 peptide containing HLA-A*0201 tetramer complexes (gp100/A2) and anti-CD8 Ab. The y-axis indicates the frequency of g209 peptide-specific CD8+T cells expressing the molecules indicated on the x-axis. C, Cell surface expression of the lymphoid homing molecule CD62L (L-selectin) by g209 peptide-specific CD8+T cells for therapy (shaded histogram), compared with isotype Ab staining (open histogram). D, Tumor Ag-specific CD8+T cells were actively proliferating at the time of ACT. PBMC for treatment were incubated in BrdU for ~5 h, subsequently stained with gp100/A2 tetramer, and CD8, CD4, CD3, and BrdU Abs to assess T cell subset proliferation. The frequency of proliferating T cells is indicated by the percentage of cells expressing BrdU. BrdU incorporation is shown for individual patient cells as well as the mean for all patient cells.
Transferred T cells from four representative patients (patients 2, 5, 8, and 9) were analyzed for phenotypic characteristics. g209-specific, tetramer-positive CD8+T cells for transfer were generally characterized by an activated effector phenotype based upon positive expression of CD45RO, CD25, CD38, CD69, HLA-DR, as well as the effector molecules, granzyme B and perforin, albeit at a varying frequencies (Fig. 1B). Lower frequencies of g209-specific CD8+T cells also expressed CD27 and CD28 costimulatory receptors. Expression of CD62L (L-selectin), a lymph node homing molecule not found on TIL samples (17), was detected on 20–56% of g209-specific CD8+T cells (Fig. 1C).
To further characterize the status of the transferred T cell population, proliferation of T cell subsets in the cell population used for therapy was measured by incubating cells for ~5 h in culture medium containing 10 μM BrdU without IL-2 and measuring BrdU incorporation as an indicator of DNA synthesis/cell cycling by flow cytometry (Fig. 1D). At the time of cell transfer, proliferation of CD8+, CD4+, and g209/HLA-A*0201 tetramer-positive and negative CD8+T cells was detectable in vitro; however, CD8+T cells preferentially proliferated compared with CD4+ cells (p = 0.002) and g209/HLA-A*0201 tetramer-positive CD8+T cells preferentially proliferated compared with tetramer-negative CD8+T cells (p = 0.009). Control PBMC from healthy donors did not incorporate BrdU. Thus, large fractions of transferred g209-specific CD8+T cells (46–9%) were actively proliferating at the time of cell infusion.
The reactivity of the transferred cells is shown in Table II. Infused cells generally recognized the native peptide down to 1 nM concentrations, although cells from patient 2 recognized even lower peptide concentrations. Except for patient 6, all PBMC reacted specifically with HLA-A2+ melanoma cell lines. In addition to IFN-γ, transferred T cells generally produced TNF-α, GM-CSF, IL-8, and IL-13 cytokines in response to g209 peptide stimulation (data not shown). Comparison of the absolute IFN-γ release in response to tumor for the highest reactive subcultures of peptide-stimulated PBMC (2172 ± 963 pg/ml; mean ± SEM; n = 9) or TIL (8318 ± 1579 pg/ml; n = 35) used for transfer revealed significantly lower IFN-γ release by PBMC (p2 < 0.005) than reported for TIL (12). This suggests that TIL might have an increased functional avidity compared with vaccine-induced PBMC.
Table II.
Reactivity of peptide-stimulated PBMC used for adoptive immunotherapya
| Melanoma Cell Lines
|
T2 Cells Pulsed with Peptide (μM)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2+ |
A2− |
|||||||||||
| Patient | None | 526 | 624 | 888 | 938 | MART-1 (1.0) | g2M (1.0) | g209 (1.0) | g209 (0.1) | g209 (0.01) | g209 (0.001) | g209 (0.0001) |
| 1 | 73 | 266 | 276 | 53 | 29 | 24 | 6035 | 1782 | 1722 | 1121 | 218 | 52 |
| 2 | 24 | 2014 | 2327 | 316 | 45 | 78 | 42464 | 28708 | 17008 | 13516 | 12460 | 9216 |
| 3 | 68 | 2270 | 3110 | 115 | 93 | 156 | 14330 | 9660 | 8030 | 3540 | 706 | 204 |
| 4 | 14 | 558 | 391 | 17 | 13 | 61 | 1990 | 1580 | 1345 | 618 | 153 | 71 |
| 5 | 0 | 2630 | 2330 | 18 | 45 | 78 | 12160 | 7680 | 6920 | 2670 | 392 | 136 |
| 6 | 99 | 94 | 138 | 46 | 38 | 41 | 3610 | 1395 | 1443 | 990 | 262 | 78 |
| 7 | 57 | 287 | 298 | 64 | 35 | 278 | 5733 | 5316 | 5963 | 4892 | 2177 | 586 |
| 8 | 42 | 1173 | 2270 | 55 | 18 | 71 | 9250 | 5460 | 4680 | 1910 | 283 | 103 |
| 9 | 16 | 706 | 724 | 30 | 24 | 112 | 10110 | 5030 | 3230 | 897 | 204 | 123 |
Cells used for therapy were measured for secretion of IFN-γ after tumor or peptide-specific stimulation. 1e5 peptide-stimulated PBMC used for cell infusion were cocultured overnight with an equal number of HLA-A2+ (526 and 624) or HLA-A2- (888 and 938) melanoma cell lines or T2 APC pulsed with either 1μM irrelevant MART-1:26–35(27L) peptide (MART-1), modified gp100:209–217(210M) peptide (g2M), or titered concentrations of native gp100:209–217 peptide (g209), and assessed for IFN-γ (pg/ml) by standard ELISA assay. Values ≥200 pg/ml and twice background are bolded and underlined.
PBMC used for infusion were next evaluated for the capacity to specifically lyse g209 peptide-pulsed T2 cells and melanoma cell targets by standard 4 h chromium release assay. The results from analyses of cells from patients 4, 5, 8, and 9 were similar, as shown in Fig. 2. PBMC for infusion lysed g209 peptide-pulsed T2 cells at E:T ratios below 1:1. Nonspecific lysis of unpulsed T2 cells by patients 4, 5, and 8 PBMC was observed at high E:T ratios, but not to the degree of g209 peptide-specific target lysis. Peptide-stimulated PBMC used for infusion also lysed HLA-matched gp100-expressing melanoma cell lines, albeit less than that seen against peptide-pulsed cells. PBMC for infusion exhibited similar lytic activity against HLA-matched melanoma cells as did TIL (data not shown).
FIGURE 2.
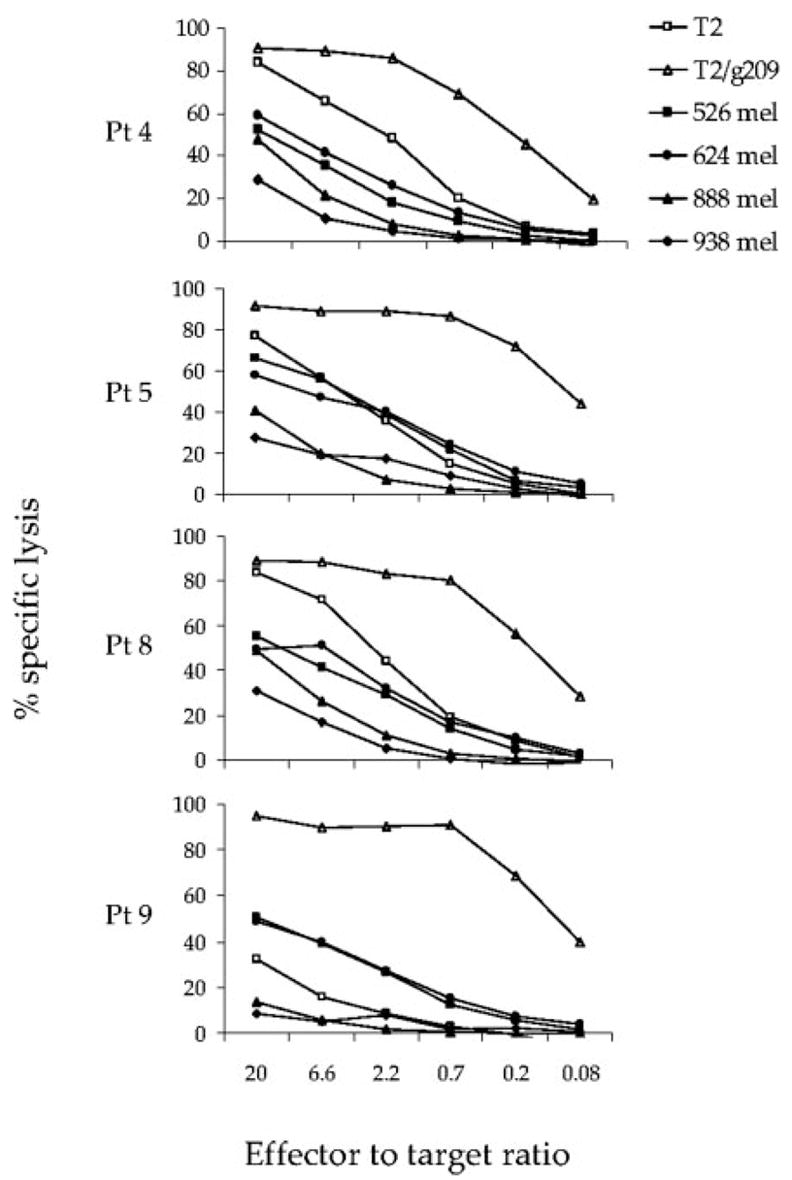
Cytotoxic activity of in vitro-stimulated, g209 peptide-specific CD8+T cells used for adoptive immunotherapy. PMBC and TIL containing gp100-reactive T cells were cultured overnight in CM containing 300 IU/ml IL-2. Effector cells were washed twice and cocultured for 4 h with chromium-labeled T2 cells alone or pulsed with 1 μM gp100:209–217 peptide, or HLA-A2+ (526 and 624) or HLA-A2− (888 and 938) melanoma cell lines in the absence of IL-2. Values represent the mean percentage of lysis of duplicate wells at the indicated E:T ratio.
CD4+CD25+FOXP3+T cells in the cell infusion
Because CD4+ CD25+ Treg cells are reportedly overexpressed in certain cancers (26, 27) and can mediate the inhibition of antitumor immune responses (28), we assayed for the presence of Treg cells, in the cell infusion. CD25 (IL-2 receptor-α-chain) was expressed by CD4+ T cells in the cell infusion with a mean positive expression of 31.6 ± 5.5% (Fig. 3A). This expression was significantly higher than that expressed by pretreatment CD4+ cells (p < 0.015), and similar to the levels measured at the peak of lymphocyte rebound (about 1 wk after transfer; p < 0.171) in vivo, although culture in IL-2 can up-regulate CD25 expression. To further test for the presence Treg cells, CD4+T cells were isolated by negative magnetic bead separation from PBMC used for infusion and measured for the expression of FOXP3, the surrogate Treg cell marker, by RT-PCR assay. FOXP3 was readily detected in CD4+ cells from all time points (Fig. 3B). FOXP3 expression by CD4+T cells from cell infusion samples was not significantly different from that measured in pre- (p < 0.390) or posttreatment CD4+T cells (p < 0.559).
FIGURE 3.
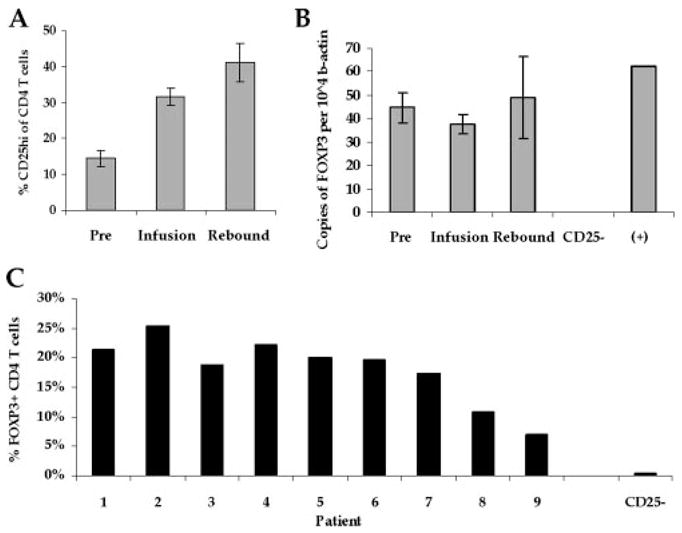
CD4+CD25+FOXP3+T cells are present in cells used for transfer and posttransfer PBMC. A, Infused CD4+T cells have increased CD25 cell surface expression compared with pretreatment levels. Pretreatment PBMC (Pre), cells used for transfer (Infusion), and PBMC collected ~1 wk after infusion (Rebound) were stained with CD3, CD4, and CD25-specific Abs, and measured for CD25 expression on CD4+T cells. Mean CD25high (CD25hi) expression for all patient samples ± SEM is shown. B, Infused CD4+T cells express FOXP3. RNA was isolated from the same cell samples following CD4+ cell enrichment, and measured for FOXP3 expression. RNA isolated from CD25-depleted PBMC from a patient donor (CD25+) and positive control RNA (+) was included. Results represent the mean relative number of FOXP3 copies per 104 copies of β-actin ± SEM. C, FOXP3 expression is detectable in CD4+T cells from in vitro-stimulated PBMC used for ACT. PBMC used for therapy, and CD25-depleted PBMC from a patient donor (CD25+) were stained with CD3, CD4, CD8, and FOXP3 Abs. The percentage of CD4+T cells expressing FOXP3 is shown on the y-coordinate. Data are representative of two independent experiments.
To assess FOXP3 protein expression by the in vitro-stimulated PBMC used for therapy, intracellular FOXP3 Ab staining was performed. FOXP3 was expressed by CD4+T cells from in vitro-stimulated PBMC (18.0 ± 1.9%; mean ± SEM; Fig. 3C). This was similar comparable to levels found in four tested TIL used for adoptive cell therapy (27.9 ± 5.3%; data not shown). The overall frequency of CD4+T cells in TIL (21.7 ± 4.3%; n = 35) and PBMC (31.2 ± 6.5; n = 9) was not significantly different (p = 0.303). With CD4+T cell frequencies > 20% of the infused PBMC population from all patients except patients 3, 5, and 9, the mean frequency of FOXP3+CD4+ Treg cells in the total infused lymphocyte population was 5.7% and ranged from 0.7 to 10.9%. These data indicate that PBMC used for therapy contained substantial frequencies of CD4+ CD25+FOXP3+ Treg cells.
Early fate of adoptively transferred peptide-stimulated PBMC in vivo
Lymphocytes were generally detectable in the peripheral blood of treated patients beginning 3–5 days after ACT (Fig. 4). Peak absolute lymphocyte counts (ALCs) for all patients ranged from ~900 cells/μl in patient 7 to nearly 60,000 cells/μl in patient 5. Lymphocytosis, defined as the upper limit of normal (4.9 × 103 cell/μl), was seen in 5 of 9 patients and was not associated with the nature of the administered vaccine (2 of 3 g209-2M peptide and 3 of 6 fowlpox virus recipients). This level of lymphocytosis was observed in 5.7% (2 of 35) of patients that had received adoptive TIL therapy (12).
FIGURE 4.
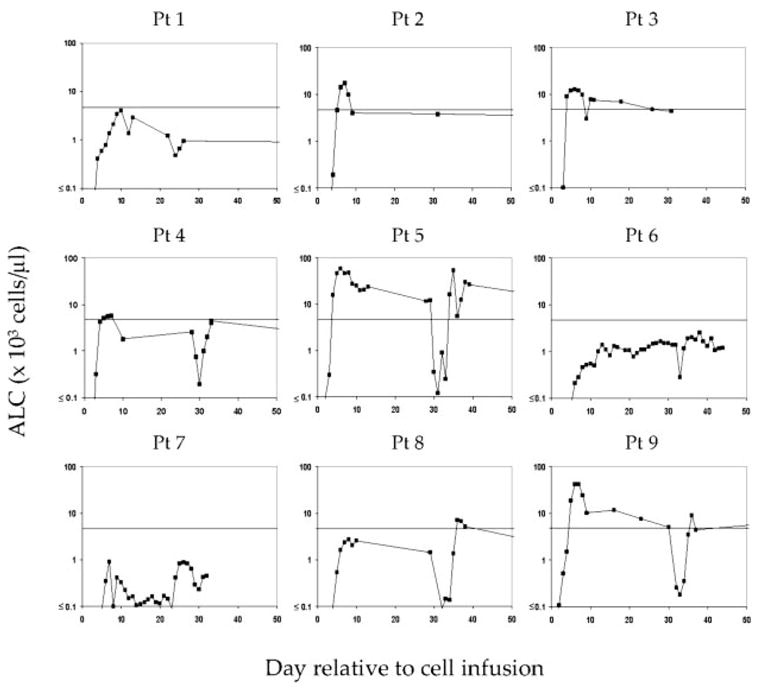
Absolute numbers of lymphocytes in the peripheral blood of treated patients following ACT of g209 peptide-stimulated T cells. Numbers of lymphocytes in the peripheral blood generally peaked 1 wk following cell transfer and contracted to normal levels in most patients. Lymphocytosis was designated as an ALC > 4.7 × 103 cells/μl blood. Lymphocytosis was noted in five of nine patients receiving adoptive immunotherapy. The x-axis represents days relative to cell transfer, with day 0 representing the day of cell infusion.
For patients receiving viral vaccination (patients 4–9), repeat fowlpox vaccination and IL-2 administration at 1 mo induced a secondary lymphocytosis in 3 of 6 patients (patients 5, 8, and 9); patients 5 and 9 had experienced lymphocytosis after primary vaccination. Administration of virus alone initially induced a transient reduction in ALC that was also seen when IL-2 was administered. For example, patient 5 had a lymphocyte count of 12,240 cells/μl 29 days after cell infusion, immediately before secondary viral vaccination. Recombinant virus vaccination alone resulted in a reduction in ALC to 345 cells/μl on day 30, which continued during IL-2 administration. Reduced ALCs were measured until day 35 when IL-2 treatment was withdrawn and a secondary lymphocytosis occurred.
Shortly after cell infusion, lymphocytes from the peripheral blood of treated patients had a blastic appearance with large diffuse amorphic nuclei (Fig. 5A). Direct ex vivo culturing of day 3 peripheral blood cells from patient 3 in CM containing tritiated thymidine demonstrated increased global proliferation, compared with resting PBMC from a healthy donor (Fig. 5B). To evaluate the kinetics of CD8+T cell proliferation, direct ex vivo BrdU-labeling was performed on serial peripheral blood samples from the same patient beginning 3 days after cell infusion, and 24 h after the last dose of IL-2. At this early time point, nearly 99% of circulating lymphocytes were CD8+T cells. Longitudinal evaluation of CD8+T cell proliferation was measured as BrdU incorporation by flow cytometry. Three days after transfer, when the ALC was only 100 cells/μl, ~20% of circulating CD8+T cells were actively proliferating (Fig. 5C). The following day, a dramatic increase in ALC was observed, up to 9,100 cells/μl. With reduced CD8+T cell proliferation by day 5, lymphocyte counts plateaued. By day 7 after transfer, a contraction in ALC was paralleled by a loss of detectable BrdU incorporation by CD8+T cells in peripheral blood. At early time points after infusion, proliferation by tumor Ag-specific CD8+T cells was also detectable (Fig. 5D); however, g209-specific CD8+T cells did not proliferate significantly more or less than non-g209-specific CD8+T cells (p = 0.423; Fig. 5E). The frequency of proliferating CD8+T cells trended toward being higher than for CD4+T cells, although the difference was not significant (p = 0.098). Thus, pan lymphocyte proliferation might reflect homeostatic proliferation in the lymphoablated host, IL-2 administration, and/or the induction of virus-specific T cell responses. Support for the latter was observed in the increased frequency of proliferating non-g209-specific CD8+T cells following secondary viral vaccination and IL-2 administration, compared with g209-specific CD8+T cells (data not shown).
FIGURE 5.

g209 peptide-stimulated T cells proliferate in the peripheral blood of patients following ACT. A, A peripheral blood smear shows the blastic, leukemic appearance of lymphocytes from patient 3, obtained 3 days after transfer (magnification, × 100). B, Peripheral blood cells from treated patients proliferate in vivo. Fresh PBMC obtained from patient 3, 3 days after transfer, proliferated overnight while incubated in medium containing [3H]thymidine, whereas PBMC from a normal healthy donor did not. Cultures were performed in quadruplicate; mean proliferation is represented in cpm ± SEM. C, Lymphocyte counts plateau as cell proliferation diminishes in vivo. Fresh PBMC obtained from patient 5, 3 days after transfer, were directly incubated in BrdU containing CM for ~5 h and measured for incorporation. The greatest increase in ALC is seen following peak BrdU incorporation by freshly collected CD8+T cells 3 days after transfer. ALCs plateaued as proliferation of CD8+T cells temporally diminished. D, g209 peptide-specific CD8+T cell proliferation is detectable from the peripheral blood of patient 5, 3 days after transfer, as measured by incorporation of BrdU. Proliferation was not noted in normal healthy donor PBMC. E, Multiple lymphocyte subsets proliferated in the blood after cell infusion. PBMC were incubated in BrdU for ~5 h, subsequently stained with gp100/A2 tetramer, and CD8, CD4, CD3, and BrdU Abs to assess T cell subset proliferation. The frequency of proliferating T cells is indicated by the percentage of cells expressing BrdU. Proliferation was assessed for the following patients on the indicated day relative to cell transfer and last IL-2 dose; patient 2 (day 5; 3 days after IL-2); patient 3 (day 4; 1 day after IL-2); patient 4 (day 5; 3 days after IL-2); patient 5 (day 3; 1 day after IL-2); patient 8 (day 7; 4 days after IL-2); patient 9 (day 5; 2 days after IL-2).
Persistence of tumor Ag-specific CD8+T cells in vivo
One month posttransfer, CD8+T cell numbers returned to physiologic levels in most patients (Fig. 6A). On average, CD4+T cell numbers were also back to normal levels (412 ± 126 cells/μl; range of 18 to 1310 cells/μl), with 5 of 9 patients having levels at or above the normal range (Fig. 6C). The persistence of transferred tumor Ag-specific CD8+T cells was noted by the continued detection of g209-specific cells in the blood, ranging from 1 to 78% of CD8+T cells (Fig. 6B). The calculated number of circulating tumor Ag-specific T cells at 1 mo ranged from ~10 to 8,500 native gp100-specific T cells per μl of blood (Fig. 6B).
FIGURE 6.
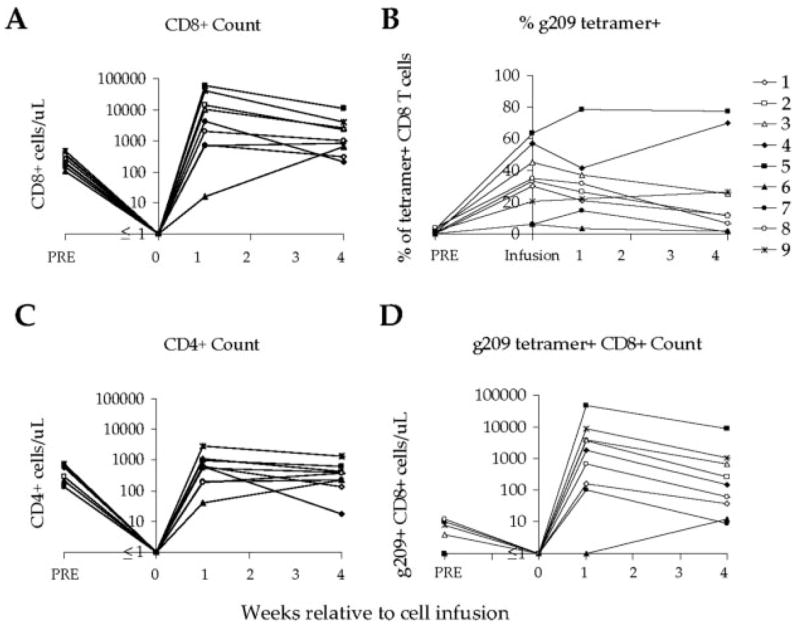
g209 peptide-specific CD8+T cells persist in vivo after ACT. A, CD8+T cell numbers peaked ~1 wk after cell infusion. One month after treatment, CD8+T cell counts remained slightly high compared with pretreatment counts but had diminished toward normal homeostatic levels. B, The frequency of g209 peptide-specific CD8+T cells in the peripheral blood after transfer largely reflected the level observed in cell infusion samples. In all cases, these levels were higher than that observed in pretreatment PBMC. C, CD4+T cell numbers generally had returned to normal levels 1 wk after cell infusion. D, High numbers of g209 peptide-specific CD8+T cells persisted in the blood 1 mo after cell infusion. The number of circulating g209 peptide-specific T cells was higher after therapy for most patients. g209 peptide-specific CD8+T cell number was determined as CD8+T cell number multiplied by the percentage of g209/A2 tetramer-positive CD8+T cells.
Longitudinal clonal composition analysis was performed on the g209-specific CD8+T cell populations from patients 3 and 5 cell samples (Table III). CD8+T cells, enriched by negative magnetic selection, were stained with PE labeled-g209/HLA-A*0201 tetramers and positively selected using anti-PE beads. Purities of tetramer+ CD8+T cells were > 80%. Using 5′ RACE analysis for detection of the TRBV gene usage, the clonal composition of gp100-specific CD8+T cells from various time points were assessed. Cell infusion samples contained multiple clones that were also detected in the pretreatment PBMC. After transfer, some clones, such as the 10-2(12S3) clone 1 in patient 5, expanded and persisted, whereas other clones, such as the 14S1 clone 1, that predominated early did not persist. Similar findings were observed for g209-specific CD8+T cells from patient 3. In both patient samples, gp100-specific CD8+ clones, detected in pretreatment PBMC and in the cell infusion, persisted one or more months after transfer.
Table III.
Clonal persistence of tumor Ag-specific CD8+ lymphocytes after ACT of peptide-stimulated PBMCa
| PBMC Sample Relative to Cell Infusion
|
|||||
|---|---|---|---|---|---|
| BV Family | Pre (%) | Infusion (%) | 1 wk (%) | 1 mo (%) | 2 mo (%) |
| Patient 5 | |||||
| 10-2(12S3) clone 1 | 18 | 6 | 28 | 52 | 67 |
| 10-2(12S3) clone 2 | 3 | 4 | 9 | 3 | 3 |
| 5-1(5S1) | 1 | 3 | < 1 | 10 | 1 |
| 15(24S1) | 5 | 1 | 1 | 1 | < 1 |
| 19(17S1) | < 1 | 6 | 2 | < 1 | 1 |
| 20-1(2S1) | 9 | 1 | 8 | 2 | < 1 |
| 27(14S1) clone 1 | 10 | 9 | 27 | < 1 | < 1 |
| 27(14S1) clone 2 | 1 | 7 | < 1 | 2 | < 1 |
| Patient 3 | |||||
| 2(22s1) | < 1 | < 1 | < 1 | 16 | n.t. |
| 6-1(13S3) | < 1 | 5 | < 1 | < 1 | n.t. |
| 7-9(6S5) | 38 | 61 | 65 | 48 | n.t. |
| 12-3(8S1) clone 1 | 5 | 7 | 4 | 10 | n.t. |
| 12-3(8S1) clone 2 | 30 | 5 | 8 | < 1 | n.t. |
| 12-4(8S2) | 4 | 7 | 8 | 2 | n.t. |
| 19(17S1) | < 1 | < 1 | 6 | 3 | n.t. |
TCRBV usage by gp100:209–217 specific CD8 T cells was analyzed by 5′ RACE analysis. The BV family name is indicated according to IMGT genomic designation, which precedes the traditional published nomenclature in parentheses. Unique clones from the same BV family are further distinguished by clone number. The percentage of distinct gp100:209–217 peptide HLA-A2 tetramer-positive CD8+ lymphocyte clones, which are expressed at ≥5% at any time point during the treatment schedule, are shown. gp100 peptide-specific CD8 T cells were isolated from cell samples using a “no touch” CD8+ cell magnetic microbead enrichment, followed by staining with a PE-labeled gp100 peptide-containing tetramer and subsequent separation using an anti-PE microbead-positive magnetic selection. Purity of isolated cell populations were verified by flow cytometry. Values are rounded to nearest whole number. n.t., Not tested.
Because g209-specific CD8+T cells persisted, we were able to evaluate phenotypic changes that occurred within the persisting T cell population in vivo. Fig. 7 shows the frequency of g209-specific T cells from four treated patients that express the indicate phenotypic marker at time points relative to cell infusion. After cell infusion, costimulatory molecules CD28 and CD27 were detectable at low to intermediate frequencies, with CD27 expression increasing slowly with time. CD45RA expression was similarly up-regulated on g209-specific T cells after 1 mo. Markers associated with activation and effector status, including HLA-DR and CD38, were elevated after 1 wk and decreased by 1 mo posttransfer. CD25 and CD69 levels were generally low on g209-specific T cells in vivo at both time points. Although the lymph node homing marker CCR7 was not detected after transfer, CD62L was continually expressed at different frequencies for individual patients. One month after infusion, cellular senescence-associated markers, CD57 and KLRG-1 (data not shown), were expressed by most persisting tumor Ag-specific CD8+T cells, as were the effector molecules perforin and granzyme B. Collectively, g209 peptide-specific CD8+T cells persisting 1 mo after infusion were similar in phenotype to pretreatment cells.
FIGURE 7.
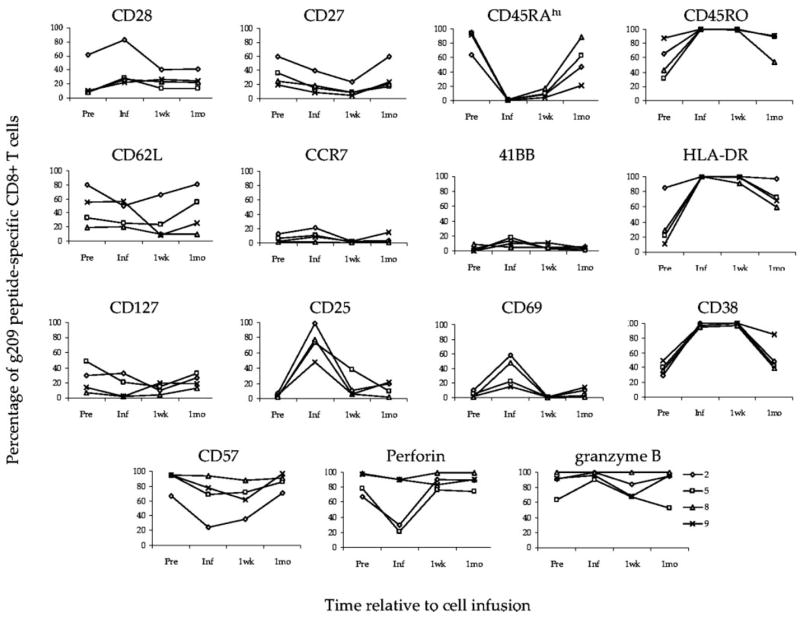
Adoptively transferred g209 peptide-specific CD8+T cells that persist in vivo assume their pretreatment phenotype. Pretreatment PBMC (Pre), cells used for infusion, and 1 wk (1wk) or 1 mo (1mo) posttransfer PBMC from patients 2, 5, 8, and 9 were stained using gp100:209–217 peptide containing HLA-A*0201 tetramer complexes, CD8 Ab, and Abs specific for the indicated molecules. Graphs show the frequency of g209 peptide-specific CD8+T cells that express the indicated molecule (y-axis) in relation to the time of cell infusion (x-axis).
Functional activity of persistent tumor Ag-specific CD8+T cells
Although continued expression of effector-associated molecules was detected, the loss of effector function by the transferred T cell population may have affected the therapy. Therefore, we evaluated whether cells persisting in the circulation after adoptive transfer maintained the ability to secrete cytokine in response to stimulation with tumor or peptide-pulsed target cells in vitro (Table IV). Cryopreserved, postinfusion PBMC were thawed, rested overnight in 300 IU IL-2/ml, and washed before establishment of cocultures with tumor or peptide-pulsed target cells. One week and 1 mo postinfusion, PMBC from five evaluated patients secreted significant amounts of IFN-γ following overnight stimulation with 1 μM g209 or g209-2M peptides, compared with stimulation by the control MART-1 peptide. Cytokine secretion by 1 mo posttransfer PBMC was detected in response to g209 peptide concentrations down to 1 nM in 4 of 5 patient samples. Similar to peptide, postinfusion PBMC recognized and secreted cytokine in response to gp100-expressing HLA-A2+ melanoma cell lines, but not cells lacking HLA-A2 expression. PBMC that were rested overnight in the absence of IL-2 were generally reactive to g209 peptide at 100–10 nM concentrations, but not to melanoma cell lines (data not shown). Similar results have been observed in the study of PBMC from patients that responded to adoptive TIL transfer (11). Posttransfer PBMC from patients 4, 5, and 9 rested overnight in the absence of IL-2 also exhibited potent and specific cytolytic function against g209 peptide-pulsed APCs, but little to no lysis of HLA-matched melanoma cell targets with or without overnight culturing in IL-2 (data not shown).
Table IV.
Maintained tumor and peptide-reactivity by PBMC after adoptive immunotherapya
| g209 Peptide (μM)
|
g209-2M | MART-1 | HLA-A2+ |
HLA-A2+ |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | PBL Sample | 1 | 0.1 | 0.01 | 0.001 | 0.0001 | 1 | 1 | 526mel | 624mel | 888mel | 938mel |
| 3 | Pre | 427 | 395 | 401 | 355 | 324 | 470 | 315 | 108 | 101 | 130 | 81 |
| Infusion | 9660 | 8030 | 3540 | 706 | 204 | 14330 | 156 | 2270 | 3110 | 115 | 93 | |
| 1 wk | 2270 | 1800 | 621 | 241 | 119 | 3500 | 94 | 854 | 676 | 24 | 28 | |
| 1 mo | 2730 | 1800 | 758 | 241 | 81 | 4320 | 52 | 1467 | 1324 | 21 | 23 | |
| 4 | Pre | 16 | 15 | 22 | 3 | 11 | 16 | 27 | 21 | 29 | 15 | 25 |
| Infusion | 1580 | 1345 | 618 | 153 | 71 | 1990 | 61 | 558 | 391 | 17 | 13 | |
| 1 wk | 1258 | 1139 | 309 | 87 | 29 | 1790 | 31 | 299 | 291 | 9 | 42 | |
| 1 mo | 2100 | 1409 | 1073 | 301 | 70 | 2490 | 45 | 1376 | 576 | 4 | 13 | |
| 2 mo | 717 | 723 | 405 | 141 | 26 | 946 | 43 | 457 | 149 | 33 | 37 | |
| 5 | Pre | 32 | 27 | 14 | 32 | 22 | 50 | 20 | 21 | 12 | 0 | 1 |
| Infusion | 7680 | 6920 | 2670 | 392 | 136 | 12160 | 78 | 2630 | 2330 | 18 | 45 | |
| 1 wk | 3710 | 3050 | 931 | 198 | 63 | 6340 | 58 | 1340 | 1700 | 2 | 17 | |
| 1 mo | 3000 | 1399 | 647 | 236 | 54 | 6870 | 42 | 2020 | 1404 | 0 | 31 | |
| 2 mo | 2990 | 2290 | 1061 | 303 | 93 | 6410 | 87 | 2060 | 1700 | 45 | 87 | |
| 8 | Pre | 84 | 42 | 37 | 13 | 24 | 97 | 33 | 47 | 47 | 10 | 10 |
| Infusion | 5460 | 4680 | 1910 | 283 | 103 | 9250 | 71 | 1173 | 2270 | 55 | 18 | |
| 1 wk | 1370 | 1539 | 787 | 225 | 44 | 2210 | 59 | 1194 | 1063 | 46 | 25 | |
| 1 mo | 1016 | 733 | 559 | 168 | 106 | 795 | 69 | 688 | 438 | 44 | 26 | |
| 9 | Pre | 91 | 81 | 37 | 42 | 50 | 198 | 48 | 23 | 25 | 10 | 10 |
| Infusion | 5030 | 3230 | 897 | 204 | 123 | 10110 | 112 | 706 | 724 | 30 | 24 | |
| 2 mo | 2190 | 1455 | 771 | 163 | 47 | 6280 | 50 | 1139 | 576 | 18 | 7 | |
IFN-γ secretion (pg/ml) from patient pretreatment PBMC, peptide-stimulated cells used for infusion, and postinfusion PBMC stimulated with HLA-A2+ (526 and 624) or HLA-A2+ (888 and 938) melanoma cell lines, or T2 APC pulsed with gp100:209–217 (g209), gp100:209–217(210M) (g209-2M), or MART-1:26–35(27L) (MART-1) peptide at the indicated concentration. Cryopreserved cells were thawed and rested overnight in CM and recombinant human IL-2 (300 IU/ml) before establishment of cocultures. Underlined, bold face values represent specific cytokine secretion specified as > 2-fold increase compared to controls and > 100 pg/ml concentration.
Maintained expression of gp100 and HLA-A2 by tumor after treatment
Of nine treated patients, four had pre- and posttreatment tumor biopsy specimens available for evaluation of MHC class I molecule and tumor Ag expression by immunohistochemical (IHC) staining. Analysis of fine needle aspirate biopsy specimens taken from the same lesion revealed maintained expression of HLA-A2 and gp100 in tumor after treatment from three patients, 1, 4, and 9, demonstrating that Ag loss did not account for the observed lack of clinical response in these patients. gp100 expression was not detectable in pre- or posttreatment tumor biopsies from patient 3. Preexistent Ag loss may have thus rendered this patient incapable of responding to the applied therapy. The expression of gp100 was also undetectable in posttreatment tumor specimens from patients 2 and 5; however, in the absence of pretreatment specimens, we could not evaluate whether Ag loss was therapy-related. Evaluation of the patient-2 tumor sample was further complicated by a high degree of necrosis within the analyzed lesion.
Discussion
The use of cancer vaccines for the treatment of solid tumors is an attractive immunotherapeutic approach, because vaccines are protective against viral diseases, effective in the treatment of certain hematological malignancies (29), and are easily administered. Cancer vaccines alone can induce the in vivo generation of high frequencies of self/tumor Ag-specific CD8+T cells in patients at high risk for recurrence of melanoma (8, 9). Nevertheless, tumor progression can occur in the presence of high levels of tumor Ag-specific T cells (9). Furthermore, the overall objective response rate in melanoma patients receiving cancer vaccines in our and other (10) clinical vaccine trials have been disappointing (3.3%). Thus, the presence of circulating self/tumor Ag-specific T cells alone appears insufficient to provide protection from recurrence or actively mediate tumor regression in most vaccinated patients. Accordingly, improvement of current immunotherapeutic modalities is necessary.
Although immunotherapy with IL-2 (30) or anti-CTLA4 Ab (31, 32) can mediate tumor regression in ~15% of treated melanoma patients, adoptive transfer of autologous, ex vivo-activated TIL following nonmyeloablative but lymphodepleting chemotherapy can mediate the regression of bulky, vascularized metastatic disease in ~50% of patients refractory to IL-2 administration (11, 12). The striking difference in the clinical effectiveness of TIL cell transfer compared with the lack of clinical effectiveness of immune cells generated endogenously by vaccines stimulated us to attempt to understand whether the differences were due to the properties of the cells or the lymphodepleting preparative regimen used in the TIL transfer protocol. In addition, vaccine-induced T cells collected by apheresis are logistically and technically less difficult to generate for adoptive immunotherapy than TIL. We thus isolated vaccine-derived cells from the peripheral blood, activated and expanded them ex vivo, and returned them to patients after administering the exact same lymphodepleting regimen used in our TIL transfer trial.
Of the nine patients treated in this trial, none experienced an objective clinical response. Two patients experienced autoimmunity following therapy. Patient 2 experienced a transient induction of uveitis, which was controlled with steroid eyedrops. Autoimmune induction was also observed in patient 1, who developed pronounced vitiligo after therapy. The lack of objective clinical responses in these nine vaccine-induced PBMC recipients represents a significant difference in therapeutic effectiveness as compared with the 51% (18 of 35) response rate reported for patients receiving TIL therapy (p = 0.006; two-tailed Fisher’s exact test) (12). The precise mechanisms that underlie this disparity in clinical effectiveness are not known but may be manifold.
Tumors may elude immune recognition via numerous routes (33). In this study, data concerning tumor escape were inconclusive; however, tumor escape due to Ag loss does not appear to have contributed to the lack of objective responses in all treated patients because MHC class I and gp100 expression was maintained by lesions from three evaluable patients with both pre- and posttreatment lesions available. gp100 expression was not detectable by IHC analysis in posttransfer lesions from two other patients, but in the absence of evaluable pretreatment lesions, pre-existent Ag loss could not be discerned from treatment-induced changes. Indeed, ~20% of melanoma lesions appear to lack gp100 expression, and gp100-specific vaccination may marginally increase this frequency (34, 35). This underscores the importance of tumor-associated Ag expression prescreening to the interpretation of clinical outcome during Ag-specific treatments. However, αβT cells are exquisitely sensitive to even a single peptide/MHC complex on the surface of an APC (36–38). In contrast, the affinity of most Abs reactive with tumor or MHC Ag is quite variable and can be weak. Thus, negative Ag expression by IHC-staining analysis may not necessarily indicate a loss of tumor recognition by T cells. Furthermore, screening for tumor Ag expression was not performed before either TIL or vaccine-induced PBMC cell therapy, yet resulted in drastically different clinical outcomes. It remains possible that isolation and expansion of TIL from excised melanoma lesions may serve to enrich for tumor Ag-specific T cells of appropriate specificity, avidity, homing capacity, and function for mediating successful adoptive immunotherapy compared with vaccine-induced T cells derived from the peripheral blood.
By virtue of their generation and expansion, vaccine-induced g209-2M peptide-specific T cells used for therapy possess highly restricted Ag specificity. By contrast, infused bulk TIL populations are often comprised of multiple tumor-reactive T cells with heterogenous Ag specificity (11, 12). Nevertheless, patients receiving bulk TIL comprised almost entirely by a single clone or multiple clones of a single specificity have experienced objective clinical responses, indicating that transferred T cell-mediated tumor regression may occur even in the absence of diverse Ag specificity (11, 12).
Whether the g209 epitope is a good immunotherapeutic target for patients with melanoma requires further clinical investigation. Analogous to the present study, lymphodepletion followed by autologous ACT of in vitro-stimulated MART-1:26–35(27L) peptide-specific PBMC from five nonvaccinated melanoma patients did not induce tumor regressions in vivo despite persistence of the transferred MART-1:26–35(27L)-specific T cell population after infusion, suggesting that the lack of clinical efficacy of the current approach may not be limited entirely by the selection of gp100 as the target Ag (D. J. Powell, Jr., and S. A. Rosenberg, unpublished data). In TIL therapy, gp100-reactive T cells can be isolated from excised melanoma deposits and identified in TIL cultures that mediated tumor regression; however, to date, no objective cancer regressions have been observed in patients receiving ACT of TIL that were solely gp100-reactive (2, 3, 11, 12). Evaluating the therapeutic activity of g209-specific TIL in adoptive immunotherapy for melanoma is further complicated by the modest numbers of TIL treatment cultures that contain g209-reactive cells and the observation that g209-reactive TIL cultures used for transfer often exhibit multiple Ag specificities (11, 12).
In our TIL transfer trial, tumor regression correlated with the in vivo persistence of transferred TIL clonotypes in the peripheral blood (11–13). In earlier ACT studies, no patient experienced tumor regression after the transfer of highly expanded, tumor Ag-reactive T cell clones that failed to persist, whether or not the patient had received lymphodepleting preconditioning (39, 40). Together, these data suggested that the inability of tumor Ag-specific T cells to engraft after transfer may limit the efficacy adoptive immunotherapy. In the current study, g209-specific CD8+T cells persisted well in the blood of all treated patients after infusion as assayed by tetramer staining, TRBV gene, or Ag-stimulated cytokine secretion analyses. The therapeutic inadequacy of the current approach thus lies beyond the inability of tumor Ag-specific T cells to persist and highlights the point that transferred T cell persistence or the mere presence of tumor Ag-reactive T cells in the blood cannot be used as a “surrogate marker” for successful immunotherapy.
Transfer of g209-specific T cells to lymphodepleted patients, combined with Ag specific vaccination and IL-2, resulted in robust proliferation and the induction of primary lymphocytosis in 55.5% (5 of 9) of patients, followed by secondary lymphocytosis in 3 of 6 patients receiving a second cycle of viral vaccine and IL-2. Based on the same criterion, the frequency of patients experiencing lymphocytosis after cell infusion was notably different from that observed in TIL recipients (2 of 35; 5.7%). This was not the result of transferred cell number because the mean number of cells administered in this (6.6 ± 1.2 × 1010) and our TIL (6.3 ± 0.7 × 1010) trial (12) was not significantly different (p = 0.84). Whereas CD4+T cell counts are often depressed in TIL recipients 1 mo after administration of therapy, CD4+T cells were normal, on average, in the peripheral blood of PBMC recipients (12). This was not the result of a disparity between the transferred populations because CD4+T cells were present in vaccine-induced PBMC used for therapy at frequencies not different from those found in TIL (31.2 ± 6.5 and 21.7 ± 4.3%, respectively; p = 0.303) (12). At 1 mo after infusion, normal CD4+T cell counts were observed in four of the five patients that experienced lymphocytosis, although this trend was not statistically significant (p = 0.206). Vaccination alone might not account for the difference in the incidence of lymphocytosis experienced by PBMC and TIL recipients, because 18 of 35 patients treated with TIL also received peptide vaccine in combination with cell transfer, suggesting that peptide-stimulated PBMC possess a superior proliferative potential compared with TIL. Whether this disparity in proliferative potential reflects a difference in culture condition is not known; however, peptide-stimulated PBMC are cultured for less time (mean = 12.3 ± 0.6 vs 47.0 ± 2.3 days), with less IL-2 (50 vs 1000 cU/ml) and a different stimulus (peptide on autologous PBMC vs anti-CD3 Ab and irradiated allogenic feeder cells) (12).
Surprisingly, longitudinal gp100-specific tetramer frequency analysis and BrdU incorporation studies demonstrated that circulating g209-specific CD8+T cells did not proliferate significantly more than the remaining CD8 T cell population despite administration of an Ag-specific cancer vaccination. The pan lymphoproliferation observed in the peripheral blood may result from IL-2 administration, homeostatic proliferation, and/or the induction of fowlpox virus-specific T cell responses. Selective proliferation or retention of tumor Ag-specific T cells in lymphoid organs, where T cell proliferation is known to largely occur, was not evaluated due to the difficulty in obtaining samples from these sites.
Mouse studies have suggested that the acquisition of a terminal effector phenotype by tumor-reactive T cells in vitro through repeated stimulation can impair in vivo antitumor efficacy (15, 41). In addition, subfractionation of early effector T cells by expression of CD62L identified CD8+T cells with superior in vivo antitumor properties (15, 41). In our study, most peptide-stimulated PBMC had only received a single in vitro stimulation with g209-2M peptide, and up to half of g209-2M peptide-stimulated CD8+T cells expressed CD62L in some patients, albeit at low levels and in a unimodal fashion. Although CD62L expression can facilitate T cell homing to lymphoid tissues, consequent activation by professional APC and a resultant increase in proliferation, transfer of peptide-stimulated T cells with a less differentiated phenotype, and increased proliferative potential did not elicit significant tumor responses in vivo. Whether CD62L expression by PBMC resulted in preferential homing and/or retention in lymphoid organs, compared with tumor, was not assessed. By contrast to vaccine-induced PBMC, TIL transferred to patients that experienced objective responses express little to no CD62L (17).
The critical factors contributing to the differential therapeutic efficacy of TIL and vaccine-induced PBMC are not known. A positive stimulation feedback loop might account for the enhanced antitumor effectiveness of transferred TIL in contrast to PBMC in vivo because TIL are 1) primed and imprinted by naturally processed Ag in the tumor bed or the local draining lymph node leading to their preferential retention in these sites, 2) ex vivo expanded and isolated from excised tumor fragments in the continued presence of autologous tumor cells, stimulating TIL activation and division, and 3) can expand, persist, and mediate immune responses against the same stimulating tumor in vivo after transfer. By contrast, the basis for the reduced therapeutic efficacy of g209-2M peptide-induced CD8+T cells might reflect the generation of a corrupted CD8+T memory cell population induced by frequently boosting with high doses of immunogen presented by somatic cells rather than tumor and in the absence of adequate T cell help (8, 42, 43).
g209-2M peptide-stimulated T cells may also possess a suboptimal functional avidity compared with TIL. Transferred gp100-specific CD8+T cells from all patients recognized g209 peptide-pulsed cells down to 1 nM concentrations, the minimum level reported to correlate with tumor recognition (44). Only PBMC from patient 2 recognized g209 peptide at a lower concentration. Transferred PBMC from one patient did not specifically recognize HLA-matched tumor cells, and others recognized tumor at low but significant levels. Comparatively, the highest absolute IFN-γ release in response to tumor for subcultures of peptide-stimulated PBMC (2172 ± 963 pg/ml; mean ± SEM) was significantly lower than reported for TIL (8318 ± 1579 pg/ml; p2 < 0.005) (12), although differences in Ag-specificity and tumor-reactive T cell frequency make such direct comparisons difficult to interpret. Notably, no TIL from responding patients were exclusively gp100-reactive (12). Peptide-stimulated cells used for transfer did, however, lyse HLA-matched melanoma cell lines in vitro, similar to TIL. Furthermore, posttransfer PBMC maintained peptide and tumor reactivity, as reported for posttransfer PBMC from TIL recipients (11).
Study of the TCR repertoire from vaccine-induced vs naturally arising NY-ESO-1-specific CD8+T cells revealed highly conserved but distinct structural TCR features, which result in decreased functional avidity and tumor reactivity by cells induced by synthetic peptides (45). Synthetic peptides might not adequately mimic their naturally processed counterparts; therefore, gp100-vaccine-derived cells may possess TCRs with poor affinity for naturally processed and presented tumor Ag in vivo. Alternatively, tolerating mechanisms such as clonal deletion, ignorance, anergy, or suppression in the host likely eliminate or reduce the highest-affinity T cells reactive against self/tumor Ags, such as gp100 and MART-1, thus dampening the ability of cancer vaccines to induce and expand such cells in vivo. Novel strategies, such as TCR transduction technology, can be used to circumvent host tolerance and distinctively generate peripheral blood T cells with high-affinity TCRs for ACT (46). High-affinity, tumor-specific TCRs from TIL that have mediated tumor regression (47), T cells from vaccinated HLA-A2 transgenic mice (48) or allorestricted CTLs (49), or generated through TCR mutagenesis (50) can be isolated and used for transduction of peripheral blood T cells, thereby increasing the therapeutic potential of transferred PBMC (51).
Other factors, both T cell and tumor-related, may have contributed to reduced antitumor responses in vivo. CD4+T cells can influence CD8+T cell effector function, memory and maintenance; however, distinct CD4+T cell subsets may have diverse influences on adoptive immunotherapy. In mice, cotransfer of CD25+ CD4+ Th cells with tumor/self-reactive CD8+ T cells and vaccination into CD4+ T cell-deficient recipients resulted in autoimmunity and regression of established tumor (28). By contrast, transfer of CD25+ Treg cells alone or combined with CD25+ CD4+ Th cells inhibited effective immunotherapy. CD4+ CD25+ FOXP3+ T cells were detectable in PBMC for treatment. In study of four TIL infusion products, CD25+ FOXP3+ CD4+ T cells were present at similar if not higher frequencies than PBMC, indicating that Treg cell-mediated immune suppression might not account for the difference in the therapeutic efficacy of these two populations. The frequency, number, and function of Treg cells in transferred TIL cell products is currently investigation. Nevertheless, selective depletion of Treg cells from cell infusion samples before transfer may augment the effectiveness of clinical approaches, particularly those that incorporate a lymphodepleting preconditioning (23).
Whether transferred g209-reactive T cells migrated to tumor was not directly assessed, although g209-reactive T cells were detected in tumors excised after PBMC transfer from 3 (patient 1, 4, and 5) of 4 patients. TIL isolated from an excised tumor from patient 3, whose tumor had preexistent gp100 Ag loss, did not recognize g209 peptide or HLA-matched allogeneic melanoma cell lines, but exhibited unique recognition of autologous tumor.
Our study demonstrates that adoptive transfer of vaccine-induced PBMC after lymphodepletion can result in the persistence in vivo of high numbers of g209 peptide-specific, tumor-reactive CD8+ T cells in patients with metastatic melanoma. The lack of objective clinical responses in these vaccine-induced PBMC recipients may be multifactoral. Based upon these clinical findings, we hypothesize that the limited effectiveness of cancer vaccines in immunocompetent individuals may be due to the generation of inadequate cells because vaccine-induced cells did not mediate tumor regression even in the face of host lymphodepletion, exogenous cytokine, and Ag activation in vivo.
Acknowledgments
We thank Paul F. Robbins, Yong Li, Laura Johnson, Thomas Shelton, and Michelle Langhan for technical assistance, as well as Arnold Mixon and Shawn Farid from the Surgery Branch FACS laboratory facility.
Footnotes
This work was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Abbreviations used in this paper: TIL, tumor-infiltrating lymphocyte; ACT, adoptive cell transfer; CM, complete medium; TRBV, TCRβ-chain V region; Treg, regulatory T; ALC, absolute lymphocyte count; IHC, immunohistochemical.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 2.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–3968. [PubMed] [Google Scholar]
- 4.Skipper JC, Gulden PH, Hendrickson RC, Harthun N, Caldwell JA, Shabanowitz J, Engelhard VH, Hunt DF, Slingluff CL., Jr Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int J Cancer. 1999;82:669–677. doi: 10.1002/(sici)1097-0215(19990827)82:5<669::aid-ijc9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salgaller ML, Marincola FM, Cormier JN, Rosenberg SA. Immunization against epitopes in the human melanoma antigen gp100 following patient immunization with synthetic peptides. Cancer Res. 1996;56:4749–4757. [PubMed] [Google Scholar]
- 8.Powell DJ, Jr, Rosenberg SA. Phenotypic and functional maturation of tumor antigen-reactive CD8+ T lymphocytes in patients undergoing multiple course peptide vaccination. J Immunother. 2004;27:36–47. doi: 10.1097/00002371-200401000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins PF, Dudley ME, Wunderlich J, El Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Khong HT, Dudley ME, El Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28:258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Topalian SL, Sherry RM, Restifo NP, Wunderlich JR, Seipp CA, Rogers-Freezer L, et al. Recombinant fowlpox viruses encoding the anchor-modified gp100 melanoma antigen can generate antitumor immune responses in patients with meta-static melanoma. Clin Cancer Res. 2003;9:2973–2980. [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins S, Gritz L, Fedor CH, O’Neill EM, Cohen LK, Panicali DL. Formation of lentivirus particles by mammalian cells infected with recombinant fowlpox virus. AIDS Res Hum Retroviruses. 1991;7:991–998. doi: 10.1089/aid.1991.7.991. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, El Gamil M, Dudley ME, Li YF, Rosenberg SA, Robbins PF. T cells associated with tumor regression recognize frame-shifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol. 2004;172:6057–6064. doi: 10.4049/jimmunol.172.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powell DJ, Jr, Parker LL, Rosenberg SA. Large-scale depletion of CD25+ regulatory T cells from patient leukapheresis samples. J Immunother. 2005;28:403–411. doi: 10.1097/01.cji.0000170363.22585.5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoof E, Girstl M, Frobenius W, Kirschbaum M, Dorr HG, Rascher W, Dotsch J. Decreased gene expression of 11β-hydroxysteroid dehydrogenase type 2 and 15-hydroxyprostaglandin dehydrogenase in human placenta of patients with preeclampsia. J Clin Endocrinol Metab. 2001;86:1313–1317. doi: 10.1210/jcem.86.3.7311. [DOI] [PubMed] [Google Scholar]
- 25.Purbhoo MA, Boulter JM, Price DA, Vuidepot AL, Hourigan CS, Dunbar PR, Olson K, Dawson SJ, Phillips RE, Jakobsen BK, et al. The human CD8 coreceptor effects cytotoxic T cell activation and antigen sensitivity primarily by mediating complete phosphorylation of the T cell receptor ζ chain. J Biol Chem. 2001;276:32786–32792. doi: 10.1074/jbc.M102498200. [DOI] [PubMed] [Google Scholar]
- 26.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 27.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 28.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002;99:1517–1526. doi: 10.1182/blood.v99.5.1517. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Lotze MT, Yang JC, Aebersold PM, Linehan WM, Seipp CA, White DE. Experience with the use of high-dose in-terleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210:474–484. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, et al. Auto-immunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043–6053. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cormier JN, Hijazi YM, Abati A, Fetsch P, Bettinotti M, Steinberg SM, Rosenberg SA, Marincola FM. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int J Cancer. 1998;75:517–524. doi: 10.1002/(sici)1097-0215(19980209)75:4<517::aid-ijc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 35.Riker A, Cormier J, Panelli M, Kammula U, Wang E, Abati A, Fetsch P, Lee KH, Steinberg S, Rosenberg S, et al. Immune selection after antigen-specific immunotherapy of melanoma. Surgery. 1999;126:112–120. [PubMed] [Google Scholar]
- 36.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- 37.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 38.Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996;4:565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]
- 39.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, Schwartzentruber DJ, Hwu P, Marincola FM, Sherry R, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 40.Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang LX, Huang WX, Graor H, Cohen PA, Kim JA, Shu S, Plautz GE. Adoptive immunotherapy of cancer with polyclonal, 108-fold hyperexpanded, CD4+ and CD8+ T cells. J Transl Med. 2004;2:41. doi: 10.1186/1479-5876-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 43.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dudley ME, Ngo LT, Westwood J, Wunderlich JR, Rosenberg SA. T-cell clones from melanoma patients immunized against an anchor-modified gp100 peptide display discordant effector phenotypes. Cancer J. 2000;6:69–77. [PubMed] [Google Scholar]
- 45.Le Gal FA, Ayyoub M, Dutoit V, Widmer V, Jager E, Cerottini JC, Dietrich PY, Valmori D. Distinct structural TCR repertoires in naturally occurring versus vaccine-induced CD8+ T-cell responses to the tumor-specific antigen NY-ESO-1. J Immunother. 2005;28:252–257. doi: 10.1097/01.cji.0000161398.34701.26. [DOI] [PubMed] [Google Scholar]
- 46.Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci USA. 1995;92:11993–11997. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Savage P, Gao L, Vento K, Cowburn P, Man S, Steven N, Ogg G, McMichael A, Epenetos A, Goulmy E, et al. Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood. 2004;103:4613–4615. doi: 10.1182/blood-2003-11-3903. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 51.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. doi: 10.1126/science.1129003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]