

Abstract
Caspase-11, a member of the murine caspase family, has been shown to be an upstream activator of caspase-1 in regulating cytokine maturation. We demonstrate here that in addition to its defect in cytokine maturation, caspase-11–deficient mice have a reduced number of apoptotic cells and a defect in caspase-3 activation after middle cerebral artery occlusion (MCAO), a mouse model of stroke. Recombinant procaspase-11 can autoprocess itself in vitro. Purified active recombinant caspase-11 cleaves and activates procaspase-3 very efficiently. Using a positional scanning combinatorial library method, we found that the optimal cleavage site of caspase-11 was (I/L/V/P)EHD, similar to that of upstream caspases such as caspase-8 and -9. Our results suggest that caspase-11 is a critical initiator caspase responsible for the activation of caspase-3, as well as caspase-1 under certain pathological conditions.
Keywords: caspase-11, initiator caspase, stroke, middle cerebral artery occlusion, apoptosis
Introduction
Mammalian caspases are a family of cysteine proteases involved in regulating cytokine maturation and apoptosis (Cryns and Yuan 1998). Caspases can be classified according to their amino acid sequence homology into caspase-1, -3, and -9 subfamilies. The caspase-1 subfamily includes caspase-1, -4, -5, and -11. Since the predominant defect of caspase-1 knockout mice is the inability to process prointerleukin-1β (pro–IL-1β), the major function of caspase-1 is believed to be regulating cytokine maturation (Kuida et al. 1995; Li et al. 1995). Caspase-11 is a murine caspase that shares the highest homology with human caspase-4 (60% identity). The expression of caspase-11 is undetectable in healthy mice and highly inducible upon injection of lipopolysaccharide (LPS; Wang et al. 1996). Caspase-11 does not process pro–IL-1β directly. Nevertheless, caspase-11 mutant mice are deficient in the processing and secretion of IL-1β (Wang et al. 1998). We showed that caspase-11 can physically interact with caspase-1 to promote its activation. Based upon these data, we proposed that elevated expression of caspase-11 under pathological condition is directly responsible for activation of caspase-1, and thus, caspase-11 is an upstream regulator of caspase-1 (Wang et al. 1998).
The caspase-3 subfamily includes caspase-3, -6, -7, -8, and -10 (Cryns and Yuan 1998). Among this family, caspase-3 shares highest homology with caspase-7 and both have short prodomains; whereas caspase-6, -8, and -10 have long prodomains. Caspase-3 has been shown to be a major execution caspase that acts downstream in the apoptosis pathway and is involved in cleaving important substrates such as ICAD (inhibitor of caspase activated DNase), which activates the apoptotic DNA ladder-forming activity of CAD (caspase activated DNase; Enari et al. 1998; Sakahira et al. 1998). The major route of activating short prodomain caspases is through direct proteolytic processing. Two known pathways that can activate procaspase-3 are through proteolytic cleavage by caspase-8 and -9. In the Fas apoptosis pathway, the activation of Fas receptor induces the formation of death inducing signaling complex (DISC) which recruits and activates caspase-8 (Kischkel et al. 1995; Muzio et al. 1996). In Fas type I cells, activated caspase-8 directly activates procaspase-3 by cleavage (Scaffidi et al. 1998). In apoptosis, where mitochondria play a critical role, the release of cytochrome c from damaged mitochondria induces the formation of cytochrome c/Apaf-1/caspase-9 complex (Li et al. 1997). Apaf-1 is a mammalian homologue of Caenorhabditis elegans cell death gene product Ced-4 (Zou et al. 1997). The interaction of cytochrome c, Apaf-1, and caspase-9 triggers the activation of caspase-9 that in turn activates procaspase-3 through direct proteolytic processing (Li et al. 1997). Thus, caspase-8 and -9 have been known as the two major upstream activators of caspase-3. Caspase-8 and -9 mutant mice exhibit severe defects in developmental apoptosis, suggesting that they are critical for activation of apoptosis and caspase-3 during development (Kuida et al. 1998; Varfolomeev et al. 1998).
Caspases may be involved in both acute and chronic neurodegenerative diseases (Friedlander and Yuan 1998). Irreversible caspase inhibitors, zVAD.fmk and zDEVD. fmk, protected brains from ischemic injury and improved neurological deficits in both mouse and rat (Hara et al. 1997b). Caspase-3–like enzyme activity and the activated caspase-3 subunit are detected in ischemic brain samples (Namura et al. 1998). Intraventricular injection of DEVD.fmk significantly reduced middle cerebral artery occlusion (MCAO)-induced apoptosis, suggesting that caspase-3 may play a critical role in ischemic brain injury (Hara et al. 1997b). As caspase-3 is a short-prodomain caspase, it is usually activated by an upstream caspase through direct proteolytic cleavage. Although caspase-3 has been implicated in a number of pathological conditions, including brain ischemia, the identity of the upstream caspase(s) is not known. We show here that caspase-11 fits all the criteria to be this upstream caspase in activation of caspase-3 under pathological conditions. Caspase-11 mutant mice are defective in apoptosis and caspase-3 activation induced by brain ischemic injury. Activated caspase-11 is an efficient activator of caspase-3 in vitro. Combinatorial library analysis showed that the preferred cleavage sequence of caspase-11 is (I/L/V/P)EHD, similar to that of caspase-8 and -9 (Thornberry et al. 1997). In addition, procaspase-11 can process itself in vitro. We propose that caspase-11 is an upstream caspase responsible for the activation of caspase-3, as well as caspase-1, under pathological conditions.
Materials and Methods
Animals
Caspase-11 knockout mice have been described previously (Wang et al. 1998). In brief, mutant ES cell clones carrying a null mutant caspase-11 allele were injected into C57BL/6J blastocysts. The resulting chimeric males were then mated with C57BL/6J × DBA2 F1 females to obtain germline transmission of the mutant allele. Chimera of clone 444 produced germline transmitted mutant mice. The offsprings from clone 444 were backcrossed to C57BL/6J four times. Heterozygous littermates of this F4 were mated and the resulting offspring were used in this study. To ensure genetic background homogeneity, littermates of wild-type and caspase-11−/− mice were used in each group. The genotype was determined by Southern blot analysis as described previously (Wang et al. 1998).
To induce ischemia brain injury, 6–8-wk-old male mice were anesthetized with 1.5% halothane and maintained in 1% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical). Permanent ischemia was induced with an 8.0 nylon monofilament coated with a silicone resin-hardener mixture (Xantopren and Elastomer Activator; Bayer Dental) as described previously (Hara et al. 1997b). The filament was introduced into the left common carotid artery, advanced into the anterior cerebral artery, and left there until the animal was killed for immunohistochemistry and TUNEL assay.
Construction of Plasmid
The construction of expression plasmid of caspase-11 was made by PCR amplification using pJ667 as a template (Wang et al. 1998). The primers used are: SY6, GAGGATCCCATGGCTGAAAACAAACACCCT; and mNO/BH2, TTGGATCCGTCAGTTGCCAGGAAAGAGGT. The PCR fragment was cloned into the BamH1 site of pET-15b and named pS15.
The cleavage site mutation of caspase-3, D175A, was created by PCR using human caspase-3 as a template. The primers used for amplifying the 5′ half of caspase-3 cDNA were: SJ1, ATGGAGAAGACTGAAAACTCAG; and SJ2, CAACACCAGTGGCTGTCTCAA. The primers for amplifying the 3′ half of caspase-3 cDNA were: SJ3, TTGAGACAGCCAGTGGTGTTG; and SJ4, CTTTAGTGATAAAAATAGAGTTC. The 5′ and 3′ half of caspase-3 cDNA fragments were used as templates for the third PCR with SJ1 and SJ4 as primers. The resulting 850-bp fragment was cloned into pCDNA3 at EcoRV site. The D175A mutation was verified by sequencing and used for in vitro translation.
Preparation of Purified Recombinant Caspase-11 Protease
The expression constructs were transformed into Escherichia coli strain BL21 (DE3).
To make bacterial lysates, 10 ml of overnight culture from a single colony was diluted 1:100 and grown for 2.5 h until OD600 reaches 0.5–1.0. Then, caspase-11 was induced by adding 0.3 mM of isopropyl-1-thio-β-d-galactopyranoside (IPTG) and grown for another 2.5 h in a 37°C shaker. The bacteria were harvested and sonicated in 1 ml lysis buffer containing 50 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.5 mM sucrose, and 5% glycerol. Protease inhibitors (1 mM PMSF, 5 μg/ml pepstatin, and 10 μg/ml leupeptin) were also added. After centrifugation at 10,000 g the supernatant was mixed with 2 ml of nickel-charged resin (Qiagen) and then incubated for 2 h at 4°C with rotation. The beads were then washed serially with 20 ml of lysis buffer plus 1 M NaCl and lysis buffer alone. The bound caspase-11 was eluted with 5 ml lysis buffer containing 125 mM imidazole and verified by SDS-PAGE and Coomassie blue staining.
In Vitro Cleavage Assay
In vitro translation of 35S-labeled proteins was made by using TNT-coupled transcription/translation kit (Promega) in the presence of [35S]methionine. 35S-labeled proteins were incubated with 0.5 μg of purified caspase-11 in the presence of 50 μM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.5 mM sucrose, and 10 mM DTT together with protease inhibitors in a total volume of 10 μl. The mixture was incubated at 30°C for 2 h. The reaction was terminated by addition of equal volume of 2× SDS protein sample buffer (60 mM Tris-HCl, pH 6.8, 2% SDS, 5% β-mercaptoethanol, and 0.01% bromophenol blue) and then analyzed by SDS-PAGE and autoradiography.
Cell Culture
L929 (mouse fibrosarcoma cell line) cells were maintained in DME supplemented with 10% FCS.
Immunoblot and Immunoprecipitation Assay
Generation of mAb against caspase-11 (17D9, hybridoma culture supernatant) was described previously (Wang et al. 1996). For immunoblotting, tissue samples were ground in liquid N2 and the resulting tissue powder was solubilized in 0.7 ml of IP lysis buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1% NP-40) with protease inhibitors (1 mM PMSF, 10 μg/ml leupeptin, and 5 μg/ml pepstatin). After incubating on ice for 10 min, samples were centrifuged at 13,000 rpm in a microfuge at 4°C for 20 min. Protein concentration was measured using BioRad protein assay reagent. 60 μg of protein was subjected to SDS-PAGE on a 15% gel. Proteins were then transferred onto immobilon-P membrane (Millipore) and incubated with blocking solution containing 5% skim milk in TBST (50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. The membrane was then incubated with primary antibody in blocking solution at 4°C overnight. After washing three times with TBST for 10 min each, the membrane was incubated with HRP-conjugated anti-rat or anti–rabbit IgG (Jackson ImmunoResearch) for 40 min at room temperature. After washing three times, the bound antibody was revealed using the ECL Western blotting reagent (Amersham Pharmacia Biotech).
For immunoprecipitation, half of the mouse brain was ground in liquid N2 and solubilized in 700 μl of lysis buffer. The resulting lysate supernatant (total 1 mg of protein) was incubated with 50 μl 17D9 overnight at 4°C with rotation, followed by incubation with protein G-coupled agarose beads for 40 min. The immune complexes were resolved by SDS-PAGE and analyzed by immunoblotting using 17D9.
Immunohistochemistry
After MCAO or LPS challenge, mice were anesthetized and transcardially perfused with cold PBS and then with 4% paraformaldehyde in PBS. Brains were dissected, embedded in OCT medium, and were then frozen on top of dry ice. The frozen blocks were kept in −80°C until use. 5-μm sections were made in a Leica CM3000 cryostat. Sections were then postfixed in ice cold ethanol/acetic acid (2:1) solution for 5 min. After brief washing with PBS, endogenous peroxidase activity was quenched by incubating sections with 0.3% hydrogen peroxide in PBS for 30 min when TSA signal amplification kit (NEN Life Science Products) was used for visualization. Sections were then incubated with 10% normal goat serum in PBS containing 0.1% Triton X-100 for 1 h at room temperature. When secondary antibody is biotin/avidin system, samples were first blocked with Vector Laboratories' Biotin/Avidin blocking kit before incubating with normal goat serum. After blocking, sections were incubated with drops of anticaspase-11 monoclonal (undiluted 17D9), anticaspase3p20 (CM1) rabbit polyclonal (1:2,000), anti-NeuN mouse monoclonal (1:1,000; Chemicon International), or anti-GFAP (glial fibrillary acidic protein) rabbit polyclonal (1:500; DACO) antibody in PBS containing 1% normal goat serum and 0.1% Triton X-100 at 4°C overnight in a humid chamber. For detection of microglial cells, biotin-conjugated B4 isolectin (5 μg/ml) was used. Samples were then washed four times with PBS containing 0.1% Triton X-100 for 5 min each, followed by incubation with secondary antibodies in PBS containing 1% normal goat serum and 0.1% Triton X-100 for 30 min at room temperature. For the detection of caspase-11, HRP-conjugated anti-rat goat IgG (Jackson ImmunoResearch) and TSA signal amplification kit were used following manufacturer's protocol. For the detection of caspase-3, NeuN, and GFAP, matching secondary antibodies conjugated with biotin were used. After washing, biotin-decorated samples were incubated with Texas red- or FITC-conjugated streptavidin (Jackson ImmunoResearch) for 30 min at room temperature. Tissue sections were then counterstained with Hoechst 33258 dye (1 μg/ml in PBS; Sigma Chemical Co.) for 10 min at room temperature. After washing, slide glass samples were mounted with mounting medium (1 mg/ml p-phenylendiamine in PBS containing 90% glycerol). Samples were examined using an Axiovert 135 microscope (Carl Zeiss, Inc.) and images were recorded using Northern Exposure software.
For TUNEL assay, ethanol/acetic acid postfixed samples were briefly washed and then treated with proteinase K (20 μg/ml in PBS) for 15 min at room temperature. After washing with PBS, samples were processed using Apoptag kit (Intergen) according to the manufacturer's manual. Samples were also counterstained with Hoechst 33258.
Enzyme Activity Assay
To see if caspase-11 can activate procaspase-3 or -1, 15 μg of purified recombinant caspase-11 (pS15) and/or 10 μg of recombinant procaspases were added into Hepes buffer (50 mM, pH 7.4) containing 2 mM DTT and 12 μΜ fluorogenic peptide substrates (acDEVD.amc or acYVAD.amc) in a total volume of 300 μl. Fluorescence as a result of caspase activation was measured at 10-s intervals using Aminco Bowman Series 2 Luminescence spectrometer (excitation wavelength = 380 nm, emission wavelength = 460 nm).
Substrate Specificity Screening
The design, synthesis, and validation of the PS-SCL have been described elsewhere (Rano et al. 1997). Each of the 60 samples (20 amino acids × 3 sublibraries) of the PS-SCL was prepared as a stock of ∼10 mM in DMSO. To determine protease specificity, 2.3 nM caspase-11 was added to reaction mixtures containing 100 μM substrate mix, 100 mM Hepes, 10% sucrose, 0.1% CHAPS, 10 mM DTT, pH 7.5, in a total volume of 100 μl. Under these conditions, the final concentration of each individual compound is ∼0.25 μM. Production of AMC was monitored continuously at room temperature in a Tecan Fluostar 96-well plate reader using an excitation wavelength of 380 nm and an emission wavelength of 460 nm.
Results
Protection of Apoptotic Cell Death in Caspase-11–deficient Mice
Previous studies suggesting a role of caspase-11 as an essential activator of caspase-1 (Wang et al. 1996, Wang et al. 1998) and a potential role of caspase-1 in ischemic brain injury (Friedlander et al. 1997; Hara et al. 1997a,Hara et al. 1997b; Schielke et al. 1998) led us to investigate the involvement of caspase-11 in apoptosis induced by MCAO, a mouse model of stroke. Caspase-1 knockout mice showed resistance to ischemic brain injury (Schielke et al. 1998). However, since we failed to detect the LPS stimulated caspase-11 induction in caspase-1 knockout mice generated both by Kuida et al. 1995 and Li et al. 1995(Fig. 1 A), we cannot distinguish whether loss of caspase-1 or caspase-11 expression is directly responsible for the protection against ischemia-induced apoptosis. The expression of caspase-1 in caspase-11 mutant mice, on the other hand, is completely normal (Wang et al. 1998). To determine whether caspase-11 is critical for ischemic brain injury, we subjected caspase-11 mutant mice and their littermate wild-type controls to MCAO. Blood flow was blocked in the territory of the middle cerebral artery of the left hemisphere by inserting a nylon filament into the artery and 12 h later, the control and injured brain samples were processed for TUNEL staining as a marker of apoptotic cell death. Caspase-11 knockout mice exhibited markedly reduced population of apoptotic cells induced by permanent occlusion of middle cerebral artery at the level of striatum and hippocampus (Fig. 1b and Fig. c). This reduction was not due to the genetic background effect in the knockout mice since wild-type C57BL/6J and 129SvImJ mice showed no significant difference in the number of TUNEL-positive cells after MCAO (data not shown).
Figure 1.


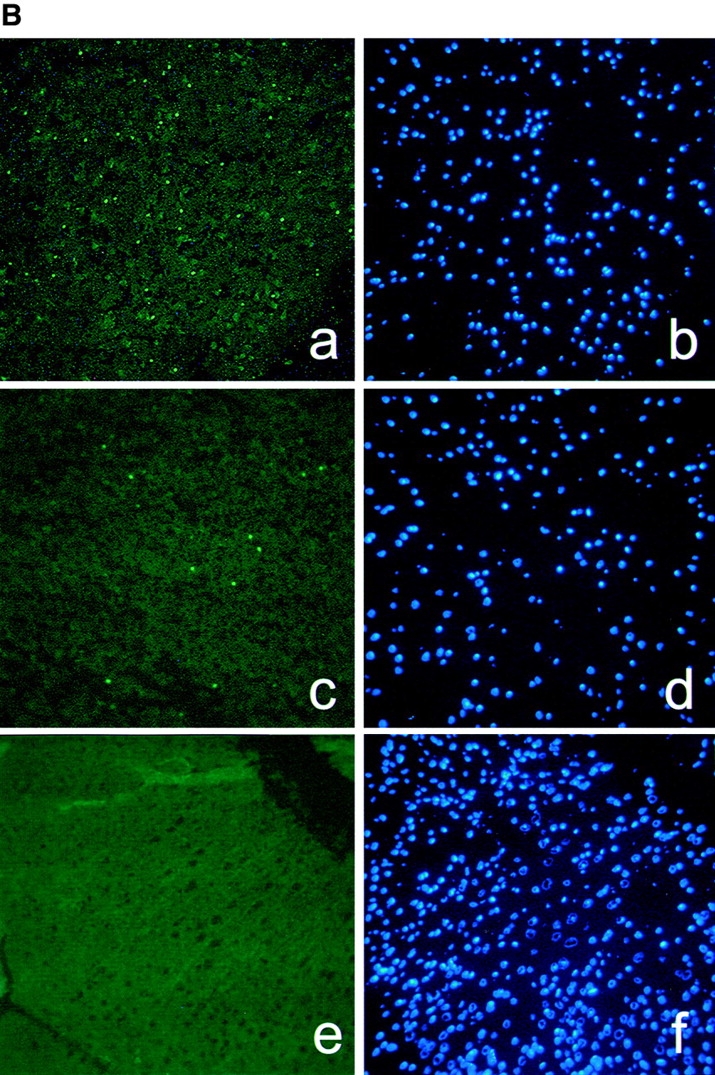
Reduction of apoptotic cell numbers after ischemic brain injury in caspase-11−/− mice. A, Western blot of spleen lysate from 3 wild-type (+/+) and 4 caspase-1 (−/−) mice, probed by anticaspase-11 (top), anticaspase-1 (middle) and antitubulin (bottom). L929 cell lysate (L929) was used as a positive control. B, TUNEL (a, c, and e) and Hoechst dye (b, d, and f) staining of wild-type (a, b, e, and f) and caspase-11−/− (c and d) brains. a–d are from ischemic brains and e and f are from untreated control brain. Permanent ischemia was induced by occlusion of the left middle cerebral artery using monofilament as described by Hara et al. 1997a. 12 h after the occlusion, brains were processed for TUNEL. C, Quantification of TUNEL-positive cells in the wild-type and caspase-11−/− ischemic brain. The percentages of TUNEL-positive cells were determined by the number of TUNEL-positive cells divided by the number of Hoechst dye-positive cells. TUNEL- and Hoechst dye-positive cells were quantified using the Zeiss Axiovert microscope equipped with Northern exposure software. Mean of the reading from five brains is shown. Error bar represents standard error.
Induction of Caspase-11 by Pathological Stimuli
Caspase-11 expression in normal healthy mice is undetectable by Western blot (Wang et al. 1996). We examined if caspase-11 is upregulated by MCAO using immunoblot and immunocytochemistry methods. Caspase-11 mutant and wild-type control littermates were subjected to the permanent MCAO for 12 h and the expression of caspase-11 was examined (Fig. 2 A). As the majority of the cells was not dying in these ischemic brains, and therefore only a small portion of cells was expressing caspase-11, we had to use immunoprecipitation to see caspase-11. We have shown that the caspase-11 locus encodes two proteins of 38 and 43 kD (Wang et al. 1996). Interestingly, the 43-kD species was induced only in the ischemic side of the brain, whereas the 38-kD species was upregulated both in ipsilateral and contralateral sides of the brain; in contrast, both were absent in control brains. In the ischemic brain samples, immunoreactivity was strong in the cortical area (Fig. 2 B, a and b) and localized in the cytoplasm with occasional filamentous or patchy condensation of the immunoreactivity (Fig. 2 B, e–g) and such staining was absent in caspase-11 mutant mice (Fig. 2 B, c and d). This result showed that caspase-11 is upregulated in a subpopulation of cells in brain after ischemic injury.
Figure 2.

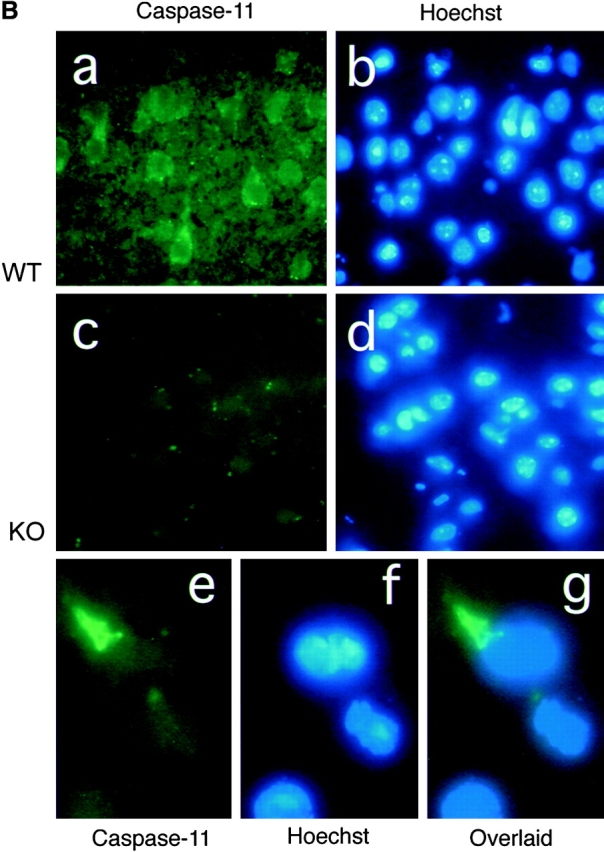

Induction of caspase-11 by ischemic brain injury. A, Permanent ischemia was induced by 12-h occlusion of the left middle cerebral artery. Wild-type ischemic (Isch), ipsilateral (L), and contralateral (R), or sham-operated (con), ipsilateral (L), and contralateral (R) sides of brain lysates were immunoprecipitated with monoclonal anticaspase-11 antibody and Western-blotted with anticaspase-11 mAb. IgG H and IgG L indicate IgG heavy and light chain, respectively. Anticaspase-11 stains a weak background band slightly smaller than 38 kD sometimes in brain samples. B, Wild-type and caspase-11−/− ischemic brains were immunostained with anticaspase-11 (a, c, and e) and counterstained with Hoechst dye (b, d, and f) 12 h after MCAO. e and f are overlaid in g. C, Wild-type ischemic brain 12 h after the occlusion was double-stained for caspase-11 (a) and anti-NeuN as a neuronal marker (b); caspase-11 (d) and B4-isolectin as a microglial cell marker (e). Counterstainings with Hoechst dye are shown in c and f.
To determine the cell types that express caspase-11 under ischemic conditions, we carried out double-immunostaining on sections of ischemic brain samples using mAb against caspase-11 and cell type marker antibodies. Caspase-11 staining was found in cells positive for neuN (Fig. 2 C, a and b), a neuronal marker (Mullen et al. 1992), as well as B4-isolectin (Fig. 2 C, d and e), a microglial marker (Streit and Kreutzberg 1987). Double-positive cells for GFAP and caspase-11 were not detected (data not shown). From these results, we concluded that caspase-11 is upregulated in subpopulations of neurons and microglial cells after ischemic brain injury.
Inhibition of Caspase-3 Activation in Caspase-11 Mutant Mice
Since caspase-3 has been shown to be a major downstream caspase in apoptosis, we set out to test if resistance of caspase-11 knockout mice to apoptosis induced by brain ischemia is correlated with a deficiency in the activation of caspase-3. Mice were challenged by MCAO and brains were processed for immunohistochemistry. Antibody to activated caspase-3, CM1, clearly labels a subpopulation of cells in wild-type cortex after ischemic injury (Fig. 3, a and b). This staining was significantly reduced in caspase-11−/− cortex that has been subjected to the same ischemic treatment (Fig. 3c and Fig. d). Untreated control wild-type brains showed little staining with CM1 (Fig. 3e and Fig. f). These results suggest that caspase-11 is essential for the activation of caspase-3 in a subpopulation of cortical neurons and microglial cells after ischemic injury.
Figure 3.
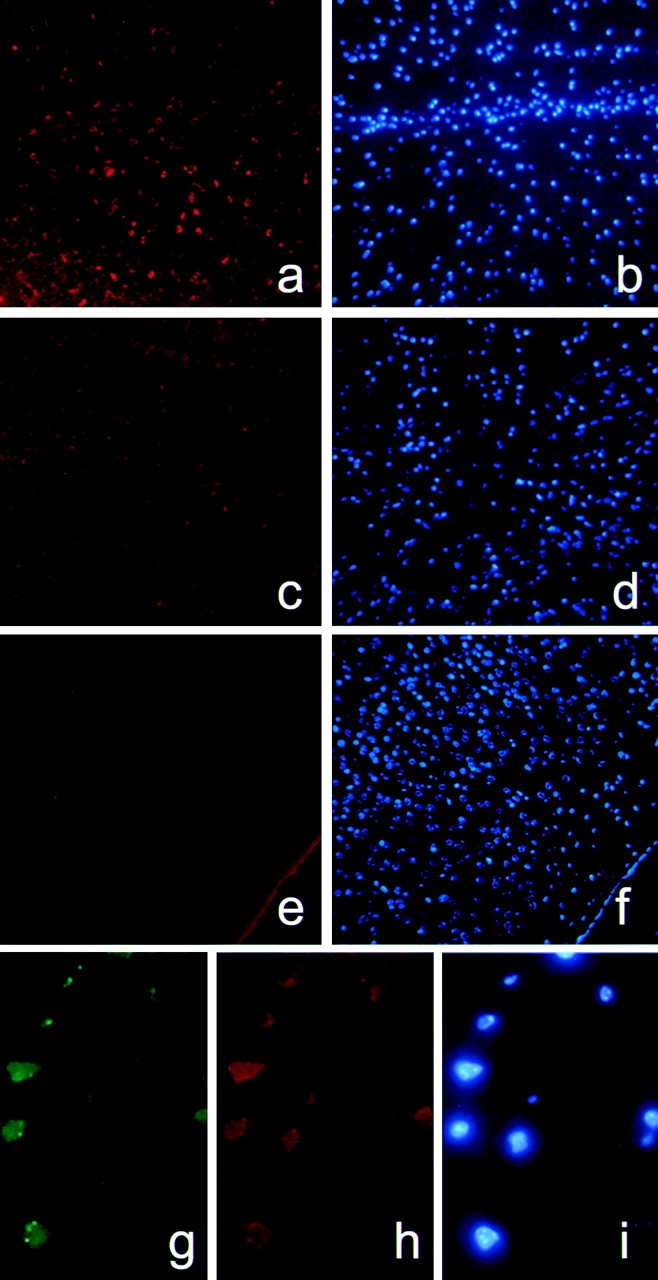
Inhibition of caspase-3 activation in caspase-11−/− ischemic brain and costaining of caspase-11 and activated caspase-3 in wild-type ischemic brain. 12 h after MCAO, wild-type (a, b, and g–i) and caspase-11−/− (c and d) brains were double-stained with CM1 (a and c) and Hoechst dye (b and d), or triple-stained with CM1 (g), anticaspase-11 antibody (h), and Hoechst dye (i). As a control, untreated brain samples from wild-type animal were stained with CM1 (e) and Hoechst dye (f). Sections from wild-type and caspase-11−/− brains were attached side by side to the slides and incubated in the same drop of antibody solution to ensure the identical experimental condition. Images were exposed for the same amount of time using Phase 3 imaging system and Northern exposure software.
Since caspase-11 regulates the activation of both caspase-1 and caspase-3, in ischemic tissues caspase-11 may activate caspase-3 through direct proteolytic processing or indirectly through regulating cytokines. We reasoned if caspase-11 acts directly to activate caspase-3, we should be able to detect the upregulated caspase-11 and activated caspase-3 in the same cells. To test this possibility, we coimmunostained ischemic brain samples with anticaspase-11 and CM1, and we found that a significant portion of caspase-11–positive cells are also positive for CM1 (Fig. 3, g–i). Among 400 cells counted in the ischemic area, 27% of the cells were positive for both caspase-11 and CM1, 7.3% were positive for caspase-11 only, and 2.3% were positive for CM1 only. Importantly, we did not find any caspase-11 single-positive cells that were right next to CM1 single-positive cells (data not shown), thereby making it highly unlikely that activated caspase-3 was mediated by cytokines released from neighboring cells with activated caspase-11. These results suggest that in the cells that are positive for both caspase-11 and activated caspase-3, the activation of caspase-3 is most likely mediated through direct processing of procaspase-3 by caspase-11.
Activation of Caspase-3 by Caspase-11 In Vitro
Since caspase-11 appears to be able to activate caspase-3 in vivo independent of its ability to regulate cytokine processing, we tested if caspase-11 can cleave procaspase-3 directly. Purified recombinant caspase-11 was incubated with in vitro translated 35S-labeled caspase-3 and the resulting products were visualized by autoradiography. As shown in the Fig. 4 A, caspase-3 was found to be an excellent substrate for caspase-11. Caspase-11 cleaved procaspase-3 much more readily than any of the other procaspases (data not shown). To examine if caspase-11 cleaves caspase-3 at Asp175 between the large and the small subunit of caspase-3, Asp175 of caspase-3 was mutated to Ala, which changed IETD175 to IETA175. This mutation completely blocked the cleavage of caspase-3 by caspase-11 (Fig. 4 A). These results suggest that caspase-11 may activate caspase-3 through direct proteolytic cleavage at IETD175.
Figure 4.




The cleavage specificity of caspase-11. A, Cleavage of caspase-3 by caspase-11. 35S-labeled in vitro translated caspase-3 wild-type or D175A mutant substrates as indicated at the bottom were incubated at 30°C for 2 h in the presence (+) or absence (−) of recombinant caspase-11 (1.16 μM) as indicated at the top. The cleavage of wild-type and mutant caspase-3 by caspase-11 was analyzed by SDS-PAGE and autoradiography. There is a nonspecific protein product from the vector right below caspase-3 product that cannot be recognized by anticaspase-3 antibody (data not shown). B, Activation of procaspase-3 by caspase-11. Purified recombinant procaspase-3 (indicated as C3 in the left panel, 1.04 μM), procaspase-1 (C1 in the right panel, 1 μM) or active caspase-11 (C11, 1.16 μM) was incubated with acDEVD.amc for caspase-3 activation and acYVAD.amc for caspase-1 activation individually or together as indicated. DEVDase or YVADase activity was followed by fluorogenic activity reading at 10-s interval at λex = 380 nm, λex = 460 nm. Representative data are shown out of four independent experiments. Note the significantly higher magnitude of activation of procaspase-3 compared with procaspase-1. C, The substrate specificity of caspase-11 was determined by PS-SCL method as in Rano et al. 1997. The y axis represents the rate of AMC production expressed as a percentage of the maximum rate observed in each experiment. The x axis shows the positionally defined amino acids. The result indicates that caspase-11 prefers (I/L/V/P)EHD.
To examine if purified caspase-11 can activate the proteolytic activity of caspase-3, we incubated purified caspase-11 with purified procaspase-3. While both active caspase-11 and procaspase-3 were unable to cleave DEVD. amc, a preferred substrate for caspase-3, incubation of procaspase-3 with active caspase-11 resulted in cleavage of DEVD.amc (Fig. 4 B, left). These results showed that purified caspase-11 can activate procaspase-3 efficiently through proteolytic cleavage. As a comparison, we incubated equal amount of active caspase-11 as in Fig. 4 A with equal amount of procaspase-1 as procaspase-3 in Fig. 4 A; although incubating active caspase-11 with procaspase-1 can also result in the activation of caspase-1, as indicated by YVAD.amc fluorogenic substrate, the activation of procaspase-1 by caspase-11 was much less effective (Fig. 4 B, right).
A positional scanning synthetic combinatorial library was developed to determine the specificity of caspases (Thornberry et al. 1997). This method made use of the fact that caspases cleave after an aspartic acid residue and can cleave tetrapeptides terminating in Asp.AMC as efficient fluorogenic substrates. The library is composed of three separate sublibraries of 8,000 compounds each, containing all the amino acid residue combinations at P2, P3, and P4 positions (P4 P3 P2 D.AMC). Interestingly, when the cleavage specificity of caspase-11 was tested in this library, we found that although caspase-11 is a member of the caspase-1 subfamily based upon its sequence homology, it prefers to cleave (I/L/V/P)EHD, which is similar to the specificity of upstream caspases, such as caspase-8 and -9 (Fig. 4 C). Since it has been well established that caspase-8 or -9 can directly activate caspase-3 through proteolytic cleavage (Muzio et al. 1996; Li et al. 1997), these results further argue that caspase-11 can directly activate caspase-3 under pathological conditions.
Processing of 43-kD Procaspase-11 by Active Caspase-11 In Vitro
The caspase-11 locus encodes two polypeptide of 43- and 38-kD (Wang et al. 1996). Since caspase-11 cDNA under the control of a heterologous promoter is transcripted and translated into the same 43- and 38-kD in vitro and in cells (Fig. 5, and data not shown), it is reasonable to assume that they are the products of alternative starts of translation. In addition, only the 43-kD product would carry the NH2-terminal tag (data not shown), which provides a further support that the 38-kD product is initiated downstream from the Met for 43 kD, and therefore is missing most of the prodomain of caspase-11. To elucidate the functional difference of 43- and 38-kD products of caspase-11, we incubated the purified bacterially expressed procaspase-11 with in vitro translated, 35S-labeled full-length caspase-11. Like all the other caspases, procaspase-11 is autoprocessed when expressed in bacteria and therefore is proteolytically active (Fig. 4 and data not shown). As shown in Fig. 5 A, purified bacterially expressed procaspase-11 can process in vitro translated full-length caspase-11 into a protein fragment of 30 kD (p30) and 10 kD (p10). Interestingly, the cleavage appears to target preferentially at the 43-kD species, whereas the 38-kD species is stable during the incubation. This result suggests that the 43-kD caspase-11 protein product may be activated early in the apoptosis pathway.
Figure 5.
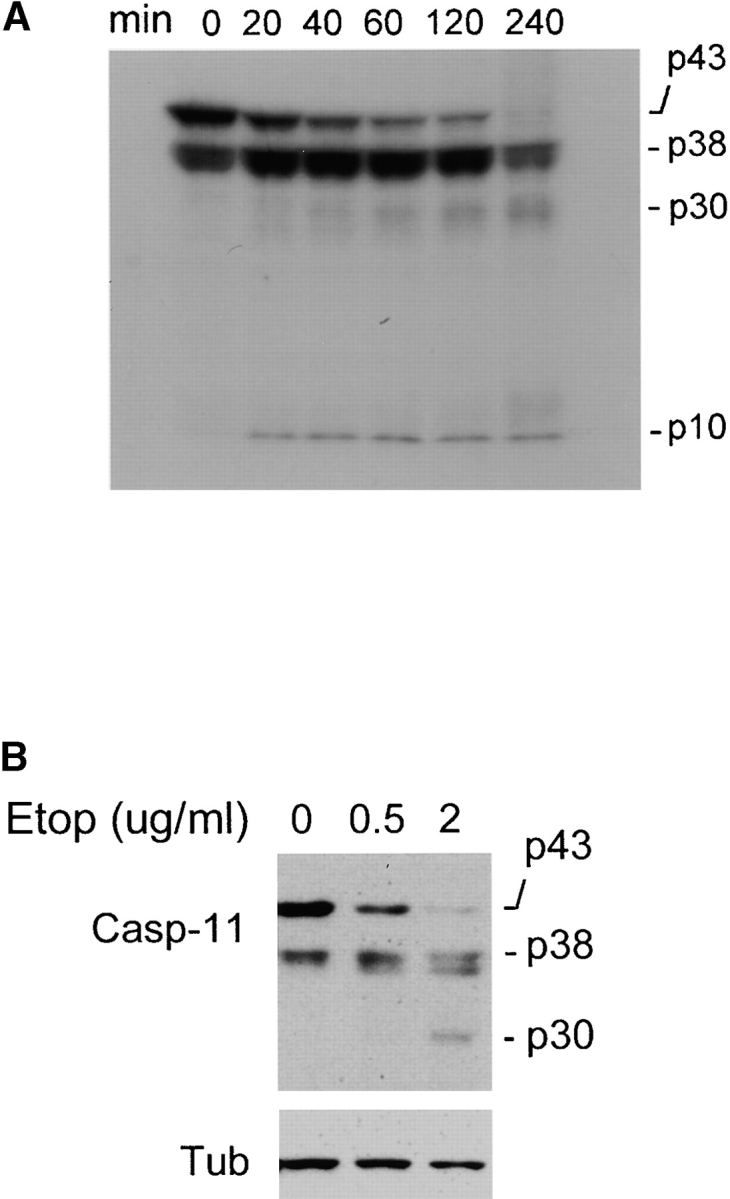
Autoprocessing of 43-kD caspase-11 in vitro. A, Cleavage of caspase-11 in vitro. 1 μl of 35S-labeled caspase-11, in vitro translated from its full-length cDNA (pS15), was mixed with 1 μg of purified procaspase-11 (pS15) in a total volume of 10 μl. The mixtures were incubated at 37°C for the time indicated at the top of the panel. The cleavage products of labeled procaspase-11 by procaspase-11 enzyme were analyzed by SDS-PAGE and autoradiography. B, Cleavage of caspase-11 in apoptosis. L929 cells were incubated with indicated amounts of etoposide overnight, which induced 50–80% of cells to die and the cleavage of caspase-11 was determined by Western blot using anticaspase-11 (17D9).
To study if caspase-11 can be activated and processed during apoptosis in culture, we chose the L929 cell line because it expresses high levels of caspase-11. Most of the cell lines express undetectable amount of caspase-11 and have to be induced with LPS to express caspase-11. The L929 cells were treated with different amounts of etoposide and incubated overnight. The cell lysates made from etoposide-treated L929 were Western blotted with anticaspase-11 mAb made against p10 of the caspase-11 (17D9). As shown in Fig. 5 B, the 43-kD product of caspase-11 was processed into p30 during etoposide-induced cell death, whereas the 38-kD remained stable. We predict that the p30 is the caspase-11 fragment removed of its N prodomain because it still can be recognized by the mAb against the p10 subunit. This result is consistent with our in vitro cleavage analysis and supports the conclusion that the 43-kD product of caspase-11 is activated more easily and earlier in apoptosis than that of the 38-kD product.
Discussion
Since our analysis of caspase-11 showed that caspase-11 may be a very important regulator of apoptosis under pathological conditions, it is critical to identify its human ortholog. The obvious candidates for caspase-11 in human are caspase-4 and -5, since they share the highest homology among all known caspases with caspase-11; however, caspase-4 expression appears to be present in unstimulated cells (Kamada et al. 1997). The preferred cleavage tetrapeptide substrates of caspase-4 and -5 are (W/L)EHD (Thornberry et al. 1997), which differs from that of caspase-11. While caspase-11 prefers LEHD as a substrate, it does not cleave WEHD to a significant degree. Thus, it is possible that either caspase-4 and -5 in human may function like caspase-11 in mouse, but with a somewhat wider specificity; alternatively, the true ortholog of caspase-11 in human remains to be identified. It will be helpful to know whether other characteristics of caspase-11 (such as its inducibility) are preserved in caspase-4 and/or -5 to distinguish these possibilities.
The expression of caspase-11 is the most stringently regulated among all the caspases identified so far. Whereas most of caspases can be detected in healthy, unstimulated cells, caspase-11 expression is below the detection limit of Northern and Western blots and its transcripts are only detectable by reverse transcriptase PCR in most tissues except intestine, where its expression can be detected in unstimulated condition (Wang, S., S. Kang, and J. Yuan, unpublished data). The expression of caspase-11 is highly inducible by a variety of apoptosis stimuli, including ischemic brain injury, systemic inflammation, head trauma, and even transfection of cultured cells (Wang, S., S. Kang, and J. Yuan, unpublished data). Whereas the activation of most caspases is mediated through recruitment into activation complexes, the activation of caspase-11 is regulated at the transcriptional and translational level. We have observed caspase-11 to form filamentous structures in cells with high levels of caspase-11 expression (Fig. 2). Overexpression of caspase-8 has been shown to form filaments that appear to be sufficient for their activation (Siegel et al. 1998). It is possible that elevated caspase-11 protein concentration in stimulated cells may be sufficient for it to form filaments which results in its activation.
The present work, together with our previous study (Wang et al. 1998), demonstrates that caspase-11 is a dual activator of caspase-1 and -3 under pathological conditions, although we cannot rule out a possibility that other caspases are also acting downstream of caspase-11, as caspase-2 and -7 are also cleaved by caspase-11 in vitro (data not shown). Caspase-7 is similar to caspase-3 in its specificity (Thornberry et al. 1997), so it is possible that caspase-7 may contribute to the downstream effect of caspase-11. Caspase-2, however, is unlikely to play a significant role in ischemic brain injury, as caspase-2 knockout mice did not show any resistance to MCAO (Bergeron et al. 1998).
Caspase-11 has at least two downstream caspases. One interesting question is whether there is cross-talk between the two downstream caspases of caspase-11. We have shown that although overexpression of caspase-1 cannot induce caspase-11−/− EF cells to die, caspase-11 induced apoptosis of caspase-1−/− EF cells efficiently (Wang et al. 1998). Thus, caspase-1 is not required for caspase-11–induced apoptosis, which supports our model that caspase-11 can activate caspase-3 directly. On the other hand, although caspase-1 is not required for caspase-11 to induce apoptosis, caspase-1 activation may contribute to apoptosis in vivo indirectly. Intraventricular microinjection of both YVAD.cmk and DEVD.fmk have been shown to reduce ischemic brain injury induced by MCAO with intriguing differences (Hara et al. 1997b). Intraventricular injection of YVAD.fmk resulted in both reduction of infarction volume and of brain swelling. Interestingly, intraventricular injection of DEVD.fmk decreased infarction volume, but had no effect on brain swelling. As we have shown that caspase-11 can mediate the activation of both caspase-1 and caspase-3, such damage may be mediated directly through proteolytic activation of caspase-3 and apoptosis, and indirectly, through activating caspase-1 and cytokine release, which causes brain swelling that may in turn initiate additional apoptosis. Thus, the regulation of both downstream caspases, caspase-1 and -3, by caspase-11 may contribute to apoptotic damage under pathological conditions.
Caspase-11 locus encodes two proteins of 38 and 43 kD (Wang et al. 1996). We observed that the 43-kD protein is the predominant protein product of caspase-11 cleaved both in vivo during apoptosis and in vitro by autoprocessing. We hypothesize that the 43-kD protein is the full-length caspase-11 that can be autoactivated, whereas the 38-kD protein may be missing most of the CARD domain that it may need to be activated by another caspase(s). Interestingly, the 43-kD species was induced only in the ischemic side of the brain, whereas the 38-kD species was upregulated both in the ipsilateral and contralateral side of the brain. Cortical ischemia has been known to modify metabolism and blood flow in nonischemic contralateral brain areas (Feendy and Baron 1986). It is possible that the concurrent upregulation of 38-kD caspase-11 product in the contralateral hemisphere may reflect activation in homologous brain regions, the consequences of which have not yet been identified.
Whereas caspase-11 can be activated by proteolytic cleavage, the cleavage may not be essential for its activation, as we only observed its cleavage when its expression levels are very high (Fig. 5; and Kang, S., S. Wang, and J. Yuan, unpublished data). In LPS-stimulated mice, although caspase-11 is induced in almost all tissues, its cleavage can only be observed in spleen where its expression is the highest (Wang et al. 1996; Kang, S., and J. Yuan, unpublished results). It has been increasingly accepted that at least long prodomain caspases can be activated without cleavage (Stennicke et al. 1999). Alternatively, it remains possible that the cleavage products of caspase-11 are unstable, and therefore, only when the levels of caspase-11 are very high can we observe its cleavage products. This is certainly possible for caspase-11 in ischemic brain. As the majority of the cells are not dying in these ischemic brains, and therefore only a small portion of cells are expressing caspase-11 that can only be observed through immunoprecipitation, we cannot find the cleavage product of caspase-11 in all cases.
Neither caspase-1 knockout line can be induced to express caspase-11, whereas our caspase-11 knockout mouse line expresses caspase-1 normally (Wang et al. 1998). This may be due to the fact that caspase-1 and -11 are genetically closely linked (Li, P., personal communication) and the disruption of genomic structure in caspase-1 has a negative effect on the expression of caspase-11. Preliminary examination of caspase-11 locus in caspase-1−/− mice by Southern blot and PCR showed that the locus of caspase-11 is intact, suggesting that inability to induce caspase-11 is not due to disruption of caspase-11 gene structure in caspase-1−/− mice (Wang, S., and J. Yuan, unpublished results). Alternatively, the cytokines regulated by caspase-1, IL-1β, IL-1α, IL-18 (Ghayur et al. 1997), and interferon γ may have a positive feedback effect on caspase-11 expression. Therefore, the loss of cytokine production in caspase-1 mutant mice may prevent the induction of caspase-11. However, we have been unable to induce caspase-11 expression by coinjecting caspase-1 knockout mice with LPS and IL-1β (Kang, S., and J. Yuan, unpublished data). The loss of caspase-11 expression in caspase-1 knockout mice complicated the interpretation as to why caspase-1 knockout mice are resistant to ischemic brain injury (Schielke et al. 1998), because the caspase-11 mutation alone has a strong effect on ischemic brain injury-induced apoptosis. Thus, it remains to be resolved whether the loss of caspase-1 expression alone is sufficient to inhibit ischemic brain injury-induced apoptosis. We predict, however, that the loss of caspase-1 and the absence of cytokines, such as IL-1 and IL-18, will have a significant impact on ischemic brain injury-induced by swelling, which ultimately contributes to the development of apoptosis.
Although our data demonstrated that caspase-11 can directly activate caspase-3, we do not exclude a role for cytokines in the regulation of cell death. Our previous studies showed that IL-1β may promote cell death (Friedlander et al. 1996). One possibility is that cytokines, such as IL-1β and interferon γ, of which processing and secretion of both are controlled by the caspase-11–caspase-1 pathway, may promote the upregulation of caspase-11 and -1. Xu et al. 1998 have shown that interferon γ can upregulate caspase-1 under certain conditions and our result showed that IL-1β can stimulate the production of caspase-11. Thus, cytokines may contribute to apoptosis by positively regulating the expression of caspases. On the other hand, there is evidence that apoptosis may play a key role in cytokine secretion. IL-1β is a cytokine that lacks an obvious signal peptide and its mechanism of secretion is still under debate. Whereas many cell types have the capacity to produce IL-1, its release has been shown to be restricted predominantly to monocytes/macrophages and associated with apoptosis of producer cells (Hogquist et al. 1991; Zychlinsky et al. 1994). Secretion of IL-1β from U937 cells is partially inhibited by Bcl-2 overexpression (Lizard et al. 1997), suggesting apoptosis may play an important role in regulating IL-1 β secretion in monocytic cells. Perhaps controlling both apoptosis and cytokine secretion through upregulation of a single caspase is advantageous as a way to ensure immediate and complete elimination of damaged cells.
Acknowledgments
We thank Drs. Masayuki Miura and Or Gozani for critical reading of the manuscript, Dr. Martin Raff for advice on cell type markers, Drs. Ping Li and Winnie Wong for providing caspase-1 knockout mice and communicating unpublished results, Drs. Keisuke Kuida and Richard Flavell for caspase-1 knockout mouse spleen, Drs. Toshiyuki Nakagawa and Honglin Li for helpful advice, Hong Zhu for providing purified procaspase-3, and Neisen Jing for technical assistance in mouse genotyping.
This work was supported in part by grants to J. Yuan from the National Institute of Aging, the National Institute of Neurological Disorders and Stroke, and the American Heart Association Established Investigatorship; and to M. Moskowitz from the National Institute of Neurological Disorders and Stroke.
Footnotes
Shin-Jung Kang and Suyue Wang contributed to this work equally.
Abbreviations used in this paper: GFAP, glial fibrillary acidic protein; IL-1β, interleukin-1β; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion; PS-SCL, positional scanning-synthetic combinatorial library; TUNEL, TdT-mediated dUTP-digoxigenin nick-end labeling.
References
- Bergeron L., Perez G.I., MacDonald G., Shi L., Sun Y., Jurisicova A., Varmuza S., Latham K.E., Flaws J.A., Salter J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V., Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Enari M., Sakahira H., Yokoyama H., Okawa K., Iwamatsu A., Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD [published erratum appears in Nature. 393:396] Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Feendy D.M., Baron J.-C. Diaschisis. Stroke. 1986;17:817–830. doi: 10.1161/01.str.17.5.817. [DOI] [PubMed] [Google Scholar]
- Friedlander R.M., Yuan J. ICE, neuronal apoptosis and neurodegeneration. Cell Death Diff. 1998;5:823–831. doi: 10.1038/sj.cdd.4400433. [DOI] [PubMed] [Google Scholar]
- Friedlander R.M., Gagliardini V., Rotello R.J., Yuan J. Functional role of interleukin 1 beta (IL-1 beta) in IL-1 beta-converting enzyme-mediated apoptosis. J. Exp. Med. 1996;184:717–724. doi: 10.1084/jem.184.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R.M., Gagliardini V., Hara H., Fink K.B., Li W., MacDonald G., Fishman M.C., Greenberg A.H., Moskovwitz M.A., Yuan J. Expression of a dominant mutant of interleukin-1 β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Hara H., Fink K., Endres M., Friedlander R.M., Gagliardini V., Yuan J., Moskowitz M.A. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein J. Cereb. Blood Flow Metab 17 1997. 370 375a [DOI] [PubMed] [Google Scholar]
- Hara H., Friedlander R.M., Gagliardini V., Ayata C., Fink K., Huang Z., Shimizu-Sasamata M., Yuan J., Moskowitz M.A. Inhibition of ICE family proteases reduces ischemic and excitotoxic neuronal damage Proc. Natl. Acad. Sci USA. 94 1997. 2007 2012b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist K.A., Nett M.A., Uanue E.R., Chaplin D.D. Interleukin-1 is processed and released during apoptosis. Proc. Natl. Acad. Sci. USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada S., Washida M., Hasegawa J., Kusano H., Funahashi Y., Tsujimoto Y. Involvement of caspase-4 (-like) protease in Fas-mediated apoptotic pathway. Oncogene. 1997;15:285–290. doi: 10.1038/sj.onc.1201192. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt S., Behrmann I., Germer M., Pawlita M., Krammer P.H., Peter M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Lippke J.A., Ku G., Harding M.W., Livingston D.J., Su M.S.S., Flavell R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1b converting enzyme. Science. 1995;267:2000–2002. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Kuida K., Haydar T.F., Kuan C.-Y., Gu Y., Taya C., Karasuyama H., Su M.S.S., Rakic P., Flavell R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase-9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Li P., Allen H., Banerjee S., Franklin S., Herzog L., Johnston C., McDowell J., Paskind M., Rodman L., Salfeld J. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lizard G., Lemaire S., Monier S., Gueldry S., Neel D., Gambert P. Induction of apoptosis and of interleukin-1beta secretion by 7beta-hydroxycholesterol and 7-ketocholesterolpartial inhibition by Bcl-2 overexpression. FEBS Lett. 1997;419:276–280. doi: 10.1016/s0014-5793(97)01473-7. [DOI] [PubMed] [Google Scholar]
- Mullen R.J., Buck C.R., Smith A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni K., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3 like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Namura S., Zhu J., Fink K., Endres M., Srinivasan A., Tomaselli K.J., Yuan J., Moskowitz M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rano T.A., Timkey T., Peterson E.P., Rotonda J., Nicholson D.W., Becker J.W., Chapman K.T., Thornberry N.A. A combinatorial approach for determining protease specificitiesapplication to interleukin-1beta converting enzyme (ICE) Chem. Biol. 1997;4:149–155. doi: 10.1016/s1074-5521(97)90258-1. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.-M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schielke G.P., Yang G.Y., Shivers B.D., Betz A.L. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J. Cereb. Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Siegel R.M., Martin D.A., Zheng L., Ng S.Y., Bertin J., Cohen J., Lenardo M.J. Death effector filamentsnovel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 1998;141:1243–1253. doi: 10.1083/jcb.141.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke H.R., Deveraux Q.L., Humke E.W., Reed J.C., Dixit V.M., Salvesen G.S. Caspase-9 can be activated without proteolytic processing. J. Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- Streit W.J., Kreutzberg G.W. Lectin binding by resting and reactive microglia. J. Neurocytol. 1987;16:249–260. doi: 10.1007/BF01795308. [DOI] [PubMed] [Google Scholar]
- Thornberry N., Rano T., Peterson E., Rasper D., Timkey T., Garcia-Calbo M., Houtzager V., Nordstrom P., Roy S., Vaillancourt J. A combinatiorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E.E., Schuchmann M., Luria V., Chiannilkulchai N., Beckmann J.S., Mett I.L., Tebrikov D., Brodianski V.M., Kemper O.C., Kollet O. Targeted disruption of the mouse caspase-8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Wang S., Miura M., Jung Y.K., Zhu H., Gagliardini V., Shi L., Greenberg A.H., Yuan J. Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J. Biol. Chem. 1996;271:20580–20587. doi: 10.1074/jbc.271.34.20580. [DOI] [PubMed] [Google Scholar]
- Wang S., Miura M., Jung Y.K., Zhu H., Li E., Yuan J. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/s0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Xu X., Fu X.Y., Plate J., Chong A.S. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosisrequirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998;58:2832–2837. [PubMed] [Google Scholar]
- Zou H., Henzel W.J., Liu X., Lutschg A., Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A., Fitting G., Cavaillon J., Sansonetti P.J. Interleukin-1 is released by murine macrophages during apoptosis induced by Shigella flexneri . J. Clin. Invest. 1994;94:1328–1332. doi: 10.1172/JCI117452. [DOI] [PMC free article] [PubMed] [Google Scholar]