

Abstract
In human epidermal keratinocytes, replicative senescence, is determined by a progressive decline of clonogenic and dividing cells. Its timing is controlled by clonal evolution, that is, by the continuous transition from stem cells to transient amplifying cells. We now report that downregulation of 14-3-3σ, which is specifically expressed in human stratified epithelia, prevents keratinocyte clonal evolution, thereby forcing keratinocytes into the stem cell compartment. This allows primary human keratinocytes to readily escape replicative senescence. 14-3-3σ–dependent bypass of senescence is accompanied by maintenance of telomerase activity and by downregulation of the p16INK4a tumor suppressor gene, hallmarks of keratinocyte immortalization. Taken together, these data therefore suggest that inhibition of a single endogenous gene product fosters immortalization of primary human epithelial cells without the need of exogenous oncogenes and/or oncoviruses.
Keywords: telomerase, stem cells, transformation, senescence, immortalization, p16INK4a
Introduction
In human epidermis, cell proliferation and differentiation are compartmentalized and tightly regulated. Keratinocytes endowed with proliferative capacity are located in the basal layer. Basal cells that become committed to terminal differentiation exit from the cell cycle, migrate upwards, and initiate the process of keratinization, which leads to the sequential formation of stratum spinosum, stratum granulosum, and stratum corneum, the latter being composed of anucleate corneocytes continuously shed into the environment. (Green 1980; Fuchs 1990; Roop 1995). For each corneocyte shed from the epidermal surface, a basal keratinocyte must undergo a round of cell division to replace it. To accomplish this self-renewal process, epidermis relies on the presence of stem and transient amplifying cells (Lavker and Sun 1983; Barrandon 1993; Watt 1998; Fuchs and Segre 2000). Stem cells can be defined as cells endowed with a high capacity for cell division and the ability to generate differentiated progeny (Lajtha 1979; Morrison et al. 1997; Fuchs and Segre 2000). Transient amplifying cells, which arise from stem cells, have a lower proliferative capacity and represent the largest group of dividing cells (Barrandon 1993). Both cell types can be cultivated (Barrandon and Green 1987; Jones and Watt 1993; Rochat et al. 1994; Pellegrini et al. 1999a), allowing the permanent coverage of large skin and mucosal defects with cohesive sheets of autologous cultured epithelium (Gallico et al. 1984; Pellegrini et al. 1997, Pellegrini et al. 1999b). Clonal evolution, the transition from stem cells to transient amplifying cells, is a continuous unidirectional process that occurs during natural ageing and wound healing, as well as during repeated keratinocyte subcultivation (Barrandon and Green 1987; Barrandon 1993; Campisi 1998; Lehrer et al. 1998; Watt 1998; Pellegrini et al. 1999a). Clonal evolution is instrumental in building up the epidermal structure, since transient amplifying cells eventually generate postmitotic terminally differentiated keratinocytes, which migrate upwards and form suprabasal epidermal layers (Barrandon 1993; Watt 1998; Fuchs and Segre 2000).
When somatic cells register a preset number of generations, they enter into an irreversible state of growth arrest, referred to as replicative senescence (Hayflick 1965; for review see Wynford-Thomas 1999). In human epidermis, senescence is determined by a progressive decline in the proportion of clonogenic and dividing cells, and its timing is controlled by clonal evolution and a rather precise cell-doubling clock (Barrandon and Green 1987; Rochat et al. 1994; Pellegrini et al. 1999a). The progressive erosion of chromosome telomeres occurring during cell replication and the activation of tumor suppressor genes, such as p53, p21CIP1/WAF1, and p16INK4a, are considered essential steps in determining the exit of senescent cells from the cell cycle (for review see Campisi 1996; Jacks and Weinberg 1998; Reddel 1998; Sedivy 1998; de Lange and DePinho 1999; Greider 1999; Lustig 1999; Wynford-Thomas 1999). This links the control of the proliferative capacity of somatic cells to the control of the cell cycle, and hence to the initial steps leading to the onset and development of cancer (see Paulovich et al. 1997; Orr-Weaver and Weinberg 1998).
14-3-3σ (also called HME1 or stratifin and hereafter referred to as σ) belongs to a large group of highly conserved and homologous dimeric proteins, expressed in every mammalian tissue, Xenopus laevis, Drosophila melanogaster, plants, and yeasts (Aitken et al. 1992). 14-3-3 proteins elicit their biochemical functions by binding to a variety of cellular proteins containing specific phosphoserine motifs (Muslin et al. 1996), and therefore have been associated with a diverse number of biochemical processes (Aitken 1996). For instance, binding of 14-3-3 to serine-phosphorylated Raf is essential for Raf kinase activity, and hence for the Ras/MAPK signaling cascade (Roberts et al. 1997; Tzivion et al. 1998), whereas 14-3-3 binding to the serine-phosphorylated death agonist, BAD, interferes with apoptosis (Zha et al. 1996). Recently, 14-3-3 proteins have been directly associated with the control of the cell cycle. In colorectal cancer cells, exposure to DNA damaging agents results in a p53-dependent induction of σ, which, in turn, arrests cells in the G2/M phase of the cell cycle (Hermeking et al. 1997). Accordingly, σ-deficient colorectal cancer cells fail to sequester cdc2-cyclinB1 complexes into the cytoplasm, and hence undergo mitotic catastrophe after DNA damage (Chan et al. 1999).
Despite the amount of molecular and biochemical data, little is know about the biological functions of 14-3-3 in normal human cells. Moreover, whereas most 14-3-3 proteins are ubiquitously expressed, σ is highly specific for stratified epithelia (Leffers et al. 1993; Dellambra et al. 1995). We now report that σ is abundantly expressed in the suprabasal layers of human epidermis and that its downregulation allows keratinocytes to escape senescence by impairing clonal evolution. Downregulation of σ is accompanied by the maintenance of telomerase activity and by a strong downregulation of the p16INK4a tumor suppressor gene, suggesting that σ downregulation fosters immortalization of primary human keratinocytes.
Materials and Methods
Cell Culture, Centrifugal Elutriation, and Cell Cycle Analysis
3T3-J2 cells (a gift from Prof. Howard Green), GP+E-86, and GP+env Am12 packaging cells were grown as described (Mathor et al. 1996).
Human keratinocytes were obtained from skin biopsies of healthy donors and cultivated on a feeder-layer of lethally irradiated 3T3-J2 cells as described (Dellambra et al. 1998). For serial propagation, cells were passaged at the stage of subconfluence (always), until they reached senescence (Pellegrini et al. 1999a). Centrifugal elutriation was performed as described (D'Anna et al. 1988).
For cell size evaluation, prints taken from cell-loaded Neubauer chambers were analyzed using a semiautomatic image analysis system (Kontron Elektronic Imaging System KS 300).
For cell cycle analysis, confluent keratinocytes were trypsinized and fixed in 70% ethanol at 4°C. Samples were rehydrated in PBS/1% FCS at room temperature for 10 min and stained with propidium iodine (50 μg/ml) plus RNase (0.25 mg/ml) for 1 h at 37°C. Flow cytometry was performed using a Beckton Dickinson FACScan, and data from 12,000 cells per sample were analysed with the ModFit cell cycle analysis software.
Colony Forming Efficiency, Cell Generations, and Population Doublings
Cells (100–1,000) from each biopsy and from each cell passage were plated onto 3T3 feeder-layers and cultured as above. Colonies were fixed 14 d later, stained with rhodamine B, and scored under a dissecting microscope. Colony forming efficiency (CFE) values are expressed as the ratio of the number colonies on the number of inoculated cells. Aborted colonies were calculated as described (Barrandon and Green 1987; Pellegrini et al. 1999a).
The number of cell generations was calculated using the following formula: x = 3.322 log N/No, where N equals the total number of cells obtained at each passage and No equals the number of clonogenic cells. Clonogenic cells were calculated from CFE data, which were determined separately in parallel dishes at the time of cell passage. Cumulative population doublings per passage were calculated as log2 (no. of cells at time of subculture/no. of cells plated) and plotted against total time in culture to assess replicative lifespan and slow growth phases.
Immunohistochemistry and Western Analysis
Confluent sheets of cultured epithelium were detached from the vessels with Dispase II (Green et al. 1979). Specimens were fixed in paraformaldehyde (4% in PBS) overnight at 4°C and embedded in paraffin. For immunohistochemistry (performed exactly as described in Pellegrini et al. 1999a), sections were stained with a σ-specific antiserum (a gift from Dr. Alastair Aitken), an mAb to human involucrin (Sigma-Aldrich), and a PCNA-specific mAb (Santa Cruz Biotechnology, Inc.). For immunoblots, confluent keratinocytes were extracted on ice with lysis Ripa buffer (Dellambra et al. 1998). Equal amounts of samples were electrophoresed on 12.5–15% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) filters (Immobilon-P; Millipore). Immunoreactions were carried as described (Dellambra et al. 1995) using polyclonal antibodies to σ and 14-3-3-ζ (a gift from Dr. Alastair Aitken), to human involucrin (BTI), and to p16INK4a (Santa Cruz Biotechnology, Inc.). Immobilon-bound antibodies were detected by chemiluminescence with ECL (Amersham Pharmacia Biotech).
Retroviral-mediated Gene Transfer, Southern and Northern Analysis, Telomerase Assay, and Telomere Length Determination
LaσSN and LsσSN were constructed by cloning a 1.4-kb fragment containing the full-length human σ cDNA (a gift from Dr. Julio E. Celis, Aarhus University, Denmark) in antisense and sense orientation, respectively, into the EcoRI/EcoRI sites of LXSN retroviral vector, as previously described (Dellambra et al. 1998; see Fig. 3 A). The Am12/LaσSN and Am12/LsσSN producer cell lines were generated by the transinfection protocol, as described (Dellambra et al. 1998). Both producer cell lines showed a viral titer 5 × 105 cfu/ml. Control amphotropic packaging cell line was generated as above, using the LXSN retroviral vector. Infections were carried out as described (Dellambra et al. 1998). In brief, subconfluent primary keratinocytes were trypsinized and seeded (5 × 103 cells/cm2) onto a feeder-layer (2.3 × 104 cells/cm2) composed of lethally irradiated 3T3-J2 cells and producer Am12/LaσSN cells (a 1:2 mixture). After 3 d of cultivation, cells were collected and plated onto a regular 3T3-J2 feeder-layer. Subconfluent cultures were used for further analysis and serial cultivation. An identical procedure was used to transduce cells with the LsσSN construct.
Figure 3.

Downregulation of σ by retroviral-mediated transfer of human σ cDNA in antisense orientation. A, Schematic map of the LaσSN provirus. Solid boxes indicate the viral long terminal repeat, open boxes represent the antisense σ and neomycin phosphotransferase (NeoR) cDNAs, and the arrowhead-shaped box represents the simian virus 40 early promoter. B, Southern blot analysis of LaσSN integration in genomic DNA from antisense σ-transduced K80 keratinocytes at passage 8 (lane 1) and 38 (lane 2) after transduction and from antisense σ-transduced K53 keratinocytes at passage 5 (lane 1) and 25 (lane 2) after transduction. DNA (20 μg) were digested with HindIII and hybridized to a Neo-specific probe. C, Northern blot analysis. 10 μg of total RNA obtained from control (V) and antisense σ-transduced K45 keratinocytes at different passages (indicated by the numbers 2–40) were separated by electrophoresis, transferred to nylon filters, and hybridized to a 32P-labeled sense σ riboprobe (AS-σ) or to a 32P-labeled β-actin probe. D, Western analysis on K45 and K80 keratinocytes. Cell extracts were prepared from control cells (-V lanes) and from antisense σ-transduced cells (-AS lanes) at different cell passages (indicated by numbers) after infection. Equal amounts of protein were fractionated on 12.5% SDS-polyacrylamide gels, transferred to PVDF filters, and immunostained with polyclonal antibodies to σ and 14-3-3ζ (ζ).
Analysis of integrated proviral genomes was performed as described (Mathor et al. 1996). For Northern analysis, cellular RNA was extracted with RNAfast (Sigma-Aldrich). 10 μg of total RNA was size-fractionated through 1% agarose/formaldehyde gels and transferred to nylon membrane (Hybond N+; Amersham Pharmacia Biotech). Blots were prehybridized at 68°C for 2 h in 50% formamide, 5× SSC, 0.02% SDS, 2% blocking reagent, and 0.1% N-lauroyl-sarcosine. Hybridization was performed overnight in the same conditions with the addition of 32P-labeled sense- or antisense σ riboprobes (2 × 106 cpm/ml). Filters were washed at high stringency in standard conditions.
Telomerase activity was detected using the PCR-based, telomeric repeat amplification protocol (TRAP) assay (Kim and Wu 1997). Telomere length was determined using the TeloQuant kit (BD Pharmigen), following manufacturer's instruction. After scanning of the signal smears with a GS 670 Imagine Densitometer (BioRad), the telomere length was calculated using the following formula: Σ(ODi)/Σ(ODi/Li), where ODi is the denditometer output and Li is the length of the DNA at position i.
Results
Expression of 14-3-3σ in Human Epidermis
In situ hybridization, performed on skin biopsies taken from healthy donors, revealed that σ mRNA was barely detectable in the epidermal basal layer, but abundantly expressed in suprabasal layers (not shown). Accordingly, low amounts of the σ polypeptide were detected in basal keratinocytes (Fig. 1 A, arrows), whereas a progressive increase of σ was observed in spinous and granular layers (Fig. 1 A, arrowheads). σ was undetectable in the stratum corneum (Fig. 1 A, asterisks). Cultured keratinocytes generate cohesive sheets of stratified squamous epithelium (Green et al. 1979), which differs from the corresponding natural epidermis mainly because it is formed by more flattened cells, has fewer suprabasal layers, and lacks the stratum corneum (Banks-Schlegel and Green 1981). As with natural epidermis, very low amounts of σ were detected in the basal layer of cultured epidermis (Fig. 1 B, arrows), whereas maximal expression of σ was observed immediately above the basal layer (Fig. 1 B, asterisks). Lower amounts of σ were detected in the uppermost epidermal layer, which contains mostly squame-like cells (Green 1980). In cultured epidermis, it is quite common to observe basal cells in transition from basal to suprabasal position. These cells usually expressed σ (Fig. 1 B, arrowheads) and keratin type 1 and 10 (not shown), the latter being considered as early markers of keratinocyte terminal differentiation (Green 1980; Roop 1995). Thus, both in vivo and in vitro, σ is expressed in areas of the epidermis where cell proliferation has ceased and where markers of terminal differentiation are synthesized.
Figure 1.
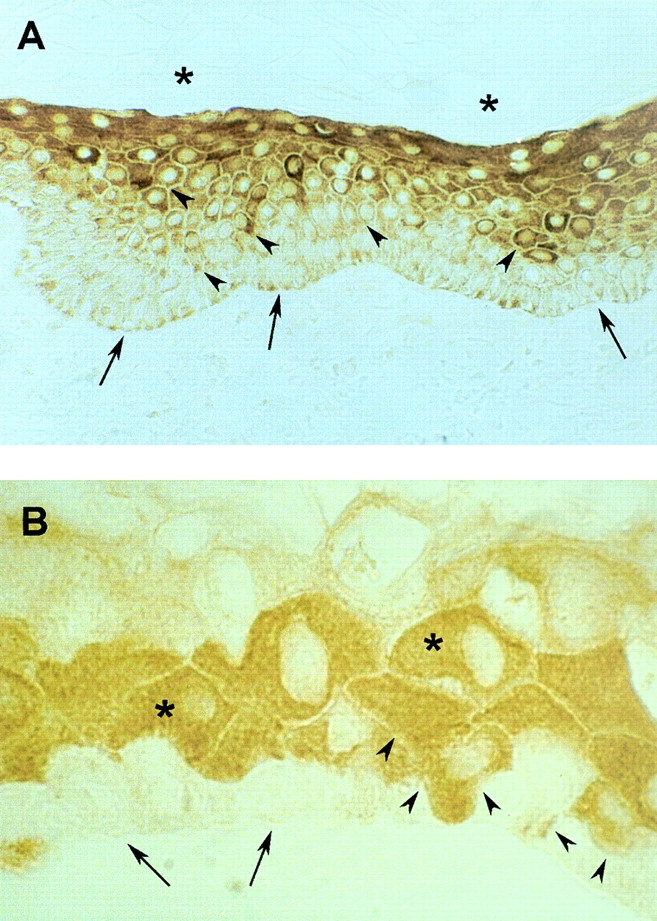
Expression of σ in natural and cultured epidermis. Sections of skin biopsies (A) or of cultured epidermal sheets prepared from primary cultures (B) were stained with σ-specific antibodies. A, σ was barely detectable in the basal layer (arrows), and its expression progressively increased in suprabasal layers (arrowheads). Asterisks mark the stratum corneum. B, σ was barely detectable in the basal layer (arrows), and abundantly expressed immediately above the basal layer (asterisks). Note a keratinocyte expressing high amounts of σ in transition from basal to suprabasal position (arrowheads).
Relation of 14-3-3σ to Keratinocyte Proliferation
Cell size is a major determinant of keratinocyte clonogenic ability (Barrandon and Green 1985) and terminal differentiation (Watt and Green 1981). Clonogenic cells capable of multiplication are the smallest cells in the epithelium (Barrandon and Green 1985) and express PCNA, a DNA polymerase δ-associated protein that is synthesized in G1 and S phases of the cell cycle and is therefore expressed by proliferating cells (Bravo et al. 1987). Upon commitment to terminal differentiation, keratinocytes increase their size and express involucrin (Watt and Green 1981), which is considered an early intermediate differentiation marker (Roop 1995).
To examine the relation between the expression of σ, the potential for multiplication, and the expression of PCNA and involucrin, confluent sheets of primary keratinocytes were trypsinized and cells were separated according to their size by centrifugal elutriation (D'Anna et al. 1988). Six fractions were collected and cell diameter increased progressively from ∼10 μm (Fig. 2 A, 1 and 2) to 30–50 μm (Fig. 2 A, 4–6). As shown in Fig. 2 B, colony-forming cells capable of multiplication were concentrated almost entirely in the first two fractions (Fig. 2 B, 1 and 2), whereas cells of the last three fractions lost virtually all colony forming ability (Fig. 2 B, 4–6). As shown in Fig. 2 C, σ was barely detectable in the first fraction and its expression increased slightly in the second fraction. A sharp increase of σ expression was observed in the third and fourth fractions, to coincide with the abrupt drop of the clonogenic ability. PCNA was present in the first two fractions and its decrease coincided with the rise of σ observed in the third fraction (Fig. 2 C). As expected, PCNA was virtually undetectable in the remaining fractions. In contrast, involucrin was absent in the first two fractions, its appearance coincided with the sharp rise of σ observed in the third fraction, and its expression remained high afterwards (Fig. 2 C). Thus, the synthesis of σ increases just before cells cease to proliferate and precedes the appearance of involucrin.
Figure 2.

Centrifugal elutriation and Western blots. Cells from confluent keratinocyte cultures were separated according to their size by centrifugal elutriation. The counterflow rate was increased stepwise and six consecutive fractions (1–6) were collected. A, Size of the cells in the elutriation fractions. Bars, 100 μm. B, Colony forming ability of cells from the elutriation fractions. C, Immunoblots of cell extracts prepared from the fractions. Samples were fractionated on 12.5% SDS-polyacrylamide gels, transferred to PVDF filters, and were immunostained with polyclonal antibodies to σ and human involucrin (INV), and with mAbs to PCNA. Equal amounts (7 μg) of cell extracts were loaded in each lane.
Relation of 14-3-3σ to Keratinocyte Senescence
The availability of gene transfer protocols allowing selective and stable transduction of stem and transient amplifying keratinocytes (Mathor et al. 1996; Dellambra et al. 1998) prompted us to investigate the biological effects of antisense mediated downregulation of σ in clonogenic primary keratinocytes. Infections with defective retrovirus carrying a full-length human σ cDNA in antisense orientation (Fig. 3 A) were performed on three different strains (K45, K53, and K80) of primary keratinocytes obtained from adult healthy donors. Clonogenic cells were transduced with an efficiency of 80–100%. Southern analysis showed multiple bands (Fig. 3 B, lane 1) resulting from numerous proviral integrations in a heterogeneous transduced cell population (see also Mathor et al. 1996). Northern analysis revealed abundant expression of antisense σ transcripts in antisense σ-transduced keratinocytes (Fig. 3 C, lane 2), but not in cells transduced with an empty vector (Fig. 3 C, lane V). Similar results were obtained in all transduced strains.
To investigate the proliferative capacity of antisense σ-transduced keratinocytes, cells were serially cultivated and the number of cell generations was calculated. As shown in Fig. 4A, Fig. C, and Fig. E, closed squares, K45, K80, and K53 cells transduced with an empty vector underwent 192.2, 189.2, and 111.9 cell generations before senescence, which eventually occurred after 25, 24, and 14 cell passages, respectively. Similar values were obtained with untransduced control cells (not shown). In contrast, antisense σ-transduced keratinocytes bypassed replicative senescence (Fig. 4A, Fig. C, and Fig. E, open circles) and continued to divide at a rate of young keratinocytes. To date, K45, K80, and K53 cells underwent 576.3, 562.9, and 500.2 cell doublings, respectively, have been passaged 80, 72, and 83 times, respectively, and have been serially cultivated for 469, 450, and 488 d, respectively. All transduced cells are still in culture and, to date, do not show any sign of senescence nor any decrease of their growth rate.
Figure 4.

Proliferative potential of antisense σ-transduced cells. Primary K45 (A and B), K80 (C and D), and K53 (E and F) keratinocytes transduced with antisense σ cDNA (open circles) or with an empty LXSN vector (closed squares) were serially cultivated. The number of cell doublings (A, C, and E) and the cumulative population doublings (B, D, and F) were calculated as described in Materials and Methods. Note that control cells (closed squares) reached replicative senescence, whereas antisense σ-transduced cells proliferated indefinitely (open circles). Arrows indicate keratinocyte infection.
We also calculated the proliferative capacity as number of cumulative population doublings. As shown in Fig. 4B, Fig. D, and Fig. F, closed squares, K45, K80, and K53 cells transduced with a control vector underwent 119.5, 112.1, and 54.1 population doublings before senescence, respectively, whereas antisense σ-transduced keratinocytes (Fig. 4C, Fig. D, and Fig. F, open circles) continued to divide indefinitely. To date, K45, K80, and K53 keratinocytes underwent 409.3, 361.4, and 385.3 population doublings, respectively. The K53 strain was transduced twice in two separate experiments and similar results were obtained (see Fig. 8). It is worth noting that we did not observe a crisis (Bond et al. 1999), nor a slow growth phase (Fig. 4), suggesting that most transduced cells contributed to the lifespan extension.
Figure 8.
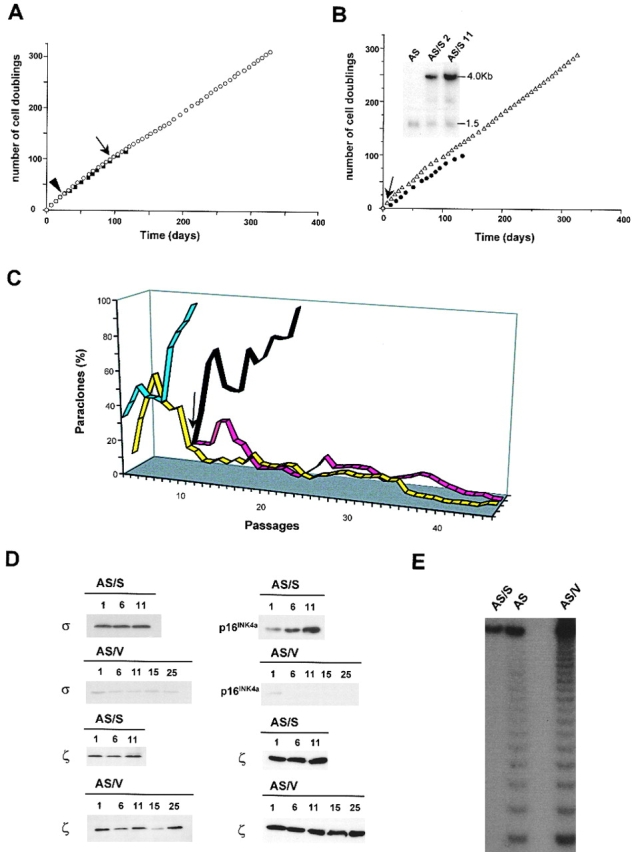
Reversion of the σ-dependent phenotype. A, Primary K53 keratinocytes transduced with antisense σ cDNA (open circles) or with an empty LXSN vector (closed squares) were serially cultivated. The number of cell doublings was calculated as for Fig. 4. Note that control cells (closed squares) reached replicative senescence, whereas antisense σ-transduced cells proliferated indefinitely (open circles). Arrowhead indicates infection of primary keratinocytes with antisense σ cDNA. Arrow indicates infection of antisense σ-transduced keratinocytes with σ cDNA in sense orientation. B, Antisense σ-transduced K53 keratinocytes transduced with sense σ cDNA (closed circles, AS/S cells) or with an empty LXSN vector (open triangles, AS/V cells) were serially cultivated. Note that AS/V cells continued to proliferate indefinitely, whereas AS/S cells underwent replicative senescence. Arrow indicates the time of keratinocyte infection with sense σ cDNA. Inset, 10 μg of total RNA obtained from antisense σ-transduced K53 keratinocytes (AS) and AS/S cells at passage 2 (AS/S2) and 11 (AS/S11) after superinfection with sense σ cDNA were separated by electrophoresis, transferred to nylon filters, and hybridized to a 32P-labeled antisense σ riboprobe. Exogenous (4.0 Kb) and endogenous (1.5 Kb) σ mRNA are shown. C, The percentage of paraclones, expressed as the ratio of aborted colonies on the total number of colonies, was calculated for each cell passage during serial cultivation of empty vector-transduced control cells (blue line), antisense σ-transduced primary keratinocytes (yellow line), AS/V cells (red line), and AS/S cells (black line). Arrow indicates infection with sense σ cDNA. D, Western analysis was performed on cell extracts prepared from AS/V and AS/S control cells at different cell passages (indicated by numbers) after infection with sense σ cDNA. Equal amounts of protein were fractionated on 12.5–15% SDS-polyacrylamide gels, transferred to PVDF filters, and immunostained with polyclonal antibodies to σ, 14-3-3ζ (ζ), and p16INK4a. Note that both σ and p16INK4a were almost undetectable in AS/V cells, whereas they were reexpressed in AS/S cells. E, TRAP assay was performed on antisense σ-transduced K45 cells at the time of σ or empty vector infection (AS), on AS/V cells at cell passage 25 after empty vector infection, and on AS/S cells close to replicative senescence (cell passage 11 after σ-transduction; AS/S).
During the last ten years, we have evaluated the long term proliferative capacity of 142 normal human epidermal and ocular keratinocyte strains, and 54 epidermal keratinocyte strains stably transduced with either empty retroviral vectors or with retroviral vectors carrying various cDNA(s). All 196 strains senesced after a variable number of cell doublings, with the exception of the three cell strains stably transduced with the human σ cDNA in antisense orientation.
Southern analysis performed after bypass of senescence (Fig. 3 B, K80 and K53, lane 2) showed multiple bands, many of which were initially observed after infection (Fig. 3 B, K80 and K53, lane 1). The disappearance of some bands and the enrichment of others suggest that a certain percentage of transduced keratinocytes (probably those with low amounts of antisense σ mRNA) were unable to overcome senescence. Northern blot analysis, performed on total RNA extracted from transduced cells at different cell passages (Fig. 3 C, lanes 2–40), showed that antisense σ transcripts were expressed constantly during serial cultivation. A slight increase of mRNA was observed at bypass of senescence (mRNA levels remained constant afterwards), further suggesting that keratinocytes expressing the highest amount of antisense σ transcripts were selected for the extension of the lifespan. Western blot analysis, performed on control cells at selected passages (indicated by the numbers in Fig. 3 D), showed that σ was detected during serial cultivation (Fig. 3 D, σ, V lanes) and that levels of σ increased when cells approached replicative senescence (Fig. 3 D, σ, K45-V, lanes 17 and 19; K80-V, lane 18). In contrast, antisense σ-transduced cells showed a sharp decrease of σ, already one passage after transduction (Fig. 3 D, σ, AS lanes). Low levels of σ were then detected during serial cultivation and after bypass of replicative senescence (Fig. 3 D, σ, AS lanes). It is worth noting that the amount of 14-3-3ζ was comparable in control cells and in antisense σ-transduced keratinocytes (Fig. 3 D, ζ), and that downregulation of σ was accompanied by a strong decrease of the expression of involucrin (not shown, see Fig. 5). Similar results were obtained in all transduced cell strains.
Figure 5.
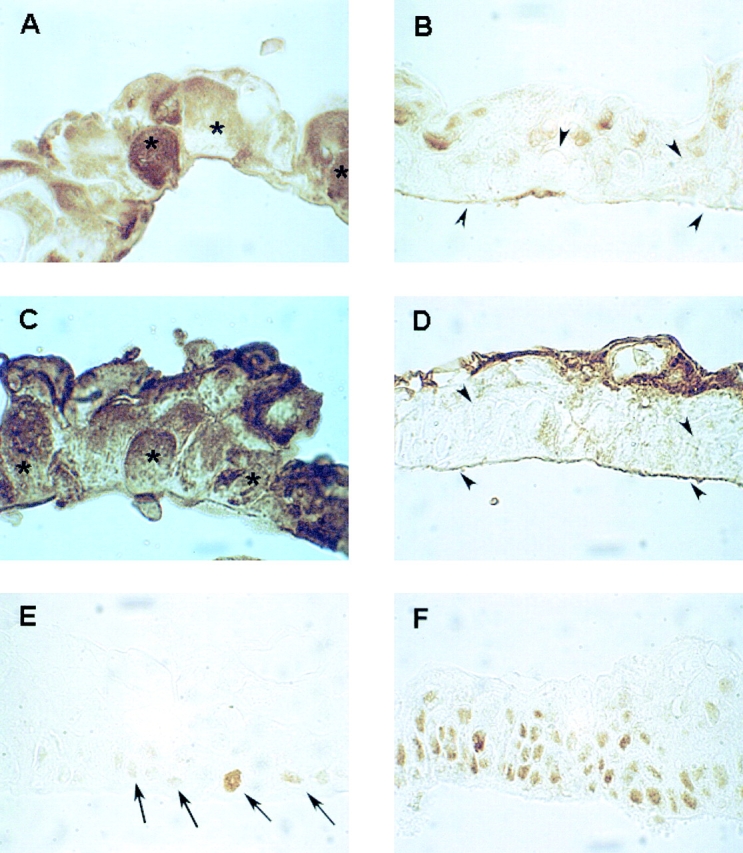
Immunohistochemical analysis on antisense σ-transduced cells. Cultured epidermal sheets were prepared from control cells approaching senescence (A and C) and from antisense σ-transduced K45 keratinocytes (B and D) 23 passages after bypass of senescence. Paraffin-embedded sections were stained with polyclonal antibodies to σ (A and B) and to human involucrin (C and D). Note that the large basal cells in control sheets (asterisks) are σ- and involucrin-positive, whereas the small basal and suprabasal cells (arrowheads) in antisense σ-transduced cells are σ- and involucrin-negative. Cultured epidermal sheets prepared from primary untransduced control cells (E) and from antisense σ-transduced K45 keratinocytes (F) were stained with a PCNA-specific mAb. Note that PCNA was expressed in few basal control cells (E, arrows) and abundantly expressed in all basal and most suprabasal antisense σ-transduced cells (F).
These data were confirmed by immunohistochemical analysis. As shown in Fig. 5, high amounts of σ (Fig. 5 A) and involucrin (Fig. 5 C) were detected in basal and suprabasal layers of cultured epidermal sheets prepared from control cells approaching senescence. The basal expression of σ and involucrin is expected since, at this stage, keratinocyte clonal evolution has been almost completed and the entire epithelial sheet is formed by large cells (Fig. 5, asterisks) already committed to terminal differentiation. In contrast, cultured epidermal sheets prepared from antisense σ-transduced keratinocytes after bypass of senescence showed very low amounts of σ (Fig. 5 B) and involucrin (Fig. 5 D), both in basal and suprabasal layers. PCNA was significantly expressed only in a few cells located in the basal layer of control cultured epidermal sheets (Fig. 5 E, arrows). In contrast, high amounts of PCNA were detected in most basal and suprabasal cells of epithelial sheets prepared from antisense σ-transduced keratinocytes after bypass of senescence (Fig. 5 F), further suggesting a severe alteration of keratinocyte growth control.
Growth Rate, Growth Requirements, and Karyotype
We did not observe any significant difference between the growth rates of control cells (at early cell passages) and of antisense σ-transduced keratinocytes (after bypass of senescence). Antisense σ-transduced keratinocytes had the same growth requirements of control cells: they needed FCS, EGF, hydrocortisone, and cholera toxin for optimal growth. Depletion of each of these components had a similar effect on control and antisense σ-transduced cells (not shown). Growth of both control and antisense σ-transduced keratinocytes was anchorage-dependent (not shown). Detailed G-banding performed on antisense σ-transduced K45, K53, and K80 keratinocytes after bypass of senescence revealed that cells had the normal complement of 46 chromosomes and no obvious translocation or abnormalities (data not shown). It is worth noting, however, that while K45 and K80 cells remained diploid even after 50 cell passages (∼400 cell doublings), K53 cells developed numerous chromosome abnormalities (mainly aneuploidy and polyploidy) at later cells passages.
Relation of Bypass of Senescence to Clonal Evolution, Phenotype, and Cell Cycle
Each keratinocyte colony is formed by single epidermal cells (Rheinwald and Green 1975) endowed with different capacities for multiplication, referred to as holoclones, meroclones, and paraclones (Barrandon and Green 1987). The holoclone is the smallest colony-founding cell, has the highest proliferative capacity, and is considered the surface epithelial stem cell (Barrandon and Green 1987; Jones and Watt 1993; Rochat et al. 1994; Watt 1998; Pellegrini et al. 1999a). Meroclones and paraclones are considered young and old transient amplifying cells, respectively (Pellegrini et al. 1999a). In particular, the paraclone is the largest colony-forming keratinocyte and generates aborted colonies containing only terminal cells (Barrandon and Green 1987; Jones and Watt 1993). Epidermal clonal evolution is characterized by a progressive increase of paraclones (Barrandon 1993). In turn, senescence occurs when all stem cells (holoclones) have completed clonal evolution and have generated only paraclones (Barrandon 1993; Mathor et al. 1996; Pellegrini et al. 1999a). Therefore, the progressive increase of paraclones occurring during serial cultivation is a direct measurement of clonal evolution, and hence, of progressive stem cell depletion.
This said, we have analyzed CFE and clonal evolution of antisense σ-transduced keratinocytes. As shown in Fig. 6 (B, yellow bars), cells infected with an empty vector showed a progressive decrease of their CFE during serial cultivation. When cells approached replicative senescence, the CFE was very low (Fig. 6 B, yellow bars) and colonies were small and irregular (Fig. 6 A, K45-V, 19). The decrease of CFE was accompanied by a continuous clonal evolution, as demonstrated by the progressive increase of the percentage of paraclones (Fig. 6 C, yellow line). 100% of paraclones developed at cell passage 20 after empty-vector infection (Fig. 6 C, yellow line) to coincide with replicative senescence. Similar values were obtained with untransduced control cells (not shown).
Figure 6.
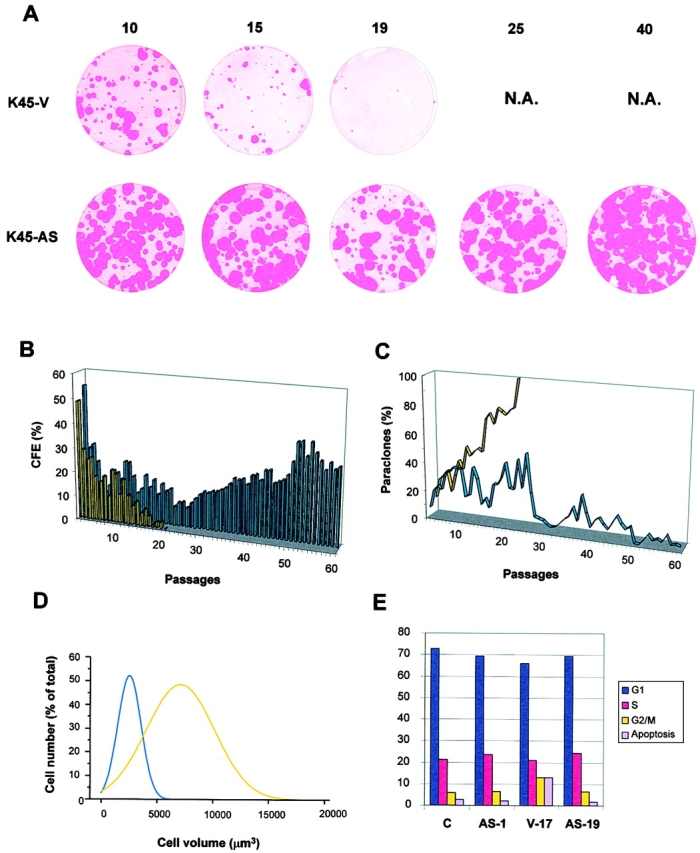
Clonogenic and cell cycle analysis of antisense σ-transduced cells. K45 keratinocytes transduced with antisense σ cDNA (K45-AS, blue) or with an empty LXSN vector (K45-V, yellow) were serially cultivated. CFE assays and evaluation of aborted colonies were performed at each cell passage (see Materials and Methods). A, Selected CFE performed at cell passages indicated by numbers. N.A. indicates not available, since vector-transduced cells reached replicative senescence. B, CFE values are expressed as the ratio of the total number of colonies on the number of inoculated cells. All colonies were scored whether progressively growing or aborted. C, Aborted colonies values are expressed as the ratio of aborted colonies on the total number of colonies. D, Statistical analysis of cell size was calculated using KS 300, a semiautomatic image analysis system, and data fell into a Gaussian distribution. Antisense σ-transduced K45 keratinocytes (50 cell passages after infection) are indicated by the blue line. Vector-transduced K45 cells (3 passages before senescence) are indicated by the yellow line. E, Cell cycle analysis of control untransduced keratinocytes in secondary culture (C), antisense σ-transduced keratinocytes at one passage after infection (AS-1) and at bypass of senescence (AS-19), and empty vector-transduced cells approaching replicative senescence (V-17).
In contrast, after an initial decrease of CFE, probably due to the progressive disappearance of poorly transduced cells, antisense σ-transduced keratinocytes showed a progressive increase of CFE (Fig. 6 B, blue bars) at levels that tended to remain constant during serial cultivation. Antisense σ-transduced keratinocytes did not show the progressive increase of paraclones observed in control cells (Fig. 6 C, blue line), and developed only large colonies, which remained large and smooth after bypass of senescence (Fig. 6 A, K45-AS). Of note, impairment of clonal evolution was observed already five to six passages after infection, namely several passages before the onset of replicative senescence of control cells (Fig. 6 C, compare yellow and blue lines).
Statistical analysis of cell size was performed using KS 300, a semiautomatic image analysis system, and data fell into a Gaussian distribution. As shown in Fig. 6 D, antisense σ-transduced keratinocytes (blue) were threefold smaller than vector-transduced cells (yellow; 2,668 μm3 vs 7,678 μm3), further confirming that cell size is a major determinant of keratinocyte clonogenic ability (Barrandon and Green 1985). Similar results were obtained with all transduced strains (see also Fig. 8). Taken together, these data suggest that downregulation of σ allows keratinocytes to bypass replicative senescence by impairing clonal evolution, and hence, by forcing keratinocytes to stay in the stem cell compartment.
Since σ has been shown to be involved in G2/M checkpoint in DNA-damaged epithelial cells (Hermeking et al. 1997; Chan et al. 1999), we sought to investigate whether σ downregulation was accompanied by premature G2/M entry. As shown in Fig. 6 E, the cell cycle profiles of antisense σ-transduced keratinocytes one passage after transduction (K45-AS1) and at bypass of senescence (K45-AS19) were virtually identical to cell cycle profiles of untransduced (K45-C) or empty-vector-transduced (not shown) keratinocytes at early passages. Control cells approaching replicative senescence (K45-V17) showed a slightly higher proportion of cells arrested in G2/M and/or in apoptosis. The increase of cells in G2/M is consistent with the increase of σ observed at senescence (Fig. 3) and it is not observed in antisense σ-transduced cells that bypassed senescence (K45-AS19). Similar results were obtained with all transduced strains.
Relation of Antisense σ-mediated Bypass of Senescence with Telomerase Activity and p16INK4a Expression
It has been proposed that telomere shortening, which occurs during cell divisions, is one of the molecular clocks that triggers senescence (Greider 1999), and that resumption or maintenance of telomerase activity is an essential step for cellular immortalization (Reddel 1998). In fact, keratinocytes endowed with proliferative capacity exhibit telomerase activity, and downregulation of telomerase correlates with terminal differentiation (Harle-Bachor and Boukamp 1996; Bickenbach et al. 1998). Immortalization of normal human epithelial cells requires, however, both telomerase activity and inactivation of the p16INK4a tumor suppressor gene (Kiyono et al. 1998; Dickson et al. 2000). Therefore, we examined telomerase activity and p16INK4a expression in antisense σ-transduced keratinocytes.
As shown in Fig. 7 A, telomerase activity was detected in untransduced (C-1), vector-transduced (V-1), and antisense σ-transduced (AS-1) cells at early cell passages, but was undetectable in control cells approaching replicative senescence (V-17). In contrast, antisense σ-transduced keratinocytes that bypassed replicative senescence maintained normal levels of telomerase activity (Fig. 7 A, AS-19). Of note, telomerase activity sharply increased at later cell passages after bypass of senescence (Fig. 7 A, AS-49). Maintenance of telomerase activity was accompanied by a stabilization, and, at later cell passages, elongation of the telomeres (Fig. 7 A). Identical results were obtained in all transduced cell strains (not shown).
Figure 7.

Telomerase activity and p16INK4a expression. A, TRAP assay was performed on primary untransduced keratinocytes (C-1), cells transduced with a control empty vector (V lanes) and antisense σ-transduced cells (AS lanes) at different cell passages (indicated by numbers). Note that telomerase activity is clearly detected in control untransduced cells (C-1), is undetectable in empty-vector-transduced cells close to senescence (V-17), and is abundantly expressed in antisense σ-transduced cells after bypass of senescence (AS-19 and AS-49). T.L. indicates telomere length expressed in Kb. Identical results were obtained in all transduced cell strains. B, Western analysis was performed on cell extracts prepared from control cells (V lanes) and from antisense σ-transduced cells (AS lanes) at different cell passages (indicated by numbers) after infection. Equal amounts of protein were fractionated on 15% SDS-polyacrylamide gels, transferred to PVDF filters, and immunostained with polyclonal antibodies to p16INK4a and 14-3-3ζ (ζ). Note that p16INK4a was undetectable in control cells at early passages, was abundantly expressed by control cells approaching senescence (V15-19), but barely detectable or undetectable by antisense σ-transduced keratinocytes. Identical results were obtained in all transduced cell strains.
p16INK4a was undetectable in Western blots performed on cell extracts prepared from keratinocytes transduced with an empty vector (Fig. 7 B, K45-V). p16INK4a remained undetectable for several cell passages (indicated by the numbers in Fig. 7 B, K45-V), was expressed by keratinocytes approaching replicative senescence, and reached its maximal levels in cells close to senescence (Fig. 7 B, K45-V). In contrast, p16INK4a expression was virtually absent in antisense σ-transduced cells that bypassed replicative senescence (Fig. 7 B, K45-AS). Similar results were obtained with all transduced cell strains (see Fig. 8). Thus, bypass of senescence is accompanied by maintenance of telomerase activity and by inactivation of the p16INK4a tumor suppressor gene, suggesting that downregulation of σ fosters immortalization of primary human keratinocytes.
Reversion of the σ-dependent Phenotype
In a separate experiment, subconfluent primary K53 cells were transduced with an empty vector or with antisense σ cDNA as described above. Control cells underwent 14 cell passages and 114.6 cell doublings before senescence (Fig. 8 A, closed squares) and, as expected, a progressive increase of paraclones was observed during serial cultivation (Fig. 8 C, blue line). Antisense σ-transduced cells readily escaped replicative senescence and continued to proliferate indefinitely (Fig. 8 A, open circles). Downregulation of σ determined a block of keratinocyte clonal evolution, which was observed already three to four passages after infection, namely several passages before the onset of replicative senescence of control cells (Fig. 8 C, yellow line).
We then sought to investigate whether the σ-dependent phenotype could be reversed by restoration of σ expression. When impairment of clonal evolution was almost complete (Fig. 8 C, arrow), antisense σ-transduced cells were infected with retrovirus carrying either an empty vector (AS/V cells) or a full-length human σ cDNA in sense orientation (AS/S cells). The time of σ infection is also indicated by the arrows in Fig. 8 A and B. Northern blot analysis, performed on total RNA hybridized with an antisense σ riboprobe, showed abundant levels of exogenous σ mRNA (Fig. 8 B, inset, 4.0 Kb). For comparison, endogenous σ mRNA is shown (Fig. 8 B, inset, 1.5 Kb). As expected, AS/S keratinocytes reexpressed significant amounts of σ, as compared with AS/V cells, already one passage after infection (Fig. 8 D).
AS/V cells (Fig. 8 B, open triangles) continued to proliferate indefinitely and maintained the impairment of clonal evolution (Fig. 8 C, red line). In contrast, AS/S cells resumed clonal evolution immediately after transduction (Fig. 8 C, black line) and, consequently, underwent replicative senescence (Fig. 8 B, closed circles). It is worth noting that AS/S cells were passaged 13 times and underwent 99 doublings (after superinfection) before senescence, just as the parental control cells (Fig. 8 A, closed squares). This last observation strongly suggests that antisense σ-transduced cells had zeroized their cell doubling clock by halting their clonal evolution, and that restoration of the proper clonal evolution process was crucial to restart the count of cell divisions, which eventually led to replicative senescence.
As shown in Fig. 8 D, reexpression of σ by AS/S cells was accompanied by reexpression of p16INK4a. Indeed, p16INK4a was almost undetectable in AS/V cells, was expressed by AS/S cells and its expression was maximal when AS/S cells approached replicative senescence. Moreover, while AS/V cells (Fig. 8 E, AS/V) continued to express levels of telomerase activity comparable to those of parental antisense σ-transduced cells (Fig. 8 E, AS), AS/S cells close to replicative senescence showed a dramatic decrease of telomerase activity (Fig. 8 E, AS/S). Thus, σ infection was able to revert the phenotype of antisense σ-transduced cells.
Discussion
Bypass of Senescence and Immortalization
It is widely accepted that replicative senescence represents one of multiple lifespan barriers (Bond et al. 1999) and that somatic cells need to acquire multiple genetic alterations to overcome those barriers and become immortal (see Reddel 1998; Greider 1999; Wynford-Thomas 1999). For instance, human keratinocytes can be immortalized by HPV E6 and E7 oncogenes, which are responsible for telomerase maintenance (and p53 degradation) and pRb inactivation, respectively (Reddel 1998; Greider 1999). Accordingly, Kiyono et al. 1998 reported that exogenous hTERT, i.e., the catalytic component of telomerase (which acts as a bona fide oncogene; Greider 1999), can directly immortalize p16INK4a negative keratinocytes, whereas Dickson et al. 2000 showed that hTERT-transduced keratinocytes need to bypass the p16INK4a/pRb pathway to become immortal. In both cases, rare clones of p16INK4a-negative keratinocytes arise and generate immortal cell lines.
Instead, we report here that downregulation of σ allows mass cultures of primary human epidermal keratinocytes to readily escape replicative senescence without the need of exogenous oncogenes or oncoviruses. We also report that σ downregulation results in maintenance of telomerase activity and in a dramatic reduction of p16INK4a expression. Thus, σ downregulation recapitulates the combined action of the HPV E6 and E7 oncogenes, and places σ upstream of two key events, i.e., telomerase and p16INK4a, in human keratinocyte immortalization (Kiyono et al. 1998; Dickson et al. 2000). To our knowledge, this is the first demonstration that inhibition of a single endogenous gene product can reproducibly give rise to immortal primary human cells.
The concomitance of σ and p16INK4a accumulation and telomerase inhibition; p16INK4a inactivation and telomerase maintenance after σ downregulation; and, more importantly, p16INK4a resumption and telomerase inhibition after σ reconstitution experiments (Fig. 8), suggests that these genes might be regulated by σ, either directly, or as a consequence of the block of clonal evolution (see below). This hypothesis is consistent with the observation that antisense σ-transduced keratinocytes are polyclonal in nature, maintain a normal karyotype and cell cycle profile, and do not show gross chromosomal abnormalities, with the exception of the K53 strain, which developed an altered karyotype only at later passages after bypass of senescence. In this respect, it is worth noting that human keratinocytes located in sun-exposed body sites carry mutations of tumor suppressor genes, such as p53, with high frequency (Jonason et al. 1996). Therefore, certain cell strains might be particularly prone to develop chromosomal abnormalities after lifespan extension.
Clonal Evolution
The observations that the synthesis of σ is correlated with cell size and coincides with the loss of clonogenic ability; σ downregulation prevents the transition from stem to transient amplifying cells; and resumption of σ expression reactivates clonal evolution and restores the capacity of transduced cells to undergo replicative senescence, strongly suggest that σ downregulation forces clonogenic keratinocytes to stay in the stem cell compartment. This is further suggested by the observation that antisense σ-transduced cells had zeroized their cell doubling clock, and that, after σ-reconstitution, keratinocytes underwent the same number of cell doublings as untransduced control cells, before senescence could ensue. The impairment of clonal evolution could account, per se, for the indefinite proliferative capacity of antisense σ-transduced cells (Lajtha 1979; Morrison et al. 1997; Fuchs and Segre 2000), and might also explain the concomitant maintenance of telomerase activity and p16INK4a inhibition. Therefore, σ might represent at least one of the long-sought biochemical mediators (see also Tseng and Green 1994; Gandarillas and Watt 1997; Gat et al. 1998; Zhu and Watt 1999) regulating keratinocyte clonal evolution. Recently, it has been shown that β-catenin (by means of its interaction with members of the Lef/Tcf family of DNA binding proteins) and c-myc play a role in regulating epidermal and hair follicle stem cell proliferation and differentiation (Gandarillas and Watt 1997; Gat et al. 1998; Waikel et al. 1999; Zhu and Watt 1999). However, the molecular pathways regulating these processes are largely unknown (Fuchs and Segre 2000). Given the general property of 14-3-3 proteins to interact with several signaling molecules (Aitken 1996), these data might therefore prove useful in elucidating biochemical and molecular pathways involved in the above processes.
Cell Cycle and Transformation
At a molecular level, control of senescence involves the same tumor suppressor genes regulating different cell cycle checkpoints and cancer progression (Cahill et al. 1998; Orr-Weaver and Weinberg 1998). Accordingly, σ can also act as a p53-regulated inhibitor of G2/M progression, playing a crucial role in the control of the G2/M phase of the cell cycle after DNA damage (Hermeking et al. 1997; Chan et al. 1999). Our data, however, suggest that the biological effects of σ in normal human epidermal cells seem to be independent of cell cycle checkpoint, since σ in constitutively expressed by keratinocytes, karyotype of transduced cells (all but one strain) is normal, and cell cycle profiles of control and transduced cells are similar. This confirms the concept that cell cycle, senescence, and cell differentiation can be regulated by common molecular pathways which, however, can act independently (Jacks and Weinberg 1998). For instance, p21CIP1/WAF1 inhibits murine keratinocyte differentiation independently of cell cycle control (Di Cunto et al. 1998), whereas members of the retinoblastoma family of proteins play specific roles during human epidermal differentiation (Paramio et al. 1998). Also, myogenic differentiation can be regulated by cyclin D1 and p16INK4a (Urashima et al. 1999; Zhang et al. 1999), and specific domains of the p27Xic1 Cdk inhibitor promote glial cell differentiation in Xenopus retina (Ohnuma et al. 1999).
Finally, bypass of senescence and immortalization are early key steps leading to cell transformation (Yeager et al. 1998; Wynford-Thomas 1999). Premature senescence is currently envisaged as a potent tumor suppressive mechanism that can be triggered in response to an aggressive mitogenic stimulus or to an oncogene (Serrano et al. 1997; see Wynford-Thomas 1999). Recently, it has been shown that ectopic expression of hTERT in combination with the SV40 large-T oncoprotein and an oncogenic allele of H-ras results in direct tumorigenic conversion of normal human epithelial cells (Hahn et al. 1999). Bypass of replicative senescence and immortalization were key initial steps in this process (Hahn et al. 1999). Thus, since cancers of the skin are now the most prevalent malignancy in the light-skinned population world-wide, it will be of great interest to investigate whether downregulation of σ can now allow one-step transformation of human keratinocytes by selected oncogenes and/or oncoviruses.
Acknowledgments
We are indebted to Prof. Marina Ferraro at the Department of Genetics and Molecular Biology, University La Sapienza, Medical School, Rome, Italy for G-banding and karyotype performed on our transduced cells.
This work was supported by Telethon-Italy (grant A.106), by EEC BIOMED 2 no. BMHG4-97-2062, and by Ministero della Sanità, Italy.
Footnotes
Abbreviations used in this paper: σ, 14-3-3σ; CFE, colony forming efficiency.
References
- Aitken A. 14-3-3 and its possible role in coordinating multiple signaling pathways. Trends Cell Biol. 1996;6:341–347. doi: 10.1016/0962-8924(96)10029-5. [DOI] [PubMed] [Google Scholar]
- Aitken A., Collinge D.B., van Heudsen B.P.H., Isobe T., Roseboom P.H., Rosenfield G., Soll J. 14-3-3 proteinsa highly conserved widespread family of eukaryotic proteins. Trends Biochem. Sci. 1992;17:498–501. doi: 10.1016/0968-0004(92)90339-b. [DOI] [PubMed] [Google Scholar]
- Banks-Schlegel S., Green H. Involucrin synthesis and tissue assembly by keratinocytes in natural and cultured human epithelia. J. Cell Biol. 1981;90:732–737. doi: 10.1083/jcb.90.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrandon Y. The epidermal stem cellan overview. Dev. Biol. 1993;4:209–215. [Google Scholar]
- Barrandon Y., Green H. Cell size as a determinant of the clone forming ability of human keratinocytes. Proc. Natl. Acad. Sci. USA. 1985;82:5390–5394. doi: 10.1073/pnas.82.16.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrandon Y., Green H. Three clonal types of keratinocytes with different capacities for multiplication. Proc. Natl. Acad. Sci. USA. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickenbach J.R., Vormwald-Dogan V., Bachor C., Bleuel K., Schnapp G., Boukamp P. Telomerase is not an epidermal stem cell marker and is down-regulated by calcium. J. Invest. Dermatol. 1998;111:1045–1052. doi: 10.1046/j.1523-1747.1998.00420.x. [DOI] [PubMed] [Google Scholar]
- Bond J.A., Haughton M.F., Rowson J.M., Smith P.J., Gire V., Wynford-Thomas D., Wyllie F.S. Control of replicative life span in human cellsbarriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol. 1999;19:3103–3114. doi: 10.1128/mcb.19.4.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R., Frank R., Blundell P.A., MacDonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase δ. Nature. 1987;326:515–517. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- Cahill D.P., Lengauer C., Yu J., Riggins G.J., Willson J.K.V., Markowitz S.D., Kinzler K.W., Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- Campisi J. Replicative senescencean old lives' tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- Campisi J. The role of cellular senescence in skin aging. J. Invest. Dermatol. 1998;3:1–5. [PubMed] [Google Scholar]
- Chan T.A., Hermeking H., Lengauer C., Kinzler K.W., Vogelstein B. 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- D'Anna F., De Luca M., Cancedda R., Zicca A., Franzi A.T. Elutriation of human keratinocytes and melanocytes from in vitro cultured epithelium. Hystochem. J. 1988;20:674–678. [Google Scholar]
- de Lange T., DePinho R.A. Unlimited mileage from telomerase? Science. 1999;283:947–949. doi: 10.1126/science.283.5404.947. [DOI] [PubMed] [Google Scholar]
- Dellambra E., Patrone M., Sparatore B., Negri A., Ceciliani F., Bondanza S., Molina F., Descalzi-Cancedda F., De Luca M. Stratifin, a keratinocyte specific 14-3-3 protein, harbors a pleckstrin homology (PH) domain and enhances protein kinase c activity. J. Cell Sci. 1995;108:3569–3579. doi: 10.1242/jcs.108.11.3569. [DOI] [PubMed] [Google Scholar]
- Dellambra E., Vailly J., Pellegrini G., Bondanza S., Golisano O., Macchia C., Zambruno G., Meneguzzi G., De Luca M. Corrective transduction of human epidermal stem cells in laminin-5-dependent junctional epidermolysis bullosa. Hum. Gene Ther. 1998;9:1359–1370. doi: 10.1089/hum.1998.9.9-1359. [DOI] [PubMed] [Google Scholar]
- Di Cunto F., Topley G., Calautti E., Hsiao J., Ong L., Seth P.K., Dotto G.P. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science. 1998;280:1069–1072. doi: 10.1126/science.280.5366.1069. [DOI] [PubMed] [Google Scholar]
- Dickson M.A., Hahn W.C., Ino Y., Ronfard V., Wu J.Y., Weinberg R.A., Louis D.N., Li F.P., Rheinwald J.G. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits lifespan become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E. Epidermal differentiationthe bare essentials. J. Cell Biol. 1990;111:2807–2814. doi: 10.1083/jcb.111.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E., Segre J.A. Stem cellsa new lease on life. Cell. 2000;100:143–155. doi: 10.1016/s0092-8674(00)81691-8. [DOI] [PubMed] [Google Scholar]
- Gallico G.G., O'Connor N.E., Compton C.C., Kehinde O., Green H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N. Engl. J. Med. 1984;311:448–451. doi: 10.1056/NEJM198408163110706. [DOI] [PubMed] [Google Scholar]
- Gandarillas A., Watt F.M. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997;11:2869–2882. doi: 10.1101/gad.11.21.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat U., DasGupta R., Degenstein L., Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- Green H. The keratinocyte as differentiated cell type. Harvey Lect. 1980;74:101–139. [PubMed] [Google Scholar]
- Green H., Kehinde O., Thomas J. Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proc. Natl. Acad. Sci. USA. 1979;76:5665–5668. doi: 10.1073/pnas.76.11.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider C.W. Telomerase activationone step on the road to cancer? Trends Genet. 1999;15:109–112. doi: 10.1016/s0168-9525(98)01681-3. [DOI] [PubMed] [Google Scholar]
- Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W., Weinberg R.A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Harle-Bachor C., Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis in human skin and in immortal and carcinoma-derived skin keratinocytes. Proc. Natl. Acad. Sci. USA. 1996;93:6476–6481. doi: 10.1073/pnas.93.13.6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Hermeking H., Lengauer C., Polyak K., He T.-C., Zhang L., Thiagalingam S., Kinzler K.W., Vogelstein B. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol. Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- Jacks T., Weinberg R.A. The expanding role of cell cycle regulators. Science. 1998;280:1035–1036. doi: 10.1126/science.280.5366.1035. [DOI] [PubMed] [Google Scholar]
- Jonason A.S., Kunala S., Price G.J., Restifo R.J., Spinelli H.M., Persing J.A., Leffel D.J., Tarone R.E., Brash D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.H., Watt F.M. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713–724. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- Kim N.W., Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP) Nucleic Acids Res. 1997;25:2595–2597. doi: 10.1093/nar/25.13.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyono T., Foster S.A., Koop J.I., McDougall J.K., Galloway D.A., Klingelhutz A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Lajtha L.G. Stem cell concepts. Differentiation. 1979;14:23–34. doi: 10.1111/j.1432-0436.1979.tb01007.x. [DOI] [PubMed] [Google Scholar]
- Lavker R.M., Sun T.-T. Epidermal stem cells. J. Invest. Dermatol. 1983;81:121S–127S. doi: 10.1111/1523-1747.ep12540880. [DOI] [PubMed] [Google Scholar]
- Leffers H., Madsen P., Rasmussen H.H., Honorè B., Andersen A.H., Walbum E., Vanderkerckhove J., Celis J.E. Molecular cloning and expression of the transformation sensitive epithelial marker stratifin. A member of a protein family that has been involved in the protein kinase C signalling pathway. J. Mol. Biol. 1993;231:982–988. doi: 10.1006/jmbi.1993.1346. [DOI] [PubMed] [Google Scholar]
- Lehrer M.S., Sun T.-T., Lavker R.M. Strategies of epithelial repairmodulation of stem cell and transient amplifying cell proliferation. J. Cell Sci. 1998;111:2867–2875. doi: 10.1242/jcs.111.19.2867. [DOI] [PubMed] [Google Scholar]
- Lustig A.J. Crisis interventionthe role of telomerase. Proc. Natl. Acad. Sci. USA. 1999;96:3339–3341. doi: 10.1073/pnas.96.7.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathor M.B., Ferrari G., Dellambra E., Cilli M., Mavilio F., Cancedda R., De Luca M. Clonal analysis of stably transduced human epidermal stem cells in culture. Proc. Natl. Acad. Sci. USA. 1996;93:10371–10376. doi: 10.1073/pnas.93.19.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison S.J., Shah N.M., Anderson D.J. Regulatory mechanisms in stem cell biology. Cell. 1997;88:287–298. doi: 10.1016/s0092-8674(00)81867-x. [DOI] [PubMed] [Google Scholar]
- Muslin A.J., Tanner J.W., Allen P.M., Shaw A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Ohnuma S., Philpott A., Wang K., Holt C.E., Harris W.A. p27Xic1, a Cdk inhibitor, promotes the determination of glial cells in Xenopus retina. Cell. 1999;99:499–510. doi: 10.1016/s0092-8674(00)81538-x. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver T.L., Weinberg R.A. A checkpoint on the road of cancer. Nature. 1998;392:223–224. doi: 10.1038/32520. [DOI] [PubMed] [Google Scholar]
- Paramio J.M., Lain S., Segrelles C., Lane E.B., Jorcano J.L. Differential expression and functionally cooperative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene. 1998;17:949–957. doi: 10.1038/sj.onc.1202031. [DOI] [PubMed] [Google Scholar]
- Paulovich A.G., Toczyski D.P., Hartwell L.H. When checkpoints fail. Cell. 1997;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- Pellegrini G., Traverso C.E., Franzi A.T., Zingirian M., Cancedda R., De Luca M. Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet. 1997;349:990–993. doi: 10.1016/S0140-6736(96)11188-0. [DOI] [PubMed] [Google Scholar]
- Pellegrini G., Golisano O., Paterna P., Lambiase A., Bonini S., Rama P., De Luca M. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface J. Cell Biol 145 1999. 769 782a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini G., Ranno R., Stracuzzi G., Bondanza S., Guerra L., Zambruno G., Micali G., De Luca M. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns Transplantation 68 1999. 868 879b [DOI] [PubMed] [Google Scholar]
- Reddel R. A reassessment of the telomere hypothesis of senescence. BioEssays. 1998;20:977–984. doi: 10.1002/(SICI)1521-1878(199812)20:12<977::AID-BIES3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Rheinwald J.G., Green H. Serial cultivation of strains of human epidermal keratinocytesthe formation of keratinizing colonies from single cells. Cell. 1975;6:331–344. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Roberts R.L., Mosch H.U., Fink G.R. 14-3-3 proteins are essential for RAS/MAPK cascade signaling during pseudohyphal development in S. cerevisiae . Cell. 1997;89:1055–1065. doi: 10.1016/s0092-8674(00)80293-7. [DOI] [PubMed] [Google Scholar]
- Rochat A., Kobayashi K., Barrandon Y. Location of stem cells of human hair follicles by clonal analysis. Cell. 1994;76:1063–1073. doi: 10.1016/0092-8674(94)90383-2. [DOI] [PubMed] [Google Scholar]
- Roop D. Defects in the barrier. Science. 1995;267:474–475. doi: 10.1126/science.7529942. [DOI] [PubMed] [Google Scholar]
- Sedivy J.M. Can ends justify the means? Telomeres and the mechanism of replicative senescence and immortalization in mammalian cells. Proc. Natl. Acad. Sci. USA. 1998;95:9078–9081. doi: 10.1073/pnas.95.16.9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a . Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Tseng H., Green H. Association of basonuclin with ability of keratinocytes to multiply and with absence of terminal differentiation. J. Cell Biol. 1994;126:495–506. doi: 10.1083/jcb.126.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G., Luo Z., Avruch J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 1998;394:88–91. doi: 10.1038/27938. [DOI] [PubMed] [Google Scholar]
- Urashima M., Teoh G., Yuza Y., Anderson K.C., Maekawa K. Restoration of p16INK4a protein induces myogenic differentiation in RD rhabdomyosarcoma cells. Br. J. Cancer. 1999;79:1032–1036. doi: 10.1038/sj.bjc.6690165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waikel R.L., Wang X.-J., Roop D. Targeted expression of c-myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene. 1999;18:4870–4878. doi: 10.1038/sj.onc.1203040. [DOI] [PubMed] [Google Scholar]
- Watt F.M. Epidermal stem cellsmarkers, patterning and the control of cell fate. Phil. Trans. R. Soc. Lond. B. 1998;353:831–837. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F.M., Green H. Involucrin synthesis is correlated with cell size in human epidermal cultures. J. Cell Biol. 1981;90:738–742. doi: 10.1083/jcb.90.3.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynford-Thomas D. Cellular senescence and cancer. J. Pathol. 1999;187:100–111. doi: 10.1002/(SICI)1096-9896(199901)187:1<100::AID-PATH236>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Yeager T.R., De Vries S., Jarrad D.F., Kao C., Nakada S.Y., Moon T.D., Bruskewitz R., Stadler W.M., Meisner L.F., Gilchrist K.W. Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998;12:163–174. doi: 10.1101/gad.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J., Harada H., Yang E., Jochel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhang J.-M., Wei Q., Zhao X., Paterson B.M. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:926–933. doi: 10.1093/emboj/18.4.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A.J., Watt F.M. Beta-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development. 1999;126:2285–2298. doi: 10.1242/dev.126.10.2285. [DOI] [PubMed] [Google Scholar]