

Abstract
Excessive release of glutamate and the subsequent influx of calcium are associated with a number of neurological insults that result in neuronal death. The calcium-activated intracellular signaling pathways responsible for this excitotoxic injury are largely unknown. Here, we report that PD098059, a selective inhibitor of the calcium-activated p44/42 mitogen-activated protein kinase (MAP kinase) pathway, reduces neuronal death in a cell-culture model of seizure activity. Dissociated hippocampal neurons grown chronically in the presence of kynurenate, a broad spectrum glutamate-receptor antagonist, and elevated amounts of magnesium exhibit intense seizure-like activity after the removal of these blockers of excitatory synaptic transmission. A 30-min removal of the blockers produced extensive neuronal death within 24 h as assayed by the uptake of trypan blue and the release of lactate dehydrogenase. Phospho-p44/42 MAP kinase immunoreactivity after 30 min of seizure-like activity was present in many neuronal somata and dendrites as well as some synaptic terminals, consistent with both the presynaptic and postsynaptic effects of this pathway. The addition of PD098059 (40 μM; EC50 = 10 μM) during a 30-min washout of synaptic blockers inhibited the phosphorylation of p44/42 MAP kinase and reduced both the trypan-blue staining (n = 13) and the release of lactate dehydrogenase (n = 16) by 73% ± 18% and 75% ± 19% (mean ± SD), respectively. The observed neuroprotection could be caused by an effect of PD098059 on seizure-like events or on downstream signaling pathways activated by the seizure-like events. Either possibility suggests a heretofore unknown function for the p44/42 MAP kinase pathway in neurons.
Stimulation of numerous cell-surface receptors leads to the activation of kinase cascades that integrate and amplify extracellular signals and transmit them to intracellular targets (1). Mitogen-activated protein kinases (MAP kinases), a family of serine/threonine kinases, are activated by phosphorylation on threonine and tyrosine (2, 3). One subfamily of MAP kinases, known as extracellular signal-regulated kinase (ERK), is activated strongly by mitogens, including growth factors, leading to many cellular responses including proliferation and differentiation (4, 5). Two ERK isoforms, ERK1 (p44) and ERK2 (p42), are highly expressed in the brain; p42 is particularly enriched in the dendrites and somata of discrete neuronal populations including the hippocampus (4, 6). The function of p44/42 MAP kinase in postmitotic, terminally differentiated neurons is unclear, although recent reports implicate these kinases in synaptic plasticity (7, 8).
Stimulation of glutamate receptors and influx of Ca2+ are associated with excitotoxic injury (9, 10) and lead to the phosphorylation of p44/42 MAP kinase in neurons (11–16). Several neurological insults that produce an excessive release of glutamate and neuronal death, including hypoglycemia, ischemia, and kainate- or bicuculline-induced seizures, also result in the phosphorylation of p44/42 MAP kinase in vivo (17–21). Tyrosine kinase inhibitors that block the activation of p44/42 MAP kinase as well as other kinases prevent neuronal injury in the hippocampus after ischemia and kainate-induced seizures (22, 23). These results suggest that p44/42 MAP kinase may have a role in excitotoxic injury, but they do not provide direct evidence for such a role.
The activation of p44/42 MAP kinase requires phosphorylation on both threonine and tyrosine residues by the dual-specificity MAP kinase/ERK kinase (MEK1/2; refs. 24 and 25). At present, p44/42 MAP kinase is the only known substrate for MEK1/2 (3). A synthetic inhibitor of MEK1/2, PD098059 binds to the dephosphorylated form of MEK1/2, preventing its phosphorylation, its activation, and thus the subsequent activation of p44/42 MAP kinase (26). In vitro and in vivo studies have shown that PD098059 is highly specific, with no known effects on at least 18 other kinases, including the other MAP kinase subfamilies, c-Jun amino-terminal2-terminal kinase and p38 (26–28). Here, we have examined the effects of PD098059 on neuronal injury in a cell-culture model of seizure activity.
Dissociated hippocampal neurons are grown for 6–7 wks in medium containing kynurenate, a broad spectrum glutamate-receptor antagonist, and elevated amounts of magnesium, agents that primarily block excitatory synaptic transmission. With the removal of the synaptic blockers, neurons undergo robust seizure-like activity consisting of paroxysmal depolarization shifts and sustained depolarizations (29, 30). If these cultures are placed in a balanced salt solution lacking synaptic blockers for 30 min, extensive neuronal death can be measured 24 h after the readdition of kynurenate. Adding the Na+-channel blocker tetrodotoxin during the washout of synaptic blockers prevents both the seizure-like activity and the delayed neuronal death, indicating that injury is caused by the seizure-like events (unpublished data).
We report here that the addition of PD098059 during the washout of synaptic blockers prevented both the phosphorylation of p44/42 MAP kinase and the subsequent neuronal injury in this model. The localization of phospho-p44/42 MAP kinase immunoreactivity with light and electron microscopy revealed seizure-induced phosphorylation of p44/42 MAP kinase in presynaptic and postsynaptic structures, indicating possible roles for p44/42 MAP kinase in the release of transmitters and/or postsynaptic signaling leading to neuronal injury.
METHODS
Cultures.
Cultures were prepared as reported with minor modifications (29, 31). Briefly, hippocampal formations were dissected from 1- to 2-day-old Long–Evans rats, incubated for 40 min with papain (10 units/ml, Worthington) in dissecting medium (81 mM Na2SO4/27 mM K2SO4/15.7 mM MgCl2/0.23 mM CaCl2/1 mM kynurenate/1 mM Hepes, pH 7.4), and then treated for 15 min with trypsin inhibitor (10 mg/ml, type II-O, Sigma) in dissecting medium. After being transferred to growth medium, modified L15-CO2 with 32 mM glucose (described in ref. 29), the tissue was dissociated by trituration, and aliquots of cell suspension (approximately 1.6 × 105 cells per cm2) were plated onto glass coverslips coated with poly-D-lysine (0.1 mg/ml) and laminin (20 μg/ml) (Collaborative Research).
Cultures were maintained at 37°C in a humidified, 5% CO2 (balance air) incubator. The proliferation of nonneuronal cells was inhibited after 3–6 days by irradiation in a 60Co source (1500 rad at ≈10 rad/s). Cultures were then transferred to modified L15-CO2 medium containing 1 mM kynurenate and 11.3 mM MgCl2. The medium was changed every 5–7 days until experiments were performed 6–7 wk after plating.
Cell-Death Experiments.
Cultures were pretreated with PD098059 (New England Biolabs) or vehicle (dimethyl sulfoxide) for 30 min (in two experiments) or 1 h (in five experiments) by adding stock solutions directly to the growth medium. To induce seizure-like activity, cultures were transferred to Earle’s balanced salt solution (EBSS, Sigma) containing glucose (30 mM) and sodium bicarbonate (26 mM). The activity was stopped after 30 min by adding EBSS containing 5 mM kynurenate (Sigma). Control cultures (no seizure) were kept in 5 mM kynurenate throughout. For each exchange of EBSS, cultures were washed three times with 1.5 ml of solution at 37°C and then returned to the incubator. PD098059 (0–80 μM) was added 1 h before, during, and after the washout of the blockers. In another experiment, PD098059 (40 μM) was added either throughout the experiment, only during the washout, or only after the washout of the blockers. The concentration of dimethyl sulfoxide (≈0.16% vol/vol) was constant across conditions. Twenty-four hours after the termination of seizure activity, medium was removed and later assayed for LDH activity with a colorimetric assay (Sigma procedure no. 500). Cultures were then stained for 30 min in 0.4% trypan blue dissolved in EBSS + 5 mM kynurenate, washed in PBS, and fixed for 30 min in 4% paraformaldehyde and 0.01% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Cells were counterstained in 0.05% basic fuchsin, then dehydrated and cleared before mounting coverslips onto glass slides.
Immunohistochemistry.
Cultures were transferred to either EBSS, EBSS + 5 mM kynurenate, or EBSS + 40 μM PD098059 for 30 min. Then, cultures were fixed immediately in 0.4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 30 min at room temperature, washed in PBS, and incubated for 1 h in blocking buffer with 0.3% Triton X-100. Blocking buffer consisted of 10% normal goat serum, 0.5 M NaCl, and 0.1 M phosphate buffer and was used as diluent for both primary and secondary antibodies. Cells were incubated for 2 h at room temperature with a rabbit polyclonal antibody directed against phosphorylated (Tyr-204) p44/42 MAP kinase (1:100; New England Biolabs), and immunoreactivity was visualized with the three-step ABC technique with diaminobenzidine tetrachloride as substrate (Vectastain Elite, Vector Laboratories). All cultures were postfixed (1–2% glutaraldehyde), dehydrated, cleared, and mounted onto glass slides. Some cultures were counterstained in 0.05% basic fuchsin.
Electron Microscopy.
Cultures were transferred to EBSS for 30 min and then fixed and stained for phospho-p44/42 MAP kinase immunoreactivity as described above, with the following exceptions. After fixation, cultures were treated for 5 min in 0.02% Triton X-100 in PBS, washed with PBS, and then incubated for 1 h in a blocking buffer consisting of 10% normal goat serum in PBS. The reaction product was visualized by incubation with diaminobenzidine tetrachloride substrate (Vector Laboratories) for 10 min. Cultures were then postfixed in 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 30 min, rinsed in 0.1 M cacodylate buffer, and fixed for 1 h in 2% osmium tetroxide and 1.5% potassium ferrocyanide. Cultured cells were dehydrated in ethanol and rinsed in propylene oxide. The cell layer was infiltrated slowly with a mixture of EMbed812 and Araldite 6005 diluted with decreasing concentrations of propylene oxide and embedded in full-strength EMbed812 and Araldite 6005 (Electron Microscopy Sciences, Fort Washington, PA) at 60°C for 2 days. Ultrathin (pale gold) sections were picked up onto 200-mesh copper grids and viewed unstained in a JEOL 100CX transmission electron microscope.
Western Blotting.
Cultures were transferred to EBSS without synaptic blockers for 15–60 min and then lysed to assess p44/42 MAP kinase activation. At 0 min, cultures were washed three times with EBSS + 5 mM kynurenate and immediately lysed. Additional groups were transferred to EBSS + 5 mM kynurenate or 40 μM PD098059 for 30 min. Cells were lysed in ice-cold buffer containing 10 mM potassium phosphate, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM DTT, 0.5% Nonidet P-40, and 0.1% Brij 35 (Sigma). Lysates were then clarified at 14,000 g for 10 min. Protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad, 5000–006). To test for the phosphorylation of p44/42 MAP kinase, 30 μg of total-cell lysate was run on an SDS/10% polyacrylamide gel and transferred onto Immobilon-P membrane (Millipore), and a Western blot was performed with phospho-specific p44/42 MAP kinase antibodies (1:1000, New England Biolabs). Proteins were immunodetected with enhanced chemiluminescence (Amersham).
RESULTS
Delayed Neuronal Injury After Removal of Synaptic Blockers.
Cultures transferred to EBSS lacking glutamate-receptor blockers for 30 min followed by 24 h in EBSS + kynurenate showed extensive neuronal death when assayed by trypan-blue staining and by the release of LDH into the culture medium (Figs. 1a and 2b). Control cultures transferred to EBSS + 5 mM kynurenate throughout showed minimal trypan-blue staining and release of LDH (Figs. 1b and 2b). In four separate platings, 35–54% of neurons stained for trypan blue 24 h after a 30-min washout of blockers compared with 2–13% of neurons from control cultures. Adding tetrodotoxin during the 30-min washout of blockers also prevented the increase in the release of LDH at 24 h (data not shown).
Figure 1.
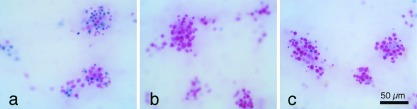
Trypan-blue staining, a marker of neuronal injury, 24 h after a 30-min washout of synaptic blockers. (a) Approximately half of the neurons stained for trypan blue after a 30-min washout of synaptic blockers. (b) Few neurons were stained for trypan blue in cultures that remained in a synaptic blocker (5 mM kynurenate) or (c) that were treated with 40 μM PD098059 during the washout of blockers. Neurons were counterstained with basic fuchsin (pink).
Figure 2.

Release of LDH, a marker of neuronal injury, 24 h after a 30-min washout of synaptic blockers. (a) PD098059, at the concentrations indicated, was added 1 h before, during, and 24 h after a 30-min washout of kynurenate. PD098059 reduced the release of LDH with an EC50 of ≈10 μM, close to the IC50 for MEK1 inhibition (mean ± SEM, three to seven cultures per dose). (b) Adding PD098059 (40 μM) only during the washout of synaptic blockers (PD During) prevented the release of LDH and was as effective as adding it before, during and after the washout (PD Before, During, & After). PD098059 did not block the release of LDH if it was added only after the washout of synaptic blockers (PD After). In this experiment, PD098059 was only slightly less effective than 5 mM kynurenate in preventing the release of LDH (mean ± SEM, three cultures per condition).
A p44/42 MAP-Kinase-Kinase Inhibitor, PD098059, Mitigates Neuronal Injury Induced by the Removal of Blockers.
In five separate platings, we added 40 μM PD098059 1 h before the washout, during the 30-min washout, and for 24 h after the reintroduction of kynurenate. In these experiments, PD098059 reduced the release of LDH induced by the washout of synaptic blockers by 75% ± 19% (mean ± SD; n = 16 cultures). Trypan-blue staining also was reduced by 73% ± 18% (n = 13 cultures; Fig. 1c). A dose–response curve was obtained in two experiments; PD098059 blocked the release of LDH with an EC50 of ≈10 μM (Fig. 2a). To determine when PD098059 exerted its protective effect, we added it either before, during, or after the washout of blockers (Fig. 2b). Adding PD098059 only during the 30-min washout provided as much protection as adding the drug before, during, and after the washout. Adding PD098059 only after the 30-min washout did not reduce the release of LDH.
p44/42 MAP Kinase Is Phosphorylated After the Removal of Blockers.
Cultures were fixed and stained for phospho-p44/42 MAP kinase immunoreactivity immediately after 30 min in EBSS (seizure) or after 30 min in EBSS + kynurenate (no seizure) (Figs. 3 and 4). More staining was observed in cultures that underwent seizure-like activity for 30 min than in those that did not. In one experiment, ≈60% of neurons stained after 30 min in EBSS compared with ≈13% of neurons after 30 min in EBSS + 5 mM kynurenate. Staining in “seizure” cultures was prominent in the processes, cytoplasm, and nuclei of many neurons. The staining of the processes was faint in “no seizure” cultures, although occasional staining of cell bodies, usually those of smaller neurons, was intense. Punctate, apparently intracellular staining was also evident in some neurons. Smaller puncta, which were observed on the surface of neurons, were more prominent in the seizure cultures. Not adding the primary antibody prevented staining.
Figure 3.

Phospho-p44/42 MAP kinase immunohistochemistry after 30 min in EBSS without synaptic blockers or after 30 min in EBSS + 5 mM kynurenate. (a and c) After the washout of blockers, staining was prominent in the processes, many somata, and some nuclei. (b and d) In cultures that remained in synaptic blockers, faint staining of the processes and rare staining of the cell bodies were observed. Punctate staining that appeared to be intracellular was observed in some neurons that remained in synaptic blockers (b). Smaller punctate staining that appeared to be on the surface of the neurons was more prominent after the washout of blockers and likely indicates phosphorylation of p44/42 MAP kinase in synaptic endings. Neurons were counterstained with basic fuchsin (pink).
Figure 4.

Effect of PD098059 on the phosphorylation of p44/42 MAP kinase after the removal of synaptic blockers. (a) In this plating, ≈60% of neurons were phospho-p44/42 MAP kinase-immunoreactive after 30 min in EBSS without synaptic blockers. (b) Sister cultures transferred to EBSS + 5 mM kynurenate showed less staining (≈13% of neurons). (c) Staining in cultures transferred to EBSS + 40 μM PD098059 was almost undetectable. Neurons were not counterstained. (d) Western blot showing the course of p44/42 MAP kinase phosphorylation over time after the removal of synaptic blockers and the inhibition by kynurenate and PD098059. The lengths of time that synaptic blockers were removed are: lane 1, 0 min; lane 2, 15 min; lane 3, 30 min; lane 4, 60 min; lane 5, 30 min (with 5 mM kynurenate); and lane 6, 30 min (with 40 μM PD098059). The phosphorylation at 0 min appears to be caused by the triplicate wash in EBSS + kynurenate immediately before cell lysis and assay (see text).
Western blotting was performed to confirm the phosphorylation of p44/42 MAP kinase by the washout of blockers and to assess the course of the effect over time (Fig. 4d). The growth medium was removed from six cultures (lanes 1–6) and replaced with EBSS with or without additional drugs (after two washes with the same solution). After different intervals, the cultures were lysed and assayed; the experiment was performed in triplicate. Washing the cultures appeared to cause transient phosphorylation of p44/42 MAP kinase (lane 1, culture assayed immediately after three washes with EBSS containing kynurenate; see refs. 32 and 33). This effect decayed over 30 min in kynurenate-containing EBSS (lane 5). However, in cultures that underwent seizure-like activity for 30 min in EBSS without kynurenate, p44/42 MAP kinase phosphorylation was increased (lane 3). The level of p44/42 MAP kinase phosphorylation was also higher after 15 min in EBSS (lane 2) than that at 0 min and remained high even after 60 min in EBSS (lane 4). Alternative explanations for the phosphorylation of p44/42 MAP kinase at 0 min (lane 1) include a high basal level in growth medium or activation by the removal of medium (e.g., withdrawal of serum). These explanations seem less likely, because the preincubation of the cultures for 60 min in EBSS + 5 mM kynurenate, followed by three washes in the same solution, still gave rise to high levels of phospho-p44/42 MAP kinase staining (data not shown). Also, cultures processed for phospho-p44/42 MAP kinase immunoreactivity directly out of growth medium showed little staining (data not shown). Nevertheless, it seems clear from the comparison of lanes 3 (EBSS for 30 min) and 5 (EBSS + kynurenate for 30 min) that the absence of kynurenate and, presumably, the resulting seizure-like activity greatly increased the phosphorylation of p44/42 MAP kinase.
PD098059 Prevents Phosphorylation of p44/42 MAP Kinase After the Removal of Blockers.
Phospho-p44/42 MAP kinase immunostaining was inhibited when 40 μM PD098059 was added 1 h before and during the 30-min washout of synaptic blockers (compare Fig. 4a and 4c). Cultures were fixed and stained immediately after the 30-min washout. Phospho-p44/42 MAP kinase immunostaining after treatment with 40 μM PD098059 was almost undetectable (Fig. 4c). The ability of PD098059 to block phosphorylation of p44/42 MAP kinase after the washout of blockers was confirmed with Western blot analysis (Fig. 4d, lane 6).
Phosphorylation of p44/42 MAP Kinase Occurs in Presynaptic and Postsynaptic Structures.
Small puncta were observed on the surface of cell bodies and processes in the immunohistochemical staining for phospho-p44/42 MAP kinase, particularly in cultures that had undergone seizures. To determine whether this staining was caused by the phosphorylation of p44/42 MAP kinase in terminal boutons, we examined some cultures with electron microscopy. Electron-dense labeling of some vesicle-filled terminals was observed (Fig. 5), in addition to the labeling of some cell bodies and dendrites (not shown).
Figure 5.

Electron micrograph of phospho-p44/42 MAP kinase immunoreactivity after 30 min in EBSS without synaptic blockers. Electron-dense label can be seen in a vesicle-filled terminal (t1) in close apposition to a small, unlabeled spine (s). The flattened disk-shaped structures in the spine are characteristic of the spine apparatus. Two other unlabeled terminals are visible (t2, t3). This section was not stained with uranyl acetate or lead citrate. (Bar = 0.5 μm.)
DISCUSSION
The main finding of our study is that the specific MEK1/2 inhibitor PD098059 sharply reduces both the phosphorylation of p44/42 MAP kinase and neuronal injury after the washout of synaptic blockers in a cell-culture model of seizure activity. PD098059 inhibited the release of LDH, a marker of neuronal injury, with an EC50 of 10 μM. MEK1 and MEK2 are inhibited by PD98059 with IC50 values of 10 and 50 μM, respectively (26–28), indicating that the inhibition of MEK1 was largely responsible for the neuroprotective effect of PD98059 in this model. PD098059 needed to be present only during the washout of synaptic blockers to obtain maximal neuroprotection. Adding it after the washout of synaptic blockers, when p44/42 MAP kinase and, presumably, MEK1/2 were phosphorylated already, did not protect neurons from delayed injury. This result is consistent with PD098059’s known mechanism of action (26).
Intracellular recordings from a large number of cultures in this model have shown that the removal of synaptic blockers results in seizure-like activity consisting of paroxysmal depolarization shifts and sustained depolarizations (refs. 29 and 30; unpublished data). Although the protective effect of PD098059 reported here suggests that the p44/42 MAP kinase pathway may have a role in seizure-induced neurodegeneration, we have not ruled out effects of this kinase cascade on the seizure activity itself. Thus, the addition of PD098059 during the washout of synaptic blockers may inhibit some aspects of the seizure-like activity, the activation of signaling pathways underlying seizure-induced neuronal death, or both. Preliminary electrophysiological observations indicate that PD098059 does not prevent the seizure-like activity, but in some cases PD098059 reduces the duration and/or amplitude of the seizure-like events (data not shown). It is not yet clear whether these reductions in seizure-like activity account for the robust neuroprotective effect of PD098059 in this model. One obvious question is whether the p44/42 MAP kinase pathway is involved in the regulation of the function of ion channels. Several ion channels are known to be regulated by phosphorylation (34), but we know of no reports suggesting that kinases in the p44/42 MAP kinase pathway have effects on channel currents in neurons. (However, see ref. 32 for effects of PD098059 on Cl− currents in astrocytes.) Another family of MAP kinases, the c-Jun amino-terminal kinases, has been implicated in kainate-induced seizures and neuronal damage. Higher doses of kainate are required to induce seizures in c-Jun amino-terminal kinase-3 knockout mice, and these mice show resistance to seizure-induced injury even when the severity of the seizure appears to be comparable to that induced in wild-type mice (35). Thus, several members of the MAP kinase family may contribute to seizures and seizure-induced injury.
Previous studies have localized p42 MAP kinase to the somata and dendrites of hippocampal neurons, in particular the CA3 pyramidal and dentate granule neurons (6). We report here phospho-p44/42 MAP kinase immunostaining in some terminal boutons as well as processes and somata in cultured hippocampal neurons after seizure-like activity. The specificity of the phospho-p44/42 antibody used in this study is suggested by the ability of PD098059 to block the immunohistochemical staining and the presence of only two bands (corresponding to p44 and p42) in the immunoblot (Fig. 4). The presynaptic localization reported here is not surprising given that synapsin I, a phospho-protein associated with synaptic vesicles, is phosphorylated by p44/42 MAP kinase in vitro and in intact neuronal preparations (36, 37) and that its phosphorylation appears to enhance neurotransmitter release (38). The activation of p44/42 MAP kinase in excitatory presynaptic terminals could therefore increase the release of glutamate, contributing to seizure activity and excitotoxic cell death. In fact, the phosphorylation of synapsin I is increased in the kindling model of seizures (39). These results suggest that the activation of p44/42 MAP kinase could lead to seizure-induced injury by both presynaptic and postsynaptic mechanisms.
A fraction of activated p44/42 MAP kinase translocates to the nucleus (40), leading to the phosphorylation of transcription factors including c-Myc, c-Fos, c-Jun, Elk-1 (2, 3), and CREB (41, 42). p44/42 MAP kinase also has cytosolic, cytoskeletal, and membrane-bound substrates that include cytoplasmic phospholipase A2, the microtubule-associated proteins MAP2 and tau, midsized and heavy molecular weight neurofilaments, synapsin I, and epidermal and nerve growth factor receptors (2, 3, 36, 37, 43). Kinases within the MAP kinase pathway are themselves substrates for p44/42 MAP kinase, including the upstream kinases c-Raf-1 and MEK1/2 as well as the downstream kinase, p90rsk (2, 3); p44/42 MAP kinase may therefore regulate gene expression, transmitter release, cytoskeletal proteins, and other intracellular signaling pathways. It remains to be determined whether some or all of the neuroprotective effect of PD098059 is caused by blocking the seizure-induced phosphorylation of any of these substrates.
MAP kinases have been implicated in both cell survival and cell death. The activation of p44/42 MAP kinase and/or the inhibition of the stress-activated protein kinase/c-Jun amino-terminal kinase families of MAP kinases have been shown to protect against cell death (44–49). Neurotrophins, which strongly activate p44/42 MAP kinase in many cells including hippocampal neurons (5, 50), are neuroprotective in several models of neuronal injury (51, 52). Thus arises the question of how to reconcile the neuroprotective effect of activating p44/42 MAP kinase reported in previous studies with the neuroprotective effect of inhibiting p44/42 MAP kinase reported here. The finding that neurotrophins can exacerbate neuronal death under some circumstances may be relevant to this question (53–55). Neurotrophins protect cultured cortical neurons against apoptotic death induced by serum withdrawal, but in the same system, neurotrophins exacerbate necrotic death induced by either glucose-oxygen deprivation or the brief application of N-methyl-d-aspartate (54). Similarly, p44/42 MAP kinase activation may protect against apoptosis but exacerbate necrosis. Based on the morphology of our damaged neurons, we suspect that neuronal death in our model is primarily necrotic. Our results, taken together with previous studies, suggest that the activation of the p44/42 MAP kinase pathway is necessary, but not sufficient, for necrotic death. It seems likely that the p44/42 MAP kinase pathway gates the activation of other cell-death signaling pathways.
Acknowledgments
We thank Dr. J. B. Little for providing access to a 60Co source and Drs. D. D. Potter and M. E. Greenberg for insightful comments. This work was supported by a fellowship from the Freudenberger Fund, Department of Neurobiology, Harvard Medical School (to B.M.) and grants from the American Heart Association (to A.A.), the Whitehall Foundation (to A.J.C.), and the National Institutes of Health (to A.J.C. and E.J.F.).
ABBREVIATIONS
- LDH
lactate dehydrogenase
- ERK
extracellular signal-regulated kinase
- MAP kinase
mitogen-activated protein kinase
- MEK1/2
MAP kinase/ERK kinase
References
- 1. Girault J-A. Neurochem Int. 1993;23:1–25. doi: 10.1016/0197-0186(93)90139-v. [DOI] [PubMed] [Google Scholar]
- 2.Davis R J. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 3.Seger R, Krebs E G. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 4.Boulton T G, Nye S H, Robbins D J, Ip N Y, Radziejewska E, Morgenbesser S D, DePinho R A, Panayotatos N, Cobb M H, Yancopoulos G D. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- 5.Segal R A, Greenberg M E. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 6.Fiore R S, Bayer V E, Pelech S L, Posada J, Cooper J A, Baraban J M. Neuroscience. 1993;55:463–472. doi: 10.1016/0306-4522(93)90516-i. [DOI] [PubMed] [Google Scholar]
- 7.Martin K C, Michael D, Rose J C, Barad M, Casadio A, Zhu H, Kandel E R. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 8.Bailey C H, Kaang B-K, Chen M, Martin K C, Lim C-S, Casadio A, Kandel E R. Neuron. 1997;18:913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 9.Choi D W. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- 10.Tymianski M, Charlton M P, Carlen P L, Tator C H. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bading H, Greenberg M E. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- 12.Fiore R S, Murphy T H, Sanghera J S, Pelech S L, Baraban J M. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 13.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Durkin J P. J Biol Chem. 1995;270:22783–22787. doi: 10.1074/jbc.270.39.22783. [DOI] [PubMed] [Google Scholar]
- 15.Xia Z, Dudek H, Miranti C K, Greenberg M E. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen L B, Ginty D D, Weber M J, Greenberg M E. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 17.Gass P, Kiessling M, Bading H. Neurosci Lett. 1993;162:39–42. doi: 10.1016/0304-3940(93)90554-x. [DOI] [PubMed] [Google Scholar]
- 18.Kim Y S, Hong K S, Seong Y-S, Park J-B, Kuroda S, Kishi K, Kaibuchi K, Takai Y. Biochem Biophys Res Commun. 1994;202:1163–1168. doi: 10.1006/bbrc.1994.2050. [DOI] [PubMed] [Google Scholar]
- 19.Campos-Gonzalez R, Kindy M. J Neurochem. 1992;59:1955–1958. doi: 10.1111/j.1471-4159.1992.tb11032.x. [DOI] [PubMed] [Google Scholar]
- 20.Hu B R, Wieloch T. J Neurochem. 1994;62:1357–1367. doi: 10.1046/j.1471-4159.1994.62041357.x. [DOI] [PubMed] [Google Scholar]
- 21.Kurihara J, Hu B-R, Wieloch T. J Neurochem. 1994;63:2346–2348. doi: 10.1046/j.1471-4159.1994.63062346.x. [DOI] [PubMed] [Google Scholar]
- 22.Kindy M S. J Cereb Blood Flow Metab. 1993;13:372–377. doi: 10.1038/jcbfm.1993.50. [DOI] [PubMed] [Google Scholar]
- 23.Ohtsuki T, Matsumoto M, Kitagawa K, Mabuchi T, Mandai K, Matsushita K, Kuwabara K, Tagaya M, Ogawa S, Ueda H, Kamada T, Yanagihara T. Am J Physiol. 1996;271:C1085–C1097. doi: 10.1152/ajpcell.1996.271.4.C1085. [DOI] [PubMed] [Google Scholar]
- 24.Anderson N G, Maller J L, Tonks N K, Sturgill T W. Nature (London) 1990;343:651–653. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 25.Alessandrini A, Crews C M, Erikson R L. Proc Natl Acad Sci USA. 1992;89:8200–8204. doi: 10.1073/pnas.89.17.8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 28.Pang L, Sawada T, Decker S J, Saltiel A R. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 29.Furshpan E J, Potter D D. Neuron. 1989;3:199–207. doi: 10.1016/0896-6273(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 30.Koroshetz W J, Furshpan E J. Neurosci Res Suppl. 1990;13:S65–S74. doi: 10.1016/0921-8696(90)90033-y. [DOI] [PubMed] [Google Scholar]
- 31.Furshpan E J, Landis S C, Matsumoto S G, Potter D D. J Neurosci. 1986;6:1061–1079. doi: 10.1523/JNEUROSCI.06-04-01061.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crépel V, Panenka W, Kelly M E, MacVicar B A. J Neurosci. 1998;18:1196–1206. doi: 10.1523/JNEUROSCI.18-04-01196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schliess F, Sinning R, Fischer R, Schmalenbach C, Häussinger D. Biochem J. 1996;320:167–171. doi: 10.1042/bj3200167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levitan I B. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 35.Yang D D, Kuan C-Y, Whitmarsh A J, Rincón M, Zheng T S, Davis R J, Rakic P, Flavell R A. Nature (London) 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 36.Jovanovic J N, Benfenati F, Siow Y L, Sihra T S, Sanghera J S, Pelech S L, Greengard P, Czernik A J. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- 38.Greengard P, Valtorta F, Czernik A J, Benfenati F. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 39.Yamagata Y, Obata K, Greengard P, Czernik A J. Neuroscience. 1995;64:1–4. doi: 10.1016/0306-4522(94)00492-n. [DOI] [PubMed] [Google Scholar]
- 40.Chen R-H, Sarnecki C, Blenis J. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ginty D D, Bonni A, Greenberg M E. Cell. 1994;77:713–725. doi: 10.1016/0092-8674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 42.Xing J, Ginty D D, Greenberg M E. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 43.Veeranna, Amin N D, Ahn N G, Jaffe H, Winters C A, Grant P, Pant H C. J Neurosci. 1998;18:4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 45.Verheij M, Bose R, Lin X H, Yao B, Jarvis W D, Grant S, Birrer M J, Szabo E, Zon L I, Kyriakis J M, et al. Nature (London) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 46.Yoon S O, Casaccia-Bonnefil P, Carter B, Chao M V. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maroney A C, Glicksman M A, Basma A N, Walton K M, Knight E, Jr, Murphy C A, Bartlett B A, Finn J P, Angeles T, Matsuda Y, et al. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan C Y I, Greene L A. J Neurosci. 1998;18:4042–4049. doi: 10.1523/JNEUROSCI.18-11-04042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kummer J L, Rao P K, Heidenreich K A. J Biol Chem. 1997;272:20490–20494. doi: 10.1074/jbc.272.33.20490. [DOI] [PubMed] [Google Scholar]
- 50.Marsh H N, Scholz W K, Lamballe F, Klein R, Nanduri V, Barbacid M, Palfrey H C. J Neurosci. 1993;13:4281–4292. doi: 10.1523/JNEUROSCI.13-10-04281.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mattson M P, Scheff S W. J Neurotrauma. 1994;11:3–33. doi: 10.1089/neu.1994.11.3. [DOI] [PubMed] [Google Scholar]
- 52.Lindvall O, Kokaia Z, Bengzon J, Elmér E, Kokaia M. Trends Neurosci. 1994;17:490–496. doi: 10.1016/0166-2236(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 53.Yankner B A, Caceres A, Duffy L K. Proc Natl Acad Sci USA. 1990;87:9020–9023. doi: 10.1073/pnas.87.22.9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koh J-Y, Gwag B J, Lobner D, Choi D W. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 55.Samdani A F, Newcamp C, Resink A, Facchinetti F, Hoffman B E, Dawson V L, Dawson T M. J Neurosci. 1997;17:4633–4641. doi: 10.1523/JNEUROSCI.17-12-04633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]