

Abstract
The Zn atom in the structure of the title compound, [ZnCl2(C5N2H8)2], exhibits a distorted tetrahedral geometry and is coordinated by two Cl and two N atoms, with Zn—Cl distances of 2.2519 (2) and 2.2531 (2) Å, and Zn—N distances of 1.9998 (7) and 2.0087 (7) Å. The angles Cl—Zn—Cl and N—Zn—N are 114.742 (9) and 108.50 (3)°, respectively.
Comment
The title complex, dichlorobis(1-ethylimidazole)zinc(II), (I), shows a distorted tetrahedral geometry around the central metal atom, which is coordinated to two Cl and two N atoms. A similar distorted tetrahedral geometry has been reported for the analogous structures of dichloro-bis (1,2-dimethylimidazole)zinc(II) (Bharadwaj et al., 1991) and dichlorobis(imidazole)zinc(II) (Lundberg, 1966).
The Zn—Cl distances in (I) are 2.2519 (2) and 2.2531 (2) Å, which are comparable with 2.243 (1) and 2.205 (1) Å in [Zn(BIPhMe)C12] (Tolman, et al., 1991); 2.2468 (8) and 2.2509 (8) Å in [Zn(1,2–Me2Im)2Cl2] (Bharadwaj et al., 1991). The Zn—N distances of 1.9998 (7) and 2.0087 (7) A in (I) are consistent with the values of 2.006 (3) and 2.008 (3) Å in [Zn(1,2–Me2Im)2Cl2] and 2.020 (11) and 1.995 (11) Å in [Zn(HIm)2Cl2] (Lundberg, 1966). The Cl—Zn—Cl angle of 114.742 (9)° in (I) is larger than that of the unbridged complexes [Zn(1,2–Me2Im)2Cl2] (114.50(3)°) and [Zn(HIm)2Cl2] (111.46(12)°); while the N—Zn—N angle of 108.50 (3)° is just between that of the six-membered ring complex [Zn(BIPhMe)C12] ((90.4(1)°) and eight-membered ring complex [Zn(SBI)Cl2] ((111.3(1)°, Bouwman, et al., 1991).
There are two mean planes made by the two imidazole five-membered rings, between which the dihedral angle is 71.45 (4)°. For the first mean plane made up from N1—C1—C2—N2—C3, the out of mean plan distances are 0.2730 (14) for Zn1, 1.1096 (19) for Cl1 and 1.6059 (19) Å for Cl2, with two Cl atoms in the same side of mean plane; for the second mean plane made up from N3—C6—N4—C8—C7, the out of mean plan distances are 0.0103 (14) for Zn1, 1.1214 (19) for Cl1 and 2.0811 (17) Å for Cl2, with two Cl atoms staying in different side of the mean plane.
Experimental
All experimental manipulation were performed under a purified nitrogen atmosphere using standard Schlenk techniques. The title compound is a by-product produced from a modified prepare of bis-imidazole “picket fence” porphyrinate [Fe(TpivPP)(1–EtIm)2] (Collman, J. P., et al., 1975). [Fe(TpivPP)Cl] was reduced by fresh zinc amalgam in toluene solution with addition of excess 1-ethylimidazole. After filtering, the red solution was layered carefully with dry hexanes. One week later, the title complex was harvested as several suitable block crystals.
| Crystal data | |
| C10H16Cl2N4Zn | Mo Kα radiation |
| Mr = 328.54 | λ = 0.71073 Å |
| Monoclinic | Cell parameters from 9241 reflections |
| P21/n | θ = 2.5960–33.1704° |
| a = 11.3901 (3) Å | μ = 2.066 mm−1 |
| b = 10.0587 (3) Å | T = 100 (2) K |
| c = 13.2143 (4) Å | Block |
| β = 108.408 (2)° | Dark Purple |
| V = 1436.49 (7) Å3 | 0.42 × 0.22 × 0.20 mm |
| Z = 4 | |
| Dx = 1.519 Mg m−3 | |
| Dm not measured | |
| Data collection | |
| Bruker Smart Apex CCD area detector diffractometer | 5032 reflections with >2sigma(I) |
| φ and ω scans | Rint = 0.0244 |
| Absorption correction: | θmax = 33.00° |
| multi-scan (SADABS 2.10; Sheldrick, 2004) | h = −17 → 17 |
| Please give reference | k = −15 → 15 |
| Tmin = 0.4774, Tmax = 0.6827 | l = −20 → 20 |
| 70286 measured reflections | ? standard reflections |
| 5418 independent reflections | every ? reflections intensity decay: ?% |
| Refinement | |
| Refinement on F2 | |
| R[F2 > 2σ(F2)] = 0.0182 | where |
| wR(F2) = 0.0510 | (Δ/σ)max = 0.008 |
| S = 1.056 | Δρmax = 0.578 e Å−3 |
| 5418 reflections | Δρmin = −0.257 e Å−3 |
| 156 parameters | Extinction correction: none |
| H-atom parameters constrained | Scattering factors from International Tables for Crystallography (Vol. C) |
All H atoms were initially located in a difference Fourier map. The methyl H atoms were then constrained to an ideal geometry with C—H distances of 0.98 Å and Uiso(H) = 1.5Ueq(C), but each group was allowed to rotate freely about its C—C bond. The position of the amine H atom was refined freely along with an isotropic displacement parameter. All other H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms with C—H distances in the range 0.95–1.00 Å and Uiso(H) = 1.2Ueq(C).
Data collection: APEX2 (Brucker, 2004). Cell refinement: APEX2/SAINT (Brucker, 2004). Data reduction: SAINT (Brucker, 2004). Program(s) used to solve structure: SHELXS-97 (Sheldrick, 1990). Program(s) used to refine structure: SHELXL97 (Sheldrick, 1997). Molecular graphics: ORTEP3 (Farrugia, 1997). Software used to prepare material for publication: SHELXL97 (Sheldrick, 1997).
Supplementary Material
Supplementary data for this paper are available from the IUCr electronic archives (Reference: PREVIEW). Services for accessing these data are described at the back of the journal.
Fig. 1.
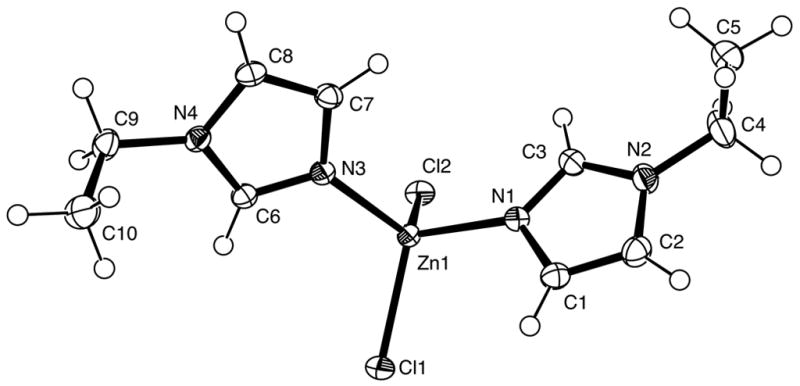
View of (I) showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms are represented by circles of arbitrary size.
Fig. 2.
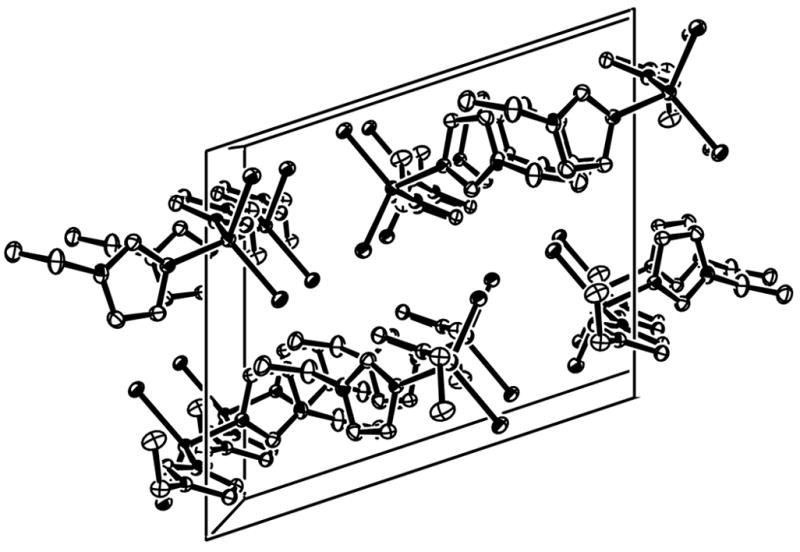
The molecular packing of (I) viewed along the b axis. H atoms bonded to C atoms have been omitted for clarity.
Table 1.
Selected geometric parameters (Å, °)
| Cl1—Zn1 | 2.2519 (2) | C5—C4 | 1.5091 (15) |
| Cl2—Zn1 | 2.2531 (2) | C1—N1 | 1.3803 (11) |
| Zn1—N3 | 1.9998 (7) | C7—N3 | 1.3822 (10) |
| Zn1—N1 | 2.0087 (7) | C3—N1 | 1.3270 (11) |
| C10—C9 | 1.5142 (14) | C6—N3 | 1.3281 (11) |
| N3—Zn1—N1 | 108.50 (3) | N1—Zn1—Cl2 | 108.47 (2) |
| N3—Zn1—Cl1 | 107.83 (2) | Cl1—Zn1—Cl2 | 114.742 (9) |
| N1—Zn1—Cl1 | 106.10 (2) | C3—N1—C1 | 106.28 (7) |
| N3—Zn1—Cl2 | 110.95 (2) | C6—N3—C7 | 106.31 (7) |
References
- Bharadwaj PK, Schugar HJ, Potenza JA. Acta Cryst C. 1991;47:754–757. doi: 10.1107/s0108270190011726. [DOI] [PubMed] [Google Scholar]
- Bouwman E, Driessen WL, Koolhaas GJAA, van Steenbergen AC, Reedijk J, Dartmann M, Krebs B. Inorg Chim Acta. 1991;189:253–257. [Google Scholar]
- Bruker-Nonius AXS. APEX2. Bruker-Nonius AXS; Madison, Wiscousin, USA: 2004. [Google Scholar]
- Bruker-Nonius AXS. SAINT. Bruker-Nonius AXS; Madison, Wiscousin, USA: 2004. [Google Scholar]
- Collman JP, Gagne RR, Reed C, Halbert TR, Lang G, Robinson WT. J Am Chem Soc. 1975;97:1427–1439. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- Farrugia LJ. ORTEPIII. University of Glasgow; England: 1997. [Google Scholar]
- Lundberg BKS. Acta Cryst. 1966;21:901–909. doi: 10.1107/s0365110x66004171. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM. Acta Cryst A. 1990;46:467–473. [Google Scholar]
- Sheldrick GM. SHELXL97. University of Göttingen; Germany: 1997. [Google Scholar]
- Sheldrick GM. SADABS 2.10. University of Göttingen; Germany: 2004. [Google Scholar]
- Tolman WB, Liu Shuncheng, Bentsen JG, Lippard SJ. J Am Chem Soc. 1991;113:152–164. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data for this paper are available from the IUCr electronic archives (Reference: PREVIEW). Services for accessing these data are described at the back of the journal.