

Abstract
Most transformed cells have lost anchorage and serum dependence for growth and survival. Previously, we established that when serum is absent, fibronectin survival signals transduced by focal adhesion kinase (FAK), suppress p53-regulated apoptosis in primary fibroblasts and endothelial cells (Ilić et al. 1998. J. Cell Biol. 143:547–560). The present goals are to identify survival sequences in FAK and signaling molecules downstream of FAK required for anchorage-dependent survival of primary fibroblasts. We report that binding of the SH3 domain of p130Cas to proline-rich region 1 of FAK is required to support survival of fibroblasts on fibronectin when serum is withdrawn. The FAK–p130Cas complex activates c-Jun NH2-terminal kinase (JNK) via a Ras/Rac1/Pak1/MAPK kinase 4 (MKK4) pathway. Activated (phospho-) JNK colocalizes with FAK in focal adhesions of fibroblasts cultured on fibronectin, which supports their survival, but not in fibroblasts cultured on collagen, which does not. Cells often survive in the absence of extracellular matrix if serum factors are provided. In that case, we confirm work of others that survival signals are transduced by FAK, phosphatidylinositol 3′-kinase (PI3-kinase), and Akt/protein kinase B (PKB). However, when serum is absent, PI3-kinase and Akt/PKB are not involved in the fibronectin-FAK-JNK survival pathway documented herein. Thus, survival signals from extracellular matrix and serum are transduced by FAK via two distinct pathways.
Keywords: fibroblast, anchorage-dependent survival, focal contacts, MAP kinase, apoptosis
Introduction
Interactions of cells with the extracellular environment regulate many basic cellular functions, including differentiation, migration, cell growth, and programmed cell death (apoptosis). Many such signals from the extracellular matrix (ECM), including those required for cell survival, are relayed to the intracellular signaling machinery by integrins, heterodimeric transmembrane receptors with overlapping specificity toward ECM components (Damsky and Werb 1992; Hynes 1992; Meredith et al. 1993; Howlett et al. 1995; Giancotti and Ruoslahti 1999). One way to study the effects of ECM signals independently of signals from other extracellular sources has been to deprive cells of soluble factors from serum and then to analyze the effects of specific ECM ligands on such functions as adhesion, migration, and survival and on activation of specific intracellular signaling pathways. Using this approach, we confirmed that fibronectin (FN) is particularly effective in providing survival signals for several cell types, and that these survival signals are transduced by focal adhesion kinase (FAK) (Frisch et al. 1996; Hungerford et al. 1996; Ilić et al. 1998). We then demonstrated that the FN/FAK survival signal suppresses an apoptosis pathway regulated by the tumor suppressor p53 in fibroblasts and endothelial cells (Ilić et al. 1998).
FAK is a novel type of nonreceptor protein tyrosine kinase. Its central kinase domain is similar to that of other nonreceptor protein tyrosine kinases (e.g., Src family kinases). The COOH-terminal region of FAK can also exist as an independently transcribed protein (termed FAK-related nonkinase [FRNK]). Both FAK and FRNK contain a COOH-terminal focal adhesion targeting (FAT) domain. FAT mediates the association of FAK and FRNK with sites of cell–substratum interactions (Hildebrand et al. 1993; Richardson and Parsons 1996). At these sites, FAK and FRNK colocalize with integrins and other focal adhesion components such as paxillin and vinculin. FRNK also contains two proline-rich regions. Proline-rich region 1 (PR-1) (residues 712–718) is NH2-terminal to the FAT domain and provides a binding site for the adaptor protein p130Cas (Cas). PR-2 (residues 873–876) is within the FAT site and interacts with Graf (GTPase regulator associated with FAK), and to a lesser extent with Cas (Hanks and Polte 1997). The region of FAK NH2-terminal to its kinase domain contains the autophosphorylation site (Y397), which becomes phosphorylated in response to integrin cross-linking, thus providing a binding site for the SH2 domain of Src-family kinases and for phosphatidylinositol 3′-kinase (PI3-kinase) (Chen et al. 1996). The NH2-terminal domain also contains a region with a significant homology to band 4.1 (Girault et al. 1999) that may interact directly or indirectly with the cytoplasmic domains of integrin β subunits (Schaller et al. 1995). Disruption of the fn and fak genes results in very similar embryonic lethal phenotypes in mice, which suggests that FAK is a principal mediator of signals induced by the binding of cells to FN (George et al. 1993; Furuta et al. 1995). FAK has also been implicated in signaling initiated by the binding of receptors for various growth factors, neuropeptides, and cytokines (Hanks and Polte 1997; King et al. 1997; Schlaepfer and Hunter 1998). In fact, recent data indicate that FAK can interact, at least indirectly, with receptors for EGF and PDGF (Sieg et al. 2000). Thus, FAK likely coordinates signals from multiple inputs.
FAK has been proposed to couple integrins and cytoskeletal proteins to multiple signaling pathways. Several lines of evidence suggest that integrin activation of PI3-kinase, c-Jun NH2-terminal kinase (JNK), and extracellular signal–regulated kinase (ERK) signaling pathways require FAK (King et al. 1997; Dolfi et al. 1998; Zhao et al. 1998; Schlaepfer et al. 1999). However, data from other groups suggest that integrins are able to activate at least some of these pathways independently of FAK (Wary et al. 1998; Giancotti and Ruoslahti 1999; Oktay et al. 1999).
A high level of apoptosis results if FAK function is eliminated, by blocking its interaction with integrin β1 cytoplasmic domain (Hungerford et al. 1996), by gene targeting, or by displacement of FAK from focal contacts after introduction of the FAT domain, which acts as a dominant-negative (DN) for the FAK survival function (Ilić et al. 1998). Interestingly, we found that cells survive if they overexpress FRNK, even though FRNK, like FAT, lacks a kinase domain. Together, these data suggest that (a) assembly of FAK-containing molecular complexes at focal adhesion sites is required for survival of anchorage-dependent, serum-deprived cells, and that (b) a region of FAK (and FRNK) NH2-terminal to FAT contains a site important for transduction of survival signals.
The goals of this study are twofold. The first is to identify the mechanism by which FAK conveys survival signals from FN in primary fibroblasts after serum withdrawal. We now report that both recruitment of Cas to the PR-1 region of FAK located in focal adhesion sites and phosphorylation of Cas are required to support survival. The second goal is to determine the pathway downstream of FAK that transmits the survival signals from FN. We now report that FN survival signals conveyed by the FAK–Cas complex require Ras. Downstream of Ras, signals that activate ERK1/2 through the Raf1/MAPK kinase (MEK1) cascade are not essential for survival of anchorage-dependent primary fibroblasts on a FN matrix. In contrast, activation of a pathway involving Rac1, Pak1, MAPK kinase 4 (MKK4), and JNK1/2 is required. Furthermore, activated (phospho–) JNK, but not activated ERK, is present in focal contacts in cells plated on FN, which supports survival, but not in cells on collagen I, which does not. Thus, activation of JNK by MKK4 and its recruitment to focal contacts appear to be critical for supporting anchorage-dependent survival in primary fibroblasts in the absence of survival signals in serum. Finally, we confirm that when serum is present and cells are in suspension or are poorly spread, FAK is also required. However, survival signals are conveyed by a distinct pathway involving PI3-kinase and Akt/protein kinase B (PKB) as reported previously (Khwaja et al. 1997). PI3-kinase and Akt/PKB are not required for the FN-FAK-JNK survival pathway reported here.
Materials and Methods
Cells
Isolation of primary rabbit synovial fibroblasts (RSF) was described previously (Werb et al. 1989). Primary cultures were expanded up to passage 3 in DME containing 10% FCS supplemented with glutamine, nonessential amino acids, and penicillin/streptomycin and frozen. Just before experiments, cells were thawed and used between passages 4 and 8.
MKK4-null embryo stem (ES) cells and matched wild-type ES cells (Yang et al. 1997) were grown and induced to form embryoid bodies as described previously for FAK-null ES cells (Ilić et al. 1998).
Survival Assays in Nontransfected Cells
RSF were plated in DME supplemented with sodium pyruvate, nonessential amino acids, and penicillin/streptomycin, but without serum, on FN- or collagen I–coated tissue culture plastic. Coating was done with 25 μg/ml of FN (GIBCO BRL) or fibrillar collagen (Vitrogen) in PBS overnight at 4°C, followed by washing with PBS. Survival was assayed 16 h later by fixing the cells with 4% paraformaldehyde in PBS followed by adding 10 μg/ml Hoechst 33342 (Molecular Probes) and counting condensed, bright nuclei characteristic of apoptotic cells vs. large, lightly stained nuclei of healthy cells (Ilić et al. 1998). When indicated, cultures contained 15 μM LY294002 (IC50 = 1.4 μM) or 50 nM wortmannin (IC50 = 5 nM) to inhibit PI3-kinase, or 20 μM PD98059 (IC50 = 2 μM) (Calbiochem) to inhibit MEK1/2. The LY294002 and wortmannin were added to the cells either before adhesion, while the cells were in suspension, or 1 h after plating to allow for cell attachment to the matrix substrate. The PD98059 was added before adhesion. Survival was evaluated after 16 h. All survival assays were performed at least three times, with triplicate samples recorded photographically every time. To confirm that the PD98059 was active, control cultures and cultures pretreated with 20 μM PD98059 for 15 min in suspension, were plated on FN for 30 min in serum-free medium. Cell lysates were immunoblotted for total ERK or pERK levels using anti-ERK or anti-pERK antibodies.
For analysis of apoptosis in wild-type and MKK4-null embryoid bodies, wild-type and MKK4-null ES cells were grown in suspension for 12 d in the presence of serum as described previously (Ilić et al. 1998) to permit differentiation of a variety of cell types and elaboration of endogenous ECM. Serum was then withdrawn for 24 h, after which the embryoid bodies were fixed in 4% paraformaldehyde and processed for assessment of cell death by terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) in situ staining according to the manufacturer's instructions (Boehringer Mannheim).
Plasmids
Mutagenesis of FAK proline residues 712 and 713 to alanine to generate FAK-P was performed according to the protocol outlined in the MutaGene Phagemid kit (Bio-Rad) using oligonucleotide Ala 712/713 (5′-GATGAAGCAGCAGCCAAGCCCAGC-3′), and a single-stranded DNA produced from the ClaI-SmaI FAK fragment cloned into pBluescript. Mutations were always confirmed by DNA sequencing, and the FAK fragment in pBluescript was cloned into the corresponding sites within the mouse FAK cDNA in the pCDNA3 expression vector (Invitrogen). The FRNK construct was created as described (Ilić et al. 1998). FRNK with mutated 712 and 713 residues (FRNK-P) was generated as described above for FAK-P.
To construct the green fluorescent protein (GFP)-FRNKΔFAT fusion protein, the SnaBI-XhoI fragment from the GFP-FRNK expression vector was isolated and subcloned into a new pEGFP C1 plasmid (Clontech) opened with SnaBI and XhoI. GFP-FRNK PΔFAT was generated from GFP-FRNK P in the same manner.
pSSRα Cas SH3 expression vector was generated by self-ligation of pSSRα Cas plasmid (Nakamoto et al. 1997) after KpnI digestion.
DNA fragments were isolated from agarose gels using QIAquick Gel Extraction Kit (Qiagen). All blunting and ligation reactions were performed with either Blunting or Ligation kits (Takara; imported by PanVera Corporation). Plasmid DNAs were isolated using appropriate-size Qiagen kits.
Other Reagents
The following investigators kindly provided cDNA constructs and other reagents for this study: constitutively active and DN Pak1 constructs, Dr. J. Chernoff (Fox Chase Cancer Center, Philadelphia, PA) and Dr. G. Bokach (The Scripps Research Institute, La Jolla, CA); DN and constitutively active RAS constructs, Dr. H. Bourne (University of California San Francisco, San Francisco, CA); MKK4-null embryo stem cells and DN MKK4 construct, Dr. R. Flavell (Yale University School of Medicine, New Haven, CT) and Dr. R. Davis (University of Massachusetts Medical School, Worcester, MA); full-length Cas mutant and Cas constructs, Dr. H. Hirai (University of Tokyo, Tokyo, Japan) and Dr. T. Nakamoto (University of Tokyo); pJNK antibody control peptides, Dr. B. Jarvis (Promega, Madison, WI); constitutively active and DN Rac1 constructs, and DN RhoA and CDC42 constructs, Dr. M. Symons (Picower Institute for Medical Research, Manhasset, NY).
Transfections and Apoptosis Assay
Primary RSF were plated on FN-coated wells (24-well dishes) at ∼75% confluence in DME supplemented with 10% FCS, glutamine, and nonessential amino acids and allowed to adhere and grow for 24 h on the FN-coated wells. Immediately before transfection, adherent cells were washed three times with serum-free DME supplemented with glutamine and nonessential amino acids. Cells were then transfected using Lipofectamine Plus in serum-free medium (GIBCO BRL) according to the manufacturer's instructions. In brief, a total 0.5 μg of DNA per well in 24-well culture plates was used per transfection. In cotransfection experiments, we used equimolar amounts of different plasmid DNAs adding up to a total of 0.5 μg per well. In the Cas competition experiment, the 1:1 ratio of Cas-SH3 to full-length Cas was equimolar, as described above, adding up to 0.5 μg of DNA. For 1:2, 1:3, and 1:4 ratios, Cas-SH3 was kept constant and the amount of Cas multiplied by 2, 3, or 4 respectively, thus increasing total DNA transfected. Expression of the transfected constructs was checked either visually by GFP expression or in cell lysates by Western blotting. Cultures were analyzed at the indicated times between 8 and 48 h after addition of the liposomes. For most experiments, analysis occurred at 16 h. In each experiment, three randomly chosen fields were photographed per well, and at least three separate experiments were performed, each using three wells. Photographs were taken using an inverted fluorescence microscope equipped with dual FITC/Hoechst filters (Nikon Corp.). All cells expressing GFP were counted as either alive or apoptotic. Cells not expressing GFP were not counted. The apoptotic index represents the percentage of GFP-positive cells that were scored as apoptotic after Hoechst staining (Ilić et al. 1998).
Western Blots and Kinase Assays
To examine expression levels of transfected constructs, cells were lysed in modified RIPA buffer (1% sodium deoxycholate, 0.1% SDS, 1% NP-40, 150 mM NaCl, 10 mM Tris/HCl, pH 7.4, 1 mM EDTA) containing freshly added protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mM PMSF). Lysates were precleared with protein G–Sepharose (Amercham Pharmacia Biotech) and separated by SDS-PAGE using 4–20% gradients of acrylamide. Gels were transferred to nitrocellulose and were blotted using antibodies that recognized specific domains of proteins, or hemagglutinin or Myc tags. Monoclonal anti-hemagglutinin (HA.11) was purchased from BAbCo and monoclonal anti-Myc (9E10) from Santa Cruz Biotechnology. Polyclonal anti-Cas antibody (N17; Santa Cruz Biotechnology) against an epitope within the SH3 domain was used to detect expression of the CasSH3 vs. full-length Cas. A polyclonal antibody against the COOH-terminal region of FAK (JF1; Ilić et al. 1995) was used to detect levels of transfected GFP-FAT, GFP-FRNK, and full-length FAK.
To detect matrix-dependent activation of MAP kinases (p38 mitogen-activated, dual-action kinase [MAPK], ERK1/2, MKK4, and JNK1/2), RSF were plated for 1 or 10 h on FN or collagen I in serum-free medium, and lysed in RIPA buffer containing freshly added proteinase inhibitors as described above, plus phosphatase inhibitors (1 mM Na3VO4, 1 mM NaF). The positive control for evaluating p38 MAPK activation was obtained by lysis of serum-starved RSF treated with 10 μg/ml anisomycin (Calbiochem) for 15 min. Equal amounts of proteins were run on SDS-PAGE, transferred to nitrocellulose filters, and blotted with antibodies against the indicated protein kinases or phosphorylated forms of these kinases. All phospho-specific antibodies were obtained from New England Biolabs, except anti-pJNK and its control peptide, which were purchased from Promega Corporation. Antibodies against ERK1/2, MKK4, and JNK1/2 proteins were obtained from Santa Cruz Biotechnology, and HRP-conjugated secondary antibodies were obtained from Jackson ImmunoResearch. Signals were detected by enhanced chemiluminescence (Amersham Pharmacia Biotech).
For assessing the kinase activity of JNK, RSF were cotransfected as described above with Flag-tagged JNK1 or JNK2, GFP, or GFP and FAK. 16 h after transfection, RSF were washed with PBS and lysed in buffer containing 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, with 150 mM NaCl, 1.5 mM MgCl2, 10% glycerol, 1 mM EGTA, 50 mM Hepes, pH 7.4, containing freshly added protease and phosphatase inhibitor cocktails. JNK1 and JNK2 were immunoprecipitated from the cleared lysates with anti-Flag antibodies coupled to agarose beads (Kodak) for 4 h at 4°C. The precipitates were washed two times with lysis buffer, and once with 50 mM Hepes buffer, pH 7.4, containing 150 mM NaCl, 0.5 mM NaF, and 0.5 mM Na3VO4. After washing, the immunoprecipitates were suspended in 40 μl JNK kinase buffer containing 10 μCi/sample [32P]γATP (3,000 Ci/mmol; NEN/DuPont), 10 μM ATP, and 1 μg/sample of an NH2-terminal glutathione S-transferase–c-Jun fusion protein as substrate (Santa Cruz Biotechnology), and incubated for 15 min at 30°C. The reaction was stopped by addition of 2× reducing SDS sample buffer, and the samples were separated by 12.5% SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and analyzed by autoradiography. To determine the levels of immunoprecipitated JNK, the membranes were probed with a monoclonal anti-Flag antibody and detected by enhanced chemiluminescence. All kinase assays and Western blots were performed at least three times.
Immunocytochemistry
Immunocytochemistry was carried out as described in Sieg et al. 1998. In brief, cells were plated without serum on FN-coated coverslips and fixed at indicated times with 4% paraformaldehyde/PBS. Cells were permeabilized for 10 min in cold acetone. pJNK was detected using rabbit anti-pJNK antibody, followed by a fluorescein-conjugated donkey anti–rabbit secondary antibody. To ensure specific staining of pJNK by anti-pJNK, we combined the anti-pJNK (10 μg/ml) with 100 μg/ml of the phosphopeptide used to generate the phospho-specific antibody, which should block interactions of the antibody with pJNK in RSF. Homology scanning by DNA Star software of the pJNK peptide sequence against the regions surrounding the FAK autophosphorylation site (Y397) or kinase domain (Y576/Y577) was carried out to assess the possibility that the anti-pJNK antibody might cross react with FAK. Western blotting was also used to verify that the anti-pJNK did not cross react with pFAK. In another control, we used a nonphosphorylated version of the peptide, which should permit staining of pJNK by anti-pJNK. F-actin was visualized by rhodamine-phalloidin (Molecular Probes). GFP and GFP-FAT were visualized directly in samples. pJNK immune complexes were visualized with rhodamine-conjugated secondary antibody. All immunocytochemical staining was carried out at least three times.
Results
We used transfection approaches for determining the domains of FAK that were important for transducing survival signals in RSF plated on FN, and both pharmacological and transfection approaches for determining the signaling intermediates downstream of FAK that transmitted these survival signals. For transfection experiments, RSF were initially plated on FN-coated substrates in the presence of serum. Subsequently, RSF were washed to remove any traces of serum, transfected, and cultured for the times indicated. The apoptotic index was determined as described in Materials and Methods and in Ilić et al. 1998. Furthermore, we compared the ability of nontransfected RSF plated in serum-free medium on FN, which supports survival, or on collagen I, which does not, to activate the signaling intermediates in the survival pathway downstream of FAK that were implicated by the results of the transfection experiments. Finally, we asked the extent to which the survival pathway downstream of FN overlapped with the survival pathway downstream of survival factors in serum.
FN Matrix Survival Signaling Requires the Cas-binding, PR-1 Region of FAK or FRNK and Focal Contact Localization
Expression of a GFP-FAT fusion protein in RSF triggered a high level of apoptosis within 16 h after transfection and serum withdrawal (Fig. 1 A), confirming our previous data (Ilić et al. 1998). GFP-FAT was detected initially in focal contacts of spread cells 8–10 h after transfection. The majority of GFP-FAT–expressing cells subsequently rounded up, and by 16 h they displayed condensed and fragmented nuclei, characteristics of apoptotic cells. Cells expressing GFP alone, or GFP and FAK, maintained the control level of apoptosis (∼25%) for at least 48 h. Surprisingly, expression of GFP-FRNK at a similar level did not promote apoptosis, (Fig. 1a and Fig. B; Ilić et al. 1998), although as reported previously, spreading of replated cells was slower than in cells expressing GFP-FAK (Gilmore and Romer 1996; Richardson and Parsons 1996; Ilić et al. 1998; Zhao et al. 1998).
Figure 1.



The PR-1 region of FAK/FRNK and focal contact localization are required for the transmission of FN matrix survival signals. RSF plated on FN in the presence of serum were transfected with wild-type FAK and GFP, GFP-FRNK, GFP-FAT, or GFP-FRNK with mutations in PR-1. Cells were cultured subsequently in the absence of serum for the times indicated. The apoptotic index was assessed by determining the percentage of transfected cells (GFP-positive) showing condensed or fragmented nuclei after staining with Hoechst dye. (A) Cells expressing GFP+FAK for 16 h remain spread on a FN matrix (upper left panel). Cells expressing GFP-FAT for 16 h (upper right panel) are rounded with condensed, fragmented nuclei. In cells expressing GFP-FAT for 8, 12, and 16 h (lower left panel), GFP-FAT initially localizes to focal contacts in spread cells. By 12 h, cells expressing GFP-FAT start to round. By 16 h, rounded cells have condensed nuclei as detected by Hoechst staining (arrow). Adjacent cell not expressing GFP-FAT has a normal nucleus (indicated by asterisk). In cells expressing GFP-FRNK for 16 h (lower right panel), GFP-FRNK localizes to focal contacts, but cells survive. (B) Expression levels of FAK, GFP-FRNK, and GFP-FAT in RSF 8 h after transfection. All are expressed at similar levels as shown by Western blot using a COOH-terminal FAK antibody. (C) GFP-FAT (upper row) and GFP-FRNK (lower row) both replace endogenous FAK in focal contacts. GFP, but not endogenous FAK, which is detected by an antibody against the NH2-terminal domain of FAK (N-FAK), is present in focal contacts of transfected cells (arrowheads). Nontransfected (GFP-negative) cells in the same field stain with N-FAK (arrows). (D) Diagrammatic representation of constructs used to test the importance of PR-1 in FAK and FRNK for supporting survival of RSF on a FN matrix (see text for description). (E) Apoptotic index for RSF transfected with constructs shown in D after 16 h. Mutation of PR-1 in both FRNK and FAK promotes apoptosis at levels comparable to GFP-FAT. A GFP-FRNK mutant that does not localize to focal contacts but has intact prolines 712–713 (GFP-FRNKΔFAT) is pro-apoptotic, whereas the GFP-FRNK P mutant that does not localize to focal contacts (GFP-FRNK PΔFAT) loses the ability to induce apoptosis. PBD, paxillin-binding domain.
This striking difference in survival of FRNK and FAT transfectants, despite the observation that both displace endogenous FAK from focal contact sites (Fig. 1 C), suggested that the region in FRNK NH2-terminal to FAT contains a site(s) for interaction with molecule(s) critical for transduction of survival signals from FN. To test whether the PR-1 (712–718) sequence in that region is critical, we mutated prolines 712 and 713 into alanines (P712/713A) in FRNK and FAK (GFP-FRNK P and FAK P, respectively) (Fig. 1 D). Expression of these mutant proteins resulted in high levels of apoptosis, similar to that caused by expression of GFP-FAT (Fig. 1 E). Next, we expressed a cDNA encoding only the region of FRNK NH2-terminal to FAT (GFP-FRNKΔFAT; Fig. 1 D). This also resulted in a high level of apoptosis (Fig. 1 E), presumably because this fragment, which is unable to displace endogenous FAK from focal contacts, could act as a sink for important components of the survival complex that would ordinarily bind to PR-1 in endogenous FAK. In support of this idea, cells expressing FRNKΔFAT containing P712/713A (GFP-FRNK PΔFAT) survived at levels similar to those of cells expressing GFP-FRNK or GFP alone (Fig. 1D and Fig. E).
Survival Signals from FN Mediated by FAK Require Cas with Functional SH3 and Substrate Domains
A strong candidate for a FAK- or FRNK-interacting molecule responsible for supporting survival signals from FN in fibroblasts is Cas, as it has been clearly shown that the SH3 domain of Cas binds preferentially and with high affinity to the PR-1 of FAK and FRNK (Polte and Hanks 1995). We therefore expressed the Cas SH3 domain independently, as a DN for wild-type Cas (Fig. 2 A, panel 1). Coexpression of GFP-FRNK and the Cas SH3 domain caused apoptosis at a level similar to that caused by expression of GFP-FAT (Fig. 2 A, panel 2). This could be rescued by allowing wild-type Cas to compete with Cas SH3 for interaction with GFP-FRNK in focal contacts. Thus, coexpression of wild-type Cas and the Cas SH3 fragment in a ratio of 4:1 did not cause apoptosis (Fig. 2 A, panels 2 and 3). These data suggest that in RSF expressing FRNK, Cas is required for transmission of matrix survival signals from FN via interaction with the PR-1 of FAK/FRNK.
Figure 2.

The Cas SH3 and SD are required for transmission of survival signals from FN and FAK. (A, panel 1) Diagram of the domain structure of Cas and the isolated Cas SH3 domain. (A, panel 2) Expression of isolated Cas SH3 domain with GFP-FRNK disrupts survival. Coexpression of full-length Cas and Cas SH3 in a 4:1 DNA molar ratio restores survival. (A, panel 3) Western blot of lysates of cells transfected with Cas SH3 or Cas, and with both Cas and Cas SH3 at increasing ratios of Cas cDNA. A paxillin reblot of the same membrane shows that sample loading is similar in all lanes. (B, panel 1) Diagrams of Cas cDNAs with deletions of the SH3 domain (CasΔSH3), the substrate-binding domain (CasΔSD), or the Src-binding domain (CasΔSrc-BD). (B, panel 2) Expression of CasΔSH3 or CasΔSD triggers high levels of apoptosis, whereas expression of CasΔSrcBD does not. (B, panel 3) Western blot showing that all Cas constructs are expressed in RSF at similar levels. Reblot of the same membrane with an anti-paxillin antibody shows similar loading in all lanes.
Next, we tested the consequences for survival of deleting each of the three principal functional domains of Cas. We coexpressed GFP and FAK with mutant constructs of Cas lacking the SH3 domain (CasΔSH3), the substrate domain (CasΔSD) in which Cas can be phosphorylated at multiple sites, or the Src-binding domain (CasΔSrc-BD) (Fig. 2 B, panel 1). All were expressed at similar levels (Fig. 2 B, panel 3). We observed a high level of apoptosis after expression of CasΔSH3 (Fig. 2 B, panel 2), which supports the results in Fig. 1 and Fig. 2 A and suggests that interaction of FAK with Cas is essential for the transduction of survival signals from a FN matrix. We also found that the substrate domain (SD) of Cas was required for FAK-mediated survival, whereas the Src-binding domain of Cas was not (Fig. 2 B, panel 2). All together, these data suggest that Cas binding to PR-1 and Cas phosphorylation, most likely by FAK itself, are important for transducing survival signals in cells expressing GFP and FAK.
FAK-transduced Matrix Survival Signals Converge on Ras
FAK is known to activate the Ras and MAP kinase pathways in fibroblast cell lines (Schlaepfer et al. 1994). Furthermore, Cas has been implicated in communicating with the Ras pathway via Crk (Dolfi et al. 1998). Therefore, we introduced DN or constitutively active forms of Ras into cells expressing GFP and FAK to determine whether signaling via Ras is required to support survival of RSF on FN in the absence of serum. Cotransfection of DN Ras with GFP+FAK (Fig. 3), or transfection of DN Ras alone (not shown), resulted in a high rate of apoptosis in RSF after serum withdrawal. Coexpression of DN Ras with GFP+FAK did not trigger elevated apoptosis if serum was present, indicating that DN Ras was not acting as a broad-spectrum signaling inhibitor. Confirming the importance of Ras for this anchorage-dependent survival pathway, we determined that expression of constitutively activated Ras rescued RSF from apoptosis triggered by GFP-FAT (Fig. 3).
Figure 3.

Ras is involved in the transduction of matrix-dependent survival signals from FN and FAK. Expression of GFP-FAT triggered high apoptosis in RSF cultured either in the presence or absence of serum. DN Ras promoted a high level of apoptosis when coexpressed with GFP+FAK in RSF in the absence, but not in the presence of serum. Expression of constitutively active (CA) Ras rescued GFP-FAT–mediated cell death of RSF plated on FN in the absence of serum.
FN/FAK-mediated Survival Is Linked to the JNK Pathway
Ras relays a wide variety of signals from the cell surface to the nucleus via a network of three distinct but closely related groups of MAPKs: ERK1/2, p38 MAPK, and JNK1/2 (Wary et al. 1998). We next examined whether any of these pathways was involved in FN matrix survival signaling.
The Ras–ERK pathway involves Raf1, which phosphor–ylates MEK1/2, which in turn phosphorylate ERK1/2. Coexpression of DN Raf1 or DN MEK1 with GFP and FAK resulted in only a small increase in apoptosis compared with control apoptosis levels in cells transfected with GFP and FAK (Fig. 4 A), despite the observation that cells expressing DN Raf1 or DN MEK1 showed reduced staining with pERK antibodies compared with cells in the same field that did not express these constructs (data not shown). Furthermore, survival of GFP-expressing RSF plated on FN in serum-free medium was not affected when cells were treated with 10× the IC50 concentration of PD98059, a selective and potent inhibitor of MEK1/2 (Fig. 4 A), although the activities of ERK1/2 were suppressed by this treatment, as assessed by immunoblotting lysates of control- or inhibitor-treated RSF, using an anti–pERK-specific antibody (Fig. 4 A). Using the same antibodies, we compared levels of pERK1/2 in whole lysates of nontransfected primary RSF plated either on FN, a survival-promoting matrix, or on collagen I, which does not support survival. At 1 h after plating, ERK1/2 were both activated similarly in cells on either substrate. Although at 10 h after plating the activity of both ERKs had decreased significantly, this occurred on both FN and collagen I (Fig. 4 B). Thus, ERKs are activated equally in response to matrices that do and do not support survival. The data in Fig. 4 suggest that the Ras–ERK pathway is not primarily responsible for transduction of matrix survival signals from FN in anchorage-dependent RSF.
Figure 4.
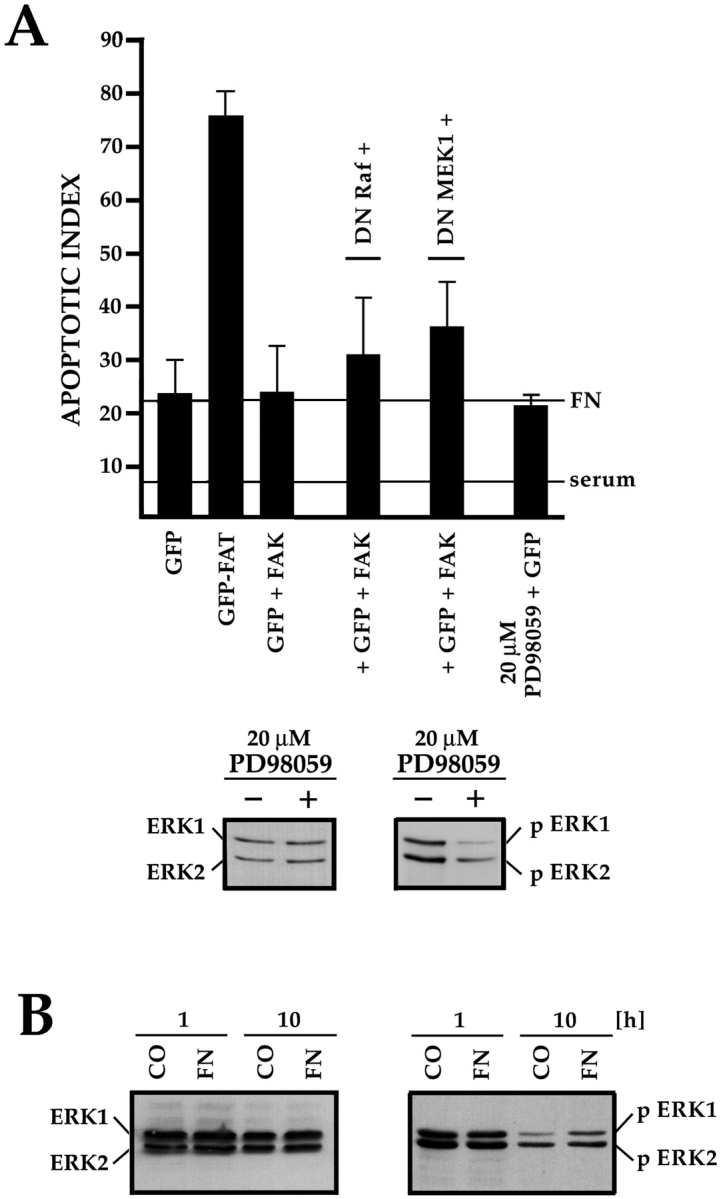
The ERK-MAPK pathway is not essential for supporting FN-FAK–dependent survival. (A) Cotransfection of DN Raf1 or DN MEK1, along with GFP+FAK, resulted in only a small increase in apoptosis compared with GFP+FAK alone. Addition of the MEK1/2 inhibitor PD98059 at 10× the IC50 to cultures of nontransfected RSF did not trigger elevated apoptosis. The PD98059 was active, as treated cultures (+) showed reduced levels of activated pERK1/2 compared with untreated cultures (−). Levels of total ERK proteins were similar in both cultures. (B) ERK1/2 were activated similarly by contact with FN, which supports survival, or collagen I (CO), which does not. Lysates of nontransfected RSF cultured in the absence of serum for either 1 or 10 h, were immunoblotted using antibodies that detect total ERK1/2, or phosphorylated pERK1/2.
Ras also transduces signals that communicate with the JNK or p38 MAPK families. These signals can be regulated further by the Rho family of small GTPases and by p21-activated serine/threonine kinases (Pak family) (Clark et al. 1998; Dolfi et al. 1998; Hall 1998; Sells et al. 1999). To establish whether the Rho family of small GTPases participates in FN survival signaling, we cotransfected GFP and FAK with the DN mutants: N19RhoA, N17Rac1, or N17cdc42. Only DN Rac1 effectively interfered with survival (Fig. 5 A), whereas the DN forms of RhoA and cdc42 did not (data not shown). Cotransfection of GFP and FAK with DN Pak1 or DN MKK4 also resulted in high rates of apoptosis, whereas constitutively activated forms of Rac1 and Pak1, expressed at the same levels as their DN counterparts, suppressed apoptosis when coexpressed with GFP-FAT (Fig. 5 A). The DN constructs did not trigger apoptosis when transfected into cells cultured in the presence of serum (Fig. 5 A). Finally, an antibody specific for an activated form of MKK4 (anti-pMKK4) produced a strong signal in immunoblots of lysates of nontransfected RSF plated in serum-free medium on FN, but not in those from cells plated on collagen I (Fig. 5 B).
Figure 5.




The JNK MAPK pathway transduces survival signals from FN-FAK via Rac1/Pak1/MKK4 after serum withdrawal. (A) Expression of DN Rac1, DN Pak1, or DN MKK4, along with GFP+FAK, triggered high levels of apoptosis in RSF cultured on FN in the absence, but not in the presence of serum. Coexpression of constitutively active forms of Rac1 (CA Rac1) or Pak1 (CA Pak1) rescued cells from GFP-FAT–mediated death. The Western blot panels show that CA and DN Rac1/Pak1 construct pairs were expressed at similar levels in RSF and that DN MKK4 was expressed at a level similar to the other constructs. (B) Active MKK4 was detected in lysates of cells plated on FN but not on collagen I. Lysates of RSF cultured in the absence of serum for either 1 or 10 h on FN or on collagen I were immunoblotted using antibodies that detect total MKK4 protein or pMKK4. MKK4 protein levels were similar to one another on both matrices at each timepoint. Significant levels of pMMK4 were detected only in lysates of RSF cultured on FN at the 1 h timepoint. (C) MKK4-null and wild-type (Wt) ES cells were grown in suspension for 12 d in the presence of serum. Serum was withdrawn for 24 h and embryoid bodies were processed to detect DNA fragmentation by TUNEL in situ staining. Only MKK4-null embryoid bodies showed increased TUNEL staining when serum was withdrawn. (D) JNK, but not p38 MAPK, was activated in response to attachment to FN. Lysates of RSF cultured without serum for 1 or 10 h on FN or collagen I (CO), were immunoblotted using antibodies that detect p38 MAPK protein or pp38, or JNK1/2 proteins or pJNK1/2. p38 MAPK was only weakly activated on FN. Lysates of anisomycin-treated primary RSF were used as a positive control (CTL). The signal for pJNK2 was particularly strong at 10 h in cells plated on FN. JNK1 and JNK2 protein levels in cells on FN and collagen were similar to one another at each timepoint. (E) Active pJNK (rhodamine) is strongly visible in cells transfected with GFP-FAK (green), but remains at low levels in adjacent untransfected cells (see nuclei with no corresponding GFP staining in the merged picture). Cells on FN in the absence of serum were fixed and stained after 16 h. (F) FAK, but not GFP, activates JNK1/2 in RSF. Flag-tagged JNK1 or JNK2, and FAK or GFP were coexpressed in RSF for 16 h after serum withdrawal. Cell lysates were incubated with anti-Flag antibody to precipitate JNK1/2. Precipitates were tested for their kinase activities using glutathione S-transferase–c-Jun as a substrate. JNK1/2 were activated strongly by FAK, but not by GFP.
If MKK4 is a critical intermediate in the FN-FAK survival pathway, we reasoned that MKK4-null cells, like FAK-null cells (Ilić et al. 1998), should show abnormally high apoptosis when cultured in the absence of serum. To test this, we generated embryoid bodies by culturing wild-type or MKK4-null ES cells in suspension culture for 12 d in the presence of serum, allowing them to elaborate an endogenous matrix. After serum withdrawal for 24 h, only the MKK4-null embryoid body culture showed elevated apoptosis (Fig. 5 C), confirming the importance of MKK4 for propagating matrix survival signals downstream of FAK when serum is withdrawn.
Both JNK1/2 and p38 MAPK can be activated by MKK4. Using antibodies specific for phosphorylated forms of these kinases, we determined that JNK2 was strongly activated and JNK1 weakly activated in nontransfected cells plated on FN, but not on collagen I, for 10 h in the absence of serum, whereas p38 MAPK was not activated significantly on either matrix (Fig. 5 D). This latter result also suggests that MKK7, another potential MAP kinase kinase that activates p38 MAPK, does not play a role in this survival pathway. To visualize the FAK activation of JNK, we transfected GFP-FAK into RSF plated on FN without serum and stained for pJNK after 16 h. Most cells transfected with GFP-FAK showed extremely bright pJNK staining relative to untransfected cells (Fig. 5 E). To assess at the biochemical level whether FAK was capable of activating JNK1/2, we cotransfected FAK or GFP along with Flag-tagged JNK1 and JNK2 (Fig. 5 F). After 16 h, we determined the activity of anti-Flag–precipitated JNK1 and JNK 2 using glutathione S-transferase–c-Jun as a substrate. JNK1 and especially JNK2 were strongly activated in cells transfected with FAK, whereas cells expressing GFP alone showed very little activation of either JNK. Together, these data suggest that JNK is activated by FAK and that activation of the JNK pathway rather than the ERK or p38 MAPK pathways, plays the major role in transducing survival signals from FN and FAK in RSF when serum survival factors are absent.
Activated JNK Is Localized in Focal Contacts and in the Nucleus in Response to FN but Not to Collagen I
Several upstream signaling molecules that our experiments have implicated in the FN survival pathway, including FAK and Cas, are localized in focal contacts. In contrast, the downstream target transcription factors of JNK are nuclear. Therefore, we hypothesized that one or more intermediate signaling molecules in the pathway, such as JNK, might have a role to play in focal contacts as well as in the nucleus. To test this possibility, we stained nontransfected RSF, plated for 10 h in the absence of serum on either FN or collagen I, with an antibody that detects dually-phosphorylated T and Y within the catalytic core of activated JNK1 and JNK2. Indeed, pJNK was detected both in the nucleus and in focal contacts in cells plated on FN (Fig. 6 A, upper left panel). In contrast, only faint, diffuse staining for pJNK was detected in the cytoplasm and nucleus of RSF plated on pro-apoptotic collagen I (Fig. 6 A, upper right panel). The specificity of the antibody for pJNK was confirmed by absorption with a phospho-JNK peptide: after absorption, no residual staining was detected (Fig. 6 A, lower left panel), whereas a nonphosphorylated version of the same peptide had no effect on staining (not shown). Antibody against whole JNK1/2 proteins also stained focal contacts and the nucleus, although more diffusely, amidst additional cytoplasmic staining (Fig. 6 A, lower right panel). These localization data correlated well with the biochemical data showing that pJNK levels were lower in RSF plated on collagen I as compared with RSF plated on FN (see Fig. 5 D). Interestingly, when matrix survival signals from FN were interrupted by expression of GFP-FAT, and the cells were observed before they started to show signs of apoptosis (8–10 h), GFP-FAT, but not pJNK, was detected in focal contacts (Fig. 6 B, upper row, arrows). Nontransfected cells in the same field showed strong focal contact staining for pJNK (Fig. 6 B, upper row, arrowheads). In contrast, cells expressing GFP-FRNK showed a similar intensity of staining for pJNK as nontransfected cells in the same field (Fig. 6 B, middle row). When levels of full-length FAK were increased by transfection of GFP-FAK, staining for pJNK overall and in focal contacts was much more intense than it was in untransfected cells, supporting the biochemical data and showing that enhanced activation is associated with focal contacts (Fig. 6 B, lower row).
Figure 6.



(A) pJNK is detected both in focal contacts and in the nucleus in cells plated on FN, but not in cells plated on collagen I. RSF plated on FN or collagen I for 10 h were fixed, permeabilized, and stained with antibodies specific for pJNK. Actin was detected by rhodamine-phalloidin. In cells on FN, strong staining for pJNK was detected in the nucleus and in focal contact sites at the ends of actin stress fibers (arrowheads). In cells plated on collagen I, stress fibers and focal contacts were poorly developed. Staining for pJNK was weak and diffuse in the cytoplasm and nucleus. As a control, the anti-pJNK antibody was added to RSF in the presence of 100 μg/ml of the pJNK peptide used to generate the antibody. No focal contact staining was detected (lower left panel). Antibodies that recognize all forms of JNK 1/2 showed a diffuse cytoplasmic staining pattern as well as faint focal contact staining (arrowheads, lower right panel). (B) Expression of GFP-FAT (upper panels) in RSF plated on FN in serum-free medium for 8–10 h (a time before appearance of apoptotic changes; see also Fig. 1), prevents pJNK location in focal contacts (arrows). Staining for pJNK was strong in focal contact sites (arrowheads) of adjacent cells that were not transfected. Expression of GFP-FRNK (middle panels) does not bar pJNK from focal contacts (arrows). pJNK staining in focal contacts of GFP-FAT–expressing cells is similar in intensity to that in focal contacts of adjacent nontransfected cells (arrowheads). Expression of GFP-FAK (lower panels) markedly increases pJNK staining in focal contacts (arrows) as well as in the cytoplasm, relative to adjacent untransfected cells (arrowheads). (C) Staining for pERK in both GFP-FAT (upper panels) and GFP-FRNK–expressing RSF (middle panels) is weak and not detected in focal contacts. pERK staining is strong in RSF expressing GFP-FAK (lower panels). However, focal contact staining is not detected.
When activation of pERK was examined using the same approach, little or no pERK staining was detected in cells expressing either GFP-FAT or GFP-FRNK. However, expression of GFP and FAK triggered intense staining for pERK, but this was not located in focal contacts. Thus, FAK can activate both ERK and JNK, but these activated kinases are located in different regions of the cells. Taken together, these data strongly suggest that recruitment and activation of JNK at focal contact sites is part of the mechanism by which survival signals originating from FN are transduced by FAK in anchorage-dependent fibroblasts plated on FN.
FAK also Transduces Survival Signals from Serum, but by a Distinct Pathway
Work from several groups has reported that PI3-kinase also binds to FAK (Chen et al. 1996) and promotes survival of epithelial and endothelial cells in the presence of serum after loss of attachment from matrix (Frisch et al. 1996; Marte and Downward 1997), the converse condition to that of the experiments described above, in which matrix is provided and serum is withdrawn. To determine the importance of PI3-kinase–mediated signaling for FN-dependent vs. serum-dependent survival, we tested the ability of the PI3-kinase inhibitors LY294002 and wortmannin to trigger apoptosis when added to cultures of RSF in the presence and absence of serum, either at the time of plating on FN or after the cells had adhered to FN for 1 h. After 18 h, these inhibitors had not promoted elevated apoptosis in RSF under any of these conditions (Fig. 7 A), indicating that the PI3-kinase pathway is not required for transducing survival signals downstream of FN. However, if these inhibitors were added to RSF adhering to collagen I, on which they cannot spread effectively within 1 h, or to RSF cultured in suspension, apoptosis levels were very high even when serum was present (Fig. 7 B), confirming conclusions of others that when matrix signals are withdrawn, serum provides survival signals that are conveyed by PI3-kinase (Khwaja et al. 1997; Marte and Downward 1997; Tamura et al. 1999). Our data that expression of GFP-FAT kills RSF plated on FN regardless of whether serum is present or not (Fig. 3), support the conclusion that FAK transduces both the matrix and serum survival signals. Coexpression of a constitutively activated form of Akt/PKB, a downstream target of PI3-kinase, was unable to rescue apoptosis triggered by GFP-FAT in RSF after serum withdrawal (Akt/PKB and GFP-FAT, 89.3 ± 13.9% apoptosis, vs. GFP-FAT alone, 86.4 ± 8.7%), supporting the conclusion that PI3-kinase is not involved in transmitting FN-FAK survival signals. To confirm that the activated Akt/PKB was functional in our experiments, we showed that constitutively activated Akt/PKB could decrease apoptosis triggered by overexpression of the FAK homologue, Pyk-2, in RSF when serum was present (from 77.2 ± 11.5% to 47.4 ± 4.5%), in agreement with the findings of Xiong and Parsons 1997.
Figure 7.

PI3-kinase is not required for transduction of survival signals if cells are spread on a pro-survival matrix (i.e., FN) whether or not serum is present, but is required to transduce serum-dependent survival signals when a pro-survival matrix is withdrawn. (A) PI3-kinase inhibitors Ly297082 and wortmannin (Wort.) have no effect on the high survival of RSF plated on FN whether or not serum is present. Inhibitors were added to cells at the time of plating or to cultures 1 h after plating. Apoptotic index was measured 18 h later. (B) PI3-kinase inhibitors promote apoptosis of cells plated on collagen I when serum is present. These inhibitors do not alter the normally high apoptotic index of cultures plated on collagen I in the absence of serum.
These apparently conflicting findings regarding the importance of PI3-kinase and Akt/PKB in transducing survival signals likely arise from the very different culture conditions and cell types used. Our studies assessed survival of primary fibroblasts attached to FN, a matrix that by itself can support survival when survival signals from serum are withdrawn. The other studies tested survival of other cell types in the presence of serum, a condition that supports survival when matrix survival signals are withdrawn. Thus, anchorage-dependent and serum-dependent survival are both regulated by FAK. However, the downstream pathways activated in the two conditions are distinct.
Discussion
Survival of anchorage-dependent cells is determined by signals from their environment. These signals are primarily derived from soluble factors present in serum and from cell interactions with ECM. When serum factors are withdrawn, cells can still survive on certain matrices, whereas they undergo apoptosis on others. Previously, we confirmed that FN is the most effective survival-promoting ECM ligand for both primary fibroblasts and endothelial cells after serum withdrawal, and showed that in conjunction with FAK, it promotes a survival pathway that suppresses p53-mediated apoptosis (Ilić et al. 1998). In this study, we have focused on dissecting the matrix-dependent survival mechanism downstream of FN and FAK in primary fibroblasts. Our data suggest several conclusions (Fig. 8): (a) recruitment of Cas to the PR-1 region of FAK located in focal contact sites, and its phosphorylation, are required for transmission of survival signals from FN; (b) signals from the FAK–Cas complex activate a Ras–JNK pathway; (c) activated JNK (pJNK) is localized to focal contacts as well as to the nucleus in cells plated on the pro-survival FN matrix; and (d) the FN-dependent survival pathway activated in RSF deprived of serum and the serum-dependent survival pathway in these cells deprived of matrix are both regulated by FAK, but are distinct from one another.
Figure 8.
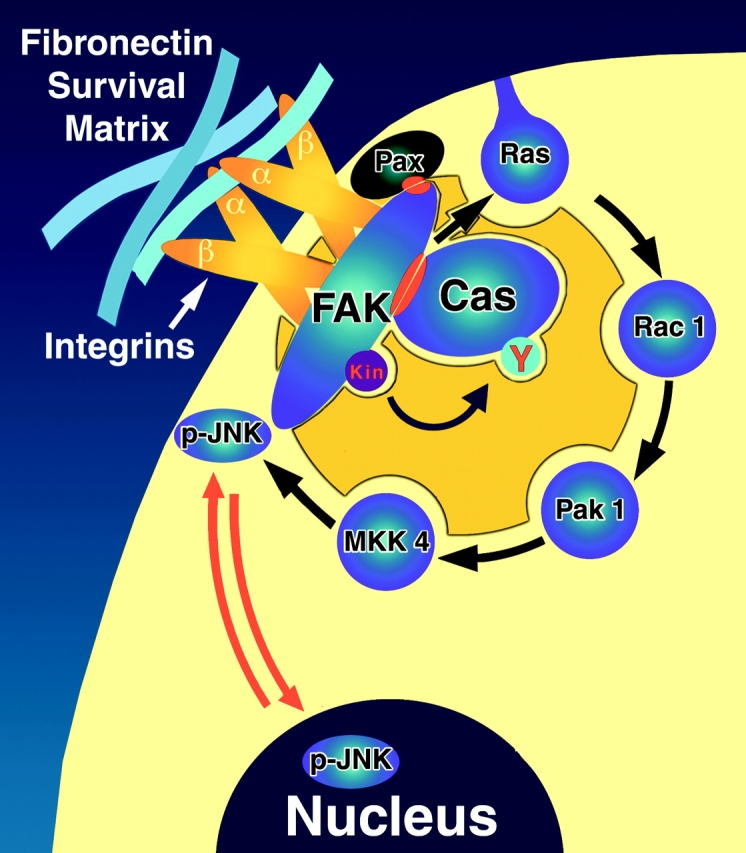
Model of the FN-dependent matrix survival pathway activated in primary RSF when serum is absent. FN supports the organization of a pro-survival signaling complex based on a FAK/Cas scaffold located at focal adhesion sites. Paxillin (Pax) is thought to participate in the binding of FAK to focal adhesion sites (Tachibana et al. 1995). Cas is recruited to the survival complex via a FAK PR-1–Cas SH3 interaction. Matrix survival signaling also requires an intact Cas SD. FN-FAK–associated survival signals activate Ras, and are propagated further via a Rac1/Pak1/MKK4/JNK signaling module, with pJNK detected in focal contacts as well as in the nucleus. This pathway is distinct from the serum-dependent survival pathway that is activated when matrix is withdrawn. The latter pathway requires FAK, but it also requires PI3-kinase, whereas the FN-FAK pathway described herein does not.
Our initial observation of the striking difference between the ability of FAT vs. FRNK transfectants to support survival on a FN matrix in the absence of serum suggested that the small NH2-terminal segment of FRNK missing in FAT contained a survival sequence. We determined that this survival sequence is the PR-1 of FAK and that it functions by binding the adaptor protein Cas via its SH3 domain, thus potentially creating an adaptor platform or scaffold with matrix-regulated kinase activity capable of recruiting and activating multiple signaling molecules at sites of contact with the ECM (see Fig. 8 and Schlaepfer et al. 1999). The strict requirement that FAK (or FRNK) be localized in focal contacts (Ilić et al. 1998; this study) (Fig. 1 C) in order for it to support survival points to its importance as a primary organizer and activator of these multimolecular signaling machines.
In its function as an adaptor protein, Cas can connect signals initiated by FN and FAK to downstream pathways. According to this model, Cas becomes phosphorylated on tyrosines when bound to FAK, creating binding sites on Cas for Crk and triggering the Cas-Crk-Ras connection (Dolfi et al. 1998). Although FAK can both bind and phosphorylate Cas, FRNK can only bind Cas. This raises the question of how survival signals from FN might be passed to the Ras pathway in cells that express FRNK rather than FAK in focal contacts and therefore form a FRNK–Cas complex. Since Cas has a Src-binding domain, one possibility is that the Src kinase substitutes for the FAK kinase in phosphorylating Cas in cells expressing FRNK, thereby generating binding sites for Crk. In support of this, we have found that RSF expressing FRNK require the Src-binding domain of Cas as well as the SH3 and SD (Ilić, D., E. Almeida, and C. Damsky, manuscript in preparation). As shown in Fig. 2 B, cells expressing FAK do not require the Src-binding domain of Cas in order to support survival. We have also found that expression of GFP-FRNK in fibroblasts derived from Src−/− mouse embryos triggers apoptosis: the effects of FRNK are indistinguishable from the effects of FAT in those cells. In contrast, GFP-FRNK does not trigger apoptosis when expressed in fibroblasts from wild-type embryos (Ilić, D., E. Almeida, and C. Damsky, manuscript in preparation). Thus, it is likely that FRNK supports survival in RSF because it can bind Cas, and because Src, which also binds Cas, can supply the kinase activity required to phosphorylate Cas.
The FN-FAK-Cas survival pathway activates Ras. Constitutively active Ras rescues death by FAT, whereas blocking Ras function using a DN approach promotes apoptosis of RSF when serum is absent, although not when serum is present. This indicates both that DN Ras is not acting as a general signaling inhibitor, and that the serum survival pathway and FN survival pathway are distinct. Ras activation, in turn, can initiate multiple signaling pathways, including the ERK, JNK, and p38 MAPK pathways. To determine whether these pathways were involved in the transmission of matrix survival signals we used several approaches. First, we determined which pathways were activated in cells that were on a pro-survival FN matrix vs. a pro-apoptotic collagen I matrix. Then we interrupted the pathways that we found were activated by using both DN and inhibitor approaches. Finally, we looked at the cellular localization of key signaling molecules involved in survival.
Using these approaches, we determined that both the ERK and JNK pathways were activated in adherent RSF. However, ERK1/2 were activated similarly on FN and collagen I, whereas JNK2 and to a lesser extent JNK1 were selectively activated on a FN matrix. Additionally, interrupting the ERK pathway either by expression of DN forms of pathway intermediate or by pathway-inhibiting drugs had a relatively small effect on FN-dependent survival, although they suppressed levels of pERK1/2. In contrast, interrupting the JNK pathway by expressing DN intermediates in RSF plated on FN triggered high levels of apoptosis, whereas expressing constitutively active forms of these intermediates at similar levels prevented death by GFP-FAT. Expression of the DN forms in cells cultured with serum did not trigger apoptosis, indicating that they were not toxic or acting as general signal inhibitors.
The importance of JNK activation for the FN-FAK survival pathway was confirmed by demonstrating that embryoid bodies derived from MKK4-null ES cells, like embryoid bodies derived from FAK-null ES cells (Ilić et al. 1998), displayed high levels of apoptosis when serum was withdrawn, whereas embryoid bodies from wild-type counterparts did not. Since MKK4 is the upstream activator of JNK1/2, these data suggest that the activation of JNK is a central event in the transmission of survival signals downstream of FN and FAK when serum is absent. Although FAK can activate the ERK pathway, this pathway alone is likely neither sufficient nor necessary for supporting matrix survival on FN. In contrast, considerable evidence suggests that FAK activation of the ERK pathway is important for regulating cell migration and cell-cycle progression (Schlaepfer et al. 1999).
Interestingly, it appears that the difference in survival signaling between FN and collagen I that is relevant to their differential ability to support survival of RSF might not be at the level of Ras activation (since ERKs are activated similarly by both matrices), but rather at the coupling of Ras with the JNK pathway. Our data indicate that under the culture conditions used, FN can couple Ras signaling to JNK activation to a much greater extent than collagen I. A key to this differential ability to activate the JNK pathway may be the ability of FN, but not collagen I, to recruit activated JNK as well as the other members of the FAK-associated survival complex or scaffold to focal contacts. In contrast, activated ERK is not detected in focal contacts in RSF plated on FN. Thus, pJNK may have a distinct role to play when located in focal contacts that are complementary to or independent of its transcriptional role in the nucleus.
Activation of JNK has been previously associated with apoptosis (Cardone et al. 1997; Frisch and Ruoslahti 1997). Other recent studies have provided evidence both in favor and against roles for JNK in caspase-mediated (Chaudhary et al. 1999; Low et al. 1999) or etoposide-mediated (Anderson et al. 1999; Jarvis et al. 1999) apoptosis. However, additional studies using the MKK4-null mouse suggest that the absence of MKK4 does not interfere with induction of apoptosis in response to etoposide, cisplatinum, adriamycin, and gamma irradiation. Furthermore, apoptosis of hepatocytes and T cells was high in the MKK4-null model, leading to death at embryonic day 12.5. These data indicate that the MKK4/JNK pathway is critical for supporting survival of at least some cell types (Ganiatsas et al. 1998; Nishina et al. 1997, Nishina et al. 1998, Nishina et al. 1999; Yang et al. 1997). Thus, there is precedence for a survival-promoting role for the MKK4/JNK pathway. Interestingly, our data show that JNK1 and 2 respond differently to plating on FN and collagen (Fig. 5 D). JNK1 becomes transiently phosphorylated in response to collagen as well as FN at 1 h, although the phosphorylation level is consistently slightly greater in response to FN. In contrast, phosphorylation of JNK2 is delayed, long-lasting and a specific response to FN in these cells. It is possible that this reflects different roles of these two JNK isoforms in promoting apoptosis vs. protecting cells from apoptosis. Although resolving specific roles for JNK 1 and 2 will require significant additional work, it is clear that in our primary fibroblast system, activation of the MKK4/JNK pathway is required for FN matrix-dependent survival when serum survival signals are absent.
Data from several groups document an important role for PI3-kinase in supporting cell survival (Frisch and Ruoslahti 1997; Marte and Downward 1997; Tamura et al. 1999). We therefore attempted to clarify conditions under which PI3-kinase is and is not critical for survival in the RSF system. Using an inhibitor approach, we confirm that a FAK–PI3-kinase/AKT pathway in RSF is required for survival when cells are cultured in the presence of serum while in suspension (Tamura et al. 1999), or when plated on an inappropriate matrix. However, our PI3-kinase inhibitor studies also showed that PI3-kinase is not required for the FAK/Cas–MKK4/JNK matrix survival pathway, which we show is activated when cells adhere to FN and serum is absent.
We confirm that FAK is required for transduction of signals from serum as well as from FN. This is most clearly shown by the fact that interruption of FAK signaling by expression of GFP-FAT promotes apoptosis in cells plated on an appropriate matrix whether or not serum is present (Fig. 3). Thus, the survival pathway activated in RSF either by serum factors when matrix is absent (PI3-kinase) or by FN when serum is absent (Ras/MKK4/JNK) shares an upstream requirement for FAK, but then diverges. The requirement for FAK in both situations is compatible with its well-documented ability to be activated by both integrins and serum components such as lysophosphatidic acid and PDGF, and its capacity to associate with a large number of cytoskeletal and signaling molecules at focal contact sites (Miyamoto et al. 1995, Miyamoto et al. 1996; Sieg et al. 2000).
Acknowledgments
We thank Drs. Diane Barber, Gary Bokoch, Henry Bourne, Jonathan Chernoff, Roger Davis, Hisamaru Hirai, Bruce Jarvis, Shigeyuki Nada, Tetsuya Nakamoto, Masato Okada, Marc Symons, Richard Flavell, and Tadashi Yamamoto for providing useful plasmids, cells, and reagents. We also thank Evangeline Leash for expert editorial assistance, Dr. Tony Hunter for helpful comments, Shinichi Aizawa, Eric Brown, and Clifford Lowell for critical reading of the manuscript, and Ana Krtolica for technical help.
This work was supported by a Biomedical Sciences Grant from the Arthritis Foundation and an American Heart Association Grant-in-Aid to C.H. Damsky, and by a Research Project Grant from the American Cancer Society to D.D. Schlaepfer. C.R. Hauck is supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft Ha 2856/1-1. E.A.C. Almeida is supported by National Institutes of Health grant T32-DE07204.
Footnotes
Eduardo A.C. Almeida and Duško Ilić contributed equally to this work.
Abbreviations used in this paper: Cas, p130Cas; DN, dominant-negative; ECM, extracellular matrix; ERK, extracellular signal–regulated kinase; ES, embryo stem; FAK, focal adhesion kinase; FAT, focal adhesion targeting; FN, fibronectin; FRNK, FAK-related nonkinase; GFP, green fluorescent protein; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated, dual-action kinase; MEK, MAPK kinase; MKK4, MAPK kinase 4; PI3-kinase, phosphatidylinositol 3′-kinase; PKB, protein kinase B; PR-1, proline-rich region 1; RSF, rabbit synovial fibroblasts; SD, substrate domain; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling.
References
- Anderson S.M., Reyland M.E., Hunter S., Deisher L.M., Barzen K.A., Quissell D.O. Etoposide-induced activation of c-jun N-terminal kinase (JNK) correlates with drug-induced apoptosis in salivary gland acinar cells. Cell Death Differ. 1999;6:454–462. doi: 10.1038/sj.cdd.4400507. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Salvesen G.S., Widmann C., Johnson G., Frisch S.M. The regulation of anoikisMEKK-1 activation requires cleavage by caspases. Cell. 1997;90:315–323. doi: 10.1016/s0092-8674(00)80339-6. [DOI] [PubMed] [Google Scholar]
- Chaudhary P.M., Eby M.T., Jasmin A., Hood L. Activation of the c-Jun N-terminal kinase/stress-activated protein kinase pathway by overexpression of caspase-8 and its homologs. J. Biol. Chem. 1999;274:19211–19219. doi: 10.1074/jbc.274.27.19211. [DOI] [PubMed] [Google Scholar]
- Chen H.C., Appeddu P.A., Isoda H., Guan J.L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Clark E.A., King W.G., Brugge J.S., Symons M., Hynes R.O. Integrin-mediated signals regulated by members of the rho family of GTPases. J. Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky C.H., Werb Z. Signal transduction by adhesion receptorscooperative processing of extracellular information. Curr. Opin. Cell Biol. 1992;4:772–781. doi: 10.1016/0955-0674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- Dolfi F., Garcia-Guzman M., Ojaniemi M., Nakamura H., Matsuda M., Vuori K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. USA. 1998;95:15394–15399. doi: 10.1073/pnas.95.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Ruoslahti E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Frisch S.M., Vuori K., Ruoslahti E., Chan-Hui P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y., Ilić D., Kanazawa S., Takeda N., Yamamoto T., Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- Ganiatsas S., Kwee L., Fujiwara Y., Perkins A., Ikeda T., Labow M.A., Zon L.I. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc. Natl. Acad. Sci. USA. 1998;95:6881–6886. doi: 10.1073/pnas.95.12.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George E.L., Georges-Labouesse E.N., Patel-King R.S., Rayburn H., Hynes R.O. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gilmore A.P., Romer L.H. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Biol. Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault J.A., Labesse G., Mornon J.P., Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem. Sci. 1999;24:54–57. doi: 10.1016/s0968-0004(98)01331-0. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hanks S.K., Polte T.R. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Hildebrand J.D., Schaller M.D., Parsons J.T. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett A.R., Bailey N., Damsky C., Petersen O.W., Bissell M.J. Cellular growth and survival are mediated by beta 1 integrins in normal human breast epithelium but not in breast carcinoma. J. Cell Sci. 1995;108:1945–1957. doi: 10.1242/jcs.108.5.1945. [DOI] [PubMed] [Google Scholar]
- Hungerford J.E., Compton M.T., Matter M.L., Hoffstrom B.G., Otey C.A. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J. Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ilić D., Almeida E.A., Schlaepfer D.D., Dazin P., Aizawa S., Damsky C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis W.D., Johnson C.R., Fornari F.A., Park J.S., Dent P., Grant S. Evidence that the apoptotic actions of etoposide are independent of c-Jun/activating protein-1-mediated transregulation. J. Pharmacol. Exp. Ther. 1999;290:1384–1392. [PubMed] [Google Scholar]
- Khwaja A., Rodriguez-Viciana P., Wennstreom S., Warne P.H., Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King W.G., Mattaliano M.D., Chan T.O., Tsichlis P.N., Brugge J.S. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol. Cell. Biol. 1997;17:4406–4418. doi: 10.1128/mcb.17.8.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low W., Smith A., Ashworth A., Collins M. JNK activation is not required for Fas-mediated apoptosis. Oncogene. 1999;18:3737–3741. doi: 10.1038/sj.onc.1202702. [DOI] [PubMed] [Google Scholar]
- Marte B.M., Downward J. PKB/Aktconnecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem. Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- Meredith J.E., Jr., Fazeli B., Schwartz M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Coso O.A., Gutkind J.S., Burbelo P.D., Akiyama S.K., Yamada K.M. Integrin functionmolecular hierarchies of cytoskeletal and signaling molecules. J. Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Gutkind J.S., Yamada K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activationroles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto T., Sakai R., Honda H., Ogawa S., Ueno H., Suzuki T., Aizawa S., Yazaki Y., Hirai H. Requirements for localization of p130cas to focal adhesions. Mol. Cell. Biol. 1997;17:3884–3897. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H., Bachmann M., Oliveira-dos-Santos A.J., Kozieradzki I., Fischer K.D., Odermatt B., Wakeham A., Shahinian A., Takimoto H., Bernstein A. Impaired CD28-mediated interleukin 2 production and proliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen-activated protein kinase kinase 4 (MKK4)–deficient T lymphocytes. J. Exp. Med. 1997;186:941–953. doi: 10.1084/jem.186.6.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H., Radvanyi L., Raju K., Sasaki T., Kozieradzki I., Penninger J.M. Impaired TCR-mediated apoptosis and Bcl-XL expression in T cells lacking the stress kinase activator SEK1/MKK4. J. Immunol. 1998;161:3416–3420. [PubMed] [Google Scholar]
- Nishina H., Vaz C., Billia P., Nghiem M., Sasaki T., De la Pompa J.L., Furlonger K., Paige C., Hui C., Fischer K.D. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development. 1999;126:505–516. doi: 10.1242/dev.126.3.505. [DOI] [PubMed] [Google Scholar]
- Oktay M., Wary K.K., Dans M., Birge R.B., Giancotti F.G. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell Biol. 1999;145:1461–1469. doi: 10.1083/jcb.145.7.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte T.R., Hanks S.K. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc. Natl. Acad. Sci. USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A., Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK [published erratum appears in Nature. 1996. 6585:810] Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Schaller M.D., Otey C.A., Hildebrand J.D., Parsons J.T. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J. Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hunter T. Integrin signaling and tyrosine phosphorylationjust the FAKs? Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hanks S.K., Hunter T., van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hauck C.R., Sieg D.J. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Sells M.A., Boyd J.T., Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 1999;145:837–849. doi: 10.1083/jcb.145.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg D.J., Ilić D., Jones K.C., Damsky C.H., Hunter T., Schlaepfer D.D. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK-cell migration. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg D.J., Hauck C.R., Ilić D., Klingbeil C.K., Schaefer E., Damsky C.H., Schlaepfer D.D. FAK integrates growth factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Tachibana K., Sato T., D'Avirro N., Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 1995;182:1089–1099. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M., Gu J., Danen E.H., Takino T., Miyamoto S., Yamada K.M. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1999;274:20693–20703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- Wary K.K., Mariotti A., Zurzolo C., Giancotti F.G. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- Werb Z., Tremble P.M., Behrendtsen O., Crowley E., Damsky C.H. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J. Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Parsons J.T. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J. Cell Biol. 1997;139:529–539. doi: 10.1083/jcb.139.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Tournier C., Wysk M., Lu H.T., Xu J., Davis R.J., Flavell R.A. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc. Natl. Acad. Sci. USA. 1997;94:3004–3009. doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.H., Reiske H., Guan J.L. Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 1998;143:1997–2008. doi: 10.1083/jcb.143.7.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]