

Abstract
cDNAs encoding TCR α- and β-chains specific for HLA-A2-restricted cancer-testis Ag NY-ESO-1 were cloned using a 5′RACE method from RNA isolated from a CTL generated by in vitro stimulation of PBMC with modified NY-ESO-1-specific peptide (p157–165, 9V). Functionality of the cloned TCR was confirmed by RNA electroporation of primary PBL. cDNA for these α- and β-chains were used to construct a murine stem cell virus-based retroviral vector, and high titer packaging cell lines were generated. Gene transfer efficiency in primary T lymphocytes of up to 60% was obtained without selection using a method of precoating retroviral vectors onto culture plates. Both CD4+ and CD8+ T cells could be transduced at the same efficiency. High avidity Ag recognition was demonstrated by coculture of transduced lymphocytes with target cells pulsed with low levels of peptide (<20 pM). TCR-transduced CD4 T cells, when cocultured with NY-ESO-1 peptide pulsed T2 cells, could produce IFN-γ, GM-CSF, IL-4, and IL-10, suggesting CD8-independent, HLA-A2-restricted TCR activation. The transduced lymphocytes could efficiently recognize and kill HLA-A2- and NY-ESO-1-positive melanoma cell lines in a 4-h 51Cr release assay. Finally, transduced T cells could efficiently recognize NY-ESO-1-positive nonmelanoma tumor cell lines. These results strongly support the idea that redirection of normal T cell specificity by TCR gene transfer can have potential applications in tumor adoptive immunotherapy.
Tumor immunology has been greatly aided by the analysis of spontaneous immune responses against defined tumor Ags in cancer patients. One of the most frequent proteins that elicit spontaneous immune responses is NY-ESO-1 (1). NY-ESO-1 is a member of the cancer/testis (CT)2 class of Ags (2, 3). CT Ags are defined as proteins that have a restricted expression in adult tissue to testis and transformed cells. NY-ESO-1 was identified by SEREX analysis from a patient with esophageal cancer (1). It is a member of a multigene family located on the X chromosome (Xq28) that includes at least two additional members, LAGE-1 and ESO-3 (4, 5). NY-ESO-1 is an attractive tumor Ag for adoptive immunotherapy because it is expressed in a high percentage (20 – 80% at the RNA level) of common tumors, including cancers of the breast, lung, bladder, liver, prostate, and ovary, and a subset of these cancer patients develop Abs to NY-ESO-1 (3). NY-ESO-1 expression and patient immune responses (mainly measured by Ab titer) can be correlated with the stage of malignancy (6 – 8).
Immunization with NY-ESO-1 can induce both Ab and CD8+ CTL responses, although little effect on cancer progression was observed in these studies (9). Adoptive immunotherapy, the transfer of lymphocytes with highly avid antitumor activity, can mediate the regression of large established tumors, but the generation of reactive lymphocytes is difficult, expensive, and labor intensive (10 –15). Tumor-infiltrating lymphocytes have been used in cell transfer therapies and have been shown to recognize a variety of melanoma tumor-associated Ags (TAA). The most commonly recognized TAA in melanoma is the MART-1, a melanocyte differentiation Ag, which is expressed in ~90% of melanomas, whereas NY-ESO-1 is expressed in ~34% of melanomas (1). In a recent trial of adoptive immunotherapy using anti-TAA-reactive T cells after nonmyeloablative lympho-depleting chemotherapy, 46% of patients with metastatic melanoma exhibited an objective regression (16). In two of these patients, repopulation of lymphocytes was associated with a distinct oligoclonal T cell expansion (as detected by TCR Vβ analysis) of MART-1-reactive cells and near total regression of their tumor burdens. These results suggest that T cells targeted to tumor Ags can mediate the regression of large solid tumors.
We recently demonstrated, using retroviral vector-mediated gene transfer, transfer of TCR genes encoding a highly avid anti-gp100 TCR into bulk populations of tumor-infiltrating lymphocytes and PBL-mediated, HLA-A2-restricted effector functions, including cytokine release and cell lysis (17). These results may have significant potential impact on the ability to treat cancer patients using adoptive immunotherapy if TCR genes to more widely expressed TAA can be identified. In this report, TCR specific to the NY-ESO-1 CT Ag were isolated and used to construct retroviral vectors, which were shown to transfer anti-NY-ESO-1 effector functions to normal primary human T cells. TCR gene vectors, directed against common tumor Ags, have the potential to be used to treat large numbers of cancer patients with their own transduced T cells without the need to identify antitumor T cells uniquely from each patient.
Materials and Methods
Patient PBMCs and cell lines
All PBMCs used in this study were obtained from metastatic melanoma patients treated at the Surgery Branch, National Cancer Institute (NCI), National Institutes of Health. The cell lines used included PG13 gibbon ape leukemia virus-packaging cell line (American Type Culture Collection (ATCC) CRL 10,686), the human lymphoid cell lines SupT1 (ATTC CRL-1942) and T2 (18), and the ecotropic packaging cell line, Phoenix Eco (provided by G. Nolan, Stanford University, Stanford, CA). SupT1 is a human T cell leukemia cell line with chromosomal translocations involving both the α and β TCR genes that prevent surface expression of the endogenous TCR complex. T2 is a lymphoblastoid cell line deficient in TAP function, whose HLA class I proteins can be easily loaded with exogenous peptides (18). Melanoma lines, 1300mel (NY-ESO-1+,HLA-A2+), 1363mel(NY-ESO-1+,HLA-A2+), 1390mel(NY-ESO-1+,HLA-A2+), 624mel(NY-ESO-1+,HLA-A2+), 624.38mel(NY-ESO-1+,HLA-A2+), 1359mel(NY-ESO-1+,HLA-A2−), 526mel(NY-ESO-1−,HLA-A2+), and 586mel(NY-ESO-1+,HLA-A2−), were generated at the Surgery Branch from resected tumor lesions, as previously described (19). Nonmelanoma tumor cell lines NCI-H345(NY-ESO-1+,HLA-A2+), NCI-H526(NY-ESO-1+,HLA-A2+), LN-18(NY-ESO-1+,HLA-A2+), Saos-2(NY-ESO-1+,HLA-A2+), MDA453s (NY-ESO-1+,HLA-A2−), and LNZTA3WT4 (NY-ESO-1−,HLA-A2+) were provided by ATCC, and TC-71(NY-ESO-1+,HLA-A2+), SKN-AS(NY-ESO-1+,HLA-A2−) were provided by S. L. Topalian (NCI) (20). All cell lines described above were maintained in R10 (RPMI 1640; Invitrogen Life Technologies) supplemented with 10% FCS (Biofluid). Culture medium (CM) for T lymphocytes was RPMI 1640 supplemented with 0.05 mM ME, 300 IU/ml IL-2 (Chiron) plus 10% human AB serum (Valley Biomedical). The HLA-A2-restricted T cell TE8-1 clone 8F (TE8-1-8F) was derived from a bulk line generated by in vitro stimulation using NY-ESO-1 157–165, 9V (p157–165V) peptide variant (21). This T cell clone recognized both p157–165 and p157–167 NY-ESO-1 peptides (22).
Peptide synthesis
The synthetic peptides used in this study were made using a solid phase method on a peptide synthesizer (Gilson) at the Surgery Branch of NCI. The quality of each peptide was evaluated by mass spectrometry (Biosynthesis). The sequences of the peptide used in this study are as follows: NY-ESO-1 p157–165, SLLMWTTQC; NY-ESO-1 p157–165V, SLLMWTTQV; NY-ESO-1 p157–168, SLLMWTTQCFLP; NY-ESO-1 p161–180, WITQCFLPVFLAQPPSGQRA (an HLA-DP4-restricted epitope); and gp100 209 –217, Mart-1 27–35 (23).
Cloning of NY-ESO-1-specific, HLA-A2-restricted TCR α and β cDNA
Total RNA was extracted with TRIzol Total RNA Isolation Reagent (Invitrogen Life Technologies) from CTL clone TE8-1-8F. One microgram of total RNA was used to clone the TCR cDNAs by a RACE method (GeneRacer Kit; Invitrogen Life Technologies). Before synthesizing the single-strand cDNA, the RNA was dephosphorylated, decapped, and ligated with an RNA oligonucleotide according to the instruction manual of the 5′RACE GeneRacer Kit. SuperScript II RT and GeneRacer oligo(dT) were used for reverse transcribing the RNA oligo-ligated mRNA to single-strand cDNAs. 5′RACE was performed using the 5′GeneRacer primer and the 3′primer of gene-specific primer TCRCAR (5′-GTTAACTAGTTCAGCT GGACCACAGCCGCAGC-3′), TCRCB1R (5′-CGGGTTAACTAGTTCA GAAATCCTTTCTCTTGACCATGGC-3′), or TCRCBR2 (5′-CTAGC CTCTGGAATCCTTTCTCTTG-3′)) as 3′ primers for TCR α-, β1-, and β2-chains, respectively. The PCR products were cloned into pCR2.1 TOPO vector (Invitrogen Life Technologies) and then transformed into One Shot TOP10 Competent Escherichia coli (Invitrogen Life Technologies). Plasmid DNAs were prepared from 32 individual clones, 16 clones from TCR α-chain cDNA and 16 clones from TCR β-chain cDNA. Full-length inserts of all 32 plasmids were sequenced to determine the vα/vβ usage in the CTL clone TE8-1-8F.
Construction of retroviral vectors
The retroviral vector backbone used in this study, pMSGV1, is a derivative of the vector pMSGV (murine stem cell virus (MSCV)-based splice-gag vector), which uses an MSCV long terminal repeat (LTR) (24) and contains the extended gag region and env splice site from vector SFGtcLuc+ITE4− (25). Vector pMSGV was generated from pMINV (26) by substituting a 756-bp SpeI/XhoI fragment with a 798-bp SpeI/XhoI fragment from SFGtcLuc+ITE4− and by replacing a 1955-bp XhoI/BamHI fragment containing a phosphoglycerate kinase (PGK)-internal ribosomal entry site (IRES)-NEO cassette with a 47-bp XhoI/BamHI polylinker containing unique XhoI, EcoRI, SalI, SacII, and BamHI sites. Vector pMSGV1 was derived from pMSGV by replacing a 43-bp PmlI/XhoI fragment of pMSGV with a 76-bp PmlI/XhoI fragment from the vector GCsap (27). The latter modification incorporates a naturally occurring Kozak sequence to enhance translational efficiency.
Two different retroviral vectors were constructed. Each expresses both chains of the TCR. TCR α-chain (plasmid clone 8FA2) and β-chain (clone 8FB6) cDNAs were amplified by PCR using the following pairs of oligonucleotide primers, for 8FA2: forward primer, 5′-CT AAGCTTGC C ATG GAA ACT CTC CTG GGA GTG TC-3; and reverse primer, 5′-CT GCG-GCCGC TCTAGA TCA GCT GGA CCA CAG CCG CAG-3′; and for 8FB6: forward primer, 5′-CT AAGCTT GCCGCC ATG GAC TCCT GGA CCC TCTG-3′; and reverse primer, 5′-CT GCGGCCGC CTCGAG TCA GAA ATC CTT TCT CTT GAC CAT G-3′, to introduce appropriate restriction enzyme sites for subcloning. Vector MSGE1AIB (AIB) or MSGE1APB (APB) was assembled by ligation of four DNA fragments: pMSGV1 (NcoI/XhoI), TCR α cDNA (NcoI/XbaI), IRES (28), or PGK promoter (XbaI/HindIII), and TCR β cDNA (HindIII/XhoI). The inserts were determined by PCR and restriction enzyme digestion. PCR-amplified TCR cDNAs and the orientation of the inserts were confirmed by DNA sequencing. pMSGIN (GIN), is derived from the pMSGV vector and contains the GFP-IRES-Neo genes.
Generation of PG13-packaging cell clones was initiated by starting a coculture of PG13 and Phoenix Eco cells. This coculture was then transfected with 50 μg of DNA for each construct, AIB and APB, using the GenePorter reagent (Gene Therapy Systems). The transgene expression was tested by intracellular staining with anti-Vβ8 Ab. Fourteen days post-transfection, constructs AIB and APB yielded 41 and 45% of cells expressing Vβ8, respectively, whereas control vector MSGIN yielded 55% GFP expression. Phoenix Eco cells were removed from PG13 cells using magnetic bead negative selection (with anti-LYT-2; Dynal Biotech) and PG13 cell clones obtained by limiting dilution. Clones were expanded, and high titer clones were selected by the dot-blot titration method (29). Clones were determined to be producing biologically active retrovirus vector by transduction of SupT1 cells and analysis of FACS data (using anti-Vβ8 and anti-CD3). Southern blot analysis of packaging cell clones was used to confirm proper vector integration and copy number.
Transduction of PBL
PBL were collected by leukopheresis, and lymphocytes were separated by centrifugation on a Ficoll/Hypaque cushion, washed in HBSS, then resuspended at a concentration of 1 × 106/ml in CM supplemented with 50 ng/ml OKT3 and 300 IU/ml IL-2. The lymphocytes were then plated at 1 × 106 cells/ml in 24-well plates (Costar). The lymphocytes were cultured in vitro for 48 h before transduction. After stimulation, lymphocytes were transduced with retroviral vectors by transfer to culture dishes that had been precoated with retroviral vectors as previously described (30). To coat culture plates with vector, non-tissue culture-treated, six-well plates (BD Biosciences) were first treated with 25 μg/ml recombinant fibronectin fragment (RetroNectin; Takara). To these plates was then added retroviral vector supernatant, and the plates were incubated at 32°C for 2 h, followed by overnight incubation at 4°C. The following day, plates were allowed to warm to room temperature, the supernatant was removed, and stimulated PBL was added to each well at 1 × 106 cells/ml (4 – 6 ml/well). Cells were then incubated overnight at 32°C, and the procedure was repeated the following day (total of two transductions), after which the cells were expanded at 37°C in a 5% CO2 incubator and split as necessary to maintain cell density between 0.5 and 4 × 106 cells/ml.
FACS analysis
Cell surface expression of Vβ8, CD3, CD4, and CD8 molecules on PBL was measured by immunofluorescence using FITC-, PE-, or PerCP-conjugated Abs, as directed by the supplier of anti-TCR Vβ8 (Immunotech) and all others (BD Biosciences). For analysis, the relative log fluorescence of live cells was measured using a FACScan flow cytometer (BD Biosciences). Intracellular cytokine staining was performed using BD FastImmune intracellular cytokine detection kits as directed by the manufacturer (BD Biosciences). An IFN-γ secretion assay detection kit (Miltenyi Biotec) was used for the cytokine secretion assay. Responder cells (5 × 105) and peptide-pulsed T2 cells (5 × 105) were cocultured in a 1-ml volume in 24-well tissue culture plates for 1 h, and the secretion of IFN-γ was detected as directed by the manufacturer. In brief, after coculture, the cells were incubated with IFN-γ catch reagent at 10 μl/1 × 106 cells on ice for 10 min, then cultured at 37°C in an excess amount of medium for 45 min to capture the secreted IFN-γ on the cell surface. The captured IFN-γ was then detected by addition of PE-anti-IFN-γ Ab to the cells, and the cells were subjected to flow cytometric analysis.
Cytokine release assays
PBL cultures were tested for reactivity in cytokine release assays using commercially available ELISA kits (IFN-γ, GM-CSF, TNF-α, IL-4, and IL-10; Endogen). T2 cells were pulsed with peptide (1 μg/ml, or as described in figures) in CM for 3 h at 37°C, followed by washing (three times) before initiation of cocultures. For these assays, 1 × 105 responder cells (PBL) and 1 × 105 stimulator cells (T2) were incubated in a 0.2-ml culture volume in individual wells of 96-well plates. In experiments in which melanoma cells served as stimulator cells, 5 × 104 tumor cells were used in the same volume. Stimulator and responder cells were cocultured for 24 h for all cytokines, except TNF-α, which was measured after 6 h of incubation. Cytokine secretion was measured in culture supernatants diluted so as to be in the linear range of the assay.
51Cr release assay
The ability of the transduced PBL to lyse HLA-A2+ melanoma targets was measured using a 51Cr release assay as described in detail previously (19). Briefly, 1 × 106 melanoma cells were labeled for 1 h at 37°C with 200 μCi of 51Cr (Amersham Biosciences). Labeled target cells (5 × 103) were incubated with effector cells at the ratios indicated in the text for 4 h at 37°C in 0.2 ml of complete medium. Harvested supernatants were counted using a Wallac 1470 Wizard automatic gamma counter (Wallac). Total and spontaneous 51Cr release were determined by incubating 5 × 103 labeled targets in either 2% SDS or medium for 4 h at 37°C. Each data point was determined as an average of quadruplicate wells. The percent specific lysis was calculated as follows: % specific lysis = ((specific 51Cr release − spontaneous 51Cr release)/(total 51Cr release − spontaneous 51Cr release)) × 100.
RT-PCR detection of NY-ESO-1-expressing cells
Total RNA isolated from 42 nonmelanoma tumor lines was subjected to RT-PCR to determine NY-ESO-1 expression using the Titan One Tube RT-PCR System (Roche). NY-ESO-1-specific primers (forward primer, 5′-ACCTCTAGAAGGCTCCGGAGCCATGCAG -3′; reverse primer, 5′-AGGGAAAGCTGCTGGAGACAG-3′) and GAPDH-specific primers (forward primer, 5′-GTCAACGGATTTGGTCGTATT-3′; reverse primer, 5′-AGTCTTCTGGGTGGCAGTGAT-3) were used. RT-PCRs were conducted according to the instructions provided with the Titan One Tube RT-PCR System. Briefly, 0.1–1 μg of total RNA was used for each 50-μl reaction, and RT-PCR was subjected to GeneAmp PCR System 9700 (Applied Biosystems) for 30 min at 50°C, followed by 2 min at 94°C and 35 cycles at 94°C for 20 s, 60°C for 20 s, and 72°C for 1 min. PCR products were visualized on 1.0% agarose gels by ethidium bromide staining. The expected PCR product sizes for NY-ESO-1 and GAPDH are 487 and 540 bp, respectively.
Results
Cloning functional NY-ESO-1-specific and HLA-A2-restricted TCR αβ cDNA
To determine Vα and Vβ usage information for CTL clone TE8-1-8F, total RNA was isolated and subjected to the 5′RACE procedure. Thirty-two individual cDNA clones for both α- and β-chains were fully sequenced from two separate PCRs. Sequence data demonstrated that Vα3.1 (TRAV17) and Vβ.2 (TRBV12-4) were the TCR genes present in the CTL TE-8-1-8F.
Before generating retroviral vector constructs, the function of the TCR αβ pair was tested by RNA electroporation of stimulated primary PBL. Greater than 90% gene expression was achieved when in vitro transcribed GFP RNA was electroporated into the PBL (Fig. 1A). Approximately 60% transgene expression was achieved (based on the detection of Vβ8 expression) when both TCR α- and β-chain RNAs were electroporated into stimulated PBL (Fig. 1B). To test the function of the TCR RNA, transfected PBLs, T2 cells, or autologous HLA-A2+ dendritic cells were pulsed with peptide and cocultured with NY-ESO-1 TCR, Mart-1 TCR, or GFP RNA-electroporated PBLs. Both NY-ESO-1 and Mart-1 Ag-specific recognitions were detected by IFN-γ secretion (Fig. 2). These functional HLA-A2-restricted and NY-ESO-1-specific TCR α- and β-chain cDNAs were used for retroviral vector construction.
FIGURE 1.
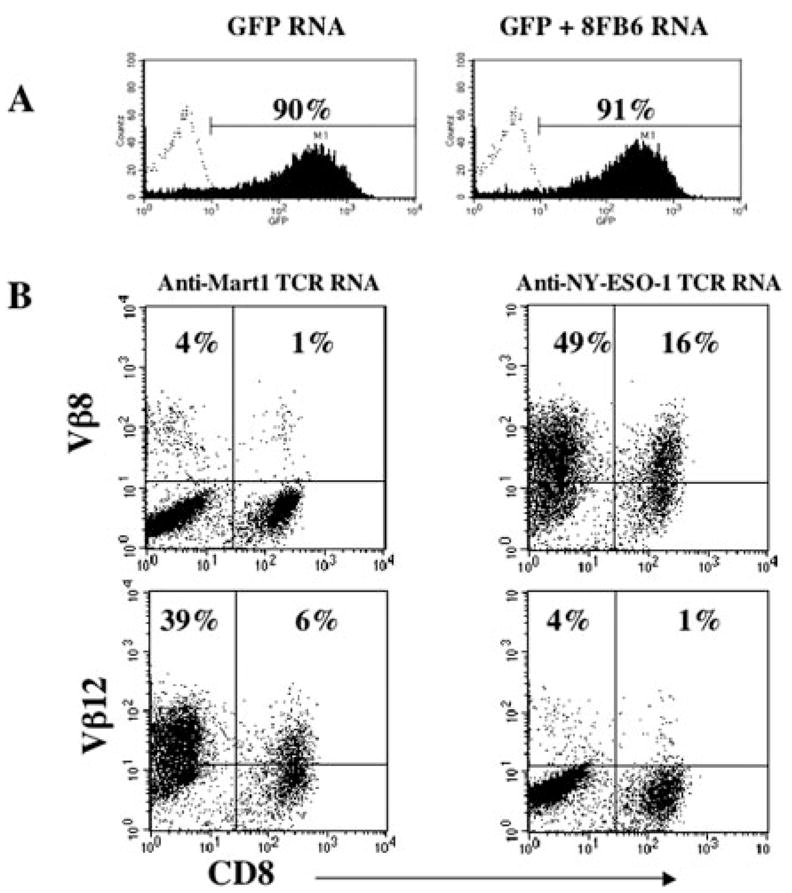
RNA electroporation of stimulated PBL. PBLs stimulated with OKT3 Ab plus IL-2 for 3 days were electroporated with in vitro transcribed RNA at 2 μg/1 × 106 cells. NY-ESO-1 and Mart-1 TCR α- and β-chain RNAs were generated by in vitro transcription of PCR-amplified templates bearing T7 promoter and poly(A) tail at the 5′ and 3′ ends, respectively (15). Twenty hours after electroporation, gene expression was determined by FACS analysis. GFP expression of RNA-electroporated PBL was compared with PBL electroporated with same amount of GFP RNA and additional β-chain RNA (8FB6; 2 μg/1 × 106; A). Mart-1 TCR and NY-ESO-1 TCR α- and β-chain RNA were electroporated into PBLs, Vβ12 (Mart-1) and Vβ8 (NY-ESO-1) expression were detected after electroporation.
FIGURE 2.
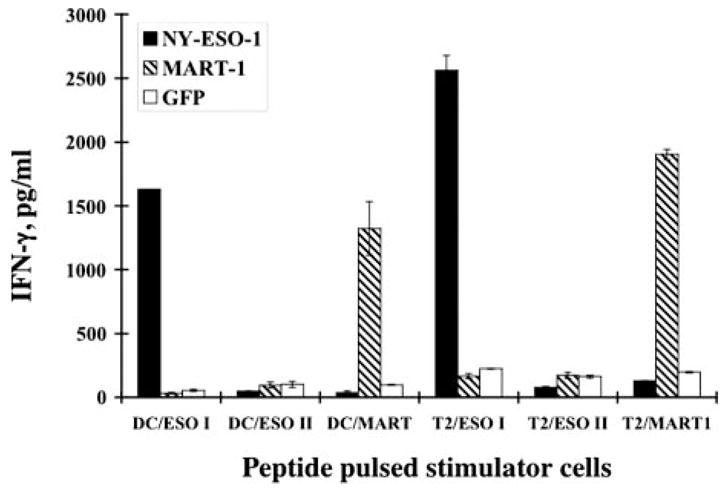
IFN-γ production of TCR RNA electroporated PBL. Three-day stimulated PBL were electroporated with NY-ESO-1 TCR RNA (NY-ESO-1), Mart1 TCR RNA (Mart-1), or GFP RNA (GFP) and cocultured with peptide-pulsed autologous dendritic cells (DC) or T2 cells. The peptides used were HLA-A2-restricted NY-ESO-1 (p157–165V; ESO-1) and Mart-1 (Mart) and HLA-DP4-restricted NY-ESO-1 (p161–180; ESO II).
Retroviral vector construction and transduction of target cell line
Two retroviral vectors, MSGE1AIB (AIB) and MSGE1APB (APB), harboring the α- and β-chain cDNAs were constructed. Both constructs used the retroviral MSCV LTR to drive the expression of α gene with the β gene governed by an IRES (AIB) or an internal PGK promoter (APB; Fig. 3). Four packaging cell clones from each construct with the highest physical titer were selected and used to transduced the human T cell line SupT1, and CD3 and Vβ8 expression were assayed by FACS analysis (Table I). CD3 and Vβ8 expression of the transduced SupT1 ranged from 37–79% and from 10–64%, respectively. The transduction efficiency was highly correlated with the titer determined by dot-blot hybridization of the packaging clones (Table I). Clones AIB38 and APB30 exhibited both high physical titer and high transduction efficiency, and these two constructs were chosen for additional study.
FIGURE 3.
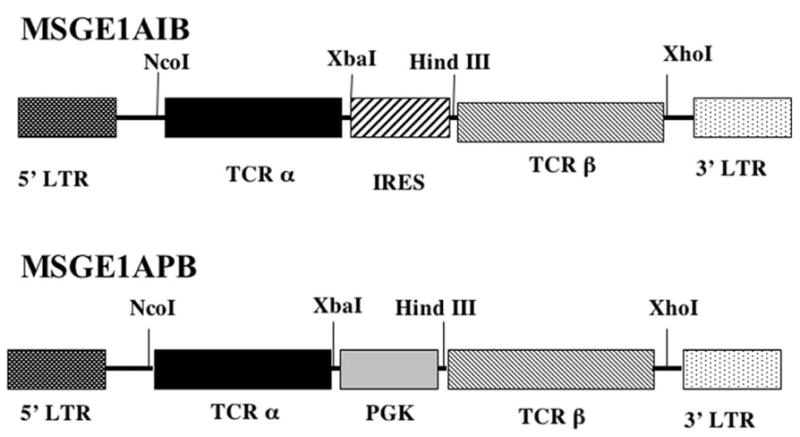
NY-ESO-1 TCR retroviral vectors. Diagrams of two retroviral vectors used to transfer and express the TCR genes from CTL clone TE8-1-8F. In vector MSGE1AIB (AIB), the expression of both α- and β-chains is mediated by the LTR, with translation coupled by the use of an IRES element. In vector MSGE1APB (APB), the expression of the α-chain is mediated by the vector LTR gene promoter, whereas β-chain expression is driven by an internal PGK gene promoter. Restriction enzymes used for cloning of the vector are indicated.
Table I.
Analysis of packaging cell clone supernatanta
| Transduced SupT1
|
|||
|---|---|---|---|
| Clone No. | Dot Blot (psl) | CD3 (%) | Vβ8 (%) |
| AIB27 | 664 | 37 | 16 |
| AIB38 | 1426 | 52 | 29 |
| AIB45 | 1046 | 37 | 22 |
| AIB68 | 798 | 40 | 23 |
| APB30 | 11926 | 79 | 64 |
| APB52 | 4632 | 59 | 10 |
| APB76 | 5889 | 73 | 47 |
| APB95 | 3213 | 70 | 29 |
Supernatant from each individual packaging cell clone was hybridized with NY-ESO-1 β-chain cDNA (8FA5). Eight packaging cell clones with the highest dot blot hybridization signal (physical titer) from the two constructs were tested by transduction of SupT1. Two days after the transduction, CD3 and Vβ8 expression on transduced SupT1 cells was determined by FACS. Dot blot results were expressed as relative phosphoimager-derived signal (psl) after subtracting the background from the negative control (supernatant from GIN packaging cells).
Functional tests of the TCR-transduced PBL
To engineer PBL with the anti-NY-ESO-1 TCR vector, we used a method of preloading vector onto culture plates, followed by the addition of stimulated T cells. Transduction was started on day 2 poststimulation, and 2 days after the last transduction, cells were harvested, stained for CD8 and Vβ8, and subjected to FACS analysis. Examples of transductions for APB and AIB are shown in Fig. 4 (background staining for untransduced PBL using anti-Vβ8 was <2%; data not shown).
FIGURE 4.
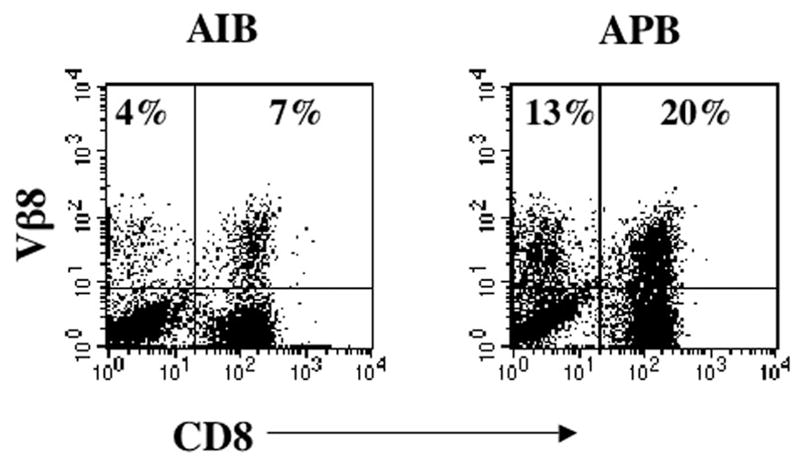
Transduction of PBLs with TCR retroviral vectors. FACS profile of PBL from a melanoma transduced with supernatant from TCR vector clone AIB38 (AIB) and clone APB30 (APB). Cells were double-stained with anti-Vβ8 and anti-CD8, and the percentage of positive cells is indicated.
To determine whether TCR-transduced PBL could mediate the release of effector cytokines, PBL were transduced with packaging cell clones AIB38 and APB30 and cocultured with T2 cells pulsed with NY-ESO-1- or gp100-specific peptide. Ag-specific IFN-γ release was detected in cocultures of peptide-pulsed T2 cells with both AIB38- and APB30-transduced PBL, with a higher level of cytokine secretion for APB30-transduced PBL (Table II). The low level IFN-γ release by AIB38-transduced PBLs was correlated with the lower transduction efficiency of AIB38 compared with APB30 (Fig. 4). Therefore, the packaging cell clone APB30 (subsequently referred to as APB) was used for additional study.
Table II.
Cytokine production of TCR transduced PBLsa
| Effector cell (IFN-γ, pg/ml)
|
||||||
|---|---|---|---|---|---|---|
| Stimulator | PBL1, AIB | PBL1, APB | PBL2, AIB | PBL2, APB | PBL1, GFP | PBL2, GFP |
| T2/p157–165v | 665 (12) | 19,309 (182) | 741 (7) | 10,842 (142) | 0 | 195 (1) |
| T2/gp100 peptide | 39 (3) | 264 (1) | 316 (11) | 186 (1) | 0 | 191 (4) |
PBL transduced with supernatant from packaging cell clones AIB38 or APB30 were cocultured with T2 cells pulsed with NY-ESO-1 (p157–165v) or gp100-specific peptide. Ag-specific IFN-γ secretion by retroviral vector-transduced PBLs from two donors was determined by ELISA. Data are the mean values (picograms per milliliter) of triplicate samples, with SD in parentheses.
Vector APB was used to transduce PBL from three different donors. The percentage of Vβ8+ cells ranged from 32– 43% (Fig. 5). To determine the avidity of the transduced PBL, APB-transduced PBLs were cocultured with T2 cells pulsed with different concentrations of peptide. Except that APB-transduced PBL2 had little detectable TNF-α release, all APB-transduced PBLs specifically released IFN-γ, GM-CSF and TNF-α (Table III). The engineered PBL populations were capable of releasing cytokines at peptide concentrations as low as 20 pg/ml.
FIGURE 5.
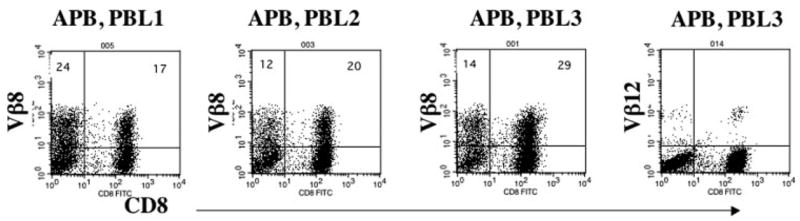
Vβ8 expression of APB-transduced PBLs. PBLs from three patients were transduced with TCR vector APB. Vβ8 and CD8 expressions of transduced PBLs from these three donors are shown; the percent positive cells is indicated. Background staining for Vβ12 in PBL3 was <1%.
Table III.
Sensitivity of cytokine secretion to dilution of NY-ESO-1 peptidea
| Transduced Effector
|
|||||||
|---|---|---|---|---|---|---|---|
| APB
|
GIN
|
||||||
| Responder T2/Peptide, nM | PBL1 | PBL2 | PBL3 | PBL1 | PBL2 | PBL3 | |
| IFN-γ (pg/ml) | |||||||
| p156–165v | 2,000 | 5,219 | 2,221 | 4,440 | 0 | 0 | 120 |
| 200 | 3,356 | 1,749 | 5,746 | 0 | 0 | 158 | |
| 20 | 1,910 | 1,234 | 4,865 | 0 | 0 | 17 | |
| 2 | 806 | 493 | 3,156 | 0 | 0 | 0 | |
| 0.2 | 102 | 96 | 1,323 | 0 | 0 | 0 | |
| 0.02 | 2 | 14 | 689 | 0 | 0 | 0 | |
| 0.002 | 0 | 0 | 188 | 0 | 0 | 0 | |
| gp100 pep. | 2,000 | 0 | 0 | 0 | 0 | 0 | 110 |
| GM-CSF (pg/ml) | |||||||
| p156–165v | 2,000 | 28,747 | 13,099 | 6,019 | 580 | 651 | 390 |
| 200 | 23,044 | 13,231 | 9,207 | 482 | 589 | 352 | |
| 20 | 19,870 | 11,858 | 7,125 | 441 | 566 | 244 | |
| 2 | 11,433 | 6,452 | 4,883 | 427 | 539 | 230 | |
| 0.2 | 4,936 | 3,345 | 2,793 | 508 | 429 | 206 | |
| 0.02 | 2,813 | 2,504 | 2,160 | 469 | 607 | 204 | |
| 0.002 | 1,198 | 1,882 | 1,780 | 457 | 561 | 236 | |
| gp100 pep. | 2,000 | 633 | 744 | 545 | 549 | 549 | 363 |
| TNF-α (pg/ml) | |||||||
| p156–165v | 2,000 | 92 | 2 | 219 | 0 | 0 | 0 |
| 200 | 33 | 0 | 179 | 0 | 0 | 0 | |
| 20 | 14 | 0 | 145 | 0 | 0 | 0 | |
| 2 | 0 | 0 | 49 | 0 | 0 | 0 | |
| 0.2 | 0 | 0 | 15 | 0 | 0 | 0 | |
| 0.02 | 0 | 0 | 5 | 0 | 0 | 0 | |
| 0.002 | 0 | 0 | 0 | 0 | 0 | 0 | |
| gp100 pep. | 2,000 | 0 | 0 | 0 | 0 | 0 | 0 |
Shown is the resultant cytokine secretion of TCR vector-transduced PBL cocultured with T2 cells pulsed with dilutions of NY-ESO-1 peptide (p157–165V) or control peptide gp100 (gp100 pep.). PBLs from three patients were transduced with either NY-ESO-1 TCR (APB) or control vector (GIN). IFN-γ, GM-CSF, and TNF-α secretion were determined by ELISA; mean values of duplicate samples are in picograms per milliliter.
To determine the relative reactivity of individually transduced PBL in the bulk transduced cell population, we subjected the transduced cells to a cytokine catch assay. Transduced PBL (40% positive by Vβ8 staining; Fig. 6A) or mock-transduced cells were incubated with peptide-pulsed T2 cells to stimulate cytokine production (Fig. 6B). IFN-γ secretion was captured at the cell surface and quantitated by FACS. Twenty-one percent of the transduced PBL was demonstrated to be actively secreting IFN-γ in this short term (45-min) assay, whereas there was no increase in IFN-γ secretion by the control cells or when transduced cells were incubated with an irrelevant peptide (Fig. 6B).
FIGURE 6.

Cytokine secretion staining of transduced CD8 T cells. A, Vβ8 staining of NY-ESO-1 TCR-transduced PBL (APB) and nontransduced PBL (NV) used in the cytokine secretion assay. B, The resultant FACS dot plots for control PBL (NV) or PBL transduced with NY-ESO-1 TCR vector (APB). Cells were cocultured with T2 cells pulsed with either HLA-A- specific gp100 peptide 209 –217 (210M) or NY-ESO-1 peptide 157–165v. Cells were gated for CD3-plus CD8-positive cells. The percentage of positive cells for IFN-γ secretion is shown.
FACS analysis (Figs. 4 and 5) and our previous observations (17) suggest TCR gene transfer into both CD8+ and CD4+ T cells. To determine the relative activities of these anti-NY-ESO-1 TCR-transduced cell types, CD4+ T cells from bulk populations of transduced PBLs were selected using CD4 magnetic beads (the negative population was used as the CD8+ control), then further depleted of CD8+ cells with CD8 magnetic beads. The purity of CD4+ T cells was 99.81% (data not shown) after this positive/negative selection procedure. Bulk PBL, CD8+ T cells, and purified CD4+ T cells (TCR- or control vector-transduced) were cocultured with NY-ESO-1 peptide-pulsed T2 cells, and cytokine production was determined (Table IV).
Table IV.
Cytokine secretion by transduced CD4 and CD8 PBLa
| Transduced Effector
|
||||||
|---|---|---|---|---|---|---|
| APB
|
Nontransduced
|
|||||
| Stimulator T2/Peptide, nM | CD4 | CD8 | PBL | CD8 | PBL | |
| IFN-γ | ||||||
| p156–165v | 1000 | 311 | 849 | 1351 | 0 | 0 |
| 100 | 219 | 1039 | 1418 | 0 | 0 | |
| 10 | 90 | 998 | 723 | 0 | 0 | |
| 1 | 18 | 262 | 219 | 0 | 19 | |
| 0.1 | 25 | 81 | 64 | 0 | 0 | |
| 0.01 | 25 | 34 | 17 | 0 | 0 | |
| 0.001 | 30 | 21 | 0 | 8 | 0 | |
| gp100 pep. | 1000 | 47 | 28 | 0 | 1 | 0 |
| GM-CSF | ||||||
| p156–165v | 1000 | 1084 | 465 | 906 | 5 | 98 |
| 100 | 901 | 634 | 965 | 9 | 62 | |
| 10 | 441 | 713 | 755 | 7 | 86 | |
| 1 | 302 | 471 | 364 | 2 | 103 | |
| 0.1 | 229 | 328 | 183 | 2 | 100 | |
| 0.01 | 241 | 347 | 162 | 8 | 97 | |
| 0.001 | 219 | 285 | 132 | 4 | 62 | |
| gp100 pep. | 1000 | 258 | 263 | 127 | 3 | 52 |
Shown is the resultant cytokine secretion of TCR vector-transduced CD4, CD8, or PBL exposed to T2 cells pulsed with dilution of NY-ESO-1 peptide (p157–165V) or control peptide gp100 (gp100 pep.). T lymphocytes from purified CD4, CD8, or unseparated PBL were transduced with NY-ESO-1 TCR (APB), untransduced CD8 or PBL was used a control. IFN-γ and GM-CSF secretion were measured. Values in picograms per milliliter.
TCR vector-transduced CD4+ T cells secreted IFN-γ in response to NY-ESO-1 peptide-pulsed T2 cells (Table IV). The response to NY-ESO-1 peptide measured by IFN-γ levels was ~100-fold lower for transduced CD4+ than the unseparated PBL or CD8+ T cells. As measured by GM-CSF production, the NY-ESO-1-specific peptide reactivity of engineered CD4+ T cells was ~10-fold less than bulk PBL or CD8+ cells. Only TCR-engineered CD4+ T cells secreted measurable amounts of IL-4 and IL-10 upon coculture with NY-ESO-1-specific, peptide-pulsed T2 cells (data not shown). These results may partially explain the different cytokine production levels in the bulk populations of transduced PBL (Fig. 5), because TCR-transduced CD4 T cells produced higher GM-CSF and lower IFN-γ levels than TCR-transduced CD8 T cells.
Recognition of melanoma cell lines by TCR-transduced PBLs
It has been reported that 34% of melanoma tumors express NY-ESO-1 (1). To determine antimelanoma reactivity, a panel of melanoma cell lines was cocultured with TCR vector-transduced PBLs or control vector-transduced PBL. Only HLA-A2 and NY-ESO-1 double-positive melanoma specifically stimulated TCR-transduced T cells to secrete IFN-γ (Fig. 7). We next measured lysis of melanoma cell lines by the engineered PBL in a 4-h 51Cr release assay. NY-ESO-1 TCR-transduced, but not control vector-transduced, PBL could efficiently kill NY-ESO-1 peptide-pulsed T2 cells (Fig. 8A) and NY-ESO-1/HLA-A2 double-positive melanoma cell lines, 624.38mel (Fig. 8B) and 1390mel (Fig. 8C) There was little or no lysis observed in gp100 peptide-pulsed T2 or non-HLA-A2 (586mel) or non-NY-ESO-1 (526mel) melanoma cell lines, and control vector-transduced cell populations were nonreactive.
FIGURE 7.
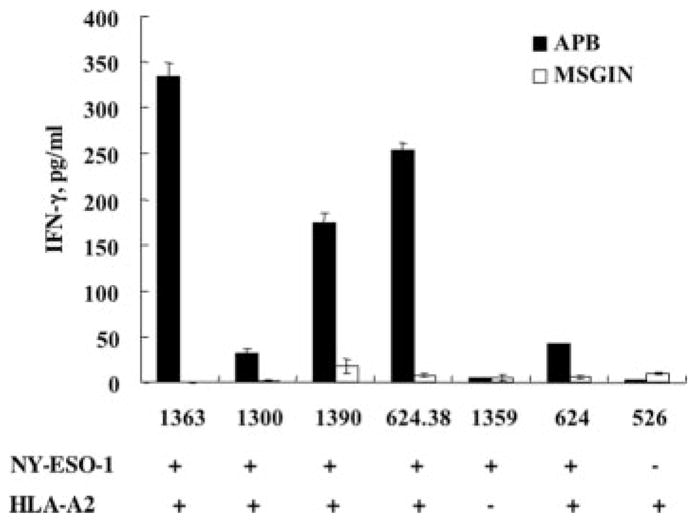
Melanoma reactivity of TCR-transduced PBL. APB TCR vector-transduced PBL were cocultured with a panel of melanoma cell lines. After coculture, IFN-γ levels were determined in culture medium from TCR vector-transduced PBL (APB) or control vector-transduced PBL (MSGIN). NY-ESO-1 and HLA-A2 expressions of the melanoma cell lines are indicated.
FIGURE 8.
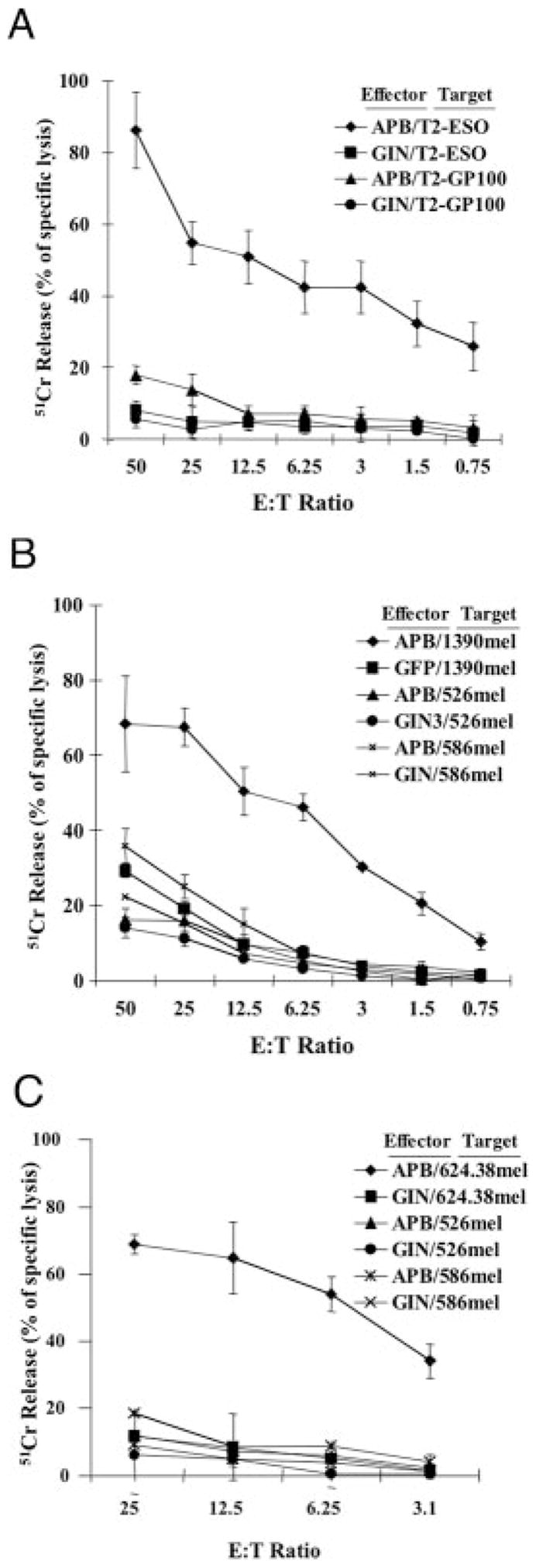
Lysis of melanoma cell lines. TCR-transduced PBL was tested in a 51Cr release assay. A, TCR- (APB) or control vector- (GIN) transduced PBL were cocultured with 51Cr-labeled, NY-ESO-1 peptide-pulsed (ESO, p157–165) T2 cells or gp100 peptide-pulsed (GP100) as a negative control. B and C, The NY-ESO-1+,HLA-A2+ melanoma cell line 1390mel (B) or 624.28mel (C) was labeled with 51Cr, then cocultured with TCR- (APB) or control vector- (GIN) transduced PBL. Two melanoma cell lines, 526mel (HLA-A2+,NY-ESO-1−) and 586mel (HLA-A2−,NY-ESO-1+), were used as negative control. Cells were incubated at the indicated E:T cell ratio for 4 h, after which the percent lysis of target cells was calculated.
Recognition of nonmelanoma tumor lines
Total RNA isolated from 42 nonmelanoma tumor lines was subjected to RT-PCR to determine NY-ESO-1 expression. Seven of these 42 lines (17%) were positive for NY-ESO-1 (data not shown). Among these seven lines, five tumor lines are HLA-A2 positive. Two HLA-A2-negative lines (SKN-AS and MDA-453s) were transduced with HLA-A2 retroviral vector and designated SKN-AS-A2 and MDA-453s-A2. TCR- or control vector-transduced PBLs were cocultured with these NY-ESO-1-positive nonmelanoma tumor lines. All the NY-ESO-1/HLA-A2 double-positive nonmelanoma tumor lines, but not the negative control, and two HLA-A2-negative tumor lines (SKN-AS and MDA-453s) were specifically recognized by TCR-transduced T cells (Fig. 9). To determine whether the nonmelanoma tumor lines were capable of being lysed by TCR-transduced T cells, breast cancer line MDA-454s-A2 and neuroblastoma line SKN-AS-A2 were used as targets in a 4-h 51Cr release assay. HLA-A2-restricted, NY-ESO-1-specific lysis was observed for these nonmelanoma tumors when cocultured with TCR-transduced PBL (Fig. 10).
FIGURE 9.
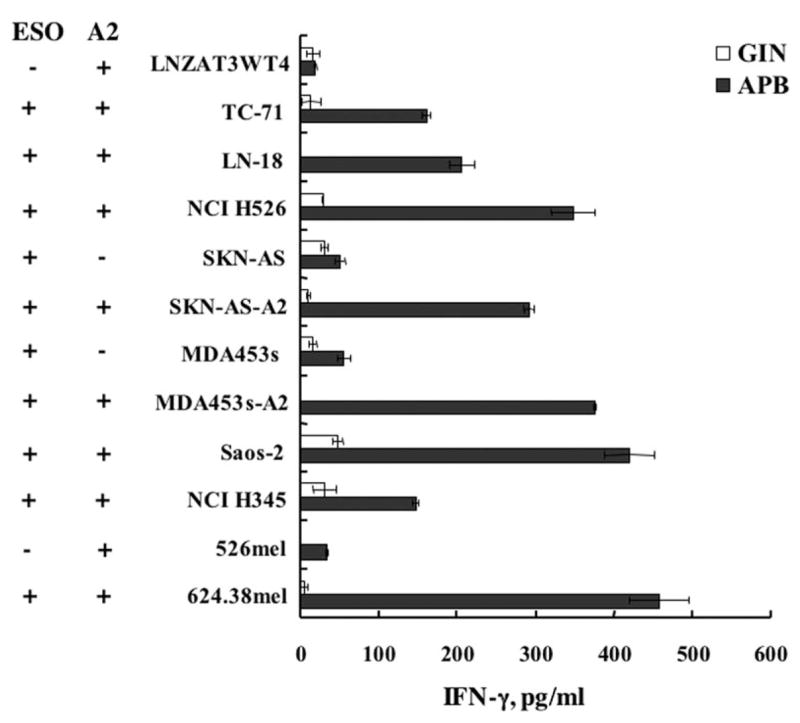
Nonmelanoma reactivity of TCR-transduced PBL. TCR-(APB) or control vector- (MSGIN, GIN) transduced PBL were cocultured with a panel of nonmelanoma tumor cell lines. IFN-γ release from the coculture supernatant was determined by ELISA. HLA-A2 (A2) and NY-ESO-1 (ESO) expressions of each cell line tested are indicated.
FIGURE 10.
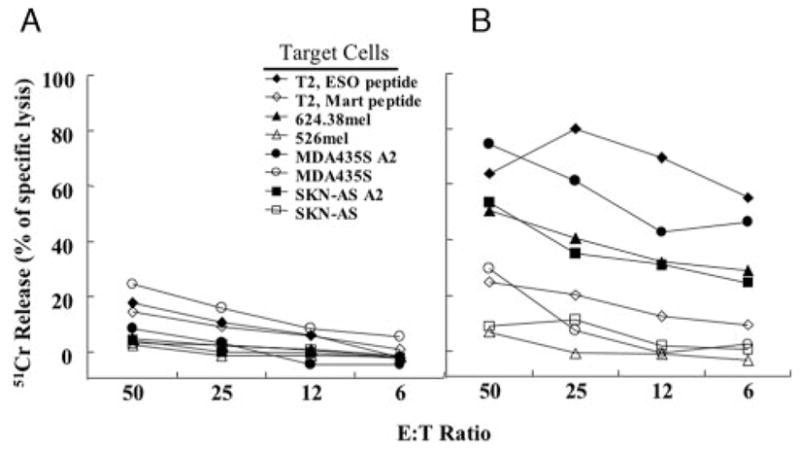
Lysis of nonmelanoma tumor lines by TCR-transduced PBL. NY-ESO-1 TCR APB-transduced PBL (B) or MSGIN-transduced PBL (A) were cocultured with 51Cr-labeled target cells as indicated. Cells were cocultured at the indicated E:T cell ratio for 4 h, after which the percent lysis of the target cells was calculated.
Discussion
Adoptive immunotherapy using melanoma reactive T cells has recently been demonstrated to mediate the regression of large established tumors in ~46% of patients (16). There are several limitations to this current therapy, including the difficulty of isolating and expanding autologous tumor-reactive T cells for each patient. Introduction of antitumor Ag TCR genes has been proposed as a method to produce antitumor lymphocytes for the immunotherapy of cancer without the need to isolate tumor-reactive T cells (31–33). This proposition requires that there exist tumor Ags common to divergent human cancers and that tumor-reactive TCR can be isolated from the appropriate T cells that recognize these natural tumor Ags.
Although adoptive immunotherapy can be effective in melanoma, it has only rarely been attempted in other tumor types because of the difficulty of isolating highly tumor-reactive T cells in nonmelanomas. The NY-ESO-1 Ag is expressed in ~30% of melanomas, but it is also expressed in a high percentage of common epithelial tumors, such as breast cancer, lung cancer, prostate cancer, and many types of sarcomas. Most tumor Ags identified to date are nonmutated self-Ags, and thus, antitumor immune responses must break immunological tolerance. In the case of CT Ags such as NY-ESO-1, expression is restricted to testicular germ cells and certain cancers. This may be one explanation for the relatively high immunogenicity of NY-ESO-1 in cancer patients (2). This is the first report of the transfer of a native TCR gene into human cells that has the potential to redirect T cells to kill a variety of common human cancers.
The TCR used in this report was isolated from PBL of a melanoma patient who received repeated in vitro immunizations with NY-ESO-1-specific peptide (21, 22). T cells transduced with retroviral vectors expressing the α- and β-chains for this receptor demonstrated HLA-A2-restricted, NY-ESO-1-specific effector T cells functions, as measured by cytokine release and cell lysis. We routinely observed that ~40% of PBL in a bulk population were transduced, as measured by the expression of anti-NY-ESO-1 TCR Vβ8. This level of gene transfer was obtained without selection and was sufficient to produce a biologically active population of cells. This level of gene transfer also minimized multiple vector integration events, a potential safety concern for clinical applications. The significance of the difference in the number of TCR Vβ8-positive (40%) vs IFN-γ capture-positive (21%) cells remains to be determined (Figs. 5 and 6). These differences could be related to technical aspects of the assays, such as differences in Ab affinities or to the fact that the capture assay is a short term (a 45-min incubation) assay. It is also possible that because of random pairing between the transduced TCR chains and the endogenous TCR chains, overall biological activity may be lessened by these nonproductive pairings. These results are similar to our previous work on anti-gp100 TCR gene transfer, demonstrating 22% positively staining cells by intracellular IFN-γ FACS analysis where ~40% of the cells were positive for Vβ staining.
TCR genetic engineering has been suggested as a way to bypass tolerance to TAA. Although many cellular components are potentially responsible for the avid recognition of TAA, the TCR is the main component of this activity (34, 35). High avidity of the anti-NY-ESO-1-transduced T cells was demonstrated by their ability to recognize T2 cells pulsed with low amounts of peptide (20 pM; Table III) and the ability to recognize naturally processed peptide found on the surface of diverse HLA-A2-positive tumor cell lines (Figs. 6 –10).
The highly avid nature of this particular TCR was also suggested by its ability to function in a CD8-independent manner. CD4+ T cells transduced with the NY-ESO-1 TCR vector were purified free of CD8 cells and were shown to produce both IFN-γ and GM-CSF cytokines after coculture with peptide-pulsed cells (Tables IV). The avidity of TCR engineered on CD4+ cells was ~2 logs lower than that of the engineered CD8+ TCR-containing T cells or unseparated PBLs, suggesting that CD8-MHC class I interactions stabilize and enhance effector function. Although recognition of peptide-pulsed T2 cells may not reflect potential in vivo situations, this result suggests that engineered CD4+ T cells can recognize the appropriate NY-ESO-1 peptide presented by class I HLA-A2 molecules in a CD8-independent fashion and is another measure of the high avidity afforded by transfer of this TCR gene.
These data demonstrate that, using retroviral vector-mediated gene transfer, we are able to transfer TCR genes for a highly avid anti-NY-ESO-1 TCR genes into a bulk population of PBL, and these cells demonstrate HLA-A2-restricted effector functions, including cytokine release and cell lysis. It is important to demonstrate functional avidity by cell lysis, because cytokine secretion and cytotoxicity are regulated independently in CD8+ T cells (36, 37). These results may have significant potential impact on the ability to treat cancer patients using adoptive immunotherapy. If this clinical approach were to be successful, TCR genes from tumor-reactive T cell clones would represent an “off-the-shelf” reagent that could be used to treat large numbers of cancer patients with their own transduced T cells without the need to identify antitumor T cells uniquely from each patient. These cells may have high specific reactivity against cancer (such as avid T cell clones) and retain the broad diversity of T cell functions and proliferative capacity of the bulk lymphocyte populations.
Using the appropriate anti-TAA TCRs, such as the anti-NY-ESO-1 TCR used in this study, makes this approach applicable to a variety of common epithelial malignancies. Additionally, it may be possible to up-regulate NY-ESO-1 expression in cancer patients by treatment with drugs that induce DNA demethylation (38, 39). Finally, we have identified MHC class II-restricted NY-ESO-1 TCR epitopes (22, 40, 41), and we are currently attempting to simultaneously engineer a population of T cells with both class I-and class II-restricted TCRs.
Acknowledgments
We acknowledge John Riley for synthesizing the NY-ESO-1 peptides used in these experiments, as well as Arnold Mixon and Shawn Farid for FACS analysis.
Footnotes
Abbreviations used in this paper: CT, cancer/testis; CM, culture medium; IRES, internal ribosomal entry site; LTR, long terminal repeat; MSCV, murine stem cell virus; TAA, tumor-associated Ag; PGK, phosphoglycerate kinase.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA. 1997;94:1914. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 3.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 4.Lethe B, Lucas S, Michaux L, De Smet C, Godelaine D, Serrano A, De Plaen E, Boon T. LAGE-1, a new gene with tumor specificity. Int J Cancer. 1998;76:903. doi: 10.1002/(sici)1097-0215(19980610)76:6<903::aid-ijc22>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 5.Alpen B, Gure AO, Scanlan MJ, Old LJ, Chen YT. A new member of the NY-ESO-1 gene family is ubiquitously expressed in somatic tissues and evolutionarily conserved. Gene. 2002;297:141. doi: 10.1016/s0378-1119(02)00879-x. [DOI] [PubMed] [Google Scholar]
- 6.Gnjatic S, Atanackovic D, Jager E, Matsuo M, Selvakumar A, Altorki NK, Maki RG, Dupont B, Ritter G, Chen YT, et al. Survey of naturally occurring CD4+ T cell responses against NY-ESO-1 in cancer patients: correlation with antibody responses. Proc Natl Acad Sci USA. 2003;100:8862. doi: 10.1073/pnas.1133324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jager E, Nagata Y, Gnjatic S, Wada H, Stockert E, Karbach J, Dunbar PR, Lee SY, Jungbluth A, Jager D, et al. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci USA. 2000;97:4760. doi: 10.1073/pnas.97.9.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jager E, Stockert E, Zidianakis Z, Chen YT, Karbach J, Jager D, Arand M, Ritter G, Old LJ, Knuth A. Humoral immune responses of cancer patients against “cancer-testis” antigen NY-ESO-1: correlation with clinical events. Int J Cancer. 1999;84:506. doi: 10.1002/(sici)1097-0215(19991022)84:5<506::aid-ijc10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 9.Jager E, Gnjatic S, Nagata Y, Stockert E, Jager D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA. 2000;97:12198. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma: a preliminary report. N Engl J Med. 1988;319:1676. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 11.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 12.Mackinnon S, Papadopoulos EB, Carabasi MH, Reich L, Collins NH, Boulad F, Castro-Malaspina H, Childs BH, Gillio AP, Kernan NA, et al. Adoptive immunotherapy evaluating escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. 1995;86:1261. [PubMed] [Google Scholar]
- 13.Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330:1185. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 14.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salter RD, Howell DN, Cresswell P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 1985;21:235. doi: 10.1007/BF00375376. [DOI] [PubMed] [Google Scholar]
- 19.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142:3714. [PubMed] [Google Scholar]
- 20.Shamamian P, Mancini M, Kawakami Y, Restifo NP, Rosenberg SA, Topalian SL. Recognition of neuroectodermal tumors by melanoma-specific cytotoxic T lymphocytes: evidence for antigen sharing by tumors derived from the neural crest. Cancer Immunol Immunother. 1994;39:73. doi: 10.1007/BF01525312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bownds S, Tong-On P, Rosenberg SA, Parkhurst M. Induction of tumor-reactive cytotoxic T-lymphocytes using a peptide from NY-ESO-1 modified at the carboxy-terminus to enhance HLA-A2.1 binding affinity and stability in solution. J Immunother. 2001;24:1. doi: 10.1097/00002371-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Zeng G, Li Y, El-Gamil M, Sidney J, Sette A, Wang RF, Rosenberg SA, Robbins PF. Generation of NY-ESO-1-specific CD4+ and CD8+ T cells by a single peptide with dual MHC class I and class II specificities: a new strategy for vaccine design. Cancer Res. 2002;62:3630. [PMC free article] [PubMed] [Google Scholar]
- 23.Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, Schwartzentruber DJ, Hwu P, Marincola FM, Sherry R, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 24.Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1:136. [PubMed] [Google Scholar]
- 25.Lindemann D, Patriquin E, Feng S, Mulligan RC. Versatile retrovirus vector systems for regulated gene expression in vitro and in vivo. Mol Med. 1997;3:466. [PMC free article] [PubMed] [Google Scholar]
- 26.Hawley RG, Lieu FH, Fong AZ, Goldman SJ, Leonard JP, Hawley TS. Retroviral vectors for production of interleukin-12 in the bone marrow to induce a graft-versus-leukemia effect. Ann NY Acad Sci. 1996;795:341. doi: 10.1111/j.1749-6632.1996.tb52687.x. [DOI] [PubMed] [Google Scholar]
- 27.Onodera M, Nelson DM, Yachie A, Jagadeesh GJ, Bunnell BA, Morgan RA, Blaese RM. Development of improved adenosine deaminase retroviral vectors. J Virol. 1998;72:1769. doi: 10.1128/jvi.72.3.1769-1774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan RA, Couture L, Elroy-Stein O, Ragheb J, Moss B, Anderson WF. Retroviral vectors containing putative internal ribosome entry sites: development of a polycistronic gene transfer system and applications to human gene therapy. Nucleic Acids Res. 1992;20:1293. doi: 10.1093/nar/20.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onodera M, Yachie A, Nelson DM, Welchlin H, Morgan RA, Blaese RM. A simple and reliable method for screening retroviral producer clones without selectable markers. Hum Gene Ther. 1997;8:1189. doi: 10.1089/hum.1997.8.10-1189. [DOI] [PubMed] [Google Scholar]
- 30.Hanenberg H, Xiao XL, Dilloo D, Hashino K, Kato I, Williams DA. Colocalization of retrovirus and target cells on specific fibronectin fragments increases genetic transduction of mammalian cells. Nat Med. 1996;2:876. doi: 10.1038/nm0896-876. [DOI] [PubMed] [Google Scholar]
- 31.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 32.Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2:512. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 33.Willemsen RA, Debets R, Chames P, Bolhuis RL. Genetic engineering of T cell specificity for immunotherapy of cancer. Hum Immunol. 2003;64:56. doi: 10.1016/s0198-8859(02)00730-9. [DOI] [PubMed] [Google Scholar]
- 34.Rubinstein MP, Kadima AN, Salem ML, Nguyen CL, Gillanders WE, Nishimura MI, Cole DJ. Transfer of TCR genes into mature T cells is accompanied by the maintenance of parental T cell avidity. J Immunol. 2003;170:1209. doi: 10.4049/jimmunol.170.3.1209. [DOI] [PubMed] [Google Scholar]
- 35.Roszkowski JJ, Yu DC, Rubinstein MP, McKee MD, Cole DJ, Nishimura MI. CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell. J Immunol. 2003;170:2582. doi: 10.4049/jimmunol.170.5.2582. [DOI] [PubMed] [Google Scholar]
- 36.Lim DG, Bieganowska Bourcier K, Freeman GJ, Hafler DA. Examination of CD8+ T cell function in humans using MHC class I tetramers: similar cytotoxicity but variable proliferation and cytokine production among different clonal CD8+ T cells specific to a single viral epitope. J Immunol. 2000;165:6214. doi: 10.4049/jimmunol.165.11.6214. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann MF, Barner M, Viola A, Kopf M. Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur J Immunol. 1999;29:291. doi: 10.1002/(SICI)1521-4141(199901)29:01<291::AID-IMMU291>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 38.De Smet C, Lurquin C, Lethe B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol Cell Biol. 1999;19:7327. doi: 10.1128/mcb.19.11.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, Fischette MR, Yu X, Chen GA, Hong JA, et al. Sequential 5-aza-2′-deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother. 2001;24:151. doi: 10.1097/00002371-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 40.Zeng G, Wang X, Robbins PF, Rosenberg SA, Wang RF. CD4+ T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci USA. 2001;98:3964. doi: 10.1073/pnas.061507398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng G, Touloukian CE, Wang X, Restifo NP, Rosenberg SA, Wang RF. Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J Immunol. 2000;165:1153. doi: 10.4049/jimmunol.165.2.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]