

Abstract
Caspases are an extended family of cysteine proteases that play critical roles in apoptosis. Animals deficient in caspases-2 or -3, which share very similar tetrapeptide cleavage specificities, exhibit very different phenotypes, suggesting that the unique features of individual caspases may account for distinct regulation and specialized functions. Recent studies demonstrate that unique apoptotic stimuli are transduced by distinct proteolytic pathways, with multiple components of the proteolytic machinery clustering at distinct subcellular sites. We demonstrate here that, in addition to its nuclear distribution, caspase-2 is localized to the Golgi complex, where it cleaves golgin-160 at a unique site not susceptible to cleavage by other caspases with very similar tetrapeptide specificities. Early cleavage at this site precedes cleavage at distal sites by other caspases. Prevention of cleavage at the unique caspase-2 site delays disintegration of the Golgi complex after delivery of a pro-apoptotic signal. We propose that the Golgi complex, like mitochondria, senses and integrates unique local conditions, and transduces pro-apoptotic signals through local caspases, which regulate local effectors.
Keywords: signaling, subcellular, substrate, coiled coil, protease
Introduction
Apoptosis is a highly regulated form of cell suicide that can be triggered by a variety of internal or external signals (for reviews see Ashkenazi and Dixit 1998; Thornberry and Lazebnik 1998). Caspases, a highly conserved family of cysteine proteases, play a critical role in apoptosis by participating in a cascade of cleavage events that result in the apoptotic phenotype. These proteases are synthesized as inactive proenzymes, which are cleaved at Asp-X sites during apoptosis, generating large and small subunits that heterodimerize to form the active enzyme. On the basis of substrate specificities, members of the caspase family can be grouped into three subfamilies: (1) group I caspases, consisting of caspases-1, -4, and -5; (2) group II caspases, consisting of caspases-2, -3, and -7; and (3) group III caspases, consisting of caspases-6, -8, -9, and -10 (Thornberry et al. 1997). The optimal substrate sequences for group III caspases resemble the cleavage sites used in several caspase proenzymes, suggesting that these proteases are the upstream components in a caspase-driven proteolytic cascade. The optimal recognition motif for group II caspases (DEXD) is similar to the cleavage sites of several downstream substrates that are cleaved during apoptosis, suggesting that members of this family act as downstream effectors of apoptosis. Proapoptotic caspases can also be grouped according to their primary structure, with caspases-2, -8, -9, and -10, containing long NH2-terminal prodomains, and caspases-3, -6, and -7, containing short prodomains. The long prodomains of initiator caspases serve a critical function in inducing dimerization and activation of caspase precursors, through interactions with similar domains in adaptor molecules (e.g., FADD, RAIDD, and Apaf-1; Muzio et al. 1996; Butt et al. 1998; Martin et al. 1998; Yang et al. 1998). In contrast, short prodomain caspases function as downstream effectors of apoptosis, which are activated when their precursors are cleaved by upstream caspases.
Despite extensive homology and similar substrate specificities among caspase subfamily members, distinct phenotypes of caspase-deficient mice demonstrate that different group II caspases contribute to apoptosis in a tissue and stimulus-specific manner (for reviews see Li and Yuan 1999; Los et al. 1999; Zheng et al. 1999). For example, caspase-2 (Nedd2/Ich-1) null mice develop normally but accumulate excess numbers of oocytes that are resistant to apoptosis induced by chemotherapeutic agents (Bergeron et al. 1998). In contrast, caspase-3 null mice have severe abnormalities limited to the brain, with enlarged brain, skull defects, and excess accumulation of neurons (Kuida et al. 1996; Woo et al. 1998). These unique phenotypes suggest that similar caspases have unique biochemical and cell biological properties, which transduce different apoptotic signals. In this regard, recent studies have demonstrated that distinct apoptotic pathways are initiated in distinct subcellular domains. For example, numerous components in the cytochrome c/Apaf-1/caspase-9/caspase-3 pathway are enriched in and around mitochondria (Liu et al. 1996; Kluck et al. 1997; Mancini et al. 1998; Samali et al. 1998; Krajewski et al. 1999; Krebs et al. 1999; Susin et al. 1999), while ligation of death receptors at the cell surface induces formation of a signaling complex that includes receptors, adaptors, and different upstream caspases (for review see Ashkenazi and Dixit 1998).
Caspase-2 is unique among the caspases in that it has features of both upstream caspases (a long prodomain) and downstream caspases (DEXD substrate specificity). The prodomain of caspase-2 has been shown to be essential for the dimerization and autoprocessing of precursor caspase-2 molecules (Butt et al. 1998). This caspase-2 prodomain also interacts with a homologous domain in the death adaptor protein RAIDD, thus, implicating caspase-2 as an upstream activator of a distinct caspase pathway (Duan and Dixit 1997). Whether caspase-2 is transducing pro-apoptotic signals from a unique subcellular domain is presently unknown. While GFP fusion constructs have localized caspase-2 to the cytoplasm and nucleus, the localization of the endogenous protease has not yet been determined (Colussi et al. 1998). Furthermore, despite significant evidence for the participation of caspase-2 in some apoptotic pathways (Kumar et al. 1994; Wang et al. 1994; Kumar 1995; Harvey et al. 1997; Li et al. 1997; Troy et al. 1997; Chen et al. 1999), no downstream substrates for this protease have yet been identified.
In the present study, we demonstrate that endogenous caspase-2 is localized predominantly to the Golgi complex and the nucleus. In addition, we identify golgin-160 as a Golgi-localized macromolecular substrate for caspase-2, and demonstrate its cleavage by caspase-2 during apoptosis. We propose that the Golgi complex transduces pro-apoptotic signals through caspase-2.
Materials and Methods
Antibodies Recognizing Caspase-2
Rabbit polyclonal antibodies to the large subunit of caspase-2 (MetAsn150-Thr435) were generated as described previously (Mancini et al. 1998) and called MF386 and MF388. The specificity of these antisera for caspase-2 was demonstrated as follows. When purified, recombinant large subunits of caspases-1–10 (excluding caspase-6) were immunoblotted with MF386 or MF388; only the large subunit of caspase-2 was recognized (data not shown). (b) Both MF386 and MF388 (but not preimmune sera) immunoprecipitated caspase-2 precursor from [35S]Met-labeled HeLa cells or generated by in vitro transcription/translation (IVTT; data not shown). MF386 and MF388 were affinity-purified against purified, recombinant p18 subunit of caspase-2 as described (Mancini et al. 1998). Anti–caspase-2 antibody sc623 and its corresponding blocking peptide were purchased from Santa Cruz Biotechnology, Inc. Anti-ICH-1L was purchased from Transduction Laboratories.
Antibodies Recognizing Golgin-160
The peptides DGASAEQDGLQEDRC (comprising the first 14 amino acids of golgin-160 after the initial Met plus a COOH-terminal Cys) and MLDQEAAFMQIQEAC (comprising amino acids 649–662, plus a COOH-terminal Cys) were synthesized and conjugated to KLH (Boston Biomolecules, Inc.). These peptides were named golgin-160(N) and golgin-160(648), respectively, and used to immunize several rabbits to generate anti–golgin-160 antisera (Covance Research Products). Specificity of both antisera for golgin-160 was confirmed by immunofluorescence, immunoblotting, and immunoprecipitation of in vitro translated golgin-160 (data not shown).
Immunofluorescence Microscopy
Immunofluorescence was performed on coverslips that were fixed with 4% paraformaldehyde and permeabilized with acetone exactly as previously described (Mancini et al. 1998; except for anti–ICH-1L staining and golgin-160 stable lines; see below). Cells were incubated with 5 μg/ml of affinity-purified MF386 or MF388, 3 μg/ml of sc623, or anti–golgin-160(N) antiserum (1:2,500; for 20 min at 4°C), followed by biotin-conjugated goat anti–rabbit IgG (for 20 min at room temperature; Organon Teknika Corp.) and Texas red–conjugated streptavidin (for 10 min at room temperature; Molecular Probes). When performing double labeling, cells were stained first with mouse monoclonal antigiantin (for 20 min at 4°C; provided by Hans-Peter Hauri, University of Basel, Switzerland), followed by incubation with fluorescein-conjugated goat anti–mouse IgG (for 20 min at room temperature; Jackson ImmunoResearch Laboratories, Inc.), and subsequently with 2% goat serum (for 20 min at 4°C). Cells were stained with anti–caspase-2 or anti–golgin-160 antibodies (as described above).
The anti-ICH-1L antibody (Transduction Labs) did not react with its epitope unless permeabilization was performed concomitant with fixation. HeLa cells stably expressing EGFP-golgin-160 (see below) were fixed in 4% paraformaldehyde containing 0.18% Triton X-100 for 10 min at room temperature, and stained with rabbit anti-GFP (Molecular Probes, Inc.) and mouse anti–ICH-1L, followed by fluorescein-conjugated goat anti–rabbit IgG and Texas red goat anti–mouse IgG (both from Jackson ImmunoResearch Laboratories, Inc.).
When specified, nuclei were visualized by labeling with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, Inc.). In all cases, coverslips were mounted on glass slides with PermaFluor (Lipshaw), and confocal microscopy was performed on a scanning confocal microscope system (model LSM 410; Carl Zeiss, Inc.).
The specificity of caspase-2 staining was confirmed as follows. Affinity-purified MF386 was preincubated with 100-fold excess of its cognate p18 antigen or with an irrelevant large caspase subunit antigen (caspase-9), and staining was performed on HeLa cells using this preabsorbed antiserum. Nuclear and Golgi staining were completely abolished by preabsorption with cognate caspase-2 antigen, whereas nuclear and Golgi staining were not affected by incubation with the caspase-9 large subunit (data not shown). Similarly, preabsorption of sc623 with its cognate peptide completely abolished nuclear and Golgi-associated staining (data not shown). (b) No nuclear or Golgi staining was observed when cells were stained with preimmune sera (data not shown). (c) Staining performed with 11 other affinity-purified antibodies to caspases-3, -4, -5, -7, -9, and -10 failed to demonstrate Golgi staining (data not shown).
The EGFP-golgin-160 stable lines were fixed with 3% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 3 min, and labeled with mouse anti-GM130 (Transduction Labs) and rabbit anti-GFP, followed by Texas red goat anti–mouse and fluorescein-conjugated goat anti–rabbit IgG.
Cell Lines and Induction of Apoptosis
HeLa cells were passaged in 10% heat-inactivated calf serum using standard tissue culture procedures. Human keratinocytes were obtained from neonatal foreskins and cultured as described (Casciola-Rosen et al. 1995). The human salivary gland cell line was a gift of Dr. Bruce Baum (National Institutes of Health, Bethesda, MD). Apoptosis was induced by irradiation with UVB (Casciola-Rosen et al. 1994) or treatment with 1 μM staurosporine (Jacobson et al. 1996).
Western Blotting
Collection and lysis of adherent and nonadherent cells, electrophoresis, transfer to nitrocellulose, and immunoblotting were performed as previously described (Casciola-Rosen et al. 1995). The densities of bands on autoradiograms were determined on a PDI Discovery densitometry system using Quantity One software (Bio-Rad Laboratories).
Cloning of Golgin-160
A cDNA clone corresponding to the open reading frame of golgin-160 and flanked by XbaI sites was generated by reverse transcriptase–PCR from human liver and subcloned into the XbaI site of pBlueScript SK+. The cDNA clone was sequenced in its entirety by dideoxy sequencing, and was identical to the published sequence with the exception of the following amino acid substitutions which for the most part were conservative substitutions: I609L, H746R, M932V, K1017R, V1281A, R1315W. More importantly, our cDNA clone contained a 5-bp insertion immediately after amino acid 1,377 that created a frameshift, resulting in a COOH-terminal tail that was 32 amino acids shorter and nonidentical to the published golgin-160 sequence (Misumi et al. 1997). We do not believe that the 5-bp insertion was introduced while amplifying the cDNA clone by PCR, as the identical 5-bp insertion is present in a human EST sequence (AL044812) and in the mouse Mea-2 protein (Kondo and Sutuo 1997). Mutagenesis of golgin-160 was carried out using the PCR-based QuikChange site-directed mutagenesis kit (Stratagene), and the mutations were confirmed by dideoxy sequencing.
In Vitro Cleavage of [35S]Methionine-labeled Substrates Generated by In Vitro Transcription/Translation
[35S]methionine-labeled golgin-160 and golgin-160 mutants were generated by coupled in vitro transcription/translation (Casciola-Rosen et al. 1996). Cleavage reactions with caspases-3 and -7 were performed in buffer A consisting of 50 mM Hepes, pH 7.4, 10% sucrose, 0.1% CHAPS, and 5 mM DTT. Reactions with caspase-3 contained 0.74 nM caspase-3, and those with caspase-7 contained 2.5 nM caspase-7. Cleavage reactions with 40 nM caspase-2 were performed in buffer B consisting of 0.1 M Mes, 10% PEG, 0.1% CHAPS, pH 5.5, and 5 mM DTT (Garcia-Calvo et al. 1999). After incubating samples at 37°C for 60 min, reactions were terminated by the addition of gel buffer and boiling, and samples were electrophoresed on 7.5% SDS-PAGE. Intact proteins and their fragments were visualized by fluorography. Catalytic constant values were determined as described in Casciola-Rosen et al. 1999 according to the formula:
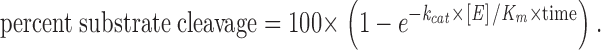 |
GFP Tagging of Golgin-160
A 350-bp segment corresponding to the 5′ end of the golgin-160 coding region was amplified by PCR, to introduce a 5′ XhoI site in-frame with the COOH terminus of GFP in the vector pEGFP-C1 (CLONTECH Laboratories, Inc.), and was digested with XhoI and SpeI. The full-length coding region of golgin-160 with the new 5′ XhoI site was subcloned into the XhoI and BamH1 sites of pEGFP-C1. The amplified region was verified by DNA sequence analysis. HeLa cells (4 × 106) were transfected with 10 μg pEGFP-golgin-160 or pEGFP-golgin-160(D59A) using Lipofectamine (Life Technologies, Inc.) following the manufacturer's directions. After selection in G418, individual G418–resistant colonies were expanded and screened for green fluorescent protein (GFP) fluorescence. One colony from each transfection was fully characterized for expression of the GFP fusion protein and used in experiments described below.
In Vivo Apoptosis Experiments with GFP-golgin-160 Stable Cell Lines
Stable cell lines expressing EGFP-golgin-160 and EGFP-golgin-160 (D59A) were plated, and when they were ∼90% confluent, they were treated with 10 mM sodium butyrate in normal growth medium for 12–15 h to increase the expression of the GFP fusion protein. In experiments where antigen detection was by immunoblotting, cells were treated with 1 μM staurosporine or left untreated for the times indicated, lysed, and processed as described (Casciola-Rosen et al. 1995). In those experiments where detection was by immunoprecipitation, cells were labeled with 100 μCi 35S-Promix (Amersham Pharmacia Biotech, Inc.) in methionine- and cysteine-free DME for 60 min at 37°C, followed by a chase in regular growth medium for 8 h in the presence or absence of 1 μM staurosporine as indicated. Cells were lysed and immunoprecipitated as described (Weisz et al. 1993), using rabbit anti-GFP serum (0.5 μl per sample; Molecular Probes, Inc.) overnight at 4°C. Immunocomplexes were collected with Pansorbin (Calbiochem), washed, electrophoresed on 10% SDS-PAGE, and visualized by fluorography.
To assess Golgi disassembly during apoptosis, stable lines plated on coverslips expressing wt or D59A EGFP-golgin-160 were untreated or treated with 1 μM staurosporine for 2.5 h. Cells were fixed, permeabilized, and labeled with rabbit anti-GFP and FITC-conjugated secondary antibodies as described above. The Golgi morphology of expressing cells was analyzed and categorized as intact or scattered. This quantitation was performed by three individuals on two different experiments. Results are presented as the mean ± SD.
Results
Caspase-2 Has a Nuclear and Golgi Localization
The subcellular localization of caspase-2 was determined by confocal immunofluorescence microscopy in HeLa cells using affinity-purified rabbit antibodies recognizing the large subunit of caspase-2 (MF386, MF388). Caspase-2 had a nuclear staining pattern, as well as a distinct juxtanuclear, reticular pattern that appeared Golgi-like (Fig. 1 a). This juxtanuclear component colocalized with giantin, a 370-kD integral Golgi membrane protein that has most of its mass facing the cytoplasm (Linstedt and Hauri 1993; Seelig et al. 1994; Fig. 1b and Fig. c). Similar Golgi staining was observed with the anti–caspase-2 antibodies after selective permeabilization of the plasma membrane with digitonin, suggesting that caspase-2 is associated with the cytoplasmic face of Golgi membranes (data not shown). Nuclear and Golgi staining was also obtained with two commercially available anti–caspase-2 antibodies: sc623, raised against an NH2-terminal peptide of caspase-2, and anti-ICH-1L, an mAb raised against residues 225–401 of caspase-2 (Fig. 1, d–i). An identical staining pattern was observed in several other cell types, including human keratinocytes and human salivary gland cells (data not shown). In preliminary subcellular fractionation experiments, 35% of the total caspase-2 was present in the nuclear fraction, 10% pelleted with the membrane, and the remainder was found in the cytosol fraction (data not shown). It is not clear if the large cytosolic pool of caspase-2 is real, or dissociates from the membranes or nuclei during fractionation. Although caspase-2 exists in two splicing isoforms (Wang et al. 1994), we were unable to detect any of the short isoform in HeLa cells by immunoblotting or immunoprecipitation (data not shown).
Figure 1.

Caspase-2 is localized to the nucleus and Golgi in HeLa cells. HeLa cells (a–f) or HeLa cells stably expressing GFP-golgin-160 (g–i) were fixed, permeabilized, and double-labeled with anti–caspase-2 (a, d, and g) and antigiantin (b and e) or anti-GFP (h). The stained cells were examined by confocal immunofluorescence microscopy. When red (caspase-2) and green (giantin or GFP) images were merged, overlapping red and green pixels appeared orange/yellow (c, f, and i). An identical caspase-2 staining pattern was observed using several different anti–caspase-2 antibodies: affinity-purified MF386, a polyclonal rabbit antiserum raised against the large subunit of caspase-2 (a–c); sc-623, a polyclonal rabbit antiserum raised against an NH2-terminal peptide of caspase-2 precursor (d–f); and anti-ICH-1L, an mAb raised against a peptide encompassing residues 225–401 of caspase-2 (g–i). MF388, a second rabbit antiserum raised against the large subunit of caspase-2, also showed identical staining (data not shown). The experiments were repeated on 11 (a–c), 5 (d–f), or 3 (g–i) separate occasions with identical results. Bars: (a–c and g-i) 10 μm; 25 μm (d–f).
Golgin-160 Is Cleaved during Apoptosis
Since caspase-2 localized to the Golgi, we investigated whether caspase-2 substrates are also located at this subcellular site. We focused our search on several Golgi autoantigens targeted in patients with systemic autoimmune diseases because proteins cleaved by caspases during apoptosis are frequently targeted as autoantigens (Casciola-Rosen et al. 1995, Casciola-Rosen et al. 1996; Casiano et al. 1996; Rosen and Casciola-Rosen 1999). These autoantigens are members of a protein family called golgins, which possess large coiled-coil domains. Some family members have been implicated in Golgi structure and membrane traffic (for review see Chan and Fritzler 1998). Using an antibody recognizing golgin-160, we immunoblotted lysates of control or apoptotic HeLa cells, in which apoptosis was induced by staurosporine (Fig. 2) or UVB irradiation (data not shown). Golgin-160 was cleaved to generate a 140-kD fragment in early apoptotic cells, as well as a less prominent 163-kD fragment. The caspase substrate PARP was also cleaved at this time point, generating an 89-kD fragment (see Fig. 4 a, lane 3).
Figure 2.

Golgin-160 is cleaved during apoptosis. HeLa cells were incubated for 2 h with 1 μm staurosporine. Lysates from control and treated cells were run on a 7.5% SDS-PAGE and immunoblotted with rabbit anti–golgin-160 antiserum. 70 μg of lysate was loaded in each lane. Results are representative of three separate experiments.
Figure 4.

Kinetics of golgin-160 cleavage during apoptosis. (a) HeLa lysates were made at 1, 2, 4, and 6 h after addition of 1 μm staurosporine, and were run on a 7.5% (top) or 10% (bottom) SDS-PAGE. 70 μg of lysate was loaded in each lane. Golgin-160 and PARP were detected by immunoblotting with anti–golgin-160(648) or patient serum recognizing PARP, respectively. Fragments are marked on the right. (b) Graph showing a decrease of the p163 fragment concomitant with an increase in the p140 fragment. Percentage of maximum fragment plotted against time in staurosporine demonstrates that the caspase-2–generated p163 band reaches a maximum at 2 h, whereas the Group II caspase-generated p140 band reaches its maximum at 4 h in staurosporine. The kinetics of the formation of the p140 fragment of golgin-160 is very similar to that for the p89 fragment of PARP.
Caspases-2, -3, and -7 Cleave Golgin-160 In Vitro and Generate Distinct Fragments
To elucidate whether golgin-160 is directly cleaved by caspases, we expressed [35S]methionine-labeled golgin-160 using IVTT and incubated the labeled golgin-160 with purified caspases in vitro. Cleavage reactions were performed using subsaturating substrate concentrations, and optimal cleavage conditions for the individual caspases (Garcia-Calvo et al. 1999). Caspases-2, -3, and -7 all cleaved golgin-160 efficiently (Fig. 3), with catalytic constant (k cat/K m) values of 3.3 × 104 M−1s−1 for caspase-2, 1.0 × 105 M−1s−1 for caspase-3, and 2.6 × 104 M−1s−1 for caspase-7. Interestingly, cleavage by the different caspases gave rise to unique fragments of golgin-160 in each case (note that intact golgin-160 migrates at 170 kD): caspase-2 generated a predominant fragment at 163 kD (p163), and another at 140 kD (p140) (Fig. 3 a, lane 2); caspase-3 generated a predominant fragment at 155 kD (p155), as well as p140 (Fig. 3 b, lane 2); and caspase-7 generated predominantly the p140 fragment (Fig. 3 c, lane 2).
Figure 3.

Caspases-2, -3, and -7 cleave golgin-160 in vitro at unique sites and generate distinct fragments. [35S]methionine-labeled golgin-160 was expressed using IVTT, and was incubated for 1 h at 37°C with purified caspases-2, -3, or -7. Intact and cleaved golgin-160 were detected by autoradiography. (a) Cleavage with caspase-2 generated a predominant fragment of 163 kD and another of 140 kD. Mutation of aspartic acid to alanine at the amino acid position 59 (D59A) specifically abolished the generation of the unique 163-kD caspase-2–generated fragment, and the D311A mutation abolished generation of the p140 fragment. (b) Caspase-3 cleaved golgin-160 to generate a predominant band of 155 kD and another of 140 kD. The unique p155 fragment was absent in the D139A mutant, and the p140 fragment was absent in the D311A mutant. (c) Cleavage of golgin-160 with caspase-7 generated a single fragment of 140 kD, which was absent in the D311A mutant. (d) Schematic depiction of cleavage of golgin-160 in vitro by caspases-2, -3, and -7. These results are representative of four separate experiments.
Identification of Caspase Cleavage Sites in Golgin-160
To determine the sites at which caspases-2, -3, and -7 cleave golgin-160, we generated several mutants of golgin-160 in which P1 aspartic acid residues at potential caspase cleavage sites were substituted with alanine, and we used these as substrates in cleavage assays in vitro. Sites were chosen based on the approximate sizes of the fragments observed in vitro, and the optimal tetrapeptide recognition motifs for individual proteases, recently defined using a combinatorial tetrapeptide library (Thornberry et al. 1997). The caspase-2 cleavage site in golgin-160 was identified as ESPD59G. A mutation of aspartic acid to alanine at amino acid position 59 (D59A) specifically abolished the unique p163 fragment generated by caspase-2 (Fig. 3 a, lane 3), whereas cleavages of the D59A mutant with caspases-3 and -7 were unchanged from wild-type golgin-160 (data not shown). The unique caspase-3 cleavage site was identified as CSTD139S using a D139A mutant for cleavage by caspase-3 in vitro. The p155 fragment generated by caspase-3 cleavage of wild-type golgin-160 was absent in the D139A mutant (Fig. 3 b, lane 3). Cleavages of D139A by caspases-2 and -7 were identical to those of wild-type golgin-160 by these proteases (Fig. 3 a, lane 5, and c, lane 3), demonstrating that CSTD139 is a site unique for caspase-3. Interestingly, this is the first substrate for caspase-3 that has cysteine in the P4 position. The caspase-7 cleavage site was identified as SEVD311G. The p140 fragment of golgin-160 generated from in vitro cleavages of IVTT-golgin-160 with caspases-2, -3, and -7 was absent with the D311A mutant (Fig. 3, a–c, lane 4). Although this site was not unique to caspase-7, it was most efficiently cleaved by this protease. Thus, although SEVD311G is shared as a cleavage site for caspases-2, -3, and -7, golgin-160 also contains unique cleavage sites for caspases-2 and -3. The cleavage data are summarized in Fig. 3 d.
In Vivo Cleavage of Golgin-160
Given the observation that golgin-160 can be cleaved by caspases-2, -3, and -7 at different sites in vitro, we sought to ascertain whether these protease cleavages, particularly caspase-2 cleavage, also occur in vivo. The fact that there is a unique caspase-2 cleavage site that is not cleaved by the other Group II caspases enabled us to use golgin-160 as a tool to probe caspase-2 activity in vivo. To study the cleavage fragments of golgin-160 generated during apoptosis, lysates of apoptotic HeLa cells were immunoblotted with rabbit anti–golgin-160 antiserum recognizing an internal epitope (peptide comprising amino acids 649–662) immediately after the NH2-terminal coiled-coil domain. A single species of 170 kD was recognized in untreated cells (Fig. 4 a, lane 1), whereas lysates from cells treated with staurosporine showed time-dependent generation of p163 and p140 (Fig. 4 a, lanes 2–5). This p163 fragment decreased at later time points, and was associated with a concomitant increase in the p140 fragment. Maximum levels of the p163 fragment were reached ∼2 h before p140, indicating that these events are kinetically distinct (Fig. 4, a and b). The kinetics of generation of the p140 fragment of golgin-160 closely approximates that of the 89-kD fragment of PARP (Fig. 4 b), which is generated by caspases-3 and -7 (Nicholson and Thornberry 1997; Salvesen and Dixit 1997). The p163 fragment was not recognized by antibodies recognizing the extreme NH2 terminus of golgin-160 (data not shown), confirming that this fragment lacks the NH2-terminal 7 kD of the protein.
To prove that this p163 fragment is indeed generated by cleavage at the unique caspase-2 site, we generated two GFP fusion constructs: one with GFP fused to the NH2 terminus of the wild-type golgin-160 coding region, and the other with GFP fused to the golgin-160(D59A) mutant. Stable HeLa lines were generated from these constructs, and localization studies showed that both GFP-golgin-160 and GFP-golgin-160(D59A) localized appropriately to the Golgi complex (Fig. 5). These HeLa cells were induced to undergo apoptosis by treatment with staurosporine, and the lysates were immunoblotted with anti-GFP to identify the NH2-terminal golgin-160 cleavage fragments (Fig. 6 a). Lysates from both sets of untreated cells showed an intact band at 200 kD (the predicted size of golgin-160 fused to 30 kD GFP; Fig. 6 a, lanes 1 and 3). Interestingly, GFP-golgin-160(D59A) migrated slightly more slowly than wild-type GFP-golgin-160 because of the more extensive phosphorylation of the mutant protein (data not shown). Upon induction of apoptosis by staurosporine, the intact wild-type GFP-golgin-160 fusion protein was cleaved, generating a new band of 38 kD (p38; Fig. 6 a, lane 2). There was also a slight increase in a 47-kD band (p47), present at very low levels in untreated cells, which comigrated with the unique fragment seen when caspase-3 was added to lysates of transfected cells (data not shown). The p38 fragment was absent in lysates from the GFP-golgin-160 (D59A) cells treated with staurosporine, which showed a concomitant increase in the level of p47 (Fig. 6 a, lane 4). In both stable lines, treatment with staurosporine resulted in the characteristic cleavage of other caspase substrates, including NuMa, PARP, and U1-70 kD (data not shown).
Figure 5.

GFP-golgin-160 and GFP-golgin-160(D59A) localize to the Golgi. Stable HeLa lines expressing either GFP-golgin-160 (GFP fused to the NH2 terminus of wild-type golgin-160) or GFP-golgin-160(D59A) (GFP fused to the NH2 terminus of the D59A golgin-160 mutant) were generated. Cells were fixed, permeabilized, double stained with anti-GM130 and anti-GFP, and examined by confocal immunofluorescence microscopy. GM-130 antibodies were visualized with goat anti–mouse IgG and assigned the color red (a and d), whereas GFP antibodies were visualized with fluorescein-conjugated goat anti–rabbit and assigned the color green (b and e). When images were merged, overlapping red and green pixels appeared orange/yellow (c and f). Experiments were repeated on four separate occasions with similar results. Bars, 10 μm.
Figure 6.

Golgin-160 is cleaved at D59 in vivo during staurosporine-induced apoptosis. (a) The stable HeLa lines expressing GFP-golgin-160 or GFP-golgin-160(D59A) were incubated with 1 μm staurosporine for 4 h. Lysates from untreated and treated cells were run on 7.5% SDS-PAGE and immunoblotted with rabbit anti-GFP. Incubation with staurosporine resulted in a decrease of the intact GFP-golgin-160 molecule in both wt and D59A mutant cells. A 38-kD fragment was detected only in staurosporine-treated golgin-160 wild-type cells and not in the staurosporine-treated golgin-160(D59A) mutant line, which instead showed an increased level of the p47 fragment. (b) The GFP-golgin-160 and GFP-golgin-160(D59A) stable HeLa lines were labeled with [35S]Met/Cys for 1 h and chased for 8 h with or without 1 μm staurosporine for different periods of time. GFP-containing fragments of golgin-160 were immunoprecipitated with rabbit anti-GFP from lysates from both untreated and staurosporine-treated cells. Staurosporine treatment resulted in the disappearance of intact GFP-golgin-160 in both wt (4 h) and D59A (6 h). A p38 fragment was detected only in staurosporine-treated GFP-golgin-160 wt cells and not in staurosporine-treated GFP-golgin-160(D59A) mutant cells. In contrast, a single fragment at 47 kD was immunoprecipitated from these D59A mutant cells. At the top is a schematic depiction showing the predicted sizes of NH2-terminal fragments of GFP-golgin-160 when cleavage occurs at the unique caspase-2 and caspase-3 cleavage sites. Experiments were performed on three (a) or five (b) separate occasions with similar results.
For a more sensitive assessment of the time course of GFP-golgin-160 cleavage during apoptosis, immunoprecipitation analysis of 35S-labeled lysates from stable cell lines was also performed. Levels of intact GFP-golgin-160 decreased in staurosporine-treated cells; this was accompanied by an increase in a p38 fragment (Fig. 6 b, wt). In contrast, this p38 fragment was absent in D59A mutants, and a single fragment of 47 kD was immunoprecipitated from these cells (Fig. 6 b, D59A). Similar to the results with endogenous golgin-160 (Fig. 4), cleavage at D59 by caspase-2 occurred sooner after staurosporine treatment than cleavage by caspase-3 at D139, since the p38 NH2-terminal fragment was present as early as 2 h (Fig. 6 b, lane 3), whereas the p47 fragment appeared only after 4 h (Fig. 6 b, lane 10). Thus, the generation of this 38-kD band in apoptotic cells, and prevention of its formation in apoptotic cells which contain golgin-160 mutated at the unique caspase-2 cleavage site, demonstrate that caspase-2 cleavage of golgin-160 occurs in vivo.
The NH2-terminal Fragment of Golgin-160 Is Released from Golgi Membranes after Cleavage by Caspase-2
We used the NH2-terminal anti–golgin-160 antibody to examine the distribution of the NH2-terminal fragment of golgin-160 in individual cells during staurosporine-induced apoptosis. Control and staurosporine-treated HeLa cells were double-stained with antigiantin and anti–golgin-160(N), and were examined by confocal immunofluorescence microscopy (Fig. 7). While control cells showed colocalization of golgin-160 and giantin (Fig. 7 d), 90% of the apoptotic cells in the staurosporine-treated sample stained exclusively for giantin (see green staining of giantin in cell with apoptotic nuclei; Fig. 7 h). These data indicate that the NH2 terminus of golgin-160 is no longer Golgi-associated during apoptosis, likely because of caspase-induced cleavage.
Figure 7.

The NH2-terminal fragment of golgin-160 is released from Golgi membranes after cleavage by caspase-2. Untreated HeLa cells (a–d) and HeLa cells treated with 1 μm staurosporine for 1 h (e–h) were fixed, permeabilized, and triple labeled with anti–golgin-160(N) (a and e), which was visualized as red; antigiantin (b and f), which was visualized as green; and DAPI (c and g), which was visualized as blue. Overlapping red and green pixels appear yellow (d and h). Arrow in h denotes an apoptotic cell, which no longer stains for the NH2-terminal portion of golgin-160. These results are representative of three separate experiments. Bars, 10 μm.
Prevention of Golgin-160 Cleavage at the Unique Caspase-2 Site (D59) Delays Golgi Disintegration during Apoptosis
The Golgi complex disassembles during apoptosis (Sesso et al. 1999), although virtually nothing is known about this process. We noticed that after inducing apoptosis with staurosporine, Golgi morphology changed from a tight, juxtanuclear reticulum to a scattered appearance before finally disappearing. To determine if cleavage of golgin-160 at its unique caspase-2 site has functional relevance during apoptosis, we compared Golgi morphology in the stable HeLa lines expressing GFP-golgin-160 (wt) or GFP-golgin-160(D59A) after inducing apoptosis. Control and staurosporine-treated cells were examined by confocal immunofluorescence microscopy, and the proportion of cells with scattered Golgi complexes was quantitated (Fig. 8). In cells expressing wt golgin-160, 44 ± 7% of the cells exhibited scattered Golgi complexes after 2.5 h of staurosporine treatment, compared with 12 ± 5% in the control cells. In contrast, only 23 ± 5% of the cells expressing the mutant golgin-160 (D59A) showed scattered Golgi complexes at this time point (P = 0.0028 wt versus D59A). This delay of Golgi disintegration in the D59A mutant indicates that cleavage at the unique caspase-2 cleavage site plays a role in inducing the apoptotic changes in Golgi morphology.
Figure 8.

Golgin-160 cleavage at D59 by caspase-2 contributes to the disintegration of the Golgi complex during apoptosis. The stable HeLa cell lines expressing GFP-golgin-160 (wt) or GFP-golgin-160(D59A) were left untreated or incubated with 1 μm staurosporine (STS) for 2.5 h. (a) GFP-golgin-160–expressing cells from these four groups were assessed for their Golgi morphology (intact versus scattered), and plotted as a percentage of total counted cells. Error bars represent the SD. **P = 0.0028 as determined by the t test (comparing percentages of scattered Golgi in staurosporine-treated wt versus staurosporine-treated D59A). (b) Representative cell with intact Golgi. (c) Representative cells with scattered Golgi. Bar, 10 μm.
Discussion
Unique Golgi Distribution of Caspase-2
The distinct phenotypes observed in animals that are deficient in caspase-2 or caspase-3 (group II caspase subfamily members that share a very similar tetrapeptide substrate specificity) suggest that unique features of individual caspases may account for distinct regulation and specialized functions. In the experiments reported here, we have demonstrated that caspase-2 has a unique subcellular distribution, and that it cleaves a macromolecular substrate at a unique site not used by other group II caspases. Indirect immunofluorescence microscopy, using four different antibodies, revealed that endogenous caspase-2 has a Golgi and nuclear distribution in several cell types. The nuclear localization of GFP-tagged caspase-2 has been observed previously; although there was also evidence of discrete cytoplasmic staining in cells overexpressing tagged caspase-2 (Colussi et al. 1998), this was not identified as Golgi. Our studies did not demonstrate any mitochondrial enrichment of caspase-2, under conditions where the mitochondrial pool of caspase-3 precursor could be clearly demonstrated (Mancini et al. 1998; Samali et al. 1998). This unique distribution of caspase-2, which differs markedly from that previously defined for other group II caspases, is in agreement with several recent observations, which indicate that different caspases are enriched in different subcellular compartments: a fraction of procaspase-3 has a mitochondrial location (Mancini et al. 1998; Samali et al. 1998; Krebs et al. 1999); and procaspase-9 is sequestered within the intermembrane space of mitochondria in some cell types (Krajewski et al. 1999; Susin et al. 1999). The available data suggest that these unique microenvironments place individual caspases in a position to interact with a unique group of upstream activators, and to cleave a discrete subset of downstream substrates enriched at these sites. Identifying the adaptors that interact with Golgi-associated caspase-2, and determining what role these adaptor molecules have in targeting or activating Golgi-associated caspase-2 is of high priority.
Caspase-2 Is Responsible for Efficient Cleavage of Golgin-160 at a Unique Site
To date, no substrates of caspase-2, other than procaspase-2 itself (Harvey et al. 1996), have been identified. Studies to define potential substrates for caspase-2 at the Golgi complex were facilitated by the observation that numerous autoantigens targeted in systemic autoimmune diseases are specifically cleaved by caspases during apoptosis (Rosen and Casciola-Rosen 1999). Golgin-160, a peripheral Golgi membrane-associated autoantigen, is cleaved by caspase-2 in vitro and during apoptosis. Caspases-3 and -7 also efficiently cleaved this substrate. Analysis of the preferred cleavage sites for caspases-2, -3, and -7 revealed that in spite of the very similar specificity of these three proteases observed using tetrapeptide substrates, each protease selected a different preferred site in golgin-160. One site (ESPD59) was uniquely susceptible to cleavage by caspase-2, and provided an essential tool to identify caspase-2 activity in intact cells. Using several different assays, we demonstrated that golgin-160 is cleaved at the unique caspase-2 site early during apoptosis, and is also subsequently cleaved at additional downstream sites. The delay in Golgi disassembly upon induction of apoptosis by staurosporine in cells expressing a mutant golgin-160 lacking the unique caspase-2 site implies that cleavage of golgin-160 is an important step in this process. It is possible that the cleavage of golgin-160 at D59 may play a more critical role in specific pro-apoptotic stimuli preferentially targeting the Golgi complex. Studies to address this issue are presently underway.
The Golgi Complex: A Novel Apoptotic Signaling Organelle?
Although the function of different golgin family members is not yet fully elucidated, some members of this family have been implicated in Golgi structure and function, particularly in vesicle traffic (Gleeson et al. 1996; Nakamura et al. 1997; Lowe et al. 1998; Sonnichsen et al. 1998). The fact that golgin-160 is a substrate for three different caspases indicates that it will also be important to determine the function of golgin-160, and the consequences of its cleavage at these different sites during apoptosis. The Golgi complex, with its central location and essential role in membrane traffic, is in an ideal position to sense and integrate information about the state of the cell. Interestingly, several signaling proteins implicated in apoptotic signaling pathways are enriched at Golgi membranes, including tumor necrosis factor receptor (TNF-R1), Fas, an isoform of protein kinase C, PI(3) kinase, and BRUCE (a novel ubiquitin-conjugating enzyme that contains a BIR [baculovirus inhibitor of apoptosis] motif) (Bennett et al. 1998; Hauser et al. 1998; Cockcroft 1999; Jamora et al. 1999; Jones et al. 1999). Golgi membranes are also likely to be the site of at least some ceramide signaling (Maceyka and Machamer 1997), which has been implicated in the induction of apoptosis (Kolesnick and Hannun 1999). The structure of the Golgi complex might be particularly conducive to sensing small changes in the lipid composition of the bilayer (Maceyka and Machamer 1997; Wright 1999). Recent studies have highlighted the clustering at distinct subcellular sites (e.g., mitochondria or plasma membrane) of multiple components (receptors, adaptors, and effectors) of the proteolytic machinery that initiates and executes apoptotic cell death. Our findings that a proportion of caspase-2 is located at the Golgi complex, and cleaves a Golgi substrate early during apoptosis, focuses attention on the Golgi complex as a potentially important site for generating and transducing unique pro-apoptotic signals.
Acknowledgments
These studies were supported by the National Institutes of Health grants AR44684 (to L. Casciola-Rosen), DE12354 (to A. Rosen), GM42522 (to C. Machamer), the SLE Foundation, and the Blum-Kovler Foundation. A. Rosen is supported by a Burroughs Wellcome Fund Translational Research Award.
Footnotes
Abbreviations used in this paper: DAPI, 4′,6-diamidino-2-phenylindole; GFP, green fluorescent protein; IVTT, in vitro transcription/translation.
References
- Ashkenazi A., Dixit V.M. Death receptorssignaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Bennett M., Macdonald K., Chan S.W., Luzio J.P., Simari R., Weissberg P. Cell surface trafficking of Fasa rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- Bergeron L., Perez G.I., MacDonald G., Shi L., Sun Y., Jurisicova A., Varmuza S., Latham K.E., Flaws J.A., Salter J.C.M. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A.J., Harvey N.L., Parasivam G., Kumar S. Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J. Biol. Chem. 1998;273:6763–6768. doi: 10.1074/jbc.273.12.6763. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Anhalt G., Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Anhalt G.J., Rosen A. DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J. Exp. Med. 1995;182:1625–1634. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Nicholson D.W., Chong T., Rowan K.R., Thornberry N.A., Miller D.K., Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repaira fundamental principle of apoptotic death. J. Exp. Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L., Andrade F., Ulanet D., Wong W.B., Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen statusimplications for initiation of autoimmunity. J. Exp. Med. 1999;190:815–825. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiano C.A., Martin S.J., Green D.R., Tan E.M. Selective cleavage of nuclear autoantigens during CD95 (Fas/APO-1)-mediated T cell apoptosis. J. Exp. Med. 1996;184:765–770. doi: 10.1084/jem.184.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.K.L., Fritzler M.J. Golginscoiled-coil-rich proteins associated with the Golgi complex. Electronic J. Biotech. 1998;1:1–10. [Google Scholar]
- Chen W., Wang H.-G., Srinivasula S.M., Alnemri E., Cooper N.R. B cell apoptosis triggered by antigen receptor ligation proceeds via a novel caspase-dependent pathway. J. Immunol. 1999;163:2483–2491. [PubMed] [Google Scholar]
- Cockcroft S. Mammalian phosphatidylinositol transfer proteinsemerging roles in signal transduction and vesicular traffic. Chem. Phys. Lipids. 1999;98:23–33. doi: 10.1016/s0009-3084(99)00015-8. [DOI] [PubMed] [Google Scholar]
- Colussi P.A., Harvey N.L., Kumar S. Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor—a novel function for a caspase prodomain. J. Biol. Chem. 1998;273:24535–24542. doi: 10.1074/jbc.273.38.24535. [DOI] [PubMed] [Google Scholar]
- Duan H., Dixit V.M. RAIDD is a new ‘death’ adaptor molecule. Nature. 1997;385:86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Rasper D.M., Vaillancourt J.P., Zamboni R., Nicholson D.W., Thornberry N. Purification and catalytic properties of human caspase family members. Cell Death Differ. 1999;6:362–369. doi: 10.1038/sj.cdd.4400497. [DOI] [PubMed] [Google Scholar]
- Gleeson P.A., Anderson T.J., Stowe J.L., Griffiths G., Toh B.H., Matheson F. p230 is associated with vesicles budding from the trans-Golgi network. J. Cell Sci. 1996;109:2811–2821. doi: 10.1242/jcs.109.12.2811. [DOI] [PubMed] [Google Scholar]
- Harvey N.L., Trapani J.A., Fernandes-Alnemri T., Litwack G., Alnemri E., Kumar S. Processing of the Nedd2 precursor by ICE-like proteases and granzyme B. Genes Cells. 1996;1:673–685. doi: 10.1046/j.1365-2443.1996.00255.x. [DOI] [PubMed] [Google Scholar]
- Harvey N.L., Butt A.J., Kumar S. Functional activation of Nedd2/ICH-1 (Caspase-2) is an early process in apoptosis. J. Biol. Chem. 1997;272:13134–13139. doi: 10.1074/jbc.272.20.13134. [DOI] [PubMed] [Google Scholar]
- Hauser H.P., Bardroff M., Pyrowolakis G., Jentsch S. A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 1998;141:1415–1422. doi: 10.1083/jcb.141.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.D., Weil M., Raff M.C. Role of Ced3/ICE-family proteases in staurosporine-induced programmed cell death. J. Cell Biol. 1996;133:1041–1051. doi: 10.1083/jcb.133.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J.R., Faulkner D.J., Malhotra V. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Jones S.J., Ledgerwood E.C., Prins J.B., Galbraith J., Johnson D.R., Pober J.S., Bradley J.R. TNF recruits TRADD to the plasma membrane but not the trans Golgi network, the principal subcellular location of TNF-R1. J. Immunol. 1999;162:1042–1048. [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kolesnick R., Hannun Y.A. Ceramide and apoptosis. Trends Biochem. Sci. 1999;24:224–225. doi: 10.1016/s0968-0004(99)01408-5. [DOI] [PubMed] [Google Scholar]
- Kondo M., Sutuo S. Cloning and molecular characterization of cDNA encoding a mouse male-enhanced antigen-2 (Mea-2)a putative family of the Golgi autoantigen. DNA Seq. 1997;7:71–82. doi: 10.3109/10425179709020154. [DOI] [PubMed] [Google Scholar]
- Krajewski S., Krajewska M., Ellerby L.M., Welsh K., Xie Z., Deveraux Q.L., Salveson G., Bredesen D.E., Rosenthal R.E., Fiskum G., Reed J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs J.F., Armstrong R.C., Srinivasan A., Aja T., Wong A.M., Abey A., Sayers R., Pham B., Vu T., Hoang K. Activation of membrane-associated procaspase-3 is regulated by Bcl-2. J. Cell Biol. 1999;144:915–926. doi: 10.1083/jcb.144.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Zheng T.S., Na S.Q., Kuan C.Y., Yang D., Karasuyama H., Rakic P., Flavell R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kumar S. Inhibition of apoptosis by the expression of antisense Nedd2. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1995;368:69–72. doi: 10.1016/0014-5793(95)00602-6. [DOI] [PubMed] [Google Scholar]
- Kumar S., Kinoshita M., Noda M., Copeland N.G., Jenkins N.A. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1β-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- Li H.L., Yuan J. Deciphering the pathways of life and death. Curr. Opin. Cell Biol. 1999;11:261–266. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- Li H.L., Bergeron L., Cryns V., Pasternack M.S., Zhu H., Shi L., Greenberg A., Yuan J. Activation of caspase-2 in apoptosis. J. Biol. Chem. 1997;272:21010–21017. doi: 10.1074/jbc.272.34.21010. [DOI] [PubMed] [Google Scholar]
- Linstedt A.D., Hauri H.P. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell. 1993;4:679–693. doi: 10.1091/mbc.4.7.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Los M., Wesselborg S., Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transuctionlessons from knockout mice. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- Lowe M., Rabouille C., Nakamura N., Watson R., Jackman M., Jamsa E., Rahman D., Pappin D.C., Warren G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94:783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- Maceyka M., Machamer C.E. Ceramide accumulation uncovers a cycling pathway for the cis-Golgi network marker, infectious bronchitis virus M protein. J. Cell Biol. 1997;139:1411–1418. doi: 10.1083/jcb.139.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M., Nicholson D.W., Roy S., Thornberry N.A., Peterson E.P., Casciola-Rosen L.A., Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distributionimplications for apoptotic signaling. J. Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.A., Siegel R.M., Zheng L., Lenardo M.J. Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHα1) death signal. J. Biol. Chem. 1998;273:4345–4349. doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- Misumi Y., Sohda M., Yano A., Fujiwara T., Ikehara Y. Molecular characterization of GCP170, a 170-kDa protein associated with the cytoplasmic face of the Golgi membrane. J. Biol. Chem. 1997;272:23851–23858. doi: 10.1074/jbc.272.38.23851. [DOI] [PubMed] [Google Scholar]
- Muzio M., Chinnaiyan A.M., Kischkel F.C., O'Rourke K., Shevchenko A., Ni J., Scaffidi C., Bretz J.D., Zhang M., Gentz R. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nakamura N., Lowe M., Levine T.P., Rabouille C., Warren G. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 1997;89:445–455. doi: 10.1016/s0092-8674(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W., Thornberry N.A. Caspaseskiller proteases. TIBS (Trends Biochem. Sci.). 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Rosen A., Casciola-Rosen L. Autoantigens as substrates for apoptotic proteasesimplications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6:6–12. doi: 10.1038/sj.cdd.4400460. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Samali A., Zhivotovsky B., Jones D.P., Orrenius S. Detection of pro-caspase-3 in cytosol and mitochondria of various tissues. FEBS (Fed. Eur. Biochem. Soc.) Lett. 1998;431:167–169. doi: 10.1016/s0014-5793(98)00740-6. [DOI] [PubMed] [Google Scholar]
- Seelig H.P., Schranz P., Schroter H., Wiemann C., Renz M. Macrogolgina new 376 kD Golgi complex outer membrane protein as target of antibodies in patients with rheumatic diseases and HIV infections. J. Autoimmunity. 1994;7:67–91. doi: 10.1006/jaut.1994.1006. [DOI] [PubMed] [Google Scholar]
- Sesso A., Fujiwara D.T., Jaeger M., Jaeger R., Li T.C., Monteiro M.M., Correa H., Ferreira M.A., Schumacher R.I., Belisario J., Kachar B., Chen E.J. Structural elements common to mitosis and apoptosis. Tissue Cell. 1999;31:357–371. doi: 10.1054/tice.1999.0042. [DOI] [PubMed] [Google Scholar]
- Sonnichsen B., Lowe M., Levine T., Jamsa E., Dirac-Svejstrup B., Warren G. A role for giantin in docking COP1 vesicles to Golgi membranes. J. Cell Biol. 1998;140:1013–1022. doi: 10.1083/jcb.140.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Brenner C., Larochette N., Prevost M., Alzari P.M., Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–393. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspasesenemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Rano T.A., Peterson E.P., Rasper D.M., Timkey T., Garcia-Calvo M., Houtzager V.M., Nordstrom P.A., Roy S., Vaillancourt J.P., Chapman K.T., Nicholson D.W. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Troy C.M., Stefanis L., Greene L.A., Shelanski M.L. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J. Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Miura M., Bergeron L., Zhu H., Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Weisz O.A., Swift A.M., Machamer C.E. Oligomerization of a membrane protein correlates with its retention in the Golgi complex. J. Cell Biol. 1993;122:1185–1196. doi: 10.1083/jcb.122.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo M., Hakem R., Soengas M.S., Duncan G.S., Shahinian A., Kägi D., Hakem A., McCurrach M., Khoo W., Kaufman S.A. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.D. Toll, a new piece in the puzzle of innate immunity. J. Exp. Med. 1999;189:605–609. doi: 10.1084/jem.189.4.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.L., Chang H.Y., Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell. 1998;1:319–325. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- Zheng T.S., Hunot S., Kuida K., Flavell R.A. Caspase knockoutsmatters of life and death. Cell Death Differ. 1999;6:1043–1053. doi: 10.1038/sj.cdd.4400593. [DOI] [PubMed] [Google Scholar]