

Abstract
Cells from Fanconi anemia (FA) patients are hypersensitive to alkylating agents and radiation traditionally used as conditioning regimens for marrow cell transplantation, and patients experience serious toxicities. In order to reduce toxicities, we used progressively lower doses of cyclophosphamide (CY) for conditioning. Here, we report the results in 43 FA patients who received marrow transplantation from HLA-matched related donors (37 siblings and 6 other relatives). Conditioning consisted of 15 mg CY/kg/day for 4 days along with Mesna. Methotrexate and cyclosporine were given for graft versus host disease (GVHD) prophylaxis. Forty patients (93%) are alive with a median follow-up of 3.7 (range 0.6 to 7.9) years. One patient with primary graft failure was successfully re-transplanted. Three of four patients with late graft failures were re-transplanted, and two of those are alive; one died before a second marrow graft. Twelve patients including three with rejection had cytogenetic abnormalities in their marrow cells before transplantation. Acute grade II-III and chronic GVHD were seen in 17% and 28.5% of patients, respectively. These results confirm and extend our previous observations that conditioning with 60 mg CY/kg allows for sustained engraftment of HLA-matched related marrow grafts in most FA patients and is associated with low toxicity, low incidences of acute and chronic GVHD, and excellent long-term survival.
Introduction
Fanconi anemia (FA) is usually associated with chromosomal instability and skeletal malformations. In addition, patients with FA have a tendency to progress to marrow failure and are at risk of developing acute myeloid leukemia, myelodysplastic syndromes (MDS), and epithelial malignancies later in life [1]. Allogeneic marrow transplantation is currently the only treatment able to restore normal hematopoiesis and improve survival of these patients [2]. A factor limiting success has been that FA cells have a unique hypersensitivity to alkylating agents which makes patients susceptible to serious regimen-related toxicities. In order to reduce toxicities, we have explored progressively lower doses of cyclophosphamide (CY) as single conditioning agent for HLA-matched related marrow grafts [3-5]. We published our early experience in FA patients who received grafts after either 80 mg/kg or 60 mg/kg of CY alone, and the results demonstrated acceptable toxicity and a high engraftment rate. A low rate of graft versus host disease (GVHD) was seen in patients conditioned with 60 mg CY/kg [6]. Here, we update the early experience in 23 patients and report data on 20 additional patients given either genotypically HLA-identical (n=37) or phenotypically HLA-matched (n=6) related marrow grafts after conditioning with 60 mg/kg CY.
Patients and Methods
Forty-three consecutive patients with FA and marrow failure received HLA-matched related marrow grafts between July 1999 and October 2006. Patient characteristics are shown in Table 1. The median interval from diagnosis to transplantation was 25 (range 1-180) months. The median number of previous blood transfusions was 6 (range 0-151). Thirty-seven patients had genotypically HLA-identical sibling donors and six had phenotypically HLA-matched donors (grandfather in two, mother in two, uncle in one, and cousin in one). Among families of these six patients, consanguinity was observed in two. Complementation groups were determined in 16 patients, of whom 12 had group A, two had group C, one had group G and one had group F. The diagnosis of FA was based on clinical features and diepoxybutane (DEB)-induced chromosomal breakage. All donors had negative DEB tests for FA. All patients were in the severe or moderately severe aplastic phase of FA, and no patient had clear evidence of myelodysplastic syndrome or acute leukemia, although twelve patients had cytogenetic abnormalities in their marrow cells. Patients were transplanted according to FHCRC protocol 1228, approved by the Institutional Review Board (IRB) of the FHCRC (Seattle-USA) and Clinics Hospital in Curitiba (Brazil). All patients signed informed consents approved by the local IRBs.
Table 1.
Characteristics of Patients
| Characteristic | |
|---|---|
| No. of patients (%) | 43 |
| Patient age, years, median (range) | 9 (5 – 29) |
| Donor type, No. of patients (%) | |
| HLA-identical siblings | 37 (86) |
| HLA-phenotypical matched relative other than siblings | 6 (14) |
| Donor/recipient gender, No. of patients (%) | |
| female/female | 12 |
| female/male | 10 |
| male/female | 8 |
| male/male | 13 |
| Months from diagnosis of Fanconi anemia to transplant, median (range) | 25 (1-180) |
| Hematological parameters before transplant, median (range) | |
| Hemoglobin g/dl | 8.8 (5.9–11.4) |
| Platelets × 103 /μL | 27,000 (6,000–72,000) |
| Neutrophils × 103 /μL | 650 (102–2,714) |
| Cytogenetic findings in marrow cells (No. of patients) | |
| Not done/no evaluable metaphases | 11 |
| Normal cytogenetics | 20 |
| Abnormal cytogenetics | 12 |
| Numbers of blood product transfusions before transplant, median (range) | 6 (0-151) |
| Nucleated marrow cells infused × 108/kg patient weight, median (range) | 4.0 (1.7 – 7.3) |
Patients received 15 mg CY/kg/day, on days -4,-3,-2,-1 before HCT, along with Mesna. GVHD prophylaxis consisted of cyclosporine, 3 mg/kg/day, beginning on day -1 and a short course of methotrexate (MTX), 15 mg/m2 on day +1 and 10 mg/m2 on days +3,+6 and + 11. All patients received bone marrow grafts and were transplanted in single or double rooms with high efficiency particulate air (HEPA). Forty-two patients were transplanted in Curitiba and one patient in Seattle. In case of major ABO incompatibility, red blood cells were removed from the marrow graft by starch sedimentation. Patients seronegative for cytomegalovirus (CMV) were transfused with CMV-negative blood or leukodepleted blood products (Curitiba) or blood products from CMV-negative donors (Seattle). Acyclovir prophylaxis for herpes simplex virus and varicella-zoster virus, trimethropim-sulfamethoxazole for Pneumocystis carinii pneumonia prophylaxis, and fluconazole for prevention of yeast infections were used in all patients. Weekly antigenemia testing for monitoring of CMV reactivation and preemptive treatment with ganciclovir were done. The median number of nucleated marrow cells infused was 4 (range 1.7–7.3) × 108 cells/kg recipient body weight. Acute GVHD, grades II to IV was defined using published criteria, and all patients surviving >21 days after HCT were considered at risk [7]. Patients surviving >90 days were considered at risk for chronic GVHD [8]. Neutrophil engraftment was defined as a neutrophil count >500 × 103/μL for at least 3 consecutive days without growth factor support. Time to platelet engraftment was defined as the first of three consecutive days on which the platelet count were >20.000 × 103/μL without transfusions for seven days. Primary graft failure was defined as failure to achieve a neutrophil count >500 × 103/μl by day 42. Secondary graft failure was defined as initial engraftment (neutrophils >500 × 103/μl) and subsequent decline to a neutrophil count <500 ×103/μl. Organ toxicity was defined according to National Cancer Institute (NCI) common toxicity criteria, and mucositis according to published standards [6]. Marrow donor engraftment was documented by ABO typing in patients with red blood cell group differences, chromosomal analysis in sex-mismatched patient/donors pairs, and PCR for variable number tandem repeats polymorphisms. Survival curves were plotted using the method of Kaplan and Meier. Results were reported as of 05/11/2007.
Results
Conditioning-related toxicities and infections
Oral mucositis was noted among all patients (Table 2). The severity of mucositis was grade I in 5, II in 12, IIIa in 20, IIIb in 6, and IV in 1 patient(s). Thirty patients (69%) received all four doses of MTX, 12 patients received three doses because of mucositis and one received only two doses of MTX due to renal and hepatic failure. One patient developed grade II hemorrhagic cystitis.
Table 2.
Outcomes after Transplantation for Patients with Fanconi Anemia Conditioned with Cy
| Outcome | No of Pts (%) |
|---|---|
| Median years follow-up (range) | 3.7 (0.6–7.9) |
| Mucositits, grade | |
| I | 4 (10) |
| II | 13 (30) |
| III | 25 (58) |
| IV | 1 (2) |
| Graft failure, - primary | 1 (2) |
| - late | 4 (9) |
| Mortality | |
| Day 100 | 1 (2) |
| 1 year | 1 (2) |
| Acute GVHD, grade | |
| 0 | 34 (81%) |
| I | 1 (2%) |
| II | 6 (15%) |
| III- IV | 1 (2%) |
| Chronic GVHD | |
| Extensive | 10 (24) |
| Limited | 2 (5) |
| None | 29 (71) |
| Secondary malignancy | 1 (2) |
No gastrointestinal toxicities were seen. Five patients developed mild to moderate hypertension while on cyclosporine. Eight patients had positive antigenemia tests for CMV but none developed CMV disease. Two patients had varicella zoster virus reactivation 1.5 years after transplantation.
One early death occurred in a 23 year-old man who had received > 150 blood transfusions and many years of androgen treatment. He died on day +27 with multiorgan failure and fungal infection; he showed neurological complications, severe hepatic toxicity and renal failure requiring dialysis. At the time of death he had evidence of neutrophil engraftment but was platelet transfusion-dependent. The autopsy showed esophageal candidiasis, fibrotic pericarditis with > 500ml of pericardial effusion, massive liver hemosiderosis without veno-occlusive disease and pulmonary hemorrhage.
Engraftment
Initial engraftment occurred in 41 of 42 evaluable patients, while one patient had primary graft failure. Median times to neutrophil and platelet engraftment were 19 (range 10-42) days and 21(range 14-33) days, respectively. Overall, 37 patients had sustained engraftment of donor cells, while four patients showed late graft rejections. One of the five patients with graft failure had mild dysplastic features in the marrow before transplantation, three had chromosomal abnormalities in marrow cells including monosomy 7, inv (3), and dup (1), all had received preceding transfusions (median 11, range 6-101 units), three had grafts from siblings, one from a grandfather and one from a cousin, and their nucleated marrow cell doses ranged from 3.4 to 64 (median 5.1) × 108 cells/kg.
The patient without engraftment was a nine year-old male given a graft from his sister. He underwent (unsuccessful) second transplantation from the same donor 150 days after the first using conditioning with fludarabine (25 mg/m2/day for five days) and CY (15 mg/kg/day for four consecutive days). On day +240 after the first transplant he received a third graft from another HLA-identical sister after conditioning with busulfan, 4 mg/kg/day for two consecutive days, 200 cGy total body irradiation, and anti-thymocyte globulin (rabbit ATG, Sangstat - Genzyme), at a total dose of 4 mg/kg over 4 days. He had complications including CMV reactivation, seizures related to cyclosporine toxicity, distal tubular acidosis and extensive chronic GVHD involving oral mucosa, liver and lungs. Thirty-three months after the first transplant the patient is well engrafted, with normal blood counts and 95% donor chimerism. Chronic GVHD disease has been well controlled, and immunosuppression is gradually being tapered.
Among the patients with late graft failures, one 29-year-old female died on day 918 with pancytopenia (the donor had declined a second marrow harvest). Another patient received a second transplant from his grandfather on day 258 after the first HCT and is alive 6 years later with a white blood cell count of 2,960/μl, a neutrophil count of 1,500/μl, hemoglobin of 17.4g/dl, and platelets of 99,000/μl. Chimerism analysis showed 100% donor marrow cells. A third patient rejected a graft from her 12-year-old cousin at 17 months, and received a successful second graft from the same donor after fludarabine, 25 mg/m2/day for five consecutive days, and CY, 15 mg/kg/day, for four consecutive days. She is alive and well 25 months after the second HCT with engraftment and 90-95% donor chimerism. The fourth patient experienced graft failure on day +365; while she had normal cytogenetics before the first HCT, her marrow cells showed complex cytogenetic abnormalities before a second HCT from her 16-year-old sister, this time using peripheral blood stem cells, after CY, 20 mg/kg/day for four consecutive days. She had rapid hematopoietic recovery, experienced grade IV acute GVHD on day +54, had viral infections with CMV, herpes simplex and EBV, and died on day +75 after the second HCT.
The median proportion of donor cells among peripheral blood leucocytes in the surviving 40 patients was 95 (range: 80-100)%. Acute GVHD, grade II, was diagnosed in six of 42 evaluable patients after their first transplantation, and grade III was seen in one patient to a total of 17% (Table 2). Twelve patients developed chronic GVHD, which was limited in two and extensive in ten patients (one with quiescent, five with “de novo,” and four with progressive onset). One additional patient developed grade IV acute GVHD after the second transplant, and another, de novo chronic GVHD after the third transplant. Six of the thirteen patients with chronic GVHD are still receiving immunosuppressive therapy.
Survival
Forty of the 43 patients (93%) are alive, with a median follow-up of 3.7 (range 0.6 -7.9) years (Fig. 1). Two patients died as a consequence of graft rejection and one due to regimen related toxicity. All six patients who received grafts from phenotypically HLA-matched relatives are alive. At last contact, the median Karnofsky performance score was 100 (range 80–100)%; specifically, only four patients had scores of <100%.
Fig. 1.
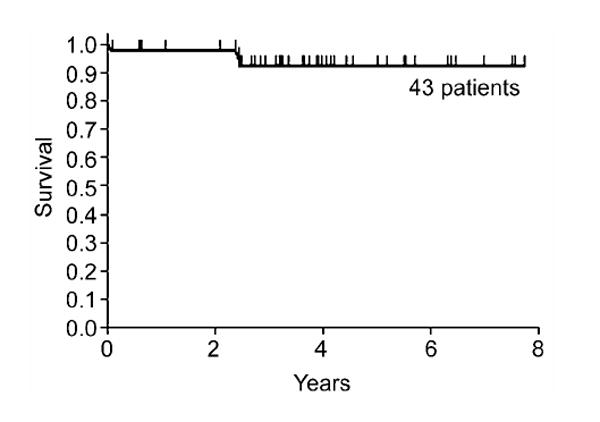
Probability of survival among 43 patients with Fanconi anemia conditioned with CY 60 mg/kg and given marrow grafts from related donors.
Discussion
Owing to its immunosuppressive properties [9-11], the fact that it is nonmyeloablative [11], and its lessened tendency to cause late malignancies compared to radiation-based regimens [12-14], CY has been a preferred conditioning regimen for patients with aplastic anemia and FA treated by marrow allografts. Initially, FA patients were conditioned with the typical aplastic anemia regimen of 200 mg CY/kg [15]; however, it was recognized soon that patients with FA experienced more serious CY-related toxicities than patients with other types of aplastic anemia [16,17], probably due to their poor ability of DNA repair after exposure to an alkylating agent. We, therefore, began a systematic study of CY dose reduction in FA patients undergoing marrow allografts [3-6].
The current and previous [6] results in 43 FA patients conditioned with 60 mg CY/kg showed that this dose was well tolerated with the major toxicity being oral mucositis (the combined result of CY and post-transplantation MTX), although only 1 patient had grade IV mucositis. Accordingly, day 100 and 1 year mortality rates were 2%, respectively. Results suggested that 60 mg CY/kg was at the lower limit of immunosuppression required for such allografts. While sustained donor engraftment was seen in 88% (37 of 42) of evaluable patients, 12% either failed to engraft (n=1) or experienced graft rejection after initial engraftment (n=4). The exact causes of graft failure/rejection in these five patients have remained unknown, but could include sensitization to non-HLA antigens by transfusions of blood products, as has been described for patients transplanted for aplastic anemia [15,18], the emergence of myelodysplastic syndromes in three of the patients with rejections, as suggested by chromosomal abnormalities in two patients, phenotypic and not genotypic HLA-identity in two of the patients, or inadequate CY-induced immunosuppression because of variations in CY metabolism in individual patients. Four of the 5 patients had successful re-transplantations, and 3 of them are surviving, while one died of acute GVHD. Perhaps patients with chromosomal changes characteristic of MDS, those with phenotypically HLA-matched donors, and those with a history of extensive transfusions might benefit from the addition of fludarabine to 60 mg CY/kg. Also, a better understanding of CY metabolism in FA patients is needed.
Others have used different conditioning regimens. Farzin et al. conditioned 30 patients with FA by combining CY, 20 mg/kg, and 400 cGy thoraco-abdominal irradiation before HLA-identical sibling grafts [19]. Five patients with chromosomal changes in their marrow and one with a 5/6 HLA-matched sibling were given more intensive conditioning with 20-40 mg CY/kg and 400-450 cGy total body irradiation. Additionally, all patients received horse ATG in the peri-transplantation period. Thirty-two patients were given marrow, and three were given cord blood (from siblings) grafts. Overall, two patients experienced graft failure, and 29 of the 35 are surviving. Bitan et al. used four different conditioning regimens, variably containing fludarabine, CY, antithymocyte globulin, alemtuzumab, and busulfan in preparation for HLA-matched grafts from family members (n=5) or unrelated donors (n=2) [20]. They encountered one graft failure, and reported that all seven patients were alive and well. Wagner et al. reviewed CIBMTR data on 98 FA patients given unrelated grafts after a multitude of conditioning regimens [21]. They reported 52% survival among patients given fludarabine-containing regimens, and 13% among those not receiving fludarabine. It was not clear from the report whether fludarabine beneficially influenced graft failure and engraftment rates. However, the authors state that day-100 platelet recovery was 74% in patients given fludarabine and 23% in those not receiving fludarabine.
Seven current patients developed grade II or III acute GVHD resulting in a cumulative incidence of 17%. The only patient who developed grade IV acute GVHD did so after the second transplant. This compared favorably to our previously reported incidences at higher Cy doses and those reported by others. Guardiola et al. [22] reported a 42% overall cumulative incidence of grades 2-4 acute GVHD, which rose to 62% in patients conditioned with 20 mg/kg CY and 5 Gy thoraco-abdominal irradiation. Ayas et al. [23] described 18% acute GVHD in patients given Cy, 60 mg/kg and antithymocyte globulin. Others found declines over time in the proportion of patients with severe acute GVHD [24]. The reasons for the very low incidence of acute GVHD in our series were not clear, but could include less regimen-related tissue damage which allowed delivering all 4 scheduled MTX doses in a majority of patients; MTX dosing has been shown previously to be important in controlling the incidence of GVHD [25]. Not surprisingly, the incidence of chronic GVHD among current patients was also low (28%), given that acute GVHD and older patient age were the most significant risk factors for the development of that complication [26]. Farzin et a. observed acute GVHD in 8 of their 35 patients, while Bitan et al. saw acute GVHD in 2 of 7 patients [19,20].
One patient with extensive chronic GVHD developed a squamous cell carcinoma of the tongue 5 years after transplant at the age of 12. The tumor was initially resected but at the time of this report the patient has persistent disease. Longer follow up is still needed to determine the incidence of malignancies in the surviving patients. Among our previously reported experience in 62 FA patients, who were conditioned with higher doses of Cy and followed for up to 22 years after HCT, there were 5 cases of squamous cell carcinoma of the tongue and one case of colon cancer occurring between 4.9 and 11.8 years after HCT [6]. Guardiola et al. [22] reported a 20% incidence of head and neck cancer in patients given Cy + TAI who were followed for 10 years and a 53% incidence in patients followed by 15 years [14,27]. Head and neck cancers have been described both in FA patients who did and did not receive HCT [1,14,27]. Farzin et al. observed two cases of squamous cell carcinoma which was fatal in one case, while the other case showed tumors both in tongue and anus; these two tumors have been successfully treated, though a new carcinoma of the skin has been diagnosed [19,20]. The CIBMTR publication [21] did not comment on late tumors.
In conclusion, 60 mg Cy/kg is a well-tolerated conditioning regimen with a sustained engraftment rate in patients with uncomplicated FA given genotypically HLA-identical HCT. Acute and chronic GVHD were relatively infrequent and overall survival excellent.
Acknowledgments
The authors are indebted to Miss Heliz R. Neves, the Brazilian data manager, for her excellent contributions and Helen Crawford for her assistance in preparing this manuscript. We thank Dr. Arlene Auerbach for performing complementation group studies in some of the patients and St. Jude Children’s Research Hospital for supporting the Fanconi Anemia Comprehensive Clinic in Curitiba (Brazil).
Supported in part by grants HL36444 and CA15704 from the National Institutes of Health (NIH), Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young NS, Alter BP. Aplastic Anemia, Acquired and Inherited. Vol. 275. WB Saunders Co.; 1994. Clinical features of Fanconi’s anemia; p. 309. [Google Scholar]
- 2.Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856–2862. [PubMed] [Google Scholar]
- 3.Flowers MED, Zanis J, Pasquini R, et al. Marrow transplantation for Fanconi anaemia: conditioning with reduced doses of cyclophosphamide without radiation. Br J Haematol. 1996;92:699–706. doi: 10.1046/j.1365-2141.1996.363898.x. [DOI] [PubMed] [Google Scholar]
- 4.Zanis-Neto J, Ribeiro RC, Medeiros C, et al. Bone marrow transplantation for patients with Fanconi anemia: a study of 24 cases from a single institution. Bone Marrow Transplant. 1995;15:293–298. [PubMed] [Google Scholar]
- 5.Medeiros C, Zanis-Neto J, Pasquini R. Bone marrow transplantation for patients with Fanconi anemia: reduced doses of cyclophosphamide without irradiation as conditioning. Bone Marrow Transplant. 1999;24:849–852. doi: 10.1038/sj.bmt.1701993. [DOI] [PubMed] [Google Scholar]
- 6.Zanis-Neto J, Flowers MED, Medeiros CR, et al. Low-dose cyclophosphamide conditioning for haematopoietic cell transplantation from HLA-matched related donors in patients with Fanconi anemia. Br J Haematol. 2005;130:99–106. doi: 10.1111/j.1365-2141.2005.05549.x. [DOI] [PubMed] [Google Scholar]
- 7.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18:295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Lee SJ, Vogelsang G, Flowers MED. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215–233. doi: 10.1053/bbmt.2003.50026. [DOI] [PubMed] [Google Scholar]
- 9.Santos GW, Owens AH., Jr Allogeneic marrow transplants in cyclophosphamide treated mice. Transplant Proc. 1969;1:44–46. [PubMed] [Google Scholar]
- 10.Storb R, Epstein RB, Rudolph RH, Thomas ED. Allogeneic canine bone marrow transplantation following cyclophosphamide. Transplantation. 1969;7:378–386. doi: 10.1097/00007890-196905000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Storb R, Buckner CD, Dillingham LA, Thomas ED. Cyclophosphamide regimens in rhesus monkeys with and without marrow infusion. Cancer Res. 1970;30:2195–2203. [PubMed] [Google Scholar]
- 12.Deeg HJ, Storb R, Prentice R, et al. Increased cancer risk in canine radiation chimeras. Blood. 1980;55:233–239. [PubMed] [Google Scholar]
- 13.Witherspoon RP, Storb R, Pepe M, Longton G, Sullivan KM. Cumulative incidence of secondary solid malignant tumors in aplastic anemia patients given marrow grafts after conditioning with chemotherapy alone (Letter) Blood. 1992;79:289–292. [PubMed] [Google Scholar]
- 14.Deeg HJ, Socié G, Schoch G, et al. Malignancies after marrow transplantation for aplastic anemia and Fanconi anemia: a joint Seattle and Paris analysis of results in 700 patients. Blood. 1996;87:386–392. [PubMed] [Google Scholar]
- 15.Storb R, Thomas ED, Buckner CD, et al. Allogeneic marrow grafting for treatment of aplastic anemia. Blood. 1974;43:157–180. [PubMed] [Google Scholar]
- 16.Gluckman E, Devergie A, Schaison G, et al. Bone marrow transplantation for Fanconi anaemia. Br J Haematol. 1980;45:557–564. doi: 10.1111/j.1365-2141.1980.tb07178.x. [DOI] [PubMed] [Google Scholar]
- 17.Deeg HJ, Storb R, Thomas ED, et al. Fanconi’s anemia treated by allogeneic marrow transplantation. Blood. 1983;61:954–959. [PubMed] [Google Scholar]
- 18.Storb R, Prentice RL, Thomas ED. Marrow transplantation for treatment of aplastic anemia. An analysis of factors associated with graft rejection. N Engl J Med. 1977;296:61–66. doi: 10.1056/NEJM197701132960201. [DOI] [PubMed] [Google Scholar]
- 19.Farzin A, Davies SM, Smith FO, et al. Matched sibling donor haematopoietic stem cell transplantation in Fanconi anaemia: an update of the Cincinnati Children’s experience. Br J Haematol. 2007;136:633–640. doi: 10.1111/j.1365-2141.2006.06460.x. [DOI] [PubMed] [Google Scholar]
- 20.Bitan M, Or R, Shapira MY, et al. Fludarabine-based reduced intensity conditioning for stem cell transplantation of Fanconi anemia patients from fully matched related and unrelated donors. Biol Blood Marrow Transplant. 2006;12:712–718. doi: 10.1016/j.bbmt.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Wagner JE, Eapen M, MacMillan ML, et al. Unrelated donor bone marrow transplantation for the treatment of Fanconi anemia. Blood. 2007;109:2256–2262. doi: 10.1182/blood-2006-07-036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guardiola P, Socie G, Li X, et al. Acute graft-versus-host disease in patients with Fanconi anemia or acquired aplastic anemia undergoing bone marrow transplantation from HLA-identical sibling donors: risk factors and influence on outcome. Blood. 2004;103:73–77. doi: 10.1182/blood-2003-06-2146. [DOI] [PubMed] [Google Scholar]
- 23.Ayas M, Al-Jefri A, Al-Mahr M, et al. Stem cell transplantation for patients with Fanconi anemia with low-dose cyclophosphamide and antithymocyte globulins withour the use of radiation therapy. Bone Marrow Transplant. 2005;35:463–466. doi: 10.1038/sj.bmt.1704787. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg PS, Alter BP, Socie G, Gluckman E. Secular trends in outcomes for Fanconi anemia patients who receive transplants: implications for future studies. Biol Blood Marrow Transplant. 2005;11:672–679. doi: 10.1016/j.bbmt.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Nash RA, Pepe MS, Storb R, et al. Acute graft-versus-host disease: analysis of risk factors after allogeneic marrow transplantation and prophylaxis with cyclosporine and methotrexate. Blood. 1992;80:1838–1845. [PubMed] [Google Scholar]
- 26.Storb R, Prentice RL, Sullivan KM, et al. Predictive factors in chronic graft-versus-host disease in patients with aplastic anemia treated by marrow transplantation from HLA-identical siblings. Ann Intern Med. 1983;98:461–466. doi: 10.7326/0003-4819-98-4-461. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg PS, Socie G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood. 2005;105:67–73. doi: 10.1182/blood-2004-04-1652. [DOI] [PubMed] [Google Scholar]