

Abstract
In nutrient-rich, vegetative conditions, the yeast Saccharomyces cerevisiae transports a resident protease, aminopeptidase I (API), to the vacuole by the cytoplasm to vacuole targeting (Cvt) pathway, thus contributing to the degradative capacity of this organelle. When cells subsequently encounter starvation conditions, the machinery that recruited precursor API (prAPI) also sequesters bulk cytosol for delivery, breakdown, and recycling in the vacuole by the autophagy pathway. Each of these overlapping alternative transport pathways is specifically mobilized depending on environmental cues. The basic mechanism of cargo packaging and delivery involves the formation of a double-membrane transport vesicle around prAPI and/or bulk cytosol. Upon completion, these Cvt and autophagic vesicles are targeted to the vacuole to allow delivery of their lumenal contents.
Key questions remain regarding the origin and formation of the transport vesicle. In this study, we have cloned the APG9/CVT7 gene and characterized the gene product. Apg9p/Cvt7p is the first characterized integral membrane protein required for Cvt and autophagy transport. Biochemical and morphological analyses indicate that Apg9p/Cvt7p is localized to large perivacuolar punctate structures, but does not colocalize with typical endomembrane marker proteins. Finally, we have isolated a temperature conditional allele of APG9/CVT7 and demonstrate the direct role of Apg9p/Cvt7p in the formation of the Cvt and autophagic vesicles. From these results, we propose that Apg9p/Cvt7p may serve as a marker for a specialized compartment essential for these vesicle-mediated alternative targeting pathways.
Keywords: aminopeptidase I, lysosome, protein targeting, vacuole, yeast
Introduction
To survive periods of starvation such as serum deprivation (Kadowaki et al. 1996; Mortimore et al. 1996; Blommaart et al. 1997) and carbon and nitrogen starvation (Takeshige et al. 1992), the autophagy pathway delivers nonessential cytoplasmic material, including proteins and organelles, to the vacuole/lysosome for subsequent digestion by resident hydrolases. The breakdown products are then recycled for various essential metabolic processes. In Saccharomyces cerevisiae, there is a genetic overlap between autophagy mutants (apg and aut; Tsukada and Ohsumi 1993; Thumm et al. 1994) and mutants in the biosynthetic cytoplasm to vacuole targeting (Cvt) pathway (Harding et al. 1996; Scott et al. 1996) indicating that these two processes are mechanistically similar.
In both pathways, mechanisms of cargo selection, transport vesicle formation, topology, and subsequent fusion and cargo release possess features that diverge from the classical tenets of vesicle transport through the secretory pathway. The Cvt pathway cargo protein, aminopeptidase I (API), is synthesized in the cytosol as a precursor enzyme (prAPI) and assembled into a higher-ordered structure called the Cvt complex (Fig. 1; Baba et al. 1997; Kim et al. 1997; Scott et al. 1997). Electron micrographs show the Cvt complex to be an electron-dense structure devoid of cytosol (Baba et al. 1997; Scott et al. 1997). Under starvation conditions, prAPI is transported to the vacuole via autophagy. The cargo in autophagy, however, includes not only prAPI in the form of the Cvt complex, but also bulk cytosolic proteins and organelles. While the recruitment of bulk cytosol in autophagy is generally nonselective, specific degradation of targeted organelles such as peroxisomes has also been reported (Tuttle and Dunn 1995; Sakai et al. 1998; reviewed in Klionsky and Ohsumi 1999).
Figure 1.

Model of Cvt and autophagy transport to the vacuole. There are many shared mechanistic features for the two pathways. 1, Cvt complex formation: prAPI is synthesized in the cytosol, oligomerizes into a dodecamer, and assembles into a Cvt complex; 2, double-membrane vesicle formation: the Cvt complex is enwrapped by a membrane source which, upon expansion and completion, forms the double-membraned Cvt vesicle (Cvt pathway) or autophagosome (autophagy); 3, vesicle targeting to the vacuole: the double-membrane vesicle is then targeted to the vacuole followed by fusion with the vacuolar membrane and release of the inner vesicle, termed the Cvt body (Cvt pathway) or the autophagic body (autophagy), into the vacuolar lumen. Subsequent breakdown of the Cvt or autophagic body releases prAPI into the lumen where it is proteolytically processed, in a proteinase A-dependent manner, to mature API. Major differences between the pathways include the following: 1, cargo selectivity: the Cvt pathway is a selective process and the Cvt vesicles appear to exclude bulk cytosolic components as cargo. In contrast, autophagy is nonselective and autophagosomes carry bulk cytosol, in addition to the Cvt complex, to the vacuole. prAPI is transported via both vesicle types. A recombinant, cytosolic marker protein, Pho8Δ60p, is transported to the vacuole via autophagy. Delivery of Pho8Δ60p to the vacuole can be monitored through cleavage of the COOH-terminal propeptide. 2, Vesicle size: Cvt vesicles are ∼150 nm in diameter, whereas autophagosomes are 300–900 nm in diameter. 3, Conditions for induction: the Cvt pathway is active in vegetative growth conditions while autophagy is induced under starvation conditions.
EM has been used to follow the sequestration process of the Cvt pathway and autophagy, and has revealed membrane structures that enwrap the Cvt complex or bulk cytosol (Fig. 1; Baba et al. 1994, Baba et al. 1997). The completion of the cargo sequestration event results in the formation of the double-membrane Cvt vesicle (130–150 nm in diameter; Baba et al. 1997; Scott et al. 1997) or autophagosome (300–900 nm in diameter; Baba et al. 1994). Therefore, compared with a typical secretory transport vesicle of 50–100 nm in diameter, a Cvt vesicle can range from 2–27 times bigger in volume, and an autophagosome can be >1,000 times as large.
The fusion of the Cvt and autophagic vesicles with the vacuole requires general vesicle fusion machinery, including the vacuolar target-SNARE (soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor) Vam3p (Darsow et al. 1997; Sato et al. 1998) and the rab GTPase Ypt7p (Kim et al. 1999; Kirisako et al. 1999; George et al. 2000). Fusion of Cvt vesicles with the vacuole requires the vesicle-SNARE Vti1p (Fischer von Mollard and Stevens 1999) and Vps18p (Scott et al. 1997). In addition, Cvt and autophagic vesicle fusion with the vacuole releases the inner vesicle (the Cvt or autophagic body) into the lumen. Maturation of prAPI and the degradation of cytosolic contents occur only after inner-vesicle breakdown. Strains defective in the vacuolar hydrolases proteinase A or B accumulate Cvt and autophagic bodies and are not able to mature prAPI (Klionsky et al. 1992; Takeshige et al. 1992). In addition, Cvt17p and/or proper vacuolar pH are required for Cvt and autophagic body breakdown, respectively (Nakamura et al. 1997; Scott et al. 1997).
A key issue that remains to be resolved is the identity of the donor compartment from which the Cvt and autophagosomal membranes are derived. Immunocytochemical studies in mammalian cells have suggested that ribosome-free regions of the ER provide the source membrane for the autophagosome (Dunn 1990; Tooze et al. 1990; Yokota et al. 1993). However, others have postulated that since the autophagosomal membrane does not resemble any known subcellular compartment, the origin of this enwrapping membrane must be derived from an equally specialized donor compartment, termed the phagophore (Seglen et al. 1996; Stromhaug et al. 1998). These and other conflicting studies demonstrate the need for a genetically tractable system in which to address this question. To that end, the autophagy/Cvt mutants from S. cerevisiae may serve as a simpler model system.
Analyses of the cvt, apg, and aut mutants have revealed defects at various stages of the Cvt and autophagy pathways. Cloning of the genes and characterization of the gene products have identified a growing number of components required for these pathways, including regulatory kinases (Matsuura et al. 1997; Noda and Ohsumi 1998), members of an essential protein conjugation system (Mizushima et al. 1998; Kim et al. 1999; Tanida et al. 1999; Yuan et al. 1999; George et al. 2000), and vesicle-associated proteins (Lang et al. 1998; Kirisako et al. 1999; Huang et al. 2000; for reviews see Klionsky and Ohsumi 1999; Kim et al. 2000). However, all of these characterized proteins are soluble or peripherally associated to membranes. Therefore, they do not represent ideal markers for the study of the subcellular compartments involved in the Cvt and autophagy processes. Here, we report the cloning and characterization of a novel protein, Apg9p/Cvt7p, the first identified integral membrane protein of the Apg/Cvt pathways. We show that Apg9p/Cvt7p does not colocalize with known endomembrane markers and forms a prominent, punctate structure proximal to the vacuole. Finally, using a temperature-sensitive mutant of Apg9p, we demonstrate its direct role in the vesicle formation step in the autophagy and Cvt pathways.
Materials and Methods
Strains
The yeast strains used in this study are listed in Table .
Table 1.
Yeast Strains Used in this Study
| Strain | Genotype | Source or reference |
|---|---|---|
| CT83-1B | MATa leu2 trp1 ura3 pho8::pho8Δ60 apg9-1 | This study |
| CTD1 | MATa leu2 trp1 ura3 apg9Δ::TRP1 | This study |
| JKY007 | MATα his3-Δ200 leu2-3,112 lys2-801 trp1-Δ901 ura3-52 suc2-Δ9 GAL apg9Δ::HIS3 | This study |
| KSL12C | MATα leu2 trp1 ura3 vps4Δ::LEU2 pho8::pho8Δ60 | This study |
| SEY6210 | MATα his3-Δ200 leu2-3,112 lys2-801 trp1-Δ901 ura3-52 suc2-Δ9 GAL | Robinson et al. 1988 |
| SKD6-1D | MATa leu2 trp1 ura3 apg6Δ::LEU2 | Kametaka et al. 1998 |
| THY154 | MATα ade2-101 his3-Δ200 leu2-3,112 trp1-Δ901 ura3-52 suc2-Δ9 GAL cvt7-1 | Harding et al. 1995 |
| TK415 | MATa leu2 ura3 ypt7Δ::LEU2 | Kirisako et al. 1999 |
| STY1 | MATa leu2 trp1 ura3 pep4Δ::URA3 | This study |
| YW5-1B | MATa ura3 trp1 leu2 | Noda et al. 1995 |
Antibodies
To prepare antiserum to Apg9p/Cvt7p, synthetic peptides corresponding to amino acids 1–15 (National Institute for Basic Biology, Center for Analytical Instruments) or 74–89 and 138–156 (Multiple Peptide Systems) were synthesized and conjugated individually to BSA or keyhole limpet hemocyanin, respectively. Standard procedures were used to generate antiserum in Japanese White or New Zealand White rabbits, respectively. Antisera against API and carboxypeptidase Y (CPY) were prepared as described previously (Klionsky et al. 1988, Klionsky et al. 1992). Antiserum against phosphoglycerate kinase (PGK) was generously provided by Dr. Jeremy Thorner (University of California, Berkeley, CA; Baum et al. 1978). Anti-HA mAb 16B12 was from Berkeley Antibody Co., Inc. FITC-conjugated secondary anti-mouse antibody was from Roche Molecular Biochemicals.
Materials
All restriction enzymes and VentR DNA polymerase were from New England Biolabs. AmpliTaq DNA polymerase was from Perkin Elmer Biosystems. Expre35S35S Protein Labeling Mix was from Dupont-New England Nuclear Research Products. Immobilon-P (polyvinylidene fluoride, PVDF) was from Millipore. ConA Sepharose and Ficoll-400 were from Pharmacia Biotech, Inc. Complete™ EDTA-free protease inhibitor cocktail and endoglycosidase H (endo H) were from Roche Molecular Biochemicals. FM 4-64 dye was from Molecular Probes. Oxalyticase was from Enzogenetics. YNB was from DifCo Laboratories, Inc. Zymolyase 100T was from Seikagakukogyo. Oligonucleotides to pBR322 were from Promega. All other oligonucleotides were synthesized by Operon Technologies, Inc. All other reagents were from Sigma Chemical Co. or Wako Chemicals.
Growth Media
Yeast cells were grown in rich medium (YPD; 1% yeast extract, 2% peptone, and 2% glucose) or various synthetic minimal media. Synthetic minimal media with carbon and nitrogen sources were: SMD, 0.67% yeast nitrogen base, 2% glucose, and auxotrophic amino acids and vitamins as needed; SD, 0.17% yeast nitrogen base without ammonium sulfate and amino acids, 0.5% ammonium sulfate, and 2% glucose; or SCD, 0.17% yeast nitrogen base without ammonium sulfate and amino acids, 0.5% ammonium sulfate, 0.5% casamino acids and 2% glucose. Synthetic minimal media lacking nitrogen and/or carbon sources were: SD(−N), 0.17% yeast nitrogen base without ammonium sulfate and amino acids and 2% glucose; and S(−N, −C), 0.17% yeast nitrogen base without ammonium sulfate and amino acids.
Isolation of Mutants
The cvt mutants were isolated on the basis of prAPI accumulation (Harding et al. 1995, Harding et al. 1996). cvt7 belongs in the group of mutants that share a genetic overlap with mutants defective in autophagy, and is in the same complementation group as aut9 and apg9 (Harding et al. 1996; Scott et al. 1996). The cvt7-1 mutant was used for subsequent analyses and cloning of the CVT7 gene as described below.
The Pho8Δ60p construct (Noda et al. 1995) was used to screen for additional autophagy-defective mutants. Pho8Δ60p is an altered form of the vacuolar hydrolase alkaline phosphatase lacking the NH2-terminal transmembrane domain. This modified protein is only delivered to the vacuole through autophagy (Fig. 1). Proteolytic cleavage of the Pho8Δ60p propeptide in the vacuole lumen generates the active form of the enzyme. Yeast strain TN124 expressing Pho8Δ60p was mutagenized with EMS and spread onto YPD plates. Approximately 36,000 colonies were replica-plated onto SD(−N) and incubated overnight at 30°C. 1% agar containing 0.1 mM α-naphthyl phosphate, 1 mg/ml Fast Red TR salt, 5 mM MgCl2, 0.1 M Tris-HCl, pH 9.0, and 1 mM PMSF was melted and kept at 60°C. Fast Red TR salt and PMSF were added to the melted agar, and 5 ml of this mixture was poured onto the plate. Colonies that were able to carry out autophagy and exhibited alkaline phosphatase activity turned brown. The colonies that did not turn brown were picked from the master plate and were subjected to a morphological observation of autophagy. Mutants that did not accumulate intravacuolar vesicles upon starvation in the presence of PMSF (Tsukada and Ohsumi 1993) were placed into complementation groups. The most abundant group was found to fit into the complementation group that was previously identified as apg9 (Tsukada and Ohsumi 1993). The apg9-1 mutant was chosen for further analysis.
Cloning of CVT7/APG9
The APG9/CVT7 gene was independently cloned by our two laboratories. Because >99% of apg9/cvt7 cells die after 6 d of nitrogen starvation, the APG9/CVT7 gene product is essential for cell viability under autophagy-inducing conditions of nitrogen deprivation. Therefore, survival in nitrogen-poor medium offered a convenient selection strategy to clone APG9/CVT7. Specifically, strain THY154 (cvt7-1) and CT83-1B (apg9-1) cells were transformed with a YCp50-based yeast genomic library (Rose et al. 1987). Approximately 60,000 transformed colonies were then subjected to nitrogen starvation for 6 d. Transformants that survived this regimen were subjected to Western blot analysis. Transformants were isolated containing genomic plasmids that complemented the prAPI processing defect of cvt7-1 or the ability to accumulate autophagic bodies in apg9-1. Partial sequences from these genomic plasmids were entered into the Saccharomyces Genome Database (SGD; http://genome-www.stanford.edu/Saccharomyces/). The full-length sequences of these plasmids contained overlapping fragments of 7–8 kb. After further manipulations, the open reading frame (ORF) of APG9/CVT7 was identified as YDL149w. For clarity, we will refer to the APG9/CVT7 gene and the gene product as APG9 and Apg9p, respectively, henceforth in this section.
Using a NotI/BamHI digestion, an APG9 cassette with 757 bp of upstream and 485-bp downstream sequences was subcloned into pRS414 and pRS424, resulting in plasmids pAPG9(414) and pAPG9(424), respectively. Both pAPG9(414) and pAPG9(424) complemented the prAPI processing defect when transformed into the cvt7 and apg9 mutants. Similarly, a 4.8-kb fragment containing a part of YCp50 cut with XbaI from plasmid p8001 encoding APG9 was subcloned into pRS316 to generate plasmid p8007. This plasmid was sufficient to suppress the autophagy defect of apg9.
Disruption of the APG9 Chromosomal Locus
The chromosomal APG9 locus was disrupted through two independent approaches. First, a PCR-based strategy was employed to disrupt the chromosomal copy of APG9. The HIS3 gene was PCR-amplified from the pRS413 vector using oligonucleotides containing vector sequences outside of the HIS3 auxotrophic marker flanked by APG9 sequences encoding regions at the beginning and end of the APG9 ORF: 5′-GAGAGAGATGAATACCAGTTACCCAACTCTCATGGGAAGTTGTAC-TGAGAGTGCACCAT-3′ and 5′-GGATGATGTACACGACACAGTCTGCCTTATCTTCCGACGTCGGTATTTCACACCGCATA-3′. The PCR product was transformed into strain SEY6210 wild-type cells and grown on minimal plates lacking histidine. Transformants were then assayed for API precursor accumulation. In addition, the apg9Δ strain was crossed with the cvt7-1 mutant, followed by tetrad analysis and Western blotting with API antiserum. All of the germinants from 12 tetrads exhibited a prAPI phenotype indicating that the APG9 gene maps to the correct locus. The apg9Δ strain was named JKY007.
In the second approach, plasmid p8007 was digested with EcoRI to remove a 700-bp fragment within the APG9 ORF. An EcoRI fragment containing the TRP1 gene was ligated into the EcoRI sites of p8007 to generate p8009. Plasmid p8009 was digested with EcoT14I/SphI and a fragment containing the TRP1 marker and flanking APG9 sequences was transformed into YW5-1B wild-type cells. Disruption at the APG9 locus was confirmed by Southern blot. The apg9Δ strain was crossed with the apg9-1 mutant. The resulting diploid retained an autophagy defect suggesting that APG9 is YDL149w.
Cell Viability Under Nitrogen Starvation Conditions
To examine the survival of various yeast strains under nitrogen starvation conditions, cells were grown to an OD600 = 1 in SMD, washed in SD(−N), and then resuspended in SD(−N) to an OD600 = 1. At the indicated times, aliquots were removed and plated onto YPD plates in triplicate. Colonies that survived the nitrogen starvation regimen were counted after 2–3 d.
Construction of Plasmids
For epitope-tagging Apg9p, a BamHI site was generated next to the initiation methionine of APG9 in plasmid pRS424 or pRS426. A DNA fragment encoding a 3×HA epitope with BamHI sites on both sides was then ligated into the BamHI site.
To construct the APG9GFP centromeric and multicopy plasmids, the APG9 gene, including 509 bp of upstream sequence, was PCR-amplified from the pAPG9(414) template using oligonucleotides that incorporated a NotI site in the 5′ primer (5′-GCAAGGTTGGGTCTATGCGGCCGCACAAATCTGG-3′) and an in-frame BamHI site immediately preceding the APG9 stop codon in the 3′ primer (5′-GACACAGTCTGCCTTAGGATCCGAC-3′). The 3.5-kb PCR product was subcloned into pCGFP(416) and pCGFP(426), which contained the green fluorescent protein (GFP) ORF, followed by actin termination sequences (Kim et al. 1999). The resulting COOH-terminal GFP fusion constructs, pAPG9GFP(416) and pAPG9GFP(426), were tested for their ability to rescue the prAPI import defect in the apg9Δ strain.
Cell Labeling
For cell labeling experiments, cells were grown to OD600 = 1 and resuspended in SMD at 20–30 OD600/ml. The resuspended cells were labeled with 10–20 μCi of 35S Express label/OD600 for the indicated times, followed by a chase reaction in SMD supplemented with 0.2% yeast extract, 4 mM methionine, and 2 mM cysteine at a final cell density of 1 OD600/ml. The labeled cells were precipitated with 10% TCA on ice, followed by two acetone washes. Crude extracts were prepared by glass bead lysis, as described previously (Harding et al. 1995).
Subcellular Fractionation
Wild-type cells were grown to midlog and incubated in 0.1 M Tris-HCl, pH 7.5, containing 40 mM 2-mercaptoethanol for 10 min at room temperature. Following a centrifugation step, the cells were spheroplasted in 1.2 M sorbitol, 50 mM Tris-HCl, pH 7.5, with 5 U/ml Zymolyase 100T at 30°C for 30–50 min. The cells were loaded on top of an equal volume of 1.8 M sorbitol and centrifuged at 2,000 rpm for 10 min. The spheroplasts were collected from the bottom of the tube. For sucrose density gradient fractionation, YW5-1B cells were grown to midlog in YPD and spheroplasted as described above. The spheroplasts were lysed in lysis buffer B (20 mM triethanolamine, pH 7.2, 12.5% sucrose [wt/vol], 1 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail) at a density of 60 OD600/ml. After removal of cell debris, the supernatant fraction (1 ml) was layered on top of a sucrose density gradient consisting of 1-ml steps of 54, 50, 46, 42, 38, 34, 30, 26, 22, and 18% sucrose (wt/wt) in 10 mM Hepes-NaOH, pH 7.5, 1 mM MgCl2, and 1 mM PMSF. The gradients were subjected to centrifugation in a Hitachi RPS40T rotor at 174,000 g for 2.5 h. Samples were collected into 16 fractions from the top of the gradients. To examine Apg9p in the ypt7Δ strain, the precleared lysate was subjected to centrifugation at 100,000 g for 1 h. The pellet was resuspended in lysis buffer B and centrifuged a second time at 100,000 g for 1 h. The pellet fraction after the second centrifugation step was resuspended in 1 ml of lysis buffer B and subjected to the same density gradient centrifugation as above for 16 h.
Endoglycosidase H Treatment
Strain CTD1 cells expressing 3×HA Apg9p were grown to midlog phase and converted to spheroplasts. The spheroplasts were lysed in 0.1% SDS, 50 mM sodium citrate, pH 5.5, 1 mM EDTA, 1 mM PMSF, protease inhibitor cocktail, and boiled for 5 min. After centrifugation at 14,000 rpm for 1 min, the protein concentration of the supernatant fraction was determined. Protein (60 mg) was diluted to 25 μl, and 25 μl 0.1% SDS, 0.1 M 2-mercaptoethanol were added, and the solution was boiled for 3 min. 15 μl of 0.5 M sodium citrate, pH 5.5, 75 μl H2O, 5 μl 100 mM PMSF, 2.5 μl endo H (1 U/ml) were then added and the mixtures were incubated overnight at 37°C, boiled for 3 min, and subjected to immunoblotting.
Generation of the apg9ts Mutant
A strategy of gap-repair PCR mutagenesis (Muhlrad et al. 1992) was employed to generate a temperature conditional mutant of Apg9p. In brief, a “gapped” APG9 plasmid was created by digestion with SpeI/BglII, which removed a 1.74-kb piece of the APG9 ORF starting 228 bp downstream of the ATG start site. A 2.23-kb PCR product of the APG9 ORF was generated by using the primers 5′-CACGGAATTATTAGGTTATGGAGAGAGATG-3′ and 5′-GTGTTATCATCAGGCTAGTGAGTTCCC-3′ and pAPG9(414) as the template. Errors were introduced in the polymerization reactions by the addition of 0.1 mM MnCl2. The resulting mutagenized PCR product shares overlapping sequences of 245 bp at the 5′ side and 240 bp at the 3′ side with the gapped APG9 plasmid. The PCR product and the gapped plasmid were cotransformed into the apg9Δ strain and grown on −Trp plates. The transformants were subjected to Western blotting analysis with anti-API antiserum after growth on −Trp plates overnight at 25 or 37°C. Out of the 400 colonies examined, four colonies exhibited a mature API phenotype at 25°C, indicative of functional Apg9p, and a prAPI phenotype at 37°C, the defective Apg9p phenotype. The four potential apg9ts colonies were analyzed by pulse/chase labeling to assess the kinetics of prAPI delivery to the vacuole at permissive and nonpermissive temperatures.
Membrane Flotation Analysis
The procedure for the membrane flotation experiments is a modification of a previously described protocol (Scott and Klionsky 1995; Babst et al. 1997; Kim et al. 1999). Spheroplasts from the apg9ts and the ypt7Δ strains were pulse-labeled with Expre35S35S label for 10 min, followed by a 30-min chase at 38°C. After centrifugation at 1500 g for 5 min, the spheroplasts were lysed in osmotic lysis buffer C (20 mM Pipes, pH 6.8, 200 mM sorbitol, 5 mM MgCl2, Complete EDTA-free protease inhibitor cocktail) at a spheroplast density of 20 OD600/ml. The lysate from 16 OD600 cell equivalents was subjected to a 5,000 g centrifugation for 5 min, resulting in low-speed supernatant (S5) and pellet (P5) fractions. The P5 fractions, which contained all of the prAPI, were resuspended in 100 μl of 15% Ficoll-400 (wt/vol) in gradient buffer (GB; 20 mM Pipes, 5 mM MgCl2, Complete EDTA-free protease inhibitor cocktail) with or without the addition of 0.2% Triton X-100. The resuspended P5 fractions were overlaid with 1 ml of 13% Ficoll-400 in GB and then 300 μl of 2% Ficoll-400 in GB. The resulting step gradients were subjected to centrifugation at 15,000 g for 10 min at 25°C in a microcentrifuge. Fractions were collected from the top. The first 500 μl was the float fraction, the remaining 900 μl was the nonfloat fraction, and the gradient pellet was considered the pellet fraction. The resulting fractions were TCA precipitated and washed twice with acetone before being immunoprecipitated with anti-API antiserum.
Protease Sensitivity Analysis
To examine the protease sensitivity of prAPI in the apg9ts and ypt7Δ strains, spheroplasts were labeled for 10 min at 38°C followed by a 30-min chase reaction at 38°C. Spheroplasts were lysed in osmotic lysis buffer C at a spheroplast density of 20 OD600/ml. The resulting lysate (20 OD600 cell equivalents per incubation condition) was separated into S5 supernatant and P5 pellet fractions by centrifugation at 5,000 g for 5 min at 4°C. The P5 pellet was resuspended in osmotic lysis buffer C in the presence or absence of proteinase K (50 μg/ml) and 0.2% Triton X-100. The resuspended P5 pellets were incubated on ice for 20 min, followed by TCA precipitation, acetone wash, and immunoprecipitation with anti-API and anti-PGK antisera.
Immunofluorescent Labeling, Immunoelectron Microscopy, and Confocal Microscopy
For immunofluorescent labeling of vegetatively growing cells, cultures were grown to midlog in YPD. To examine cells under starvation conditions, cells were grown in YPD and subsequently incubated in SD(−N) with 1 mM PMSF for 3 h. The cells were fixed in 100 mM potassium phosphate, pH 6.5, containing 5% formaldehyde for 2 h at 23°C. Following a washing step in SHA buffer (1 M sorbitol, 0.1 M Hepes-NaOH, pH 7.5, 5 mM NaN3), the fixed cells were converted to spheroplasts by incubation with 0.5 U/ml Zymolyase 100T, 24 mM 2-mercaptoethanol for 15 min at 30°C. The spheroplasts were washed in SHA buffer and mounted on polylysine-coated multiwell glass slides. The cells were permeabilized by treatment with 0.1% Triton X-100 in PBS for 10 min. Permeabilized cells were incubated with anti-HA mAb at a 1:5,000 dilution, and the FITC-conjugated secondary anti-mouse antibody was used at a 1:50 dilution. Photographs were taken using the confocal microscope Zeiss LSM510.
For EM, cells were subjected to rapid freezing and freeze-substitution fixation, and observed as described previously (Baba et al. 1997). For immunoelectron microscopy, ultrathin sections were collected onto formvar-coated nickel grids and blocked in PBS containing 2% BSA at room temperature for 15 min. Incubations were carried out by floating grids on a 20 μl drop of a 1:1,500 dilution of anti-HA mAb, 16B12, at room temperature for 1.5 h. After washing, the grids were incubated for 1 h with 5-nm gold-conjugated goat anti–mouse IgG. The grids were washed several times in PBS followed by several drops of distilled water and fixed with 1% glutaraldehyde for 3 min. The sections were stained with 4% uranium acetate for 7 min and examined.
Strains expressing the Apg9pGFP fusion protein were grown to midlog and the vacuoles were labeled with FM 4-64 dye to a final concentration of 4–8 μM and incubated at 30°C for 20 min to allow cells to take up the dye. After a wash step, the cultures were resuspended in SMD for 2 h to allow the dye to label the vacuole via endocytosis. Cells were examined on a Leica DM IRB confocal microscope. The images captured were the result of an average of eight scans of a single focal plane.
Software Programs
Hydrophobicity analysis was performed using the program TopPred II developed by M.-G. Claros (Laboratoire de Génétique Moléculaire, Paris, France).
Immunoblots were scanned using a Power Macintosh G3 and GT-9000 scanner (EPSON), and band intensities were determined using the program NIH Image 1.61.1. Alternatively, autoradiograms were quantified using the Molecular Dynamics Storm PhosphorImager and ImageQuant 5.0.
Results
Apg9p/Cvt7p Is a Shared Component of the Macroautophagy and Cvt Pathways
Autophagy is a degradative pathway used for delivery of cytoplasm to the vacuole (reviewed in Klionsky and Ohsumi 1999; Kim et al. 2000). The Cvt pathway is a biosynthetic process that targets at least one resident hydrolase to the vacuole (Klionsky et al. 1992). There are clear differences in the conditions that induce either pathway and in their kinetic parameters (Klionsky and Ohsumi 1999). Nonetheless, genetic analyses reveal a substantial overlap between the machinery employed by each process for the delivery of their corresponding cargo to the vacuole (Harding et al. 1996; Scott et al. 1996). The apg9 and cvt7 mutants were isolated by screening for starvation sensitivity or accumulation of prAPI, respectively (Tsukada and Ohsumi 1993; Harding et al. 1995). We also isolated apg9 by a third strategy in this study, by screening for mutants that were unable to deliver a recombinant cytosolic marker protein (Pho8Δ60p) to the vacuole under induced autophagy conditions (see Materials and Methods; Fig. 1).
Genetic analyses suggest that apg9 and cvt7 are in the same complementation group. Consistent with these mutations being allelic, both mutants accumulate prAPI and exhibit autophagy defects including sensitivity to nitrogen starvation. The nitrogen sensitivity phenotype was used as a basis for cloning the complementing genes. apg9-1 and cvt7-1 were transformed with a plasmid-based yeast genomic library. The plasmids from cells that survived the prolonged nitrogen starvation regimen were isolated. After further manipulations, our two laboratories independently identified YDL149w as the ORF whose gene product rescued the nitrogen sensitivity of both the apg9-1 and cvt7-1 mutants. We will refer to this ORF as APG9 hereafter in this study. Both the single copy and multicopy plasmids of APG9 rescued the prAPI processing defect in the apg9 mutants, as well as in the apg9Δ null mutant (Fig. 2 A), although overexpression of APG9 caused a slight accumulation of prAPI. Furthermore, the nitrogen starvation sensitivity was also rescued by the APG9 plasmid, showing survival rates similar to the isogenic wild-type strain (Fig. 2 B). These findings confirm that Apg9p is a required component for both the Cvt pathway and autophagy.
Figure 2.

Cloning and characterization of APG9. A, Wild-type (WT; SEY6210), apg9 (THY154), apg9Δ (JKY007), and the apg9Δ strain transformed with single copy (CEN; pAPG9(414)) or multicopy (2μ; pAPG9(424)) plasmids encoding APG9 were grown to log phase in SMD. Protein extracts were prepared and analyzed by immunoblot using antiserum to API as described in Materials and Methods. The positions of precursor and mature API are indicated. The APG9 gene complements the prAPI accumulation phenotype of the apg9Δ mutant. B, WT (SEY6210), apg9Δ (JKY007), and the apg9Δ strain transformed with pAPG9(414) centromeric plasmid were grown in SMD and transferred to SD(−N) as described in Materials and Methods. Aliquots were removed at the indicated times and spread onto YPD plates in triplicate. Numbers of viable colonies were determined after two to three days. The APG9 gene complements the starvation-sensitive phenotype of the apg9Δ mutant.
Apg9p/Cvt7p Is an Integral Membrane Protein
A search of the databases indicates that Apg9p does not share significant homology with other characterized proteins. Apg9p encodes a protein of 998 amino acids and has a predicted molecular mass of 115 kD. To further characterize the protein, antisera were generated to two sets of synthetic Apg9p peptides. Both Apg9p antisera recognized a band with a molecular mass of ∼125 kD by Western blotting analysis. The expression level of the protein was low in immunoblots prepared from wild-type yeast cell lysates and increased in a dose-dependent manner in apg9Δ cells transformed with the single copy or multicopy APG9 plasmid (Fig. 3 A). No signal was detected in the predicted mass range of Apg9p in the null mutant. These results indicate that the antisera specifically recognized the Apg9p protein. The expression level of Apg9p appeared constant under both vegetative growth and starvation conditions as examined by pulse/chase labeling and steady-state analyses (data not shown). In addition, no elevation in transcription was detected with microarray analysis during sporulation or stationary phase, conditions that are equivalent to starvation (DeRisi et al. 1997; Chu et al. 1998).
Figure 3.

Apg9p is an unglycosylated, integral membrane protein. A, Wild-type (WT; SEY6210), apg9Δ (JKY007), and the apg9Δ strain transformed with single copy (CEN) or multicopy (2μ) plasmids encoding APG9 were grown to log phase in SMD. Protein extracts were prepared and analyzed by immunoblot using antiserum to Apg9p. Apg9p is detected as an ∼125-kD protein. B, Hydrophobicity analysis of Apg9p based on the Kyte-Doolittle algorithm (Kyte and Doolittle 1982) predicts six to eight transmembrane domains. C, Cells of wild-type strain YW5-1B were grown in YPD, converted to spheroplasts, and osmotically lysed as described in Materials and Methods. After a preclearing step to remove cell debris, the lysates were incubated with or without 1% Triton X-100 on ice for 30 min, as indicated. Centrifugation at 10,000 g for 1 h separated the lysate into supernatant (S) and pellet (P) fractions, which were analyzed by immunoblots with antiserum to Apg9p. Apg9p is found in the low-speed pellet in the absence of Triton X-100. Detergent extraction completely solubilizes Apg9p and causes it to appear in the supernatant fraction. D, Strain CTD1 harboring a multicopy 3×HA APG9 plasmid was grown in YPD and converted to spheroplasts as described in Materials and Methods. A crude lysate was prepared and treated with or without endo H as indicated overnight at 37°C and analyzed by immunoblots with anti-HA antibodies or anti-CPY serum. Endo H treatment removes the N-linked glycosyl residues of CPY, causing a shift in its electrophoretic mobility. In contrast, Apg9p is not glycosylated and remains unchanged in the presence or absence of endo H treatment.
Hydrophobicity analysis of the Apg9p sequence predicts six to eight membrane-spanning regions with significant hydrophilic domains of ∼35 and ∼25 kD at the NH2 and COOH termini, respectively (Fig. 3 B). To assess whether Apg9p is an integral membrane protein, a detergent extraction procedure was performed. Wild-type cells were converted to spheroplasts, osmotically lysed in conditions that disrupt the plasma membrane but preserve vacuole membrane integrity, and incubated in a lysis buffer with or without 2% Triton X-100. After a 10,000 g centrifugation step, Apg9p appeared predominantly in the pellet fraction in the absence of detergent (Fig. 3 C). The detergent extraction procedure solubilized Apg9p, resulting in its appearance in the supernatant fraction. These results, coupled with the hydrophobicity analysis, suggest that Apg9p is an integral membrane protein.
Most integral membrane proteins enter the ER translocon complex before being directed via part of the secretory machinery to their final intracellular destinations. During transit through the secretory pathway, many integral membrane proteins undergo posttranslational modification in the ER and Golgi complex including the addition of glycosyl residues. Although the molecular mass of Apg9p did not appear to change over the course of a pulse/chase labeling experiment (data not shown), we wanted to determine if Apg9 was glycosylated. Cell lysates were treated with endo H to cleave potential N-linked oligosaccharides from Apg9p. As a control, the glycosylated protein CPY was also examined. There was a significant shift in size between untreated and endo H-treated samples of CPY (Fig. 3 D). In contrast, Apg9p did not demonstrate a change in electrophoretic mobility upon endo H treatment, suggesting that Apg9p does not contain any N-linked glycosyl modifications.
Subcellular Fractionation and Sucrose Density Analysis of Apg9p
The pelletability and detergent extraction of Apg9p suggested that it is localized to a membrane compartment. Membrane association was also supported by differential centrifugation procedures in which Apg9p appeared in both low- and high-speed pellet fractions (data not shown). The distribution of Apg9p to either the low- or high-speed pellet was partially dependent on lysis buffer conditions (data not shown). To further examine the identity of the Apg9p membrane compartment, cell lysates were analyzed by sucrose density gradients (Fig. 4). Spheroplasts from wild-type cells were osmotically lysed and, after removal of cell debris, the precleared lysates were subjected to density gradient centrifugation. The gradient was comprised of ten sucrose steps ranging from 18–54% (wt/wt). After centrifugation at 174,000 g for 2.5 h, 16 fractions were collected from the top and examined by immunoblot analysis (Fig. 4). The peak concentration of Apg9p appeared in the light fractions three to five, while vacuoles fractionate to the top of the gradients as indicated by the vacuole membrane marker alkaline phosphatase (Pho8p; Fig. 4 A). Apg9p was not detected by immunoblot analysis of a purified vacuole preparation (data not shown), confirming the sucrose gradient findings. Thus, Apg9p does not appear to be a vacuolar membrane protein. Marker proteins for other organelles including ER (Sec12p), Golgi complex (Kex2p), and the plasma membrane (Pma1p) all failed to comigrate with the peak for Apg9p (Fig. 4). The endosomal marker Pep12p showed a partial cofractionation with Apg9p, but displayed a broad distribution across the gradient (Fig. 4 B). These findings suggest that Apg9p is localized to a membrane compartment distinct from known organelles of the endomembrane system, although we cannot exclude an overlap with the endosome.
Figure 4.

Apg9p does not comigrate with typical endomembrane markers in sucrose density gradients. A and B, Strain STY1 (pep4Δ) was grown in YPD to log-phase, and osmotically lysed. Lysates were precleared, loaded on a sucrose density gradient ranging from 18–55% (wt/wt), and centrifuged for 2.5 h at 174,000 g as described in Materials and Methods. Fractions were collected from the top (fraction number 1) to the bottom (fraction number 16). Fractions were subjected to SDS-PAGE and immunoblot with antiserum against Apg9p and: in A, Kex2p (trans Golgi network), Pho8p (vacuole), and Pma1p (plasma membrane); and in B, Sec12p (ER) and Pep12p (endosome). The graphs in panels A and B are from the same gradient, but are presented separately for clarity.
The finding that Apg9p did not colocalize with known endomembrane markers suggested that it may be on the autophagosomal membrane, for which there are no known integral membrane markers. To circumvent this problem, we examined the localization of Apg9p in relation to prAPI in a strain with a chromosomal deletion of the rab GTPase Ypt7p on sucrose density gradients. A null mutation in YPT7 prevents Cvt and autophagic vesicles from fusing with the vacuole, resulting in the accumulation of prAPI inside Cvt vesicles and autophagosomes in the cytosol (Kim et al. 1999; Kirisako et al. 1999). Therefore, density gradients should resolve Apg9p and prAPI to the same peak fractions if they are colocalized on or within Cvt/autophagic vesicles in the ypt7Δ strain. Spheroplasts from ypt7Δ cells grown to midlog stage were osmotically lysed and, after removal of cell debris, the precleared lysates were subjected to centrifugation at 100,000 g to remove soluble prAPI. The resulting pellet fraction was subjected to equilibrium density gradient centrifugation. After a 16-h centrifugation, 16 fractions were collected and examined with antisera to API and Apg9p (Fig. 5). The peak for prAPI appeared in fraction 7, whereas the peak for Apg9p migrated to fractions 8 and 9. These results suggest that Apg9p does not colocalize with the transport vesicles that deliver prAPI to the vacuole.
Figure 5.

Apg9p does not comigrate with prAPI in a strain that accumulates Cvt/autophagic vesicles. Strain TK415 (ypt7Δ) was grown in YPD to log-phase, and osmotically lysed. Lysates were precleared, centrifuged at 100,000 g for 1 h, and the pellet fraction resuspended and centrifuged a second time. The resulting pellet fraction was subsequently loaded on a sucrose density gradient ranging from 18–55% (wt/wt), and centrifuged for 16 h at 174,000 g as described in Materials and Methods. 16 fractions were collected starting from the top and subjected to SDS-PAGE and immunoblot with antisera against Apg9p and API. The graph represents an average of two experiments.
Apg9p Localizes to Punctate Structures that Appear Proximal to the Vacuole
The sucrose gradient analysis indicated that the Apg9p fractionation pattern did not correspond to any endomembrane marker proteins or with Cvt vesicles. Therefore, we wanted to understand its subcellular localization from a morphological perspective. Along these lines, we first examined Apg9p by immunofluorescence microscopy. The apg9Δ strain was transformed with a multicopy plasmid encoding HA-tagged Apg9p. The 3×HA APG9 plasmid rescues the prAPI processing defect in the apg9Δ strain, indicating that the fusion protein is functional (data not shown). The apg9Δ strain harboring the 3×HA APG9 plasmid was fixed and labeled with an anti-HA mAb, followed by a FITC-conjugated anti-mouse secondary antibody as described in Materials and Methods. Under vegetative growth conditions, one or two prominent punctate structures were detected per cell. In addition, these punctate structures appeared to be adjacent to the vacuole in most cases (Fig. 6). When cells were subjected to starvation conditions in the presence of PMSF, autophagic bodies accumulated inside of the vacuole. However, the Apg9p punctate structures persisted outside of the vacuole, suggesting that Apg9p is not a constituent of the autophagic body vesicle and does not enter the vacuole (Fig. 6).
Figure 6.
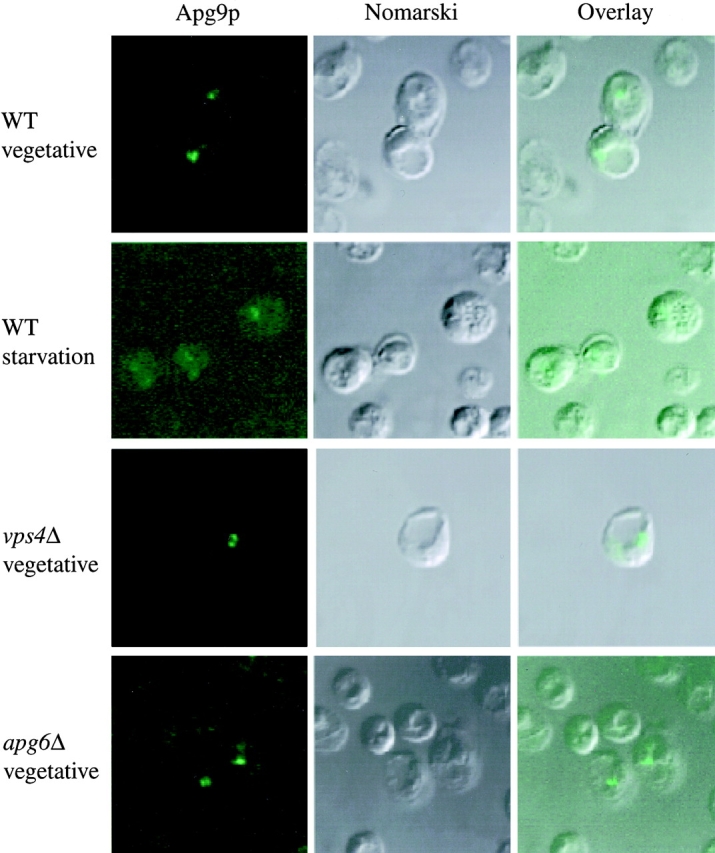
Apg9p localizes to large perivacuolar punctate structures. Immunofluorescence microscopy of strains CTD1 (apg9Δ), KSL12C (vps4Δ), and SKD6-1D (apg6Δ) transformed with the multicopy plasmid 3×HA APG9. Cells were grown in YPD to log phase (vegetative), and incubated further in SD(−N) medium with 1 mM PMSF for 3 h (starvation). The cells were fixed with formaldehyde and examined by immunofluorescence microscopy as described in Materials and Methods. Anti-HA and FITC-conjugated antibodies mark Apg9p (left), and the vacuole can be visualized by Nomarski optics (middle). An overlay is shown in the right panels.
The sucrose gradient data indicated that Apg9p shows partial cofractionation with the endosomal/prevacuolar compartment marker Pep12p. To further study this overlap, we examined the localization of Apg9p in the vps4Δ strain. The vps4Δ strain is a member of the class E vps mutants exhibiting the phenotype of accumulated endosomal structures (Babst et al. 1997, Babst et al. 1998; Finken-Eigen et al. 1997; Shirahama et al. 1997). We reasoned that if Apg9p is localized to an endosomal structure, then the accumulation of this compartment in vps4Δ would also result in a corresponding enlargement of the Apg9p punctate structures. However, no increase in the size of Apg9p-labeled punctate structures was detected (Fig. 6). In addition, we examined the localization of Apg9p in the apg6Δ mutant. The APG6 gene was identified as being essential for autophagy and was shown to be identical to VPS30 (Kametaka et al. 1998). Vps30p is required for recycling of the CPY receptor Vps10p from the endosome to the Golgi complex. The vps30 mutation results in mislocalization of the integral membrane Vps10p to the vacuole. However, there was no significant change in the immunofluorescence pattern of Apg9p in this mutant strain (Fig. 6). Taken together, these results indicate that Apg9p is localized to large punctate structures that appear to be near the vacuole, but do not overlap with the vps4Δ- or apg6Δ-enhanced prevacuolar compartment.
The in situ data obtained from indirect labeling of Apg9p by immunofluorescence microscopy indicated that Apg9p was primarily restricted to patches near the vacuolar membrane. We decided to extend our analysis of localization by examining Apg9p in vivo. To investigate the subcellular location of Apg9p in vivo, we constructed a fusion of GFP to the COOH terminus of Apg9p, and examined the localization by confocal fluorescence microscopy. Both the single copy and multicopy plasmids expressing Apg9pGFP complement the API processing defect in the apg9Δ strain, suggesting that the fusion protein is functional (data not shown). As with immunofluorescence microscopy, Apg9pGFP could only be detected reliably when expressed with the multicopy plasmid, pAPG9GFP(426).
Wild-type and apg9Δ cells transformed with pAPG9GFP(426) were grown to midlog and examined by both conventional and confocal microscopy. To evaluate the location of the Apg9pGFP structures relative to the vacuoles, FM 4-64, a lipophilic styryl dye, was used to label this organelle (Vida and Emr 1995). In nearly all cases, one to several prominent Apg9pGFP punctate structures were detected juxtaposed to the FM 4-64–labeled vacuoles (Fig. 7). However, in addition to these perivacuolar structures, smaller punctate structures could also be detected throughout the cytosol. These observations suggest that Apg9p may not be exclusively localized in large structures near the vacuole, but may also be distributed in smaller membrane compartments/vesicles throughout the cytoplasm (Fig. 7). Consistent with the biochemical and immunomicroscopy data, Apg9pGFP appears to be excluded from the vacuolar lumen.
Figure 7.
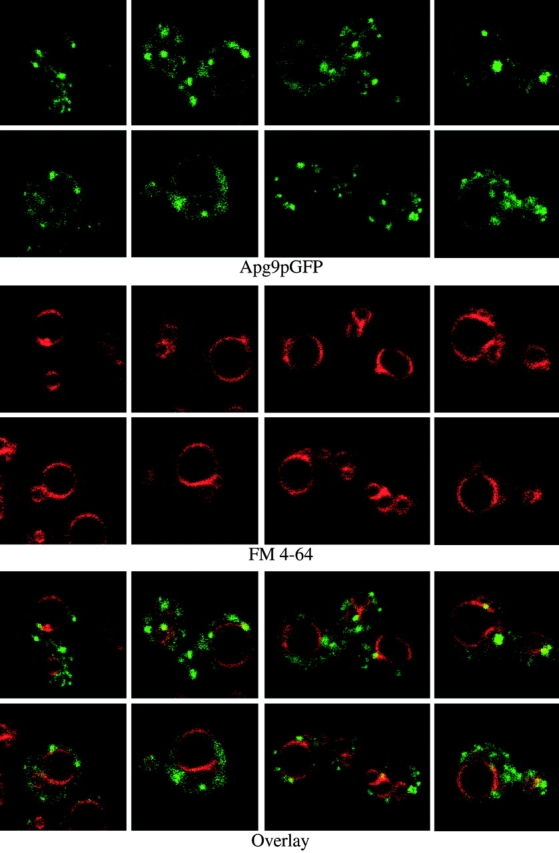
In vivo examination of the Apg9pGFP fusion protein. Wild-type (SEY6210) cells expressing Apg9pGFP from a multicopy plasmid were grown in SMD to midlog phase and labeled with FM 4-64 to stain the vacuoles as described in Materials and Methods. The labeled cells were viewed directly with a Leica DM IRB confocal microscope. Large, punctate Apg9pGFP structures appear juxtaposed to the FM 4-64–labeled vacuole membranes. In addition, smaller punctate structures appear throughout the cytoplasm.
To obtain ultrastructural information about the identity of the Apg9p structures, we examined the localization of Apg9p by immunoelectron microscopy. Thin sections of the apg9Δ strain harboring the 3×HA APG9 plasmid were labeled with an anti-HA mAb, followed by gold particle-conjugated anti-mouse secondary antibody and examined by EM. In agreement with the immunofluorescence and Apg9pGFP results, the immunogold particles were strongly concentrated to one or two structures adjacent to the vacuolar membrane (Fig. 8). Weaker immunogold labeling also appeared dispersed throughout the cell. Significantly, the immunogold labeling did not appear to encircle any vesicular structures and was not found to coincide with the autophagic bodies that accumulated in the vacuole lumen in a pep4Δ strain (Fig. 8 A) or on autophagosomes in the cytoplasm (Fig. 8 B), supporting the Apg9p density gradient analysis (Fig. 5). Taken together, these biochemical and morphological findings suggest that Apg9p may reside in a perivacuolar location that is largely distinct from known endomembrane compartments.
Figure 8.
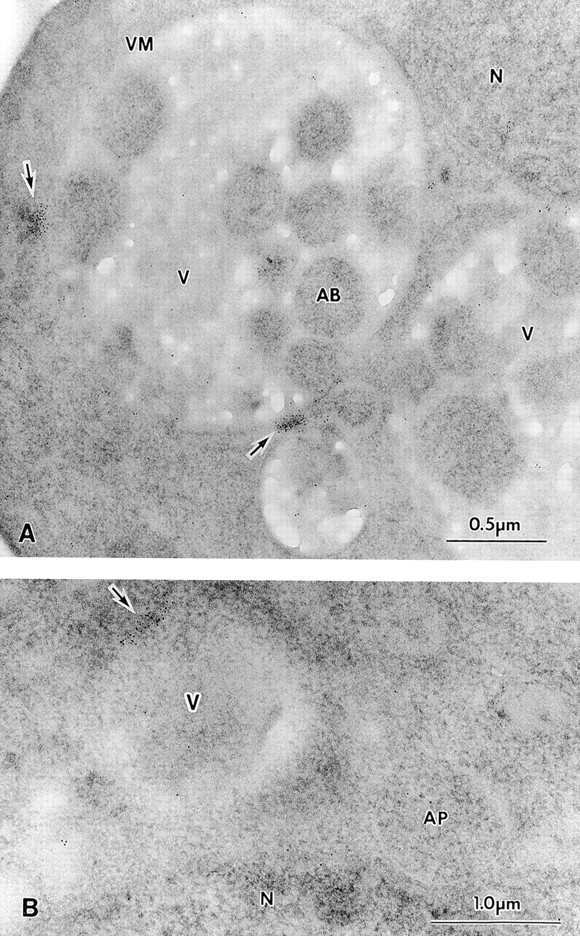
Ultrastructural localization of Apg9p for immunoelectron microscopy. Strain STY1 (pep4Δ) expressing 3×HA Apg9p was grown in YPD to log phase and transferred to SD(−N) medium for 2 h. The cells were fixed and stained with anti-HA antibody and 5-nm colloidal gold-conjugated goat anti–mouse IgG as described in Materials and Methods. A, Section showing autophagic bodies in the vacuole lumen. B, Section showing a cytosolic autophagosome. Apg9p is seen in patches near the vacuole, but not on autophagic bodies or autophagosomes. AB, autophagic body; AP, autophagosome; N, nucleus; V, vacuole; VM, vacuolar membrane. Arrows point to areas of concentrated Apg9p.
Apg9p Functions Early in the Autophagy and Cvt Pathways
We have determined that Apg9p is an integral membrane protein required for both the Cvt and autophagy pathways. It is localized to a dense membrane fraction that does not appear to comigrate with known organellar marker proteins. The subcellular localization of Apg9p appears to be in large punctate structures adjacent to the vacuole, but also within smaller punctate structures distributed throughout the cytosol. We next wanted to investigate the direct function of Apg9p in the Cvt and autophagy pathways. The first indication of the step at which Apg9p may function came from a vesicle test of the apg9Δ strain (Fig. 9 A). Under vegetative growth conditions when autophagy is not induced, both apg9Δ and wild-type cells appear devoid of autophagic bodies in the vacuole. However, when these cells are starved in the presence of PMSF, which prevents the breakdown of autophagic bodies, only wild-type cells or mutant strains defective in subvacuolar vesicle breakdown accumulate these intravacuolar vesicles. The lack of autophagic bodies in the apg9Δ strain is rescued by the plasmid-borne copy of APG9 (data not shown). This observation suggests that Apg9p acts early in the autophagy pathway, before the fusion of the autophagosomes with the vacuole and the release of the autophagic bodies into the vacuolar lumen.
Figure 9.
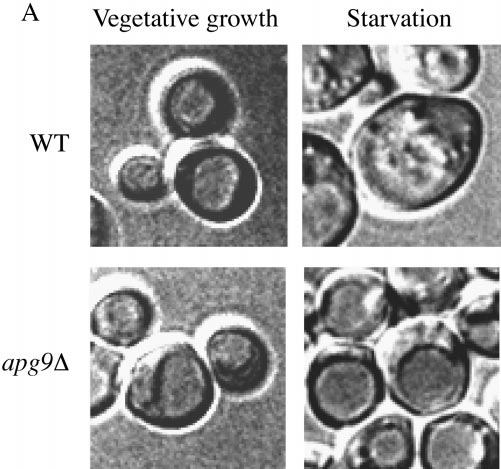

Vesicle accumulation test and kinetic analysis of prAPI import in the temperature conditional Apg9p mutant. A, Morphological analysis of apg9Δ indicates an import defect at an early step in the pathway. Wild-type (YW5-1B) and apg9Δ (CTD1) cells were grown in YPD (vegetative) or transferred to SD(−N) (starvation) for 3 h in the presence of PMSF (starvation) and examined by DIC (Nomarski) microscopy (Zeiss Axioplan). PMSF inhibits the degradation of autophagic bodies. Under starvation conditions, wild-type cells accumulate autophagic bodies when vesicle breakdown is blocked. However, under the same conditions, apg9Δ cells do not accumulate autophagic bodies, indicating that Apg9p is required for a step before vesicle fusion and release of the autophagic body into the vacuolar lumen. The apg9Δ strain transformed with the APG9 plasmid shows the same result as wild-type (data not shown). B, The apg9ts strain is tightly blocked for prAPI import at nonpermissive temperature. Wild-type (SEY6210) and apg9Δ (JKY007) cells transformed with the apg9ts centromeric plasmid were incubated at 24 and 38°C for 5 min, pulse-labeled for 10 min, and then subjected to nonradioactive chase reactions for the indicated times. Samples at each time point were immunoprecipitated with antiserum to API and resolved by SDS-PAGE as described in Materials and Methods. API-immunoprecipitated bands were quantified by a Molecular Dynamics STORM PhosphorImager and the results are presented in the graph. The percent mature API was determined by dividing the mature API value over the sum of the precursor and mature API values for each time point.
The permanent loss of function in the apg9Δ null mutant may result in phenotypes that are indirect. To determine the direct effects of Apg9p loss of function, we generated a temperature conditional mutant of Apg9p by a PCR procedure that randomly incorporated mutations in the APG9 ORF (see Materials and Methods). The temperature-sensitive apg9 mutant (apg9ts) was originally isolated based on an immunoblot procedure selecting for mutants that accumulated prAPI at nonpermissive temperature. A kinetic analysis of apg9ts was subsequently performed by radioactive pulse labeling, followed by nonradioactive chase reactions at the indicated times (Fig. 9 B). The apg9ts mutant displayed near wild-type prAPI maturation kinetics at permissive temperature. However, at nonpermissive temperature, prAPI import was tightly blocked in the apg9ts strain (Fig. 9 B). The immediate block in prAPI processing suggested that Apg9p acts directly within the Cvt pathway.
Apg9p Is Required for the Vesicle Formation Step
To determine the step of the Cvt pathway/autophagy in which Apg9p functions, we analyzed the state of prAPI in the apg9ts mutant after shift to nonpermissive temperature. In the first steps of import, prAPI oligomerizes into a dodecamer and then assembles into a higher order pelletable Cvt complex (Baba et al. 1997; Kim et al. 1997; Fig. 1). In wild-type cells, pelletability of the Cvt complex is salt dependent. Accordingly, we first examined the nature of prAPI binding and pelleting. In wild-type cells, prAPI is pelletable at 5,000 g when cell lysates are prepared in the presence of physiological salt concentrations. In contrast, prAPI is located in a soluble fraction in the absence of salt. To examine salt dependent pelletability of prAPI in the apg9ts strain, spheroplasts of apg9ts were pulse-labeled for 10 min, followed by a chase reaction of 30 min, all at nonpermissive temperature. The labeled spheroplasts were osmotically lysed in a buffer containing no salt or a physiological concentration of salt, and separated into low-speed supernatant and pellet fractions (Fig. 10 A). In lysis conditions containing no salt, the majority of prAPI is soluble, whereas it appears in the pellet fraction in the apg9ts strain when salt is introduced into the osmotic lysis conditions. This result indicates that apg9ts retains the salt dependent binding and pelleting of prAPI. In addition, the pelletable nature of prAPI suggests that prAPI is able to form the Cvt complex in the apg9ts strain.
Figure 10.


Analysis of the apg9ts strain indicates Apg9p is directly required for the vesicle formation step. A, The nature of prAPI binding and pelleting in the apg9ts strain. Spheroplasts of apg9ts were labeled for 10 min and chased for 30 min at 38°C. The spheroplasts were osmotically lysed in a buffer containing no salt (20 mM Pipes, pH 6.8) or a physiological concentration of salt (100 mM KOAc, 50 mM KCl, 5 mM MgCl2, 20 mM Pipes, pH 6.8) and pelleted at 5,000 g. All samples were immunoprecipitated with antiserum to API and the cytosolic marker PGK. apg9ts retains the salt dependent binding and pelleting of prAPI. B, prAPI in the apg9ts strain accumulates in both a membrane-associated state and a large pelletable complex. Spheroplasts of apg9ts and ypt7Δ were labeled for 10 min and chased for 30 min at 38°C. The labeled spheroplasts were then osmotically lysed and separated into supernatant (S) and pellet (P) fractions by centrifugation at 5,000 g for 5 min. An aliquot was removed for the total lysate control (T). The pellet fraction (P) was resuspended in 15% Ficoll-400 in gradient buffer (20 mM Pipes, 5 mM MgCl2, complete EDTA-free protease inhibitor cocktail) in the presence or absence of Triton X-100 and overlaid with 13 and 2% Ficoll-400 in gradient buffer. The step gradients were centrifuged at 13,000 g for 10 min. Membrane-containing float (F), nonfloat (NF), and pellet (P2) fractions were immunoprecipitated with antiserum to API as described in Materials and Methods. The position of prAPI is indicated. C, prAPI in the apg9ts strain is protease accessible. Spheroplasts isolated from apg9ts and ypt7Δ cells were pulse-labeled for 10 min and chased for 30 min at 38°C. The labeled spheroplasts were then osmotically lysed and separated into low-speed supernatant (S) and pellet (P) fractions after a 5,000 g centrifugation step. The pellet fractions were subjected to protease treatment in the absence or presence of 0.2% Triton X-100 as described in Materials and Methods. The resulting samples were immunoprecipitated with antiserum to API and PGK. The immunoprecipitated bands were quantified by a Molecular Dynamics STORM PhosphorImager. The percent protease-protected prAPI was determined by dividing the value for prAPI in the protease-treated sample by the value for total prAPI before protease treatment. The percent PGK was determined by dividing the supernatant (S) or pellet (P) value over the sum of the S and P values.
Biochemical and EM findings indicate that the Cvt complex associates with a membrane source that eventually forms the double-membrane vesicle. To determine if the Cvt complex in the apg9ts strain contained membrane constituents, we performed membrane flotation gradients. In this analysis, membrane-associated proteins would “float” up the gradient (F), whereas soluble proteins would remain in the nonfloat (NF) fraction, and proteins associated with large protein complexes would be recovered as a gradient pellet (P2). In addition to the apg9ts strain, we also examined prAPI in the ypt7Δ strain by flotation gradients.
Spheroplasts of apg9ts and ypt7Δ were pulse-labeled for 10 min, followed by a 30-min chase reaction, all at nonpermissive temperature. The labeled spheroplasts were osmotically lysed and separated into 5,000 g supernatant (S) and pellet (P) fractions. The prAPI-containing pellet fractions were then subjected to flotation gradients (see Materials and Methods). In the apg9ts strain, the majority of prAPI was found in the float fraction with a significant amount also appearing in the P2 pellet fraction (Fig. 10 B). In the presence of detergent, prAPI appeared entirely in either the nonfloat (NF) or the pellet (P2) fraction. These results suggested prAPI in the apg9ts strain associated with a large pelletable complex possessing a membrane component that causes the complex to float. Detergent treatment solubilizes this membrane component and disrupts the flotation of the prAPI-containing complex. In addition, there was an increase in the amount of prAPI in the nonfloat fraction, suggesting that detergent treatment liberated a portion of prAPI from either the float fraction or the P2 pellet fraction into the nonfloat fraction. The flotation pattern of prAPI from the ypt7Δ strain appeared nearly identical to apg9ts (Fig. 10 B). A significant portion of prAPI was found in the float fraction, as well as in the P2 pellet fraction. However, unlike apg9ts, detergent treatment during flotation in the ypt7Δ strain caused prAPI to be localized almost entirely to the P2 fraction. These results suggest that the prAPI-associated complex in the float fractions may be structurally distinct in these two mutant strains.
We recently reported that prAPI in the ypt7Δ strain is protease-protected based on steady state immunoblot analysis (Kim et al. 1999). In addition, a ypt7 mutant strain accumulates autophagosomes in the cytosol, further demonstrating that Ypt7p acts at the step of vesicle fusion with the vacuole (Kirisako et al. 1999). Given that the flotation pattern of newly synthesized prAPI in the apg9ts strain is essentially identical to ypt7Δ, we wanted to determine if the membrane-association in apg9ts reflected enclosure in a completed vesicle. This would result in protection of prAPI from exogenously added proteases. Alternatively, if Apg9p is required for the completion of vesicle formation, then prAPI would be sensitive to protease treatment and processed to the mature form in the apg9ts strain. Spheroplasts of apg9ts and ypt7Δ were pulse-labeled for 10 min, followed by a 30-min chase reaction, all at nonpermissive temperature. After osmotic lysis and low-speed centrifugation, the prAPI-containing pellet fraction was subjected to proteinase K treatment in the presence or absence of detergent. In agreement with previous steady state data (Kim et al. 1999), the pulse-labeled population of prAPI in the ypt7Δ strain was protected from exogenously added protease and was digested only after detergent treatment (Fig. 10 C). In contrast, prAPI from apg9ts was completely sensitive to proteolysis, independent of detergent addition, suggesting that membrane had not completely enwrapped the Cvt complex in this strain. The small amount of protease resistant prAPI in the apg9ts strain probably reflects incomplete spheroplast lysis based upon localization of slightly elevated levels of the cytosolic marker PGK in the pellet fraction. Taken together, these results indicate that Apg9p plays a direct role in formation of the sequestering vesicle.
Discussion
The Stages of API Import Can Be Tested Experimentally
The import of prAPI can be delineated into discrete stages (Fig. 1; Kim et al. 1999). Many of these steps can be tested using straightforward biochemical and morphological assays. Thus, the model for prAPI import offers a convenient framework through which we can assign the role of a particular component in the Cvt and autophagy pathways based on the state of API in the respective mutant background. For example, in the first step of the pathway, the ability of prAPI to properly oligomerize in a given cvt or apg mutant can be detected by glycerol gradient analysis and by native gel electrophoresis measurements (Kim et al. 1997). While newly synthesized prAPI dodecamers are soluble, the Cvt complex, which results from the salt dependent binding and assembly of prAPI into a supermolecular structure, is easily pelletable by low-speed centrifugation (Kim et al. 1997). The next step of the pathway involves the formation of the sequestering membrane around the Cvt complex. Examining the flotation competence of prAPI through a step gradient can identify the presence of an enwrapping membrane (Kim et al. 1999; George et al. 2000). The subsequent completion of the sequestering membrane to form the Cvt vesicle or the autophagosome ensures the protection of prAPI from exogenous protease treatment (Scott et al. 1997). The successful fusion of the Cvt vesicles or the autophagosomes with the vacuole can be examined by observing the lumenal accumulation of the single-membrane Cvt or autophagic bodies in the presence of inhibitors for inner vesicle breakdown (e.g., PMSF; Takeshige et al. 1992; Scott et al. 1997). Finally, if the existence of these Cvt or autophagic bodies persists, even in the absence of inhibitors, then the defect can be assigned to the final step of vesicle breakdown (Scott et al. 1997). In addition, vesicles that accumulate at the pre- and postvacuole fusion stages can be biochemically purified in their respective mutant background (e.g., ypt7 and cvt17, respectively; Scott et al. 1997).
Apg9p Functions at the Step of Vesicle Formation
Using the methods outlined above, we sought to characterize the role of Apg9p in the Cvt and autophagy pathways. prAPI oligomerization and Cvt complex assembly were both normal in the apg9 mutant backgrounds (Kim et al. 1997; Fig. 10 A), suggesting that the role of Apg9p is downstream of these early import steps. Examination of apg9Δ under starvation conditions indicated that, unlike wild-type cells, apg9Δ does not accumulate autophagic bodies in the vacuole, even when inhibitors that prevent vesicle breakdown are added (Fig. 9 A). Therefore, the role of Apg9p appears to be before vesicle fusion to the vacuole and subsequent release of autophagic bodies into the vacuolar lumen. This restricted the possible steps of Apg9p function to the membrane sequestration event and the formation of the double-membrane around the Cvt complex.
To directly assess the role of Apg9p in the Cvt and autophagy pathways, we have isolated the first temperature-sensitive allele of an integral membrane protein involved in both autophagy and Cvt pathways. By investigating the state of prAPI in the apg9ts strain, we have determined the direct consequence that is caused by an immediate loss of Apg9p function on prAPI import. The apg9ts mutant displayed near wild-type prAPI import kinetics at permissive temperature, but exhibited a strong block in prAPI maturation at nonpermissive temperature (Fig. 9 B). Biochemical analysis of apg9ts at nonpermissive temperature indicated that prAPI was localized to a low-speed pellet in a salt dependent manner (Fig. 10 A), suggesting that the Cvt complex was able to form. This prAPI-bound pellet associates with a membrane component that permits it to float through a ficoll step gradient (Fig. 10 B). Finally, protease protection analysis of prAPI at nonpermissive temperature demonstrates that the vesicles that form around the Cvt complex are incomplete, allowing prAPI to be accessible to protease digestion (Fig. 10 C). Taken together, these findings all implicate a direct role of Apg9p in the completion of the autophagosome/Cvt vesicle around prAPI.
Apg9p Does Not Appear to Colocalize with Any Known Endomembrane Markers or with Cvt and Autophagic Vesicles
In this study, we have also begun the characterization of Apg9p to gain further insight into its actual function. Apg9p is an integral membrane protein with a molecular mass of 120–125 kD that lacks N-linked glycosyl modifications (Fig. 3). Differential centrifugation indicates that Apg9p sediments in both low- and high-speed pellet fractions, a distribution pattern that appears to be salt dependent (data not shown). Sucrose gradient analysis (Fig. 4) indicates Apg9p does not appear to comigrate with marker proteins, including Pma1p (plasma membrane), Kex2p (Golgi complex), and ALP (vacuole). Apg9p does share a partial overlap with the Pep12p endosome marker (Fig. 4 B); however, due to the broad nature of the Pep12p peak, we do not hold this result as conclusive evidence for partial colocalization between these two proteins. In addition, sucrose density gradients indicate that Apg9p does not appear to peak in the fractions in which prAPI-containing cytoplasmic vesicles migrate (Fig. 5).
Subcellular localization of Apg9p by immunofluorescence microscopy and immunogold EM (Fig. 6 and Fig. 8, respectively) display similar staining patterns, consisting of one to several punctate structures proximal to the vacuolar membrane, but excluded from cytoplasmic autophagosomes and vacuolar autophagic bodies. In vivo examination of Apg9p fused to GFP also revealed large punctate structures that appear adjacent to the vacuolar membrane, in agreement with the indirect in situ observations. In addition, smaller punctate structures of Apg9pGFP could be detected throughout the cytosol (Fig. 7). This discrepancy between the in situ and in vivo observations may simply be due to differences in sensitivity among the various procedures.
Identifying the subcellular location of Apg9p would facilitate defining the actual function of the protein. However, we could not conclusively assign Apg9p to a known membrane compartment. One possibility is that Apg9p is part of the sequestering membrane of the Cvt vesicle or the autophagosome, and that defects in Apg9p function prevent the homotypic fusion events necessary to complete vesicle formation. However, this view is problematic because Apg9p does not cofractionate with prAPI by sucrose gradients (Fig. 5). In addition, when Apg9pGFP-containing punctate structures are observed in real-time over the course of one to two hours, they remain proximal to the vacuole and were never seen fusing with the vacuole (data not shown). Taken together with the immunoelectron microscopy data showing a lack of Apg9p-labeled structures on autophagosomes and autophagic bodies (Fig. 8), Apg9p does not appear to be part of either the outer or inner vesicles of the Cvt vesicle or autophagosome.
The perivacuolar localization of Apg9p by microscopy suggests that it may be localized to an endosomal compartment. The involvement of the endosome in autophagy is also supported by other lines of evidence (Liou et al. 1997; Shirahama et al. 1997). However, in the vps4Δ mutant, which accumulates endosomal structures, and in the apg6Δ/vps30Δ mutant, which is required for both autophagy and the CPY vacuolar sorting pathway (Kametaka et al. 1998), no change in the Apg9p labeling pattern was detected (Fig. 6). Sucrose gradients also indicated that the endosomal marker Pep12p does not display complete comigration with Apg9p (Fig. 4). Nevertheless, we do not consider these experiments to be conclusive because the endosome represents the convergent point for multiple routes of endomembrane traffic, and as such, may constitute a heterogeneous compartment. Thus, the Pep12p-resident endosome may be distinct from the endosomal compartment in which Apg9p is localized (Becherer et al. 1996; Holthuis et al. 1998a,Holthuis et al. 1998b).
Freeze fracture electron micrographs of autophagosomes reveal a membrane surface pattern that appears distinct from other characterized yeast organelles (Baba et al. 1995). Therefore, we postulate that the donor compartment, which contributes the membranes that form the autophagosome, may also be a specialized compartment. Since Apg9p does not appear to be part of the Cvt vesicle or autophagosomal membrane (i.e., the acceptor compartment), it may be localized to this novel (undefined) donor compartment. However, because prAPI is membrane-associated in the apg9 mutant, it is possible that Apg9p is localized to an accessory compartment that participates in a later stage of the enwrapping process.
Apg9p Acts at the Rate-limiting Step in prAPI Import
Previous kinetic analyses indicated that the rate-limiting step in prAPI import occurs after oligomerization. The events occurring after the formation of the double-membrane vesicle may be rapid because vesicle fusion with the vacuole and the breakdown of the Cvt and autophagic bodies in the vacuolar lumen are rarely detected without the aid of inhibitors or mutant strain backgrounds. Thus, the majority of the import time may be devoted to the membrane sequestration and vesiculation steps. The prAPI phenotype seen in the apg9ts mutant is similar to that seen with other apg/cvt mutant strains, including apg7Δ (Kim et al. 1999). The Apg7 protein is a component of the novel protein conjugation system required for both pathways (Kim et al. 1999; Tanida et al. 1999). By extension, the other components of the protein conjugation system may also be involved in this step of the pathway, including Apg12p, Apg5p, Apg16p, and Apg10p (Mizushima et al. 1998, Mizushima et al. 1999; Shintani et al. 1999). Our recent finding that Apg5p directly participates in the step of vesicle formation supports this hypothesis (George et al. 2000). Taken together, these findings suggest that the step of vesicle formation may constitute the rate-limiting step in the pathway, requiring the concerted action of a large number of Cvt/autophagy components, including the conjugation system and Apg9p.
This is the first report of an integral membrane protein directly involved in the formation of the Cvt vesicle and autophagosome. Future analysis of the Apg9p-localized compartment will provide a pivotal basis for investigating the mechanism by which it directly participates in the vesicle sequestration events.
Acknowledgments
The authors would like to thank C.-Y. Yu, A. Nuygyen, M. George, A. Hefner-Gravink, T. Yoshimori, and N. Ishihara for technical assistance. We are grateful to M. Osumi for the use of EM facilities.
This work was supported by Public Health Service grant GM53396 from the National Institutes of Health to D.J. Klionsky, by a National Institutes of Health Molecular and Cellular Biology training grant to J. Kim, and by Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan to Y. Ohsumi and T. Noda.
Footnotes
Takeshi Noda and John Kim contributed equally to this work.
Abbreviations used in this paper: API, aminopeptidase I; CPY, carboxypeptidase Y; Cvt, cytoplasm to vacuole targeting; endo H, endoglycosidase H; GFP, green fluorescent protein; ORF, open reading frame; PGK, phosphoglycerate kinase; prAPI, precursor API.
References
- Baba M., Takeshige K., Baba N., Ohsumi Y. Ultrastructural analysis of the autophagic process in yeastdetection of autophagosomes and their characterization. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Osumi M., Ohsumi Y. Analysis of the membrane structures involved in autophagy in yeast by freeze-replica method. Cell Struct. Funct. 1995;20:465–471. doi: 10.1247/csf.20.465. [DOI] [PubMed] [Google Scholar]
- Baba M., Osumi M., Scott S.V., Klionsky D.J., Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M., Sato T.K., Banta L.M., Emr S.D. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M., Wendland B., Estepa E.J., Emr S.D. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum P., Thorner J., Honig L. Identification of tubulin from the yeast Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA. 1978;75:4962–4966. doi: 10.1073/pnas.75.10.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherer K.A., Rieder S.E., Emr S.D., Jones E.W. Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol. Biol. Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart E.F.C., Luiken J.J.F.P., Meijer A.J. Autophagic proteolysiscontrol and specificity. Histochem. J. 1997;29:365–385. doi: 10.1023/a:1026486801018. [DOI] [PubMed] [Google Scholar]
- Chu S., DeRisi J., Eisen M., Mulholland J., Botstein D., Brown P.O., Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Darsow T., Rieder S.E., Emr S.D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi J.L., Iyer V.R., Brown P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Dunn W.A., Jr. Studies on the mechanisms of autophagyformation of the autophagic vacuole. J. Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finken-Eigen M., Rohricht R.A., Kohrer K. The VPS4 gene is involved in protein transport out of a yeast pre-vacuolar endosome-like compartment. Curr. Genet. 1997;31:469–480. doi: 10.1007/s002940050232. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G., Stevens T.H. The Saccharomyces cerevisiae vesicle-associated soluble NSF attachment protein receptor Vti1p is required for multiple membrane transport pathways to the vacuole. J. Cell Biol. 1999;10:1719–1732. doi: 10.1091/mbc.10.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George M.D., Baba M., Scott S.V., Mizushima N., Garrison B.S., Ohsumi Y., Klionsky D.J. Apg5p functions in the sequestration step in the cytoplasm to vacuole targeting and macroautophagy pathways. Mol. Biol. Cell. 2000;In press doi: 10.1091/mbc.11.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.M., Morano K.A., Scott S.V., Klionsky D.J. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.M., Hefner-Gravink A., Thumm M., Klionsky D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols B.J., Dhruvakumar S., Pelham H.R. Two syntaxin homologues in the TGN/endosomal system of yeast EMBO (Eur. Mol. Biol. Organ.) J. 17 1998. 113 126a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J.C.M., Nichols B.J., Pelham H.R.B. The syntaxin Tlg1p mediates trafficking of chitin synthase III to polarized growth sites in yeast Mol. Biol. Cell. 9 1998. 3383 3397b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.-P., Scott S.V., Kim J., Klionsky D.J. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J. Biol. Chem. 2000;In press doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- Kadowaki M., Venerando R., Miotto G., Mortimore G.E. Mechanism of autophagy in permeabilized hepatocytesevidence for regulation by GTP binding proteins. Adv. Exp. Med. Biol. 1996;389:113–119. [PubMed] [Google Scholar]
- Kametaka S., Okano T., Ohsumi M., Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae . J. Biol. Chem. 1998;273:22284–22291. doi: 10.1074/jbc.273.35.22284. [DOI] [PubMed] [Google Scholar]
- Kim J., Scott S.V., Oda M., Klionsky D.J. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1997;137:609–618. doi: 10.1083/jcb.137.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Dalton V., Eggerton K.P., Scott S.V., Klionsky D.J. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell. 1999;10:1337–1351. doi: 10.1091/mbc.10.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Scott S.V., Klionsky D.J. Alternative protein sorting pathways. Int. Rev. Cytol. 2000;In press doi: 10.1016/s0074-7696(00)98005-7. [DOI] [PubMed] [Google Scholar]
- Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Banta L.M., Emr S.D. Intracellular sorting and processing of a yeast vacuolar hydrolaseproteinase A propeptide contains vacuolar targeting information. Mol. Cell Biol. 1988;8:2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Cueva R., Yaver D.S. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lang T., Schaeffeler E., Bernreuther D., Bredschneider M., Wolf D.H., Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3597–3607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou W., Geuze H.J., Geelen M.J., Slot J.W. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997;136:61–70. doi: 10.1083/jcb.136.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A., Tsukada M., Wada Y., Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae . Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Ohsumi Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimore G.E., Miotto G., Venerando R., Kadowaki M. Autophagy. Subcell. Biochem. 1996;27:93–135. doi: 10.1007/978-1-4615-5833-0_4. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Hunter R., Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Nakamura N., Matsuura A., Wada Y., Ohsumi Y. Acidification of vacuole is required for the autophagic degradation in the yeast Saccharomyces cerevisiae . J. Biochem. 1997;121:338–344. doi: 10.1093/oxfordjournals.jbchem.a021592. [DOI] [PubMed] [Google Scholar]
- Noda T., Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Noda T., Matsuura A., Wada Y., Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae . Biochem. Biophys. Res. Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky D.J., Banta L.M., Emr S.D. Protein sorting in Saccharomyces cerevisiaeisolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Novick P., Thomas J.H., Botstein D., Fink G.R. A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Sakai Y., Koller A., Rangell L.K., Keller G.A., Subramani S. Peroxisome degradation by microautophagy in P. pastorisidentification of specific steps and morphological intermediates. J. Cell Biol. 1998;141:625–636. doi: 10.1083/jcb.141.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.K., Darsow T., Emr S.D. Vam7p, a SNAP-25–like molecule, and Vam3p, a syntaxin homolog, function together in yeast vacuolar protein trafficking. Mol. Cell. Biol. 1998;18:5308–5319. doi: 10.1128/mcb.18.9.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Klionsky D.J. In vitro reconstitution of cytoplasm to vacuole protein transport in yeast. J. Cell Biol. 1995;131:1727–1735. doi: 10.1083/jcb.131.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Hefner-Gravink A., Morano K.A., Noda T., Ohsumi Y., Klionsky D.J. Cytoplasm to vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc. Natl. Acad. Sci. USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S.V., Baba M., Ohsumi Y., Klionsky D.J. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J. Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen P.O., Berg T.O., Blankson H., Fengsrud M., Holen I., Stromhaug P.E. Structural aspects of autophagy. Adv. Exp. Med. Biol. 1996;389:103–111. doi: 10.1007/978-1-4613-0335-0_12. [DOI] [PubMed] [Google Scholar]
- Shintani T., Mizushima N., Ogawa Y., Matsuura A., Noda T., Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahama K., Noda T., Ohsumi Y. Mutational analysis of Csc1/Vps4pinvolvement of endosome in regulation of autophagy in yeast. Cell Struct. Funct. 1997;22:501–509. doi: 10.1247/csf.22.501. [DOI] [PubMed] [Google Scholar]
- Stromhaug P.E., Berg T.O., Fengsrud M., Seglen P.O. Purification and characterization of autophagosomes from rat hepatocytes. Biochem. J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige K., Baba M., Tsuboi S., Noda T., Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Mizushima N., Kiyooka M., Ohsumi M., Ueno T., Ohsumi Y., Kominami E. Apg7p/Cvt2pa novel protein-activating enzyme essential for autophagy. Mol. Biol. Cell. 1999;10:1367–1379. doi: 10.1091/mbc.10.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumm M., Egner R., Koch M., Schlumpberger M., Straub M., Veenhuis M., Wolf D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae . FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- Tooze J., Hollinshead M., Ludwig T., Howell K., Hoflack B., Kern H. In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. J. Cell Biol. 1990;111:329–345. doi: 10.1083/jcb.111.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M., Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae . FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Tuttle D.L., Dunn W.A., Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris . J. Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Vida T.A., Emr S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota S., Himeno M., Roth J., Brada D., Kato K. Formation of autophagosomes during degradation of excess peroxisomes induced by di-(2-ethylhexyl)phthalate treatment. II. Immunocytochemical analysis of early and late autophagosomes. Eur. J. Cell Biol. 1993;62:372–383. [PubMed] [Google Scholar]
- Yuan W., Stromhaug P.E., Dunn W.A. Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol. Biol. Cell. 1999;10:1353–1366. doi: 10.1091/mbc.10.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]