

Abstract
The varicella-zoster virus (VZV) genes ORF47 and ORF66 are predicted to encode serine/threonine protein kinases, which are homologs of herpes simplex virus 1 (HSV-1) UL13, and US3. When mutants were constructed by inserting stop codons into ORF47 and ORF66, the recombinants ROka47S and ROka66S, as well as intact ROka replicated in tissue culture. In contrast, inoculation of human thymus/liver or skin implants in SCID-hu mice showed that ORF47 protein was required for viral growth in human T cells and skin. Eliminating ORF66 expression inhibited VZV infectivity for T cells partially but did not impair replication in skin compared with ROka. Infectivity for T cells and skin was restored when ROka47S virus was complemented by insertion of ORF47 into a distant, noncoding site. The ORF47 gene product is the first VZV protein identified as necessary for T cell tropism. It also is essential for skin infectivity in vivo, as is glycoprotein C. Expression of ORF66 did not compensate for the absence of the ORF47 protein. The requirement for ORF47 expression in T cells and skin indicates that this gene product, which is dispensable in vitro, has a critical role within differentiated cells that are essential targets for VZV pathogenesis in vivo.
Varicella-zoster virus (VZV) is a human α-herpesvirus that causes chickenpox and herpes zoster (shingles). The α-herpesviruses contain two highly conserved genes that are predicted to encode protein kinases by sequence homology to eukaryotic serine/threonine kinases (1, 2). In VZV, these genes are encoded by ORF47, in the unique, long region of the genome, and by ORF66, in the unique, short region. ORF47 encodes a 54-kDa phosphoprotein found in the cytoplasm and nucleus of infected cells and in the virion capsid/tegument fraction. ORF66 kinase is a 48-kDa phosphoprotein located only in the cytoplasm (3). Homologs of ORF47 are found in α-, β- and γ-herpesviruses, but ORF66 homologs are specific to the α-herpesviruses (1, 2, 4–9).
The ORF47 putative kinase phosphorylates itself and ORF62, the major immediate early (IE) transactivator, uses both ATP and GTP as phosphate donors (10, 11). The other VZV IE proteins encoded by ORF4, ORF61, and ORF63, and the major glycoprotein E (gE), are not phosphorylated by ORF47 kinase in vitro (3, 10–12). VZV ORF47, like related kinases, is dispensable for replication in vitro (6, 12–14). The HSV-1 UL13 kinase is autophosphorylated and necessary for the posttranslational modification of HSV-1 regulatory proteins infected cell protein (ICP)22, homolog of VZV ORF63, and ICP0, homolog of VZV ORF61 but not ICP4, the ORF62 homolog (13, 15–17). The HSV-1 UL13 gene also is required for the virion host shutoff effect (14). More recently, UL13 kinase has been shown to phosphorylate the HSV-1 glycoprotein E/I Fc receptor complex and the cellular protein elongation factor 1δ (18, 19).
Like the ORF47 protein, the ORF66 putative kinase is not required for phosphorylation of the IE genes and is not essential in tissue culture. Although neither mutant has a phenotype in vitro, a double mutant lacking both ORF66 and ORF47 proteins grows to a reduced titer (20). Similarly, replication of pseudorabies virus (PRV) US3− and UL13/US3− mutants was reduced by 10-fold in vitro (6). HSV-1 US3 is not required in tissue culture but is involved in posttranslational processing of an essential phosphoprotein, UL34 (21, 22). HSV-2 US3 phosphorylates the UL12 alkaline nuclease in vitro (23). In some cell lines, US3 blocks apoptosis induced by HSV-1 infection (24, 25).
ORF47 and UL13 are evolutionarily similar to CKII, although only the cellular enzyme is inhibited by heparin (10, 26, 27). The α-herpesvirus protein kinases such as ORF66 and US3 clearly constitute a separate family and have no obvious cellular homolog. The kinases that give the best score in a pairwise comparison to US3 are CDC28 of Saccharomyces cerevisiae, which is involved in regulation of cell division, and the human enzyme cdc2 (1). Common structural features of these viral kinases are a highly variable noncatalytic N-terminal domain and an extraordinarily acidic region (26).
Since VZV is a highly species specific virus, the molecular analysis of VZV genes involved in cell tropism and virulence has been hindered by the limitations of small animal models. By using human thymus/liver implants in a SCID-hu model, we have defined VZV as a lymphotropic virus that infects human CD4+ and CD8+ T cells (28). The SCID-hu skin model was valuable for proving that glycoprotein C was critical for pathogenesis, even though it is dispensable in tissue culture (29). Most importantly, infection of human tissue implants composed of different cell types makes it possible to distinguish specific genes required for VZV replication in the various target cells involved in its pathogenic cycle. A comparison of the live, attenuated varicella vaccine strain, V-Oka, with its parental clinical isolate, P-Oka, showed that V-Oka was deficient in infectivity for skin but retained its T cell tropism (29).
The purpose of these experiments was to assess the contributions of ORF47 and ORF66 gene products to VZV replication in intact human tissues containing differentiated skin and T cells. Our results indicate that ORF66 is not required for growth in skin or T cells, although the ROka66S mutant replicates less efficiently in T cells. ORF47, like glycoprotein C, is necessary for skin infectivity. Most importantly, ORF47 is the first VZV gene that we have found to be required for T cell tropism, which is critical for VZV pathogenesis.
MATERIALS AND METHODS
SCID-hu Mice.
Thymus/liver or skin implants were engrafted in male homozygous C.B-17 scid/scid mice (29). Human fetal tissues were obtained with informed consent according to federal and state regulations. Animals were cared for according to guidelines of the Animal Welfare Act PL 94–279 and the Stanford University Administrative Panel on Laboratory Animal Care.
Recombinant Viruses.
Recombinant viruses were isolated in human melanoma cells, passaged onto MRC-5 lung fibroblasts, and stored at −70°C in tissue culture media (MEM, Mediatech, Washington, D.C.) supplemented with 10% fetal calf serum (Tissue Culture Biologicals, Tulare, CA), 2 mM l-glutamine (GIBCO), antibiotics, and 10% DMSO. Other cell lines used were HFF (human foreskin fibroblasts) and Vero cells (African green monkey kidney cells). Recombinant VZV was generated by using cosmids derived from the varicella vaccine strain (ROka) (30). VZV recombinants, ROka47S, and ROka66S were made from ROka by introducing stop codons into ORF47 at the 166th codon and into ORF66 at the 15th codon (12, 20). A repaired strain, ROka47SR, that expressed ORF47 protein kinase was constructed by cloning the ORF47 gene, including the putative promotor region and the polyadenylation site, into an unique AvrII restriction site at nucleotide 112,852 in the cosmid MstII A (30–32). The ORF47 gene was amplified by PCR by using the oligonucleotides, 5′-CTTCTCCTAGGCGTAACCGT-3′ and 5′-ATGTCTTCCCTAGGGTGTCT-3′ (codons 82, 744 and 84, 817) to introduce AvrII sites at each end. The amplified ORF47 gene was ligated into MstII A at the AvrII site, and a clone, MstIIA47rev, containing ORF47 in the native, 5′ to 3′ orientation (upper strand) was selected. This cosmid and cosmid NotI-B47S (12) were used to generate the VZV strain, ROka47SR, which contains the original stop mutation in the ORF47 gene and the intact ORF47 gene at the AvrII site.
To sequence ROka47SR, genomic DNA was isolated from infected cells and a 3.2-kb fragment spanning the AvrII site between ORF65 and ORF66 was amplified by using primer pairs, which map to codons 112,580 and 113,686 (31). Primers, which map to codons 81,118 and 85,186, were used to amplify the inserted gene and the region of ROka47S containing the stop codons for sequencing. The coding strand of amplified product was sequenced by using the Applied Biosystems automated sequencing apparatus (Model 373A, Version 2.0.1S).
DNA was isolated from infected thymus/liver and skin implants to verify the genotypes of all infectious VZV strains. An AscI site, introduced into ORF66 when the stop codons were inserted to create ROka66S, was used to identify this recombinant. A 1.3-kb fragment flanking the AscI site was amplified by using the PCR primers, which map to codons 112,083 and 113,368. The PCR products and a control plasmid were purified by using a QIAquick PCR Purification Kit (Qiagen), digested with AscI, and separated by agarose gel electrophoresis. Implants infected with ROka and ROka47SR were analyzed by PCR using the primers that flank the AvrII site. The PCR product from ROka was a 1.1-kb fragment whereas insertion of ORF47 at the AvrII site generated a 3.2-kb fragment from ROka47SR.
Replication of VZV Strains in Vitro.
The replication of VZV strains, ROka, ROka47S, ROka66S, and ROka47SR, in vitro was compared by using a growth curve assay (33). Cells were trypsinized from replicate wells on days 1, 2, 3, and 4, and titers were determined in an infectious focus assay (29).
Infection and Analysis of Thymus/Liver and Skin Implants.
Implants were harvested weekly, from 1 to 4 weeks after inoculation or mock infection; tissues were analyzed by infectious focus assay, flow cytometry, in situ hybridization, and Western blot (28, 29). The antibodies used for flow cytometry were human VZV-immune or nonimmune polyclonal IgG and goat anti-human-fluorescein isothiocyanate (FITC) conjugated F(ab′)2 fragments (CalTag Laboratories, South San Francisco, CA). T cell markers were detected with the mouse monoclonal anti-CD3-phycoerythrin (Becton Dickinson). Cell suspensions were analyzed with a Becton Dickinson FacScan apparatus. For in situ hybridization, tissue sections were probed with a 12.9-kb biotinylated plasmid, pVZV-C, that consists of the HindIII fragment C of VZV genomic DNA in pBR322; the negative control probe was pBR322 vector alone (28). Hybridization was detected with a streptavidin-alkaline phosphatase conjugate and visualized with NBT/BCIP (nitroblue tetrazolium salt and 5-bromo-4-chloro-3-indoyl phosophate p-toluidine salt).
For Western blot analysis, tissue extracts were prepared from skin implants, separated by SDS/PAGE in 8% gels, and transferred to Immobilon-P poly(vinylidene difluoride) membranes (Millipore, Bedford MA). The amount of total protein in 15 μl of each sample was equivalent as verified by amido black stain. VZV proteins were detected with a high-titer polyclonal human immune serum, and a secondary goat anti-human IgG horseradish peroxidase conjugate was used for chemiluminscent detection of bound antibodies (Amersham).
Immunoprecipitation.
MeWo cells infected with ROka, ROka47S, or ROka47SR, were metabolically labeled with 500 μCi [35S]methionine and [35S]cysteine (Pro-mix, Amersham) for 4 hr. The cells were lysed and ORF47 protein and gE were precipitated by using the Boehringer Mannheim Immunoprecipitation kit (Protein G) according to the instructions. The antibodies used were polyclonal rabbit antiserum against ORF47 protein (10) (a generous gift of Charles Grose, University of Iowa) and mouse mAb 7G8, directed against the C terminus of gE (a generous gift of Baghar Forgani, California State Department of Health). Precipitated proteins were separated on a 10% SDS/PAGE gel; the gel was dried and analyzed by autoradiography.
RESULTS
Effects of the Functional Deletion of ORF47 and ORF66 on the T Cell Tropism of VZV.
The ROka, ROka47S, and ROka66S recombinants were indistinguishable by growth kinetics, plaque morphology, and virus yields in vitro, confirming that blocking the expression of ORF47 and ORF66 does not interfere with VZV replication in tissue culture (12, 20) (Fig. 1A). The ROka, ROka47S, and ROka66S reached peak titers of ≈5.0 × 105 by day 2. These VZV recombinants had identical growth patterns in MRC-5, HFF, and Vero cells (data not shown).
Figure 1.

Replication of VZV in MeWo cells and thymus/liver implants. (A) MeWo cells were inoculated on day 0 with an infected-cell inoculum of either ROka (□), ROka47SR (○), ROka66S (•), or ROka47S (■). Aliquots were harvested daily for 4 days, and infectious foci were determined by titration on Vero cell monolayers. (B) Thymus/liver implants were inoculated with VZV-infected MRC-5 cells on day 0 (inoculum sizes shown on y-axis) and harvested weekly. T cell suspensions were prepared and titrated in the infectious foci assay. Each point represents the mean of at least three implants from two separate experiments.
In contrast to its infectivity for cultured cells, ROka47S did not grow in T cells in SCID-hu thymus/liver implants. Thymus/liver implants infected with 2.4 × 103 colony-forming units (cfu) of ROka47S yielded no virus on days 7 or 14 after inoculation (Fig. 1B). In two of three experiments, no virus was recovered from thymus/liver implants 21 days after inoculation with ROka47S by using an infectious focus assay, which detects one infected T cell per 106; in one experiment, a single plaque of infectious ROka47S was isolated at day 21. Although ROka66S replicated in T cells, VZV infectivity was impaired in the absence of the ORF66 protein. Infectious virus was not recovered from thymus/liver implants until 14 days after inoculation of 3.0 × 103 cfu of ROka66S, whereas high titers of ROka were detected 7 days after inoculation of 3.6 × 103 cfu (Fig. 1B). Infectious virus was detected on days 14 and 21 after ROka66S inoculation, but the mean peak titer was 4.1 × 102 cfu, compared with 5 × 103 cfu in implants inoculated with ROka. On day 14, ROka yielded a range from 1.8 × 103 to 1.0 × 104 cfu/implant whereas ROka66S produced 3.8 × 102 to 8.0 × 102 cfu/implant in infected thymus/liver implants.
Flow cytometry analysis of three ROka-infected implants harvested on day 7 showed that 5.0 ± 0.7% (mean ± SE) of CD3+ cells expressed VZV proteins, which was consistent with the recovery of infectious virus from lymphocyte suspensions (Fig. 2). The percentage of VZV-positive T cells decreased on days 14 and 21 due to the depletion of target cells in the infected implants. Implants infected with ROka66S contained significantly fewer VZV-positive T cells, ranging from 0.7 to 2.1%, than the ROka-infected T cells on days 7 and 14 (Student’s t test, P ≤ 0.05). The percentage of VZV-positive T cells ranged from 0.2 to 0.9% in a total of 10 implants infected with ROka47S and tested over the 21-day experiment, which is equivalent to the background for this assay.
Figure 2.

Flow cytometry analysis of VZV-infected T cells. Flow cytometry analysis was performed on lymphocytes harvested on days 7, 14, and 21 from implants infected with either ROka (black bars), ROka66S (shaded bars) or ROka47S (white bars). The cells were treated with antibodies and fluorescent conjugates to CD3 and VZV proteins, and the percentages of VZV-positive T cells were measured. The bars represent the mean and SE of three implants.
To establish that the failure of ROka47S to replicate in T cells was due to the absence of the ORF47 protein kinase, ORF47 expression was restored by inserting the gene, including promotor sequences and a poly(A) site, into a noncoding region between ORF65 and ORF66. The genotype of this recombinant, ROka47SR, was confirmed by PCR and sequencing, demonstrating that the stop codons were retained in the native ORF47 gene and that the intact gene was present in the AvrII site in the short unique region. Immunoprecipitation with polyclonal rabbit antiserum against the ORF47 protein showed that it was expressed by ROka and ROka47SR, yielding bands of equivalent intensity from infected cell lysates, and was absent when cells were infected with ROka47S. Immunoprecipitation with a mAb directed against gE, the major glycoprotein of VZV, was used as a control for viral protein synthesis; no differences in gE expression by ROka, ROka47S, and ROka47SR strains were detected (data not shown). Replication of ROka47SR in melanoma cells had kinetics similar to ROka and ROka47S, indicating that the construction process and isolation of ROka47SR did not impair growth in tissue culture (Fig. 1A).
When thymus/liver implants were inoculated with 5.1 × 103 cfu of ROka47SR, the kinetics of replication paralleled the ROka strain, reaching a peak titer of 9.9 × 103 cfu on day 14 (Fig. 1B). Flow cytometry analysis of an implant infected with ROka47SR showed that 4.6% of the T cells expressed VZV proteins, which was similar to the numbers of VZV-positive T cells in ROka-infected implants (Fig. 2). The growth patterns of ROka and ROka47SR in T cells in these experiments were equivalent to those observed in our previous comparisons of V-Oka, its parent strain P-Oka, and other wild-type clinical isolates of VZV (28). The inability of ROka47S to infect thymus/liver implants and the recovery of a normal growth phenotype by using the ROka47SR strain, indicated that the ORF47 protein was required for replication in T cells. Viral DNA was extracted from T cells infected with ROka, ROka66S, and ROka47SR and analyzed by PCR and sequencing. The genotypes of all strains were consistent with the input virus and no reversion or contamination was detected (data not shown).
Analysis of Infected Thymus/Liver Implants by Histology and in Situ Hybridization.
Immunohistologic staining and in situ hybridization revealed extensive viral protein and viral DNA in tissue sections from implants infected with ROka, ROka66S, or ROka47SR at day 7 after infection (Fig. 3). The in situ hybridization signal for viral DNA was restricted to areas of the implants in which T cells remained intact; little viral DNA was detected in areas that showed the most severe necrosis histologically. At day 21, implants inoculated with ROka, ROka66S, or ROka47SR were involuted and viral DNA was detected in the least damaged areas. The histologic results paralleled the observation that viral replication in implants infected with ROka or ROka47SR peaked by day 14 whereas infectious virus titers after inoculation with ROka66S peaked later and remained lower. No VZV proteins or viral DNA were detectable in implants infected with ROka47S at 7, 14, or 21 days after inoculation.
Figure 3.
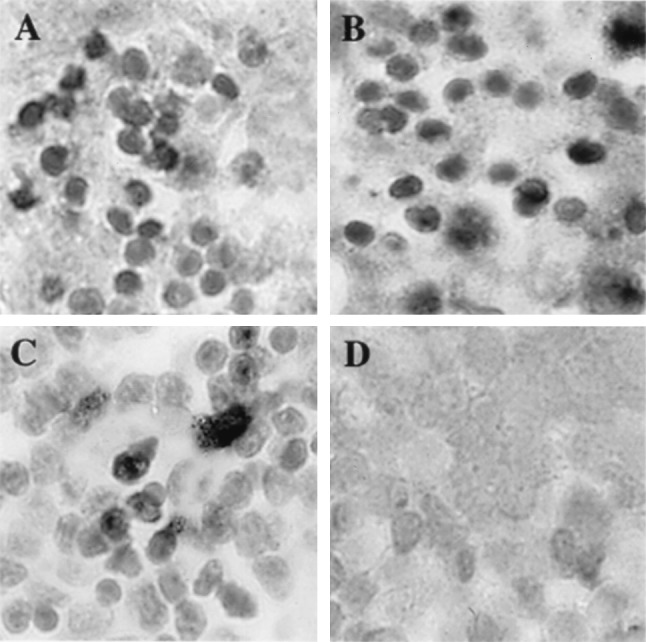
Histological analysis of VZV-infected thymus/liver implants. On day 7 after infection, thymus/liver implants infected with ROka (A), ROka47SR (B), ROka66S (C), or ROka47S (D) were fixed in paraformaldehyde, paraffin embedded, and cut into 3-μm sections before in situ hybridization was performed. A light hematoxylin counterstain was applied to reveal tissue histology. Darkly stained cells indicate VZV DNA in T cells. T cells containing VZV DNA were visible in all implants except those infected with ROka47S. (Magnification, ×786.)
Effects of the Functional Deletion of ORF47 and ORF66 on VZV Replication in Skin.
VZV DNA in the cosmids used to generate ROka and the ORF47 and ORF66 mutants was derived from V-Oka, the vaccine strain of Oka. In previous experiments, we found that V-Oka was attenuated for replication in human skin when compared with P-Oka or VZV-S, another low passage clinical isolate (29). Inoculation of skin implants with V-Oka yielded infectious virus in about half of the tissue specimens and maximal growth was not reached until 28 days. In the present experiments, the infectivity of ROka was similar to V-Oka, indicating that VZV made from V-Oka cosmids has the same phenotype of attenuation for skin as V-Oka. The ROka66S mutant showed no further attenuation in skin than ROka. Infectious virus was recovered from approximately half of the implants inoculated with ROka and ROka66S after 14 days. In contrast, no infectious virus was recovered from 24 implants inoculated with ROka47S in three separate experiments.
VZV protein synthesis in skin was assessed by Western blot on day 21 after inoculation. Viral protein synthesis was detected in 3 of 4 implants inoculated with ROka and in 3 of 4 implants infected with ROka66S; no viral protein was present in 4 of 4 specimens inoculated with ROka47S in three separate experiments. The specificity of the role of the ORF47 gene product for replication in skin was supported by the recovery of infectious virus from 3 of 4 implants inoculated with ROka47SR. Viral protein synthesis was observed in 3 of 4 implants and was equivalent to ROka by Western blot analysis (Fig. 4).
Figure 4.

VZV protein synthesis in skin implants. Implants were inoculated with ROka, ROka66S, ROka47SR, or ROka47S and harvested on day 21. Mock-infected skin was used as a negative control. Protein was extracted from the implants, separated by SDS/PAGE, and transferred to nylon membranes. Western blot analysis was performed; VZV proteins in the 90–105 kDa range were detected with a high-titer human polyclonal serum and enhanced chemiluminescence, and then the blots were exposed to x-ray film. No VZV proteins synthesis was detected in ROka47S-infected implants.
Analysis of Infected Skin Implants by Histology and in Situ Hybridization.
Infection of skin with ROka and ROka66S caused large necrotic lesions that resembled those produced by V-Oka (29). Viral DNA was detected by in situ hybridization in epithelial cells of the epidermis as well as in the glandular cells and fibroblasts of the dermis (Fig. 5). Implants infected with ROka47S showed no evidence of viral infection when stained for viral proteins or tested by in situ hybridization. Although these implants retained their normal cellular organization and no viral DNA was detectable, skin inoculated with ROka47SR showed viral DNA in many cells and necrotic changes that were similar to those caused by ROka.
Figure 5.
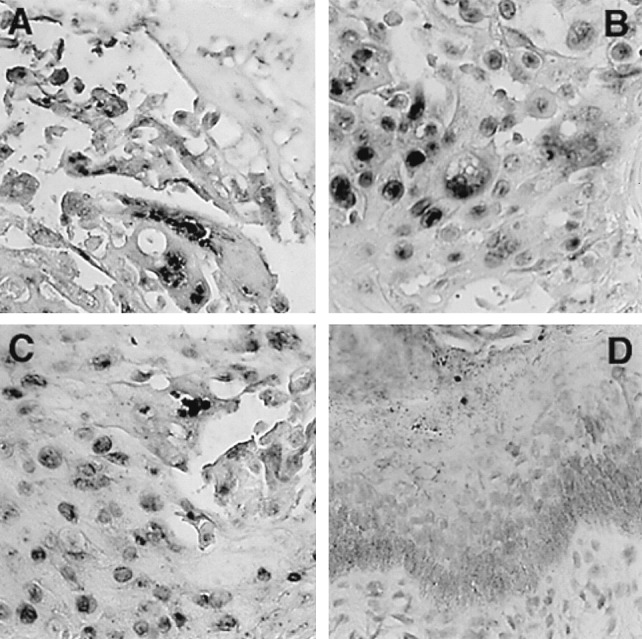
Histological analysis of VZV-infected skin implants. On day 21 after infection, skin implants infected with ROka (A), ROka47SR (B), ROka66S (C) or ROka47S (D) were fixed in paraformaldehyde, paraffin embedded, and cut into 3-μm sections before in situ hybridization was performed. A light hematoxylin counterstain was applied to reveal tissue histology. Darkly stained cells indicate VZV DNA in squamous epithelial cells. Normal skin structure is represented by ROka47S-infected implants where keratinocytes are visible in the epidermis and no VZV DNA was detected (D). (Magnification, ×314.)
DISCUSSION
These experiments in the SCID-hu model indicate that the ORF47 protein is required for VZV replication in human T cells as well as skin and that the ORF66 protein is necessary for normal growth kinetics in T cells. Since mutant VZV strains deficient in the ORF47 or ORF66 putative kinases have no identifiable phenotype in vitro (12, 20), the contributions of these genes to VZV pathogenesis could be documented only by investigating their infectivity using the SCID-hu mouse model. Based on these observations, the high degree of conservation of the viral-predicted kinases during the evolution of herpesviruses can be attributed to their importance for replication in differentiated cells within intact tissues of the natural host.
Our analysis of ROka47S and ROka47SR in the SCID-hu model strongly suggests that the ORF47 kinase is an essential determinant of virulence. The tropism of VZV for human CD4+ and CD8+ T cells exemplifies the targeting of cells of the immune system, which is characteristic of herpesviruses, many of which infect T cells, B cells, or monocytes during acute or persistent infection (34). T cell infection is critical for the viremic phase of VZV pathogenesis (35). This fundamental VZV tropism was blocked completely in the absence of ORF47 expression. The ROka47S mutant could not be detected in T cells analyzed by infectious focus assay, flow cytometry, or in situ hybridization whereas ROka47SR exhibited complete reversion to the wild-type phenotype for T cell replication. The recovery of T cell tropism after insertion of the ORF47 gene at the nonnative AvrII site in the short unique region strongly suggests that no unknown, confounding mutations were introduced during construction of the original ROka47S mutant from cosmids. Since ROka47S and ROka47SR were in separate cosmid reconstructions, there is a remote possibility that unidentified mutations account for their different phenotypes. Studies of VZV molecular pathogenesis are constrained by this need to use cosmid-generated recombinant viruses because of the cell-associated nature of VZV replication. However, the expression of the ORF47 gene from a nonnative site of the genome, the fact that our endpoint was the restoration rather than impairment of infectivity, and the observation that infectivity was restored in two types of differentiated cells, i.e., T cells and skin, constitute persuasive evidence that the ORF47 gene product provides an essential function. In contrast to ORF47 gene product, the ORF66 protein is not essential for T cell infection but it enhances VZV replication in these cells. The ROka66S mutant exhibited a slower rate of growth in T cells, with no recovery of virus until 14 days after inoculation of thymus/liver implants, and virus titers were low compared with ROka. VZV proteins were expressed by ≈2% of cells after 7 days, suggesting that production of infectious virions in T cells is delayed relative to viral protein synthesis in the absence of ORF66.
In addition to its role in T cell infectivity, these experiments demonstrate that ORF47 is absolutely required for VZV replication in cutaneous epithelial cells, which is necessary for transmission of the virus to other susceptible individuals. Viral DNA and protein synthesis were undetectable after inoculation of skin implants with ROka47S whereas infectivity for dermal and epidermal skin cells was restored fully by expression of ORF47 in ROka47SR. In contrast to ROka47S, the ROka66S mutant caused skin lesions that were indistinguishable in size and viral protein content from those produced by ROka infection. The analysis of the ROka66S mutants in the SCID-hu model revealed a differential effect of blocking ORF66 expression on infectivity for skin and T cells. The absence of ORF66 reduced T cell infectivity but did not alter skin tropism whereas our earlier studies showed that VZV mutants lacking glycoprotein C expression do not infect skin but retain their T cell tropism (29). These experiments with ORF66 and glycoprotein C mutants confirm the value of the SCID-hu system for identifying genes that constitute virulence factors for specific target cell types involved in different phases of VZV pathogenesis.
ORF66 is the homolog of HSV-1 US3. HSV-1 US3 acts to protect infected cells from undergoing apoptosis, whether triggered by the virus or by other inducers, and with effects that vary depending on the cell type (25). If ORF66 serves a similar function, then blocking ORF66 expression may reduce infectious virus yields when T cells that support VZV replication are eliminated more rapidly by apoptosis. Release of infectious virus also may be affected because US3 mutations in PRV cause a defect in virion egress from the nucleus of infected cells; virions accumulated in the perinuclear space, suggesting that phosphorylation of proteins by US3 was involved in budding of virus particles at the outer nuclear membrane (36).
Our observation that the ORF66 putative kinase supports optimal VZV replication in T cells is consistent with evidence that the homologous gene products of other α-herpesviruses are important in viral tropism for cells of the immune system. Mutation of HSV-2 US3 blocked replication in mouse peritoneal macrophages in vitro and in vivo (37, 38). This mutant was avirulent when given by i.p. inoculation, in which pathogenic effects depend on an initial phase of macrophage replication, but was only 10-fold less virulent than wild-type when given by ocular or intracerebral inoculation (37). US3 expression may be less critical for neurotropism because HSV-1 and HSV-2 US3 mutants were neurovirulent, although mortality was reduced, and retained the capacity to reactivate from latency (39, 40). In PRV experiments, the deletion of the US3 gene blocked infectivity for a porcine B cell line but did not alter its tropism for epithelial cells (41). In vivo, deleting the US3 kinase from PRV reduced its virulence substantially in the natural host and infection protected pigs from challenge with wild-type virus (41). Further investigation of the ORF66/US3 gene products as virulence determinants for cells of the immune system may yield a better understanding of their contributions to α-herpesvirus pathogenesis (20, 21, 37, 41, 42).
Our finding that ORF47 putative kinase was essential for VZV replication in T cells and skin was unexpected given the efficient replication of the ROka47S mutant in various cultured cells in vitro. The failure of ROka47S to infect human tissues in the SCID-hu model suggests that cultured cells produce a cellular protein or proteins that compensate for its absence and that these cellular products are not made in differentiated human T cells or skin. Known interactions between the ORF47 gene product and other VZV proteins suggest steps in VZV replication for which this putative kinase may be essential in vivo. The ORF47 gene product is located in the tegument and enters the cell with the virus. It phosphorylates IE62, the predominant tegument protein which is the major VZV-transactivating factor required for rapid induction of viral protein synthesis (3, 11, 43–45). Since IE62 can be phosphorylated by CKII, high concentrations of CKII in tissue culture cells may be needed to compensate for the absence of ORF47, even though some differentiated cells produce this kinase (11, 46). In contrast to its homologs in other α-herpesviruses, the VZV gE is an essential glycoprotein that is phosphorylated in infected cells and can be phosphorylated by CKII in vitro (32, 47, 48). The ORF47 protein may be necessary to phosphorylate gE, as has been shown for the HSV-1 homologs, gE and UL13. UL13 binds to gE and phosphorylates both gE and gI, which form the HSV-1 Fc receptor complex (19). In previous studies, the mobility of immunoprecipitated gE was unchanged in tissue culture cells infected with the ROka47S, ROka66S, or ROka47S/66S double mutants, suggesting that phosphorylation was mediated by a cellular kinase (20). ORF47 protein may have a gE phosphorylating function in differentiated cells that is not compensated by cellular kinases. Finally, ORF47 may interact directly with cellular proteins to induce conditions favorable for viral replication. Such effects could be essential in quiescent cell types that are typically found in differentiated tissues. The recent report describing the hyperphosphorylation of elongation factor 1δ by HSV-1 UL13 and the associated increase in the efficiency of protein synthesis in infected cells, provides evidence in support of this hypothesis (18).
Several features of the VZV-predicted kinases make them potential targets for new antiviral therapies and rational vaccine design. ORF66 is a good choice for deletion in an engineered vaccine because it is fully capable of growth when inoculated s.c. but is impaired for growth in T cells. This phenotype would allow the limited viral replication necessary to induce protective immunity to VZV infection but would be unlikely to result in viremia because the mutant virus would spread slowly, whether at all, to T cells (49). In contrast, an ORF47-deficient strain is not a good candidate for a vaccine because lack of viral growth in skin would hinder the induction of immunity. On the other hand, an antiviral agent that interfered with ORF47 function could block viral growth in T cells and thus prevent cell-associated viremia or terminate skin infection during primary VZV infection or reactivation. Acyclovir is currently used to treat varicella and zoster, but virulent TK− mutants that are resistant to acyclovir can arise after long-term antiviral therapy (50). ORF66 also could be an effective antiviral target because mutants grow slowly in T cells and blocking ORF66 kinase in vivo could help prevent the viral dissemination, which causes morbidity and mortality among high risk patients.
Acknowledgments
We thank Eu Meng Lam for technical assistance. J.F.M. was supported by a training grant from the Department of Pediatrics, Division of Developmental and Neonatal Biology (HD07249) and a National Research Service Award AI09195. The work was supported by a Public Health Service Grant AI36884 (to A.M.A.).
ABBREVIATIONS
- VZV
varicella-zoster virus
- cfu
colony-forming unit
- ICP
infected cell protein
- SCID-hu
severe combined immunodeficient-human
- IE
immediate early
- PRV
pseudorabies virus
- gE
glycoprotein E
References
- 1. McGeoch D J, Davison A J. Nucleic Acids Res. 1986;14:1765–1777. doi: 10.1093/nar/14.4.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith R F, Smith T F. J Virol. 1989;63:450–455. doi: 10.1128/jvi.63.1.450-455.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevenson D, Colman K L, Davison A J. J Gen Virol. 1994;75:317–326. doi: 10.1099/0022-1317-75-2-317. [DOI] [PubMed] [Google Scholar]
- 4.Albrecht J-C, Nicholas J, Biller D, Cameron K R, Biesinger B, Newman C, Wittmann S, Craxton M A, Coleman H, Gleckenstein B, et al. J Virol. 1992;66:5047–5058. doi: 10.1128/jvi.66.8.5047-5058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chee M S, Lawrence G L, Barrell B G. J Gen Virol. 1989;70:1151–1160. doi: 10.1099/0022-1317-70-5-1151. [DOI] [PubMed] [Google Scholar]
- 6.de Wind N, Domen J, Berns A. J Virol. 1992;66:5200–5209. doi: 10.1128/jvi.66.9.5200-5209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagesha H S, Crabb B S, Studdert M J. Arch Virol. 1993;128:143–154. doi: 10.1007/BF01309795. [DOI] [PubMed] [Google Scholar]
- 8.van Zijl M, van der Gulden H, de Wind N, Gielkens A, Berns A. J Gen Virol. 1990;71:1747–1755. doi: 10.1099/0022-1317-71-8-1747. [DOI] [PubMed] [Google Scholar]
- 9.Zhang G, Stevens R, Leader D P. J Gen Virol. 1990;71:1757–1765. doi: 10.1099/0022-1317-71-8-1757. [DOI] [PubMed] [Google Scholar]
- 10.Ng T I, Grose C. Virology. 1992;19:9–18. doi: 10.1016/0042-6822(92)90161-h. [DOI] [PubMed] [Google Scholar]
- 11.Ng T I, Keenan L, Kinchington P R, Grose C. J Virol. 1994;68:1350–1359. doi: 10.1128/jvi.68.3.1350-1359.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heineman T C, Cohen J I. J Virol. 1995;69:7367–7370. doi: 10.1128/jvi.69.11.7367-7370.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purves F C, Ogle W O, Roizman B. Proc Natl Acad Sci USA. 1993;90:6701–6705. doi: 10.1073/pnas.90.14.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Overton H, McMillan D, Hope L, Wong-Kai-In P. Virology. 1994;202:97–106. doi: 10.1006/viro.1994.1326. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham C, Davison A J, Dolan A, Frame M C, McGeoch D J, Meredith D M, Moss H W M, Orr A C. J Gen Virol. 1992;73:303–311. doi: 10.1099/0022-1317-73-2-303. [DOI] [PubMed] [Google Scholar]
- 16.Purves F C, Roizman B. Proc Natl Acad Sci USA. 1992;89:7310–7314. doi: 10.1073/pnas.89.16.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogle W O, Ng T I, Carter K L, Roizman B. Virology. 1997;235:406–413. doi: 10.1006/viro.1997.8710. [DOI] [PubMed] [Google Scholar]
- 18.Kawaguchi Y, Van Sant C, Roizman B. J Virol. 1998;72:1731–1736. doi: 10.1128/jvi.72.3.1731-1736.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng T I, Ogle W O, Roizman B. Virology. 1998;241:37–48. doi: 10.1006/viro.1997.8963. [DOI] [PubMed] [Google Scholar]
- 20.Heineman T C, Seidel K, Cohen J I. J Virol. 1996;70:7312–7317. doi: 10.1128/jvi.70.10.7312-7317.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purves F C, Longnecker R M, Leader D P, Roizman B. J Virol. 1987;61:2896–2901. doi: 10.1128/jvi.61.9.2896-2901.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purves F C, Spector D, Roizman B. J Virol. 1991;65:5757–5764. doi: 10.1128/jvi.65.11.5757-5764.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daikoku T, Yamashita Y, Tsurumi T, Nishiyama Y. Arch Virol. 1995;140:1637–1644. doi: 10.1007/BF01322537. [DOI] [PubMed] [Google Scholar]
- 24.Galvan V, Roizman B. Proc Natl Acad Sci USA. 1998;95:3931–3936. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leopardi R, Van Sant C, Roizman B. Proc Natl Acad Sci USA. 1997;94:7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leader D P. Pharmacol Ther. 1993;59:343–389. doi: 10.1016/0163-7258(93)90075-o. [DOI] [PubMed] [Google Scholar]
- 27.Daikoku T, Shibata S, Goshima F, Oshima S, Tsurumi T, Yamada G, Yamashita Y, Nishiyama Y. Virology. 1997;235:82–93. doi: 10.1006/viro.1997.8653. [DOI] [PubMed] [Google Scholar]
- 28.Moffat J F, Stein M D, Kaneshima H, Arvin A M. J Virol. 1995;69:5236–5242. doi: 10.1128/jvi.69.9.5236-5242.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moffat J F, Zerboni L, Kinchington P R, Grose C, Kaneshima H, Arvin A M. J Virol. 1998;72:965–974. doi: 10.1128/jvi.72.2.965-974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen J I, Seidel K E. Proc Natl Acad Sci USA. 1993;90:7376–7380. doi: 10.1073/pnas.90.15.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davison A J, Scott J E. J Gen Virol. 1986;67:1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- 32.Mallory S, Sommer M, Arvin A M. J Virol. 1997;71:8279–8288. doi: 10.1128/jvi.71.11.8279-8288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heineman T C, Cohen J I. J Virol. 1994;68:3317–3323. doi: 10.1128/jvi.68.5.3317-3323.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roizman B. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1996. pp. 2221–2230. [Google Scholar]
- 35.Arvin A M. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1996. pp. 2547–2585. [Google Scholar]
- 36.Wagenar F, Pol J M A, Peeters B, Gielkens A L J, de Wind N, Kiman T G. J Gen Virol. 1995;76:1851–1859. doi: 10.1099/0022-1317-76-7-1851. [DOI] [PubMed] [Google Scholar]
- 37.Nishiyama Y, Yamada Y, Kurachi R, Daikoku T. Virology. 1992;190:256–268. doi: 10.1016/0042-6822(92)91212-d. [DOI] [PubMed] [Google Scholar]
- 38.Kurachi R, Daikoku T, Tsurumi T, Maeno K, Nishiyama Y, Kurata T. Arch Virol. 1993;133:259–273. doi: 10.1007/BF01313767. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto M, Kurachi R, Morishima T, Kito J, Nishiyama Y. Microbiol Immunol. 1996;40:289–294. doi: 10.1111/j.1348-0421.1996.tb03348.x. [DOI] [PubMed] [Google Scholar]
- 40.Meignier B, Longnecker R, Mavromara-Nazos P, Sears A, Roizman B. Virology. 1988;162:251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 41.Kimman T G, de Wind N, de Bruin T, de Visser Y, Voermans J. Virology. 1994;205:511–518. doi: 10.1006/viro.1994.1672. [DOI] [PubMed] [Google Scholar]
- 42.Longnecker R, Roizman B. Science. 1987;236:573–576. doi: 10.1126/science.3033823. [DOI] [PubMed] [Google Scholar]
- 43.Inchauspe G, Nagpal S, Ostrove J M. Virology. 1989;173:700–709. doi: 10.1016/0042-6822(89)90583-7. [DOI] [PubMed] [Google Scholar]
- 44.Kinchington P R, Hougland J K, Arvin A M, Ruyechan W T, Hay J. J Virol. 1992;66:359–366. doi: 10.1128/jvi.66.1.359-366.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perera L P, Mosca J D, Ruyechan W T, Hayward G S, Straus S E, Hay J. J Virol. 1993;67:4474–4483. doi: 10.1128/jvi.67.8.4474-4483.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuazon P T, Traugh J A. In: Advances in Second Messenger and Phosphoprotein Research. Greengard P, Robison G A, editors. New York: Raven; 1991. pp. 123–164. [PubMed] [Google Scholar]
- 47.Litwin V, Jackson W, Grose C. J Virol. 1992;66:3643–3651. doi: 10.1128/jvi.66.6.3643-3651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao Z, Jackson W, Grose C. J Virol. 1993;67:4464–4473. doi: 10.1128/jvi.67.8.4464-4473.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arvin A M. J Infect Dis. 1992;166:S35–S41. doi: 10.1093/infdis/166.supplement_1.s35. [DOI] [PubMed] [Google Scholar]
- 50.Linnemann C J, Biron K, Hoppenjans W, Solinger A. AIDS. 1990;4:577–579. doi: 10.1097/00002030-199006000-00014. [DOI] [PubMed] [Google Scholar]