

Fourteen years after its initial description, the nuclear factor κB (NF-κB) signaling pathway endures as a prime example of rapidly responsive gene regulation (Sen and Baltimore 1986). Even with the complexities revealed through the elucidation of converging activation pathways and diverging downstream effectors, a generalized scheme of NF-κB signaling remains elegant in its logic: (a) external signals stimulate a kinase(s), such as NF-κB–inducing kinase (NIK), MEKK1, Akt, or TBK1/NAK, which activates the inhibitor of NF-κB protein kinase (IKK); (b) IKK then phosphorylates the inhibitor of NF-κB protein (IκB) on two critical serine residues, which targets IκB for ubiquitination and subsequent degradation by the 26S proteasome; (c) previously held captive in the cytoplasm through its association with IκB, the Rel/NF-κB dimeric transcription factor is now free to enter the nucleus, find its DNA sequence recognition motifs, and regulate transcription (reviewed in Mercurio and Manning 1999; Ozes et al. 1999; Pomerantz and Baltimore 1999; Romashkova and Makarov 1999; Tojima et al. 2000).
The ongoing examination of NF-κB signaling has revealed its everexpanding role in stress responses, apoptosis, cell survival, oncogenesis, and development. The list of potent inducers of NF-κB is also diverse and includes proinflammatory stimuli such as tumor necrosis factor α (TNFα), cytokines and interleukin 1β (IL-1β), bacterial and viral products, pro-apoptotic and necrotic stimuli ranging from ultraviolet light and γ-irradiation to oxygen free radicals, and, most recently, cell survival factors, e.g., neurotrophins (reviewed by Gerondakis et al. 1998; Foo and Nolan 1999; Li and Karin 1999; Middleton et al. 2000). While first discovered as a key regulatory factor of the immune system, NF-κB is now recognized as an important player in the functioning of many organs and cell types. In the past few years, compelling evidence has been accumulating to suggest that the diversity in the outcome of NF-κB activation may be largely a reflection of the cell type and differentiation state of the cell (Ozes et al. 1999; Romashkova and Makarov 1999; Ernfors 2000).
In the skin, NF-κB plays a particularly central role in epidermal biology. Perpetually subjected to the harmful ultraviolet rays of the sun, the proliferative cells of the epidermis may rely on NF-κB activation for protection and survival (Fisher et al. 1996; Qin et al. 1999; Seitz et al. 2000). More recent gain or loss of function studies in mice suggest an equally important role in balancing growth and differentiation in the epidermis. If correct, this would add yet another provocative function for NF-κB in signaling pathways.
Effectors: The Rel/NF-κB Family
The Rel/NF-κB transcription factor family includes five members, c-Rel, RelA (p65), RelB, NF-κB1 (p50), and NF-κB2 (p52), which function as hetero- or homodimeric transcription factors (reviewed in Foo and Nolan 1999). Although other pairings exist, the most frequently encountered dimer consists of p50/p65. A 10-bp κB consensus binding site serves as the recognition site for most if not all of the combinations of NF-κB family members, and genes that carry this sequence can serve as downstream targets for NF-κB activation. Given the known external stimuli for NF-κB activation, it is not surprising that the list of target genes includes a number of NF-κB–regulated immune genes, many of which encode acute-phase reactants to inflammation, oxidative or radiation stress, or tissue injury.
Mice have been generated that are null for each of the Rel family members. Only the p65 null embryo, which dies at embryonic day 16, displays an obvious defect external to the immune system, namely apoptosis of hepatocytes (reviewed in Gerondakis et al. 1998; Beg et al. 1995b). However, double knockouts reveal more severe defects, such as osteopetrosis and major abnormalities in B cell development (p50/p52 double null) or enhanced inflammatory defects (p50/RelB double null) (Franzoso et al. 1997; Weih et al. 1997), suggesting the existence of some functional redundancy within the Rel/NF-κB family.
No major skin defects have been noted in the single or double null animals. However, since p65 single and double null mice die by E16 at a time when epidermal stratification and differentiation is still developing, skin-specific knockouts will be needed to fully evaluate the effects of this targeting event on epidermal differentiation (Beg et al. 1995b; Grossmann et al. 1999; Horwitz et al. 1999). Additionally, given its importance to this tissue, greater redundancy might exist in the skin-specific expression of Rel/NF-κB family members than in some other organs and tissues. Thus, an understanding of the functional roles of p65 and other Rel/NF-κB family members in epidermis must await more rigorous examination of the skin of existing Rel/NF-κB null animals coupled with future skin-specific targeting of the p65 gene and matings with viable Rel/NF-κB null mice to eliminate possible redundancies.
Inhibitors: The IκB Family
As is the case for the Rel/NF-κB grouping, multiple IκB family members exist, including IκBα, IκBβ, IκBγ, IκBε, and BCL3. BCL3 is nuclear and seems to act by enhancing NF-κB activation (Franzoso et al. 1992). With this exception, the other IκB proteins retain NF-κB dimeric complexes in the cytoplasm and, thus, act as inhibitors. Of these, IκBα is perhaps the most well studied and functionally most relevant to this discussion (see mini-review, Baeuerle 1998).
Receiving Release Papers and Gaining Freedom
Mice null for IκBα are seemingly normal at birth, but they eventually runt and develop dermatitis (Beg et al. 1995a; Klement et al. 1996). Marked alterations in epidermal morphology include excessive proliferation of basal keratinocytes and a paucity of keratohyalin granules that typify the granular layer, a late-stage feature of terminal differentiation. Additionally, keratin 14, a marker of the proliferative compartment of the epidermis, and keratin 16, typically induced in a variety of hyperproliferative skin disorders, are expressed in the suprabasal compartment of IκBα2/− epidermis, normally reserved for terminally differentiating cells. While these phenotypic changes in skin are dramatic, it is unclear whether they reflect direct consequences of NF-κB elevation in the epidermis or result secondarily from immune response dysfunction, another prominent feature of the IκBα null mouse (Beg et al. 1995a). More recent studies suggest that the epidermal response may indeed be secondary, at least in part, and examination of IκBα null skin grafted onto a mouse wild-type for IκB/NF-κB function would be useful in distinguishing the features that arise due to an intrinsic defect in IκBα within the epidermis itself.
Getting a Grip on the Situation
NF-κB is differentially located in normal, unstressed epidermis. As judged by immunofluorescence, p50 translocates from the cytoplasm to the nucleus as epidermal cells exit the basal layer and undergo terminal differentiation (Seitz et al. 1998). Using an antibody to p65, the major partner of p50, Takeda et al. 1999 find additional evidence for nuclear NF-κB in a few basal cells, suggesting that NF-κB activation may occur just before or concomitant with exiting the cell cycle. To explore the possible functional significance of NF-κB activation in the skin, Seitz et al. 1998 have also sought to disrupt NF-κB signaling in the basal layer of the epidermis. The researchers used a keratin 14 promoter to target basal-specific expression of a constitutively stable form of IκBα in which the PEST sequence and IKK phosphorylation sites had been mutated. At sufficiently high levels, this IκBα inhibitor was expected to prevent nuclear translocation (and therefore transactivation) of all known NF-κB family members.
Interestingly, mice expressing the K14-IκBα mutant transgene, IκBαM, exhibited skin and hair abnormalities and by day 4 of postnatal development, runting was also evident. Additionally, the epidermis was unusually thick, displaying excessive suprabasal layers (Fig. 1 A, compare with control in Fig. 1 B). This feature was a reflection of hyperproliferation as judged by the persistence of DNA-synthesizing cells in the suprabasal layers (Seitz et al. 1998). Extending their analysis, these authors discovered that pharmacological inhibition of NF-κB signaling could also induce epidermal hyperplasia, suggesting that the observed effects are directly attributable to suppression of NF-κB activity at a time when basal cells would normally activate this transcription factor and exit the cell cycle. Intriguingly, if left unchecked, suppression of NF-κB in the skin through stable IκBα overexpression imparted an invasive character to epidermal cells, leading to the appearance of well-differentiated squamous cell carcinomas in older transgenic animals or when transgenic epidermal cells were transplanted onto the backs of immunocompromised mice (Seitz et al. 1998; van Hogerlinden et al. 1999).
Figure 1.
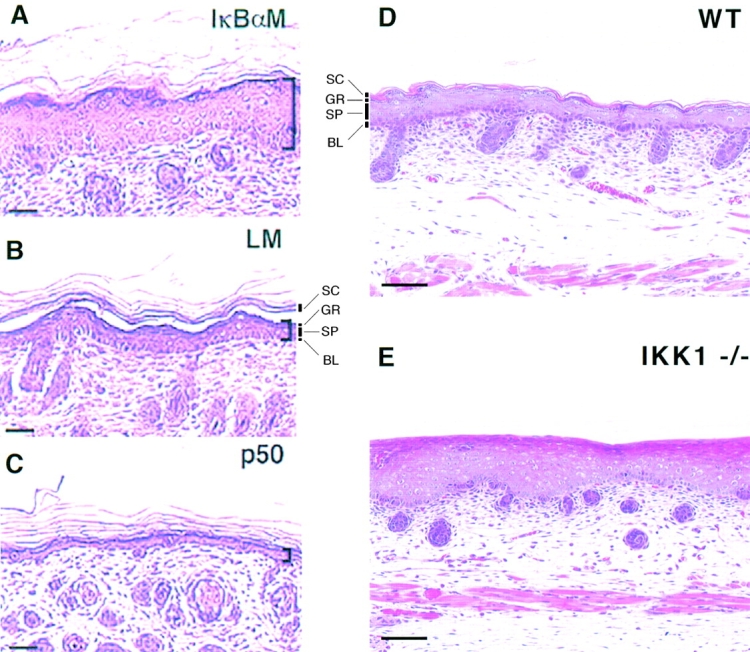
An unusual correlation between NF-κB activation and cell cycle arrest in the epidermis. (A–C). Histology of skin from transgenic mice overexpressing stabilized IκBα (A) or the p50 subunit of NF-κB (C) under the control of the basal epidermal specific human K14 promoter. Shown is a skin section from a nontransgenic littermate (LM) for comparison (B). Brackets show thickness of epidermis. (Figure courtesy of P. Khavari; reprinted with permission from Seitz, C.S., Q. Lin, H. Deng, and P.A. Khavari. 1998. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc. Natl. Acad. Sci. USA. 95:2307–2312. Copyright 1998. National Academy of Sciences, USA.) (D–E). Dorsal skin from wild-type (D) and IKK1−/− (E) embryos (E185). (Figure courtesy of S. Akira; reprinted with permission from Takeda, K., O. Takeuchi, T. Tsujimura, S. Itami, O. Adachi, T. Kawai, H. Sanjo, K. Yoshikawa, N. Terada, and S. Akira. 1999. Limb and skin abnormalities in mice lacking IKKα. Science. 284:313–316. Copyright 1999. American Association for the Advancement of Science.) SC, stratum corneum; GR, granular layer; SP, spinous layer; BL, basal layer. Bars: (A–C) 75 μm; (D–E) 30 μm.
To further examine the possible link between NF-κB signaling and cessation of epidermal proliferation, Seitz et al. 1998 also generated mice expressing a transactivating p50 or p65 under the control of the keratin 14 promoter. In this case, nuclear, i.e., activated, NF-κB was seen throughout the skin, and as predicted from the converse experiment, a thin, hypoproliferative epidermis was the outcome (Fig. 1 C, example shown is p50 overexpression). The abnormalities in epidermis that occurred as a consequence of alterations in NF-κB signaling appeared to be restricted to defects in proliferative state: neither activating nor repressing NF-κB signaling altered the suprabasal expression patterns of the differentiation-specific markers, keratin 10, involucrin, transglutaminase 1, or filaggrin.
From these data, we can begin to speculate about the role of NF-κB signaling in the epidermis. An attractive possibility is that NF-κB activation is necessary to either cause or maintain cell cycle arrest as keratinocytes commit to terminally differentiate and exit the basal layer. In this regard, the behavior of the epidermis seems to be opposite from that of the immune system, where NF-κB activity seems to play a positive role in proliferation (reviewed in Gerondakis et al. 1998). NF-κB has been associated with promotion of apoptosis in double positive thymocytes (Hettmann et al. 1999), and although controversial, NF-κB activation may promote cell death in some neurons exposed to ischemia, oxidative stress, or excitotoxicity (Ernfors 2000). Thus, while an apparently antiproliferative effect of NF-κB is less common, it is not without some precedent.
A number of issues remain unsettled. In two separate studies using similar IκB-stabilizing mutants driven by basal epidermal keratin promoters, the mice either died after 4–7 d (Seitz et al. 1998) or survived for at least 4 mo (van Hogerlinden et al. 1999), leaving open the possibility that, at least in certain cases, transgenic manipulation of NF-κB signaling using protein overexpression can lead to consequences that are not necessarily physiologically relevant. In addition, by using a basal keratin promoter to express a highly stable IκB mutant, it is likely that the inhibitor persisted in the suprabasal layers of epidermis. Hence, it remains unclear as to whether the alterations induced by perturbations in NF-κB signaling arise from transcriptional changes in basal or alternatively in suprabasal cells. This latter issue is especially important in distinguishing whether NF-κB activation might be a critical event in initiating withdrawal from the cell cycle as cells leave the basal layer or, rather, if this signal is crucial in maintaining the post-mitotic state of suprabasal cells. In the future, suprabasal-specific expression of the IκB mutant under the control of the differentiation-specific involucrin promoter will help to distinguish between these possibilities. A final curiosity is that both groups noted perturbations in hair development. The mechanism by which hair development and/or differentiation might be dependent upon the NF-κB signaling machinery is an area that also merits further investigation.
Regulators: The IKK Complex
An important feature of NF-κB regulation is the phosphorylation of the IκB inhibitor and its subsequent targeting for ubiquitin-mediated degradation. In the last several years, the elusive IκB regulatory kinase was identified and shown to consist of two catalytic subunits, IKK1 and IKK2 (DiDonato et al. 1997; Mercurio et al. 1997; Regnier et al. 1997), and a regulatory subunit, referred to alternatively as IKKγ, IKKAP-1, FIP-3, or NEMO (Rothwarf et al. 1998; reviewed in Mercurio and Manning 1999). The story appears even more complex as a recently identified kinase, termed IKK-i or IKKε, may function in a different IκB regulatory complex (Shimada et al. 2000; Peters et al. 2000). In the past year, the consequences of ablating the IKK1, IKK2, and NEMO/IKK-γ subunits were explored using gene knockout technology in mice (Li et al. 1999a; Takeda et al. 1999; Hu et al. 1999; Rudolph et al. 2000).
In the absence of IKK1, embryos died at birth, exhibiting gross abnormalities typified by a shiny epidermis, a shortened tail, and seemingly stunted limb growth. The shiny appearance of the epidermis was a consequence of an impairment in the epidermal barrier. This was accompanied by a hyperthickened spinous layer and a paucity of typical keratinized, enucleated squames (Fig. 1, D–E). In addition, expression of late-stage terminal differentiation markers, e.g., loricrin and filaggrin, was noticeably reduced.
In agreement with both IKK's role as an instigator of IκB degradation and the induction of nuclear NF-κB as normal basal cells commit to terminally differentiate, basal cells of IKK1 null epidermis displayed an increase in IκBα and IκBβ immunostaining, and a cytoplasmic restriction of NF-κB. The thickened epidermis was consistent with a model whereby failure to activate epidermal NF-κB results in hyperproliferation due to a failure of transiently amplifying basal cells to withdraw from the cell cycle. However, alterations in terminal differentiation had not been previously seen when IκB levels were elevated in the basal layer of transgenic epidermis (Seitz et al. 1998). These findings suggested that the effects of IKK ablation in the skin may go beyond mere elevation of IκB in the basal layer.
Another mystery still unsolved is how IKK1 ablation might restrict appendage outgrowth in developing mouse embryos. A clue stems from the fact that the limb and tail bones were only slightly retarded, suggesting that the limb defects could be a secondary event caused by IKK1-mediated defects in the skin. One possibility is that signaling between developing ectoderm and mesoderm in the limbs is dependent, to some degree, on IKK1 activity and, most likely, NF-κB activity. Indeed, limb outgrowth is known to involve lateral migration of keratinocytes as well as mesenchymal-epithelial interactions, and in this regard, it is likely relevant that the skin of the IKK1 null animals was taut, lacking folds or wrinkles. Additionally, haptotactic migration of keratinocytes in cell culture seems to be dependent upon functional NF-κB signaling (Benoliel et al. 1997), further pointing to the notion that defects in epidermal migration may underlie the limb abnormalities in the IKK1 null embryos. NF-κB activity. While additional studies will be necessary to assess the extent to which this hypothesis may be correct, a defect in cell migration could indirectly influence epidermal thickness and proliferation. Another factor likely to be relevant is that NF-κB activation plays a central role in cell survival through its ability to block or reduce apoptosis in various cell types including keratinocytes (Chaturvedi et al. 1999; Seitz et al. 2000). It is possible, therefore, that elimination of NF-κB activation may result in ablation of cells or altered signaling in the progress zone of the limb bud (Bushdid et al. 1998; Kanegae et al. 1998), thereby accounting at least in part for the defects observed.
This issue prompts us to revisit the question of why the phenotype of the IKK1 null mouse is markedly more severe than that of various transgenic mice expressing constitutively stabilized IκB in the basal epidermal layer. Since basal layer NF-κB appeared to be restricted to the cytoplasm in both types of mice, it seems unlikely that the differences are rooted in variations in overall levels of basal epidermal IκB. A priori, it could be that the increased severity in IKK1−/− mice is due to stabilization of IκB within all skin cells, rather than only basal epidermal cells. While this possibility seems likely, a more intriguing notion is that as yet unidentified substrates for IKK1 exist that might influence pathways extending beyond NF-κB signaling. In this regard, it may be relevant that IκB and β-catenin, another key protein in the skin, contain similar sequences that are phosphorylated as an important step in regulating their stability (Aberle et al. 1997). Interestingly, recent evidence has shown that a specificity factor in the ubiquitination pathway, referred to as Slimb in flies and β-TRcP in mammals, is involved in recognizing this phosphorylated sequence in both IκB and β-catenin (reviewed in Maniatis 1999). It is tempting to speculate that a kinase, such as IKK1, may also recognize this sequence in both proteins, although other protein domains may also contribute to kinase specificity. As β-catenin stabilization has been implicated in epidermal stem cell character and/or maintenance (Gat et al. 1998; Zhu and Watt 1999), and Wnt/Wingless signaling is likely to be involved in epidermal and/or hair follicle development/differentiation, the more severe phenotype of the IKK1−/− mice could reflect a more complex role for IKK1 in governing processes beyond the NF-κB pathway where protein stability is involved in transcriptional regulation.
Strengthening the Foundation and Looking Ahead
With the potential of IKK1 to affect multiple signal transduction pathways, additional studies will be necessary to fully understand the IKK1 null phenotype at a molecular level. This said, based upon the prior transgenic mouse studies with stabilized forms of IκB, epidermal hyperproliferation was both an expected and observed feature of IKK1 null animals. Additionally, it is notable that in the IKK1 null embryos, IL-1– and TNFα-induced activation of NF-κB was not impaired, while these responses were diminished in IKK2 and NEMO/IKKγ null embryos (Li et al. 1999b; Rudolph et al. 2000). Thus, it appears that components of IKK may have distinct roles in controlling NF-κB activation, and our understanding of the response to proinflammatory stimuli is likely not sufficient to explain in full how NF-κB might impact on balancing growth and differentiation in the epidermis.
Present literature would seem to suggest that NF-κB plays a role in either inducing an epidermal cell to withdraw from the cell cycle and/or in maintaining this state once achieved. If future studies substantiate this notion, then NF-κB would have a different role in the epidermis than it has in other cell types such as lymphocytes and neurons. NF-κB family members were first identified as the cellular equivalents of viral oncogenes and also as translocated genes in T and B cell cancers (for review, see Foo and Nolan 1999). While these observations contribute to the view that NF-κB acts to enhance proliferation, many of NF-κB's stimulatory effects have been attributed to its powerful ability to protect against apoptosis. Thus, in most situations, activation of NF-κB does not actually promote cell cycle progression.
Recent evidence indicates that, as in most other cell types, epidermal NF-κB protects against apoptosis, apparently by enhancing the expression of anti-apoptotic factors (Seitz et al. 2000). At first glance, this finding might only serve to reinforce the apparent paradox as to why antagonizing NF-κB, as with IκB stabilization and/or IKKα ablation, would lead to an increase in cellularity within epidermis, whereas in most tissues a decrease in cellularity would be expected. However, this new finding illuminates a possible role for NF-κB in epidermis that is uniquely tailored to suit the specialized properties of this tissue. Indeed, the unusual correlation between cell cycle withdrawal and activation of epidermal NF-κB is intriguing in this regard, since cell cycle arrest is emerging as a prerequisite for protection from apoptosis (Foo and Nolan 1999). Given that NF-κB seems to be prominent in the earlier stages of terminal differentiation, it could be that through protection from apoptosis, spinous cells are able to execute and complete their differentiation-specific program of transcriptional changes necessary to produce the epidermal barrier and protect our terrestrial bodies from dehydration and microbial invasion (Seitz et al. 2000). By linking cell cycle withdrawal and protection from apoptosis, NF-κB is able to coordinate and monitor both homeostasis within the epidermis, as well as the successful transition of the epidermal cell from the basal to suprabasal layers.
A model summarizing the putative functions of NF-κB in the epidermis is presented in Fig. 2. The extent to which this view is correct must now await the identification of NF-κB target genes in the epidermis and an understanding of how these transcripts differ in response to different means of NF-κB activation. As the list of NF-κB target genes in the epidermal keratinocyte and other cell types becomes comprehensive, comparisons between cell types and differentiation states should help us to understand the underlying bases for the various responses. A more thorough characterization of the particular NF-κB family members expressed in the epidermis will also be needed, and in this regard, removal of redundancies might be needed in order to further dissect the role of NF-κB from the myriad of other changes associated with the early stages of terminal differentiation. Given the vibrance and fast pace of the field of NF-κB signaling, a coherent understanding of the present mysteries and complexities of NF-κB and its many intersecting signaling crossroads will likely evolve and further contribute to our understanding of cell growth, differentiation, and death in the epidermis.
Figure 2.

NF-κB signaling in the epidermis: balancing cell cycle withdrawal, cell survival, and differentiation. Model is based upon a classical role for NF-κB in cell survival but an atypical role for NF-κB in promoting cell cycle withdrawal in the epidermis. NF-κB appears to be activated concomitant with the time at which basal epidermal cells withdraw from the cell cycle and commit to terminally differentiate. Terminal differentiation in the epidermis requires a series of transcriptional changes in the absence of proliferation to enable the epidermal barrier to be established. This barrier is essential to keep bodily fluids in and harmful microorganisms out. Terminally differentiated squames are then shed from the skin surface, continually being replaced by basal cells withdrawing from the cell cycle, committing to differentiate, and transiting outward. The convergence of cell survival and cell cycle arrest pathways, linked through NF-κB, may help to balance homeostasis in the epidermis and coordinate these processes.
References
- Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle P.A. IκB-NF-κB structuresat the interface of inflammation control. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- Beg A.A., Sha W.C., Bronson R.T., Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκB α-deficient mice Genes Dev. 9 1995. 2736 2746a [DOI] [PubMed] [Google Scholar]
- Beg A.A., Sha W.C., Bronson R.T., Ghosh S., Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB Nature 376 1995. 167 170b [DOI] [PubMed] [Google Scholar]
- Benoliel A.M., Kahn-Peries B., Imbert J., Verrando P. Insulin stimulates haptotactic migration of human epidermal keratinocytes through activation of NF-κB transcription factor. J. Cell Sci. 1997;110:2089–2097. doi: 10.1242/jcs.110.17.2089. [DOI] [PubMed] [Google Scholar]
- Bushdid P.B., Brantley D.M., Yull F.E., Blaeuer G.L., Hoffman L.H., Niswander L., Kerr L.D. Inhibition of NF-κB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature. 1998;392:615–618. doi: 10.1038/33435. [DOI] [PubMed] [Google Scholar]
- Chaturvedi V., Qin J.Z., Denning M.F., Choubey D., Diaz M.O., Nickoloff B.J. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J. Biol. Chem. 1999;274:23358–23367. doi: 10.1074/jbc.274.33.23358. [DOI] [PubMed] [Google Scholar]
- DiDonato J.A., Hayakawa M., Rothwarf D.M., Zandi E., Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Ernfors P. Nuclear factor-κB to the rescue of cytokine-induced neuronal survival. J. Cell Biol. 2000;148:223–225. doi: 10.1083/jcb.148.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G.J., Datta S.C., Talwar H.S., Wang Z.Q., Varani J., Kang S., Voorhees J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- Foo S.Y., Nolan G.P. NF-κB to the rescueRELs, apoptosis and cellular transformation. Trends Genet. 1999;15:229–235. doi: 10.1016/s0168-9525(99)01719-9. [DOI] [PubMed] [Google Scholar]
- Franzoso G., Bours V., Park S., Tomita-Yamaguchi M., Kelly K., Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- Franzoso G., Carlson L., Xing L., Poljak L., Shores E.W., Brown K.D., Leonardi A., Tran T., Boyce B.F., Siebenlist U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat U., DasGupta R., Degenstein L., Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grumont R., Rourke I., Grossmann M. The regulation and roles of Rel/NF-κ B transcription factors during lymphocyte activation. Curr. Opin. Immunol. 1998;10:353–359. doi: 10.1016/s0952-7915(98)80175-1. [DOI] [PubMed] [Google Scholar]
- Grossmann M., Metcalf D., Merryfull J., Beg A., Baltimore D., Gerondakis S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc. Natl. Acad. Sci. USA. 1999;96:11848–11853. doi: 10.1073/pnas.96.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmann T., DiDonato J., Karin M., Leiden J.M. An essential role for nuclear factor κB in promoting double positive thymocyte apoptosis. J. Exp. Med. 1999;189:145–158. doi: 10.1084/jem.189.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz B.H., Zelazowski P., Shen Y., Wolcott K.M., Scott M.L., Baltimore D., Snapper C.M. The p65 subunit of NF-κB is redundant with p50 during B cell proliferative responses, and is required for germline CH transcription and class switching to IgG3. J. Immunol. 1999;162:1941–1946. [PubMed] [Google Scholar]
- Hu Y., Baud V., Delhase M., Zhang P., Deerinck T., Ellisman M., Johnson R., Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- Kanegae Y., Tavares A.T., Izpisua-Belmonte J.C., Verma I.M. Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Nature. 1998;392:611–614. doi: 10.1038/33429. [DOI] [PubMed] [Google Scholar]
- Klement J.F., Rice N.R., Car B.D., Abbondanzo S.J., Powers G.D., Bhatt P.H., Chen C.H., Rosen C.A., Stewart C.L. IκB deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol. Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Lu Q., Hwang J.Y., Buscher D., Lee K.F., Izpisua-Belmonte J.C., Verma I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton Genes Dev 13 1999. 1322 1328a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Van Antwerp D., Mercurio F., Lee K.-F., Verma I.M. Severe liver degeneration in mice lacking the IκB kinase 2 gene Science 284 1999. 321 325b [DOI] [PubMed] [Google Scholar]
- Li N., Karin M. Is NF-κB the sensor of oxidative stress? FASEB J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- Maniatis T. A ubiquitin ligase complex essential for the NF-κB, Wnt/Wingless, and Hedgehog signaling pathways. Genes Dev. 1999;13:505–510. doi: 10.1101/gad.13.5.505. [DOI] [PubMed] [Google Scholar]
- Mercurio F., Manning A.M. Multiple signals converging on NF-κB. Curr. Opin. Cell Biol. 1999;11:226–232. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- Mercurio F., Zhu H., Murray B.W., Shevchenko A., Bennett B.L., Li J., Young D.B., Barbosa M., Mann M., Manning A., Rao A. IKK-1 and IKK-2cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Middleton G., Hamanoue M., Enokido Y., Wyatt S., Pennica D., Jaffray E., Hay R.T., Davies A.M. Cytokine-induced nuclear factor κB activation promotes the survival of developing neurons. J. Cell Biol. 2000;148:325–332. doi: 10.1083/jcb.148.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozes O.N., Mayo L.D., Gustin J.A., Pfeffer S.R., Pfeffer L.M., Donner D.B. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Peters R.T., Liao S.-M., Maniatis T. IKKε is part of a novel PMA-inducible IκB kinase complex. Mol. Cell. 2000;5:513–522. doi: 10.1016/s1097-2765(00)80445-1. [DOI] [PubMed] [Google Scholar]
- Pomerantz J.L., Baltimore D. NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.Z., Chaturvedi V., Denning M.F., Choubey D., Diaz M.O., Nickoloff B.J. Role of NF-κB in the apoptotic-resistant phenotype of keratinocytes. J. Biol. Chem. 1999;274:37957–37964. doi: 10.1074/jbc.274.53.37957. [DOI] [PubMed] [Google Scholar]
- Regnier C.H., Song H.Y., Gao X., Goeddel D.V., Cao Z., Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Romashkova J.A., Makarov S.S. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi E., Natoli G., Karin M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- Rudolph D., Yeh W.C., Wakeham A., Rudolph B., Nallainathan D., Potter J., Elia A.J., Mak T.W. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- Seitz C.S., Freiberg R.A., Hinata K., Khavari P.A. NF-κB determines localization and features of cell death in epidermis. J. Clin. Invest. 2000;105:253–260. doi: 10.1172/JCI7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz C.S., Lin Q., Deng H., Khavari P.A. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc. Natl. Acad. Sci. USA. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R., Baltimore D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- Shimada T., Kawai T., Takeda K., Matsumoto M., Inoue J., Tatsumi Y., Kanamaru A., Akira S. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Intl. Immunol. 2000;11:1357–1362. doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- Takeda K., Takeuchi O., Tsujimura T., Itami S., Adachi O., Kawai T., Sanjo H., Yoshikawa K., Terada N., Akira S. Limb and skin abnormalities in mice lacking IKKα. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- Tojima Y., Fujimoto A., Delhase M., Chen Y., Hatakeyama S., Nakayama K., Kaneko Y., Nimura Y., Motoyama N., Ikeda K. NAK is an IκB kinase-activating kinase. Nature. 2000;404:778–782. doi: 10.1038/35008109. [DOI] [PubMed] [Google Scholar]
- van Hogerlinden M., Rozell B.L., Ahrlund-Richter L., Toftgard R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-κB signaling. Cancer Res. 1999;59:3299–3303. [PubMed] [Google Scholar]
- Weih F., Durham S.K., Barton D.S., Sha W.C., Baltimore D., Bravo R. p50-NF-κB complexes partially compensate for the absence of RelBseverely increased pathology in p50(−/−)relB(−/−) double-knockout mice. J. Exp. Med. 1997;185:1359–1370. doi: 10.1084/jem.185.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A.J., Watt F.M. β-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development. 1999;126:2285–2298. doi: 10.1242/dev.126.10.2285. [DOI] [PubMed] [Google Scholar]