

Abstract
Over 130 mutations to copper, zinc superoxide dismutase (SOD) are implicated in the selective death of motor neurons found in 25% of familial amyotrophic lateral sclerosis (ALS) patients. Despite their widespread distribution, ALS mutations appear positioned to cause structural and misfolding defects. Such defects decrease SOD’s affinity for zinc, and loss of zinc from SOD is sufficient to induce apoptosis in motor neurons in vitro. To examine the importance of the zinc site in the structure and pathogenesis of human SOD, we determined the 2.0 Å resolution crystal structure of a designed zinc-deficient human SOD, in which two zinc-binding ligands have been mutated to hydrogen-bonding serine residues. This structure revealed a 9° twist of the subunits, which opens the SOD dimer interface and represents the largest inter-subunit rotational shift observed for a human SOD variant. Furthermore, the electrostatic loop and zinc-binding sub-loop were partly disordered, the catalytically important Arg143 was rotated away from the active site, and the normally rigid intramolecular Cys57-Cys146 disulfide bridge assumed two conformations. Together, these changes allow small molecules greater access to the catalytic copper, consistent with the observed increased redox activity of zinc-deficient SOD. Moreover, the dimer interface is weakened and the Cys57-Cys146 disulfide is more labile, as demonstrated by the increased aggregation of zinc-deficient SOD in the presence of a thiol reductant. However, equimolar Cu,Zn SOD rapidly forms heterodimers with zinc-deficient SOD (t1/2 ≈ 15 min) and prevents aggregation. The stabilization of zinc-deficient SOD as a heterodimer with Cu,Zn SOD may thus be a contributing factor to the dominant inheritance of ALS mutations. These results have general implications for the importance of framework stability on normal metalloenzyme function and specific implications for the role of zinc ion in the fatal neuropathology associated with SOD mutations.
Keywords: Amyotrophic lateral sclerosis, Cu, Zn superoxide dismutase, Lou Gehrig’s disease, zinc-deficient superoxide dismutase, crystal structure
Introduction
A major focus of neurodegeneration research involves the characterization of how the superoxide-scavenging enzyme Cu, Zn superoxide dismutase (SOD) is involved in amyotrophic lateral sclerosis (ALS). ALS is an adult onset disease involving the progressive death of lower motor neurons in the spinal cord and upper motor neurons in the brain stem and cortex.1 In 1993, the first clue implicating a molecular cause for ALS came when mutations to the SOD gene were identified in approximately 2–3% of ALS patients 2; 3. Since then, more than 130 different missense mutations as well as several C-terminal truncation mutations to the SOD gene have been linked to ALS (www.alsod.org).
The discovery of a relationship between SOD mutations and ALS spurred intensive structural and other biophysical analyses aimed at revealing the molecular basis for the disease. Currently over 70 Cu,Zn SOD structures have been solved from various sources, including over 23 variants of human SOD4. Human SOD is a tightly associated and unusually stable homodimer of 153 residues per subunit with each chain folding into an eight-stranded Greek key β-barrel (see Figure. 1(a)). The active site channel on each subunit is formed on the outside of the β-barrels by two long loops (known as loop IV and loop VII)5; 6. Loop IV contributes His 63, the bridging ligand between the copper and zinc sites, and the other three zinc ligands5; 6; 7. Loop IV can be divided into a dimer interface sub-loop, a disulfide sub-loop and a zinc-binding sub-loop13 (Figure 1(a)). The dimer interface sub-loop forms 38% of the contact area that builds the dimer (Figure 1(b)). Loop VII, also known as the electrostatic loop, helps attract the anionic superoxide substrate into the active site7; 8. Loop VII also contributes Arg143, whose side-chain guanidinium group provides hydrogen bonds to anchor the main chain of Loop IV 6 and orient the bound substrate 9.
Figure 1.

Key structural features of wild-type Cu,Zn SOD (PDB code 1HL5). (a) In the right hand subunit (chain A) copper (orange) and zinc (grey) are shown as spheres. Loop IV is divided into three sections; the dimerization sub-loop (residues 50-54, yellow) creates part of the dimer interface, the disulfide sub-loop (residues 55-61, raspberry) covalently attaches to the β-barrel via the Cys57-Cys146 disulfide and the zinc-binding region (residues 62-83, blue) contains His63, His71, His80 and Asp83 (shown as sticks). Copper is coordinated by residues His63, His46, His48, and His120 (orange sticks). (b) Surface buried upon dimerization is shown for chain A. The ~1400 Å2 area is divided into three regions; the area contributed by the dimerization sub-loop (yellow), the area buried by the interaction with the dimerization sub-loop of chain B (cyan with yellow ribbon showing chain B segment) and th area from the β-barrel interaction (light grey). The dimerization sub-loop is involved in ~ 75% of the total surface area buried by the dimer.
Despite a wealth of structural and biochemical knowledge, the mechanism by which SOD mutations promote ALS remains controversial. The mutations associated with ALS occur in all of the functional elements of the protein structure, including the active site, dimer interface and β-barrel and many weaken the stability of SOD.3; 10; 11; 12; 13; 14 The available X-ray structures of seven ALS-causing SOD mutants (A4V [1N1915,1UXM10], G37R [1AZV16], H43R [1PTZ11], H46R [1OZT17, 1OEZ17], I113T [1UXL10], D125H [1P1V18], S134D [134N17]) show some local distortions at the mutation sites, but are otherwise remarkably similar to the wild type Cu,Zn SOD structure, conserving the positions of the metals, the disulfide bridge, and the active site residue Arg143. However, the autosomal dominant inheritance and the development of motor neuron disease in transgenic animals over-expressing ALS SOD mutants provides strong evidence that these mutations somehow confer a toxic gain-of-function.2; 19 Yet, structural and biochemical evidence also support the original proposal that ALS mutations are likely to cause structural defects in the SOD dimer3. Besides possible destabilizing effects of mutations, the metal ion binding and disulfide status are known to profoundly alter SOD stability suggesting that these features merit significant attention. The loss of copper and zinc from SOD facilitates the reduction of the intra-subunit disulfide bond between Cys57 of the zinc-loop and Cys146 of the β-barrel. Reduction of the disulfide favors dissociation of Cu,Zn SOD into monomers, which greatly increases their propensity to form insoluble aggregates20; 21; 22. Recently, studies of SOD in which the disulfide has been removed, either by reduction or by mutation, have shown that metal loss and disulfide reduction are thermodynamically linked, with disulfide formation contributing about 1.6 kcal.mol−1 to dimer stability.20; 23 Structural studies of disulfide-free SOD in both holo (copper and zinc bound) or apo (metal-deficient) forms23; 24 indicate that the disulfide stabilizes the dimer by ordering the dimer interface sub-region of loop IV (Figure 1(a)).
SOD-containing aggregates have been found in motor neurons and astrocytes expressing ALS mutant SODs, leading to the hypothesis that aggregation confers the proposed toxic gain-of-function25; 26. Aggregation has been variously postulated to confer toxicity through disrupting mitochondrial function, interfering with chaperones,14 or by inducing dysfunction of the ubiquitin-proteasome system.12; 13; 27 Misfolded SODs may also preferentially associate with other proteins and even with RNA.28 ALS mutant proteins have been shown to have increased hydrophobic character29; 30 and associate with the anti-apoptotic protein bcl-2 in a hydrophobic manner.31 However, a major shortcoming of the aggregation hypothesis is that little evidence directly demonstrates that either SOD aggregates or apoSOD are directly toxic to motor neurons.32; 33 A long standing enigma is why the A4V mutation, which causes the most aggressive form of ALS in patients, does not produce disease when overexpressed in transgenic mice despite the presence of aggregated A4V protein in motor neurons.19 However, Deng et al.34 have recently shown that co-expression of A4V with wild-type human SOD does cause motor neuron disease in mice, suggesting a role for human SOD heterodimers in ALS.
Many SOD ALS mutants, including the A4V SOD protein, have superoxide scavenging activity comparable to that of wild-type SOD.3; 35; 36 However, ALS mutations make the SOD mutants more susceptible to the loss of their metal cofactors.35 Zinc is more likely to disassociate than copper, because SOD has approximately a seven thousand fold lower affinity for zinc than it does for copper.35 Loss of zinc impacts the redox properties of the copper, in part through their bridging His63 ligand. Zinc-deficiency dramatically increases the reduction of the active site copper by low molecular weight reductants such as ascorbate.32 Delivery of zinc-deficient SOD intracellularly into cultured motor neurons by liposomes is sufficient to induce apoptosis, associated with increased redox activity of the remaining copper.32 Furthermore, zinc-deficient wild-type SOD was shown to be just as toxic to cultured motor neurons as zinc-deficient mutant SOD.32 Thus, the loss of zinc from wild-type SOD could be involved in the 98% of ALS patients without SOD mutations. In the zinc-deficient Cu/Zn-SOD hypothesis for ALS, the mutations to SOD do not directly cause the toxic gain-in-function, but rather increase the propensity of SOD to become zinc-deficient.
The loss of zinc from SOD is potentially important for both the aggregation and zinc-deficient Cu/Zn-SOD hypotheses. Currently, structures in the context of wild-type or mutant SOD are available for apoSOD,17; 37 Zn,Zn SOD,17; 18; 38 E,Zn SOD (i.e. copper-deficient, zinc-containing SOD),17 and for an engineered obligatory monomeric Cu,Zn SOD.39 To characterize the effects of losing zinc alone, we have demetallated the zinc-binding pocket by generating mutations to create constitutively zinc-deficient SOD proteins. Here, we have solved the X-ray crystal structure of this zinc-deficient SOD protein and characterized how the loss of zinc affects dimer stability and aggregation, with general implications for understanding the role of SOD mutations in neurodegenerative disease.
Results
Crystal structure of zinc-deficient SOD
To examine the importance of the zinc site in the structure and pathogenesis of human SOD, we determined the 2.0 Å resolution crystal structure of a designed zinc-deficient human SOD, in which two zinc-binding ligands (His80 and Asp 83) have been mutated to hydrogen-bonding serine residues. These mutations not only produce a constitutively zinc-deficient SOD, but also prevent the migration of the active-site copper or trace metal contaminants into the zinc site during the expression, purification, and crystallization processes. The zinc site mutations were made in the active thermostable human SOD double mutant, which lacks free thiols (C6A, C111S)6 (see Materials and Methods). This background allows examination of the role of the zinc site without the complicating variability of the free thiols, which are known to be reactive in vitro. The resulting constitutively zinc-deficient SOD mutant H80S/D83S/C6A/C111S was used to grow crystals in a form distinct from any previously published SOD crystals(space group C2 with a=107.2 Å, b=35.9 Å, c=68.30 and β=104.8°), suggesting the possibility of an underlying structural change. The asymmetric unit contained one SOD homodimer. The structure was solved by molecular replacement and refined to 2.0 Å resolution with final R/Rfree factors of 0.19/0.25 (Table 1) with well-ordered parts of the structure having an expected coordinate accuracy of ~0.3 Å. The electron density map confirmed the presence of all four mutated side chains. Compared to either the wild-type or thermostable C111S/C6A Cu,Zn SOD, the basic fold of zinc-deficient SOD remained unchanged; the main chain conformations for the 123 residues principally forming the β-barrel core of SOD were well defined. These residues were generally similar in chains A and B (Cα rmsd=0.38 Å) and structural descriptions refer to both chains unless explicitly noted.
Table I.
Data collection and refinement statistics
| Data collection | |
| Resolution limits ( ) | 20-2.0 (2.07-2.0) |
| Unique observations | 15899 |
| Rsym | 0.061 (0.267) |
| Completeness (%) | 95.8 (94.5) |
| Refinement | |
| No. of protein molecules | 2 |
| No. of amino acid residues | 268 |
| No. of water sites | 190 |
| Average B-factor ( 2) | 36 |
| RWork | 0.187 |
| RFree (5% of data) | 0.246 |
| r.m.s.d. bond lengths ( ) | 0.016 |
| r.m.s.d. bond angles (□) | 1.84 |
Numbers in parentheses correspond to values in the highest resolution shell
Consistent with the H80S/D83S mutations causing zinc-deficiency, electron density for zinc was absent in the zinc-binding site, but density for copper was clearly defined in the copper-binding site. Surprisingly, the coordination of copper remained quite similar to that found in wild-type Cu,Zn SOD. Even His63, which is the bridging ligand for both copper and zinc in oxidized (Cu2+) wild-type SOD, remained in a nearly native position in the zinc-deficient structure. In chain A, a small fraction (~17%) of copper could be modeled at an alternate position shifted by 1.3 Å to a location commonly associated with reduced (Cu1+) SOD (Figure 2). For catalysis, wild-type Cu, Zn SOD is poised in a redox equilibrium. The proportion of Cu1+ versus Cu+2 can be influenced by crystal lattice contacts16; 40 or radiation-induced reduction of copper during X-ray diffraction data collection 9; 41; 42; 43; 44; 45, as well as by other in vitro or in vivo conditions. Only chain A of the zinc-deficient SOD dimer had evidence for the alternate, reduced (Cu1+) geometry.
Figure 2.

Position of copper in the active site of zinc-deficient SOD chain A. The 2Fo-Fc electron density is contoured at 7σ (blue mesh) and the Fo-Fc difference map is contoured at 3σ (magenta mesh) after omitting the alternate copper. The axial water (red sphere) and histidine residues 46, 48, 63, 120 coordinate Cu2+ (orange sphere) with a distorted square planar geometry. The magenta positive density peak (peak height 5.7σ) indicates some of the copper (light yellow sphere) is in the alternate copper site, implying that it is in the Cu1+ state.
The most striking change in the zinc-deficient SOD subunits was the lack of electron density for part (residues 68-78) of the zinc-binding section of loop IV, as might be expected from the loss of zinc coordination to its four ligands (His63, His71, His80 and Asp83) in this loop. The disruption extended to residues 132-139 of the electrostatic loop VII, which makes contacts with the zinc-binding sub-loop. The combined disordering of the electrostatic and zinc-binding loops substantially disrupted and widened the active-site channel, resulting in greater accessibility of the copper to solvent (Figure 3).
Figure 3.

Increases copper accessibility in zinc-deficient SOD. (a) Access to the active site copper (orange) of wild-type Cu,Zn SOD (green) is limited by the electrostatic and zinc-binding loops. The red dashes illustrate the dimensions of the active site channel, which narrows from ca. 12 Å to ca. 4 Å. (b) The active site of zinc-deficient SOD (blue) is more open and accessible due to the disorder of the electrostatic and zinc-binding loops. Although the disordered residues in these loops may transiently restrict the active-site channel, their flexibility allows small molecules greater access to the copper.
Asymmetry in the Zn-deficient SOD dimer
The dimer interface of zinc-deficient SOD was substantially opened compared to wild-type Cu,Zn SOD. Chain A was rotated 9° relative to chain B (Figure 4(a)) and exhibited subtle differences that affected the disulfide and interface sub-loop sections of loop IV (Figure 4(b)). The ca. 1 Å shift of the disulfide sub-loop into the dimer interface of chain A involves 5–15° shifts in the phi/psi angles of residues 53-59 and was accompanied by a new hydrogen bond between Thr58N and Ala55O. Together the changes in residues 53-59 of chain A result in a more defined 310 helix. The shift in the disulfide sub-loop was also associated with a 0.6 Å shift of the guanidinium group of Arg143 away from the copper active site in chain A (Figure 4(b) and (d)). In the new conformation exhibited by chain A, Arg143 maintained hydrogen bonds to the carbonyl of Cys57 and two water molecules, but lost hydrogen bonds to the carbonyl of Gly61 and instead formed a hydrogen bond to the carbonyl of Thr58. Coinciding with these shifts was the presence of two distinct conformations for the Cys57-Cys146 disulfide bridge (Figure 4(c)). The predominant conformation (occupancy=0.65) was the canonical left-handed spiral conformation found in nearly all SOD structures; the second conformation involved a significant shift of only one atom, Cys146-Sγ, to yield a right-handed hook, the second major conformation found for disulfide bridges in proteins. The conformations of Arg143 and the disulfide subloop in chain B remained similar to those seen in wild-type Cu,Zn SOD.
Figure 4.

Origin of the shift in dimer orientation. (a) The zinc-deficient SOD dimer (blue ribbon) was overlaid with C6A, C111S Cu,Zn SOD (gray ribbon) based on the left hand subunit (chain A). The shift in the right hand subunit orientation of 9.3° can be seen in the misalignment of the β-barrel in chain B. The average shift between zinc-deficient SOD and five different human SOD structures is 9.3°± 1.4°(std) (n=21 dimers)[PDB codes 1PU0, 1N18, 1HL5, 1HL4,1OZT]. (b) Close up view of the shift in the disulfide sub-loop of chain A towards the dimer interface. Thr58 prevents the shift in Arg143 due to clashes that would be created between the Nη and Cα of Thr58. The shift in the disulfide sub-loop is ~1 Å as measured at the Cα atom of Thr58. (c) The native left-handed spiral conformation (Sγ-Sγ distance 2.2 Å) found in Cu,Zn SOD and the zinc-deficient structure is shown as orange spheres. The right-handed hook conformation (Sγ-Sγ distance 2.1 Å) observed as an alternate conformation in chain A of zinc-deficient SOD is shown as yellow sphere with omit Fo-Fc density contoured to 3σ (magenta mesh). (d) Stereo image of the altered position of Arg143 in Chain A. In wild type Cu,Zn SOD (PDB code 1HL5) Arg143 H-bonds (gray dashes) to three water molecules (gray spheres), to the carbonyl oxygen of Gly61 and to the carbonyl oxygen of Cys57. Conversely, Arg143 in chain A of zinc-deficient SOD no longer makes H-bonds to Gly61-O, but forms a new H-bond to Thr58-O (red dashes).
In chain A, residues 124-131 of the electrostatic loop deviated substantially from other SOD structures, due in part to crystal packing interactions made between these residues and the equivalent residues of a symmetry-related chain B subunit (Figure 5). Also, the observed asymmetry in the disulfide sub-loop of chain A was stabilized by crystal contacts with chain B of a symmetry mate (Figure 5). Two hydrogen bonds linked Arg143 of chain A with residues Lys91Bsym and Asp92Bsym, while Thr58A made two hydrogen bonds with Lys91Bsym and Glu40Bsym.
Figure 5.

Crystal contacts in the zinc-deficient SOD structure. Chain B of a symmetry-related molecule (grey; Sym1) contacts both chains A and B (blue) of the asymmetric unit. The inset shows the symmetry mate makes four hydrogen bonds (red dashes) with chain A. The Sym1 symmetry mate also makes a single salt bridge with chain B. The electrostatic loop residues 124-131 remain ordered due to an interaction across a twofold with the same residues of another (Sym2) symmetry-related molecule.
An important aspect of the zinc-deficient SOD structure is the asymmetry of the dimer. None of the other human SOD crystal structures show asymmetry of this magnitude: an unprecedented 9° subunit rotation and ~1 Å shift in the disulfide loop. The somewhat different conformations between chain A and chain B are related to the structural changes in the dimer interface. In the crystal, the asymmetry observed in chain A was stabilized by interactions with symmetry-related molecules of chain B (see above). Although the observed asymmetry could be inherent to zinc-deficient SOD or could have been induced by crystal packing interactions, we favor the hypothesis that the asymmetry reflects alterations to the dimer that could also occur in solution. In either case, the perturbation of chain A in the crystal structure of Zn-deficient SOD indicates a fundamental destabilization of the structure to an extent not seen for other ALS SOD mutants characterized to date.
Conformational changes in conserved Arg143 and the disulfide bond
To assess the novelty of various structural features seen in zinc-deficient SOD, an overlay was carried out with all human SOD crystal structures in the Protein Data Bank4 with 2.0 Å or better resolution. Fifteen structures comprising 83 SOD chains meet these criteria. These comparisons show that the Arg143 conformation seen here was unusual, with only the A4V/C6A/C111S mutant having a similar positioning and the artificially engineered monomeric form of human SOD having a conformation that is even more dramatically shifted from its canonical position (Figure 6(a)). The shift in the position of Arg143 in A4V/C6A/C111S is stabilized by two a hydrogen bonds, one to a water and the second to Glu77 of a symmetry related molecule. However, the conformation of Arg143 in the artificially engineered monomer was not stabilized by crystal contacts.
Figure 6.

Deviation of the conserved conformation of Arg143 in human Cu,Zn SOD. (a) Comparison of 83 independent Arg143 side chains from 15 human SOD structures with resolution of 2.0 Å or better (PDB codes 1AZV, 1HL4, 1HL5, 1MFM, 1N18, 1N19, 1OZU, 1P1V, 1PTZ, 1PU0, 1UXL, 1UXM, 2C9S, 2C9U, 2C9V). The overlay was based on the main chain and Cβ atoms of Arg143. Thicker models of Arg143 indicate chain A of zinc-deficient SOD (blue), chain B of A4V/C6A/C111S (red, 1N1915), and monomeric SOD (green, 1MFM39), which was designed in a collaboration of Tainer and Getzoff with Robert A. Hallewell and Guy T. Mullenbach.. (b) Overlay excluding the Arg143 of A4V/C6A/C111S, monomeric and zinc-deficient SOD to confirm that no other conformations were hidden by the sticks in panel A.
The incomplete occupancy of the left-handed spiral conformation of the disulfide observed in this study is unusual among SODs. A search of all SODs in the PDB (n=76) revealed that only three structures exhibited partial occupancy for the left-handed spiral conformation: In the human monomeric Cu,Zn SOD and “as-isolated” human Zn,Zn SOD (2C9U), the second conformation was not a right-handed hook, but instead showed evidence of partial reduction, with long Sγ-Sγ distances of ~2.7 Å. The recently determined Schistosoma mansoni Cu,Zn SOD(1TO4) provides on additional example of partially occupied alternate conformation of the disulfide. Similar to the structure reported here, the Schistosoma mansoni disulfide had a partially occupied right-handed hook with Sγ-Sγ distances of ~2.1 Å indicating that the disulfide remains oxidized 46.
Thiol-dependent aggregation and heterodimer formation
The propensity of zinc-deficient SOD to aggregate was studied using analytical ultracentrifugation (Figure 7(a)). In the absence of the reductant dithiothreitol (DTT), all SODs tested, whether Cu,Zn-replete or zinc-deficient, were resistant to aggregation (<10% aggregation after 72 hours at 20°C). In the presence of DTT, ~50% of both the D124N and D83S forms of zinc-deficient SOD aggregated, whereas < 5% of wild-type and C111S Cu,Zn SODs aggregated. Surprisingly, the DTT-induced aggregation of both zinc-deficient SOD mutants was prevented by the addition of equimolar amounts of wild-type Cu,Zn SOD (Figure 7(a)). Sedimentation velocity experiments yielded S20,w values of 2.8 for Cu,Zn SOD and 3 for zinc-deficient SOD. To determine whether heterodimers could form between wild-type Cu,Zn SOD and D83S SOD, two techniques were employed: fluorescence resonance energy transfer (FRET) and native gel electrophoresis (Figure 7(b) and 7(c)). Both approaches showed that subunit exchange between wild-type Cu,Zn SODs and D83S occurs readily at 37°C to create heterodimers; the measured half-lives were 17 ± 4 min and 12.5 ± 0.6 min for native gel and FRET experiments, respectively.
Figure 7.
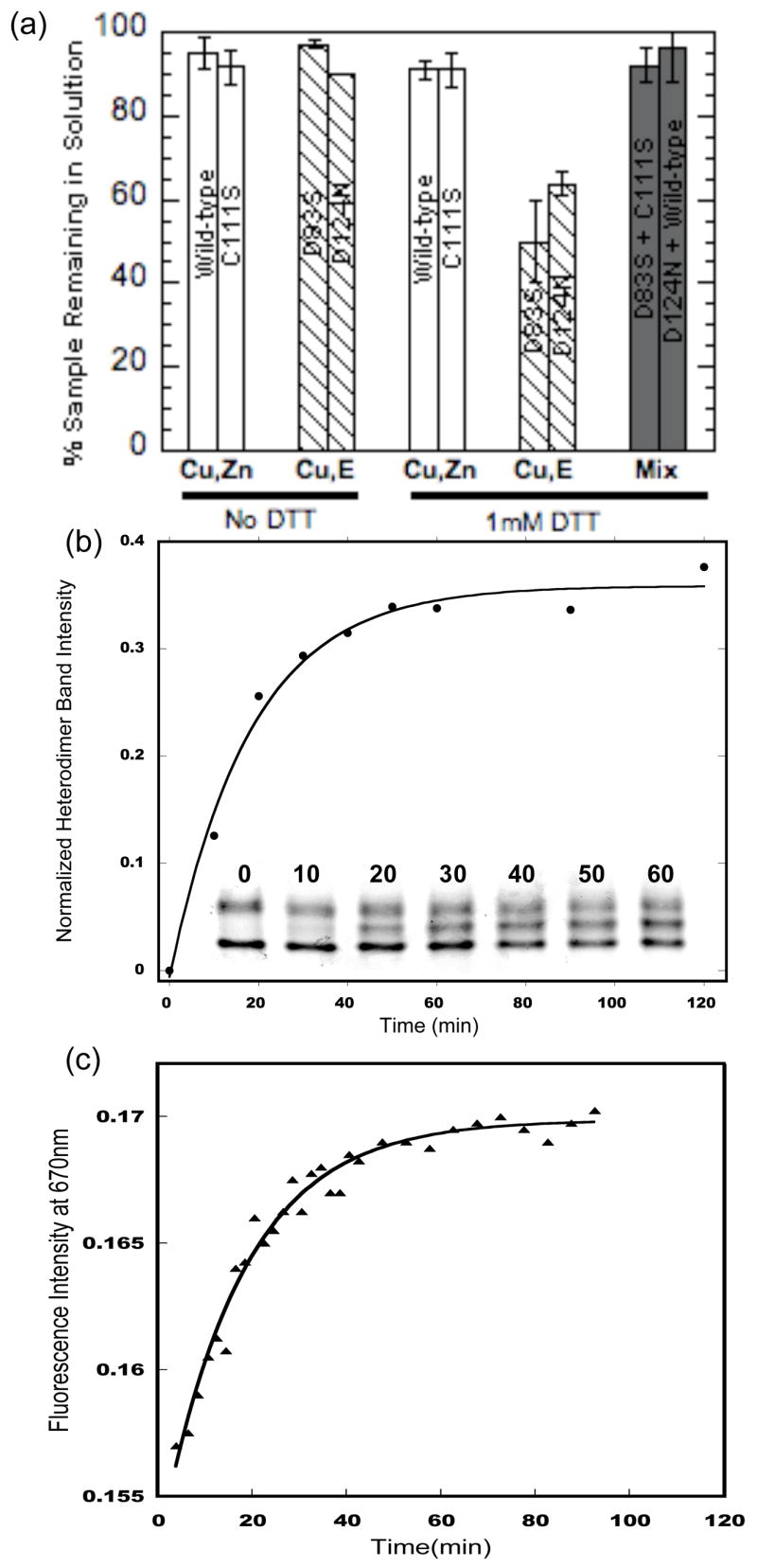
Aggregation and heterodimer formation of zinc-deficient SOD. (a) The stability of the protein against aggregation is measured as the amount of protein remaining in solution at the end of a 72 h analytical ultracentrifuge equilibrium run. Copper and zinc containing SOD (open bars), zinc-deficient SOD (hashed bars) and zinc-deficient SOD mixed with Cu,Zn SOD (filled bars) were all measured with and without DTT as indicated (n=2). (b) SOD native gel electrophoresis shows the time course of the formation of heterodimer as monitored by the appearance of an intermediate band. The half-life of heterodimer formation determined using densitometry was 17 ± 4 min (s.d.,n=3). (c) Increase fluorescence at 670nm resulting from mixing Alexa 647 conjugated zinc-deficient SOD(D83S) and Alexa-594 conjugated Cu,Zn SOD. Heterodimer formation was measured as an increase in fluorescence resonance energy transfer as indicated by the increase in the acceptor (Alexa 647) fluorophore fluorescence at 670nm. The half-life of heterodimer formation was measured to be 12.5 ± 0.6 min (s.d.,n=3).
Discussion
Zinc-deficient SOD can be an important intermediate in both the aggregation and pro-oxidant models for ALS progression. The present structure reveals that the loss of zinc alters the structure of human SOD far more than any ALS mutation that has been crystallographically characterized to date. Overall, the zinc-deficient SOD structure reveals features that help explain both its greater redox activity and increased susceptibility to aggregation. The most dramatic structural change is the disorder of the loop IV that forms the zinc-binding pocket containing all four zinc ligands. The electrostatic loop also becomes disordered because it makes significant contacts with the zinc loop. The disorder of these two loops is comparable to the disorder caused by the lack of both copper and zinc (Table II), and to that seen in molecular dynamic simulations of apo or partially metallated SOD.47 According to NMR48; 49 and molecular dynamic structures47 both metals are needed for full localization of the electrostatic and zinc-binding loops.
Table II.
The loss of zinc leaves the β-barrel and the active-site copper geometry essentially unchanged from oxidized wild-type Cu,Zn SOD. His63, which normally coordinates with both copper and zinc, also assumes a conformation similar to that found in Cu,Zn SOD even though much of the rest of loop IV is disordered. The remarkably similar geometries of the active site are consistent with the ability of oxidized (Cu+2) zinc-deficient SOD to scavenge superoxide at a rate that is nearly identical to the rate of Cu,Zn SOD.50 However, the second step in catalysis, the oxidation of Cu+1 by superoxide, is slower and becomes pH dependent upon the loss of zinc from the enzyme.50; 51
An important consequence of the disruption of loops IV and VII is the opening of the narrow 4 Å wide channel that normally restricts small molecules from accessing the catalytic copper. The increased accessibility to the copper in zinc-deficient SOD explains why copper in zinc-deficient SOD is more than a thousand-times more reactive than the wild-type holo- Cu,Zn SOD with intracellular reductants such as ascorbate and glutathione (Eqn. 1).32 Although the reduced copper will continually be reoxidized by molecular oxygen to produce superoxide (Eqn. 2), the copper will tend to be maintained in a reduced state in vivo by oxidizing low molecular weight antioxidants to their respective free radical intermediates (Eqn. 1).
| (Equation 1) |
| (Equation 2) |
With respect to motor neurons and ALS, the depletion of ascorbate has been shown to be lethal in guinea pigs on a tocopherol-depleted diet.52 Thus, the consumption of ascorbate might also contribute to motor neuron death induced by zinc-deficient SOD observed by Estevez et al.32
Zinc-loss leads to an altered dimer
Rearrangements in loop IV twist the dimer interface and result in a nine-degree rotation in the relative orientation of the two subunits (Figure 4). This is the largest shift reported for the interface of any SOD structure. The disorder in the normally rigidly held disulfide bond and the opening of the dimer interface suggest that the subunit structure is itself more loosely packed and flexible, and that the dimer interface is also weakened. These observations are consistent with the loss of zinc being an initial step in the process of forming apo enzyme and consequent disulfide reduction, dimer dissociation, unfolding and aggregation. Except for the disorder of loops IV and VII, chain B closely resembles the structure of Cu,Zn SOD, while chain A shares features that are intermediate between a genetically engineered monomeric form of SOD and Cu,Zn SOD. The rotation of the guanidinium moiety of Arg143 away from the active site copper and movement of the disulfide region of loop IV are consistent with observations seen in the recently solved structures of the metal depleted, C57S, C146S disulfide mutants of SOD.23 These changes in Arg143 emphasize the interconnections between the active site, the disulfide and the dimer interface.
Curiously, Mycobacterium tuberculosis expresses a membrane-bound, copper containing form of SOD (MtSOD) that does not have a zinc-binding site.53 This MtSOD is up-regulated and expressed on the extracellular surface of the mycobacterium upon entry into human macrophages, where superoxide levels are expected to be high.54 Despite the absence of zinc, MtSOD has an enzymatic rate comparable to that of most Cu, Zn SODs.53 Consistent with zinc-deficient human SOD, the rate of superoxide dismutation of M. tuberculosis is pH dependent.53 When exposed to high superoxide fluxes, as will occur in a macrophage phagosome, the rate of reoxidation (Eqn. 2) of Cu+1 by oxygen would be inconsequential. Because the host limits zinc availability within a macrophage, M. tuberculosis would have a selective advantage to express an efficient SOD lacking zinc55. To accommodate the absence of zinc, the dimer interface of MtSOD involves the electrostatic loop and the addition of a unique dimerization loop.53 The dimer interface in MtSOD occludes more surface area and involves over 30 hydrogen bonds, compared to only a few hydrogen bonds in human SOD.53 The increased stability of the dimer interface may be a mechanism to compensate for the loss in structural stability conferred by zinc in traditional copper and zinc containing SODs.
Aggregation properties and insights into the mechanisms of disease
Dissociation of SOD subunits is now recognized to be a major step in the formation of insoluble aggregates (Figure 8).22; 56; 57 In the present study, we found that the thiol-reducing agent DTT strongly promoted aggregation of zinc-deficient SOD. As noted above, the alternate conformation seen for the Cys57-Cys146 disulfide in chain A of the zinc-deficient SOD structure is particularly noteworthy in this regard, as it implies a greater level of mobility that would be expected to correlate with greater ease of reduction by small molecular weight thiol reductants. Remarkably, the thiol-dependent aggregation of zinc-deficient SOD was almost entirely prevented by the addition of Cu,Zn SOD (Figure 7(a)). As we have shown that heterodimers form in minutes (Figure 7(b) and (c)), we propose that Cu,Zn SOD prevents the thiol-dependent aggregation of zinc-deficient SOD through the formation of more stable heterodimers with zinc-deficient SOD. Structurally this makes sense because the Cu,Zn subunit would stabilize the more native-like conformation of the disulfide section of loop IV in the zinc-deficient SOD subunit. A more native conformation would make the disulfide less susceptible to reduction by DTT and thereby decrease the rate of formation of aggregation-prone monomers in the presence of DTT.
Figure 8.

The loss of zinc from Cu,Zn SOD will initially form a heterodimer that can eventually lose a second zinc to yield a zinc-deficient homodimer. The loss of zinc increases accessibility of small molecules to copper and weakens the dimer interface. The C57-C146 disulfide bridge in zinc-deficient homodimers is more susceptible to reduction and the monomeric proteins will readily lose copper and form insoluble aggregates stabilized by intersubunit disulfides. Although the dimer interface of Cu,Zn SOD is more stable, the formation of heterodimers from Cu,Zn and zinc-deficient SOD monomers greatly reduces the susceptibility of zinc-deficient SOD to aggregation in the presence of disulfide reducing agents due to the more stable interface from the Cu,Zn subunit.
An intriguing aspect of this work is the implications for how heterodimer formation could influence disease progression. Cu,Zn and zinc-deficient SOD heterodimer formation significantly inhibits aggregation of the zinc-deficient enzyme (Figure 7(a)). The two hypotheses of pro-oxidant toxicity and aggregation toxicity would predict different disease outcomes resulting from stabilization as heterodimers. Under the aggregation hypothesis, heterodimer formation would be expected to decrease aggregation and therefore slow disease. According to the pro-oxidant SOD hypothesis (Figure 8), heterodimer formation would lead to a more rapid disease course because decreased aggregation would increase the lifetime of zinc-deficient mutant SOD, resulting in an increased consumption of ascorbate and increased oxidation by SOD. Crossing mice overexpressing wild-type SOD with FALS SOD mice has been shown to accelerate development of motor neuron disease.34; 58 Fukada et al.58 attributed the accelerated disease progression to the stabilization of mutant SOD via the formation of heterodimers. In humans, FALS is an autosomal dominant disease, which allows for the formation of heterodimers of wild-type and mutant SOD. As point mutations have been described in all five exons of SOD-1, involving almost one-third 43 of the 153 residues, hypotheses about the toxic role of mutant SOD-1 in the pathogenesis of ALS must account for this striking molecular diversity. In accordance with the results presented here, a fundamental aspect of all the ALS mutant dimers as well as ALS SOD heterodimers with wild-type SOD will be structural defects expected to influence zinc ion binding. The involvement of the Zn ion may explain why an ALS-like Ile-to-Thr mutation in the tetrameric human mitochondrial MnSOD fails to cause any known degenerative diseases in humans despite destabilizing the protein fold, as this MnSOD contains a single active site metal ion, so there is no comparable Zn site loss to amplify the defect59. The zinc-deficient SOD structure therefore provides new insights into how the formation of heterodimers with Cu,Zn wild-type SOD would stabilize zinc-deficient mutant SOD, a species known to be toxic to motor neurons, and thus contributes to the dominant inheritance of SOD mutations.
Materials and Methods
Description of SOD mutants
In the course of this work, we used five different forms of human Cu,Zn SOD with metal content assessed using the 4-pyridylazaresorcinol (PAR) assay35: wild-type (wild-type), C111S (C111S, 1.07 copper per monomer, 1.18 zinc per monomer), H80S/D83S/C6A/C111S (H80S/D83S), D124N/C111S (D124N,0.93 copper per monomer, 0.03 zinc per monomer), and D83S/C111S (D83S, 0.93 copper per monomer, 0.08 zinc per monomer). The previously characterized C111S and C6A mutations facilitate expression of soluble, stable protein from E. coli and do not significantly alter the structure, metal binding, or catalytic superoxide dismutase activity of SOD.6; 11; 60 Mutation of the zinc ligands His80 and Asp83 makes SOD constitutively zinc-deficient. The D124N mutation was shown to yield a zinc-deficient SOD by disrupting hydrogen-bonds networks that stabilize the Zn ligands His46 and His7161. For the purposes of this study, we have grouped these variants into two categories: the normally “Cu, Zn” containing enzymes, wt SOD and C111S SOD, and the constitutively zinc-deficient (Cu, E) enzymes D124N SOD, D83S SOD and H80S/D83S SOD.
Expression and purification of SOD
Zinc-deficient SOD was created by mutating the zinc-binding residues Asp83 and His80 to Ser or by mutating Asp124 to Asn using mutagenic PCR as previously described.15; 61 All SOD proteins used in this study were expressed and purified from E. coli as previously described.15; 60 Copper and zinc content was determined using the 4-pyridylazaresorcinol (PAR) assay.35 Protein concentrations were based on ε280= 5,810 M−1cm−1 for the denatured protein that was calculated from the amino acid composition.62
Crystallization
Crystals were grown at room temperature in less than one week using a reservoir of 2.45 M ammonium sulfate, 200 mM NaCl in 50 mM Tris, pH 7.5, and a drop made from 2 μL of 15.7 mg/mL protein in 50 mM Na/K phosphate pH 7.7 mixed with 2 μL of reservoir solution.
Data collection and refinement
Diffraction data were collected at the Advanced Light Source (Lawrence-Berkeley National Laboratory, USA) at −170° C (1°oscillations; λ= 1.01 Å), and were processed using programs DENZO and SCALEPACK (Table I)63. The crystals formed in space group C2 with unit cell axes a=107.2 Å, b=35.9 Å, c=68.30 and β=104.8°. All crystallographic refinement was done using the Crystallography and NMR System software (v1.1)64. A random 5% of the data were selected for cross-validation. The structure of the C6A, C111S SOD mutant (PDB code 1N18) succeeded as a search model to carry out molecular replacement against data from 20 to 5 Å resolution. Rigid body refinement yielded R/Rfree values of 0.31/0.35, consistent with a correct solution. The 2FO – FC electron density map displayed well-defined density for residues 1-67, 79-131 and 140-153, however there was no clear density for atoms of the zinc binding sub-loop residues 68-78 or the electrostatic loop residues132-139 in either chain A or B. Hence, these residues were removed from the model for successive refinements. Residues 1-2, 23-28, 66-67 and 131 had weak backbone density but sufficient to allow them to be modeled. Local positional and B-factor refinement extending to 2.0 Å resolution, rapidly dropped the R-factors to R= 0.226 and Rfree= 0.273. Manual rebuilding of the model using the program O 65 was guided by 2FO – FC and FO – FC electron density maps. Water molecules were added both manually and using the CNS utility Water-Pick with the following criteria: (1) a minimum 3σ peak in FO – FC maps and (2) a reasonable hydrogen-bonding geometry. At the later stages of refinement, the largest positive difference peak corresponded to an alternate position for the Sγ atom of residue Cys 146. Occupancy of the Cys146-Sγ alternate position was selected as the value that yielded equivalent B-factors for the two positions 66: final B-factors of ~30 Å3 resulted from occupancies of 0.65 for the native-like Cys146-Sγ and 0.35 for the alternate conformation of Cys146-Sγ. Waters were numbered such that water 1 had the strongest electron density and water 193 had the weakest density. Waters with 2FO-Fc density of <1σ were removed from the model. Potential atomic clashes and erroneous side chain rotamers were identified with Molprobity 67. Refinement was considered complete when the largest difference map peaks were not interpretable and R and RFree had converged. Final statistics are reported in Table I. In addition, 100% of theφ,ψ angles are in the allowed regions of the Ramachadran plot.
Root-mean-square deviation calculations and structural overlays were performed using the program HOMOLOGCORE (P.A. Karplus). The program HBPLUS v2.24 was used to define hydrogen bonds within the zinc-deficient model 68. Structural figures were made using PyMOL69.
Native gel electrophoresis
Wild-type Cu, Zn SOD (40 μM) and D83S (40 μM) were combined in 100 mM sodium phosphate, pH=7.4, and 100 mM sodium chloride and incubated at 37° C for the time indicated. After incubation samples were flash frozen with liquid N2. Samples were prepared with native sample buffer (Bio-Rad) and resolved using 7.5 % acrylamide/bis-acrylamide (37.5:1) resolving gel and a 3% stacking gel. Electrophoresis was carried out at 4° C for 120 min. at 130 V. The gel was then stained with Coomassie Blue and quantified with NIH Image (http://rsb.info.nih.gov/nih-image/). Heterodimer formation was quantified as density of heterodimer band divided by the total density of all three bands.
Fluorescent resonance energy transfer (FRET)
SOD protein samples were conjugated to primary amines via succinamide esters of either donor fluorphore Alexa594 or acceptor fluorophore Alexa647 as described by the manufacturer (Invitrogen, Eugene, OR). Briefly a 10-fold molar excess of reactive dye was allowed to react with SOD for 2 h at pH=8.0 and 4° C. Conjugated SOD was purified from free dye using a Sephadex G25 column (30 cm × 0.75 cm) pre-equilibrated with 100 mM phosphate buffer, pH = 7.4. The degree of labeling ranged between 0.8 and 1.3 conjugates per SOD subunit. Fluorescent measurements were carried out at 37° C. Protein concentrations were 100 nM of each the acceptor and donor labeled protein. Fluorescence was measured by irradiating the sample with 586 nm excitation and monitoring the emission at 670 nm. To correct for background fluorescence, separate measurements were taken of the acceptor-labeled or donor-labeled SOD.
Analytical ultracentrifugation
Equilibrium experiments were performed in a Beckman Optima XL-A analytical ultracentrifuge. Buffer densities and viscosity corrections were made according to Laue et al.70. The partial specific volume of SOD (0.725 cm3/g) was estimated from the protein sequence71. The buffers were 100 mM potassium phosphate, pH 7.4, 100 mM NaCl with or without 1 mM DTT. Sedimentation velocity runs were performed at 20°C using a four hole AN-60Ti rotor and double-sector charcoal/Epon filled centerpieces. 10 μM SOD samples (A230=0.8) were centrifuged at 42,000 rpm. Aggregation of the protein was measure using two methods: (1) the difference in absorbance between baseline and after the sample had reached equilibrium (2) after the completion of the equilibrium run the samples were agitated and then the baseline absorbance was taken and compared to the initial baseline measurement. Scans were collected using absorbance optics and analyzed by the method of van Holde and Weischet72, or an enhanced method as implemented in the UltraScan software73; 74. This analysis yields the integral distribution G(s) of diffusion-corrected sedimentation coefficients across the sedimentation boundary. In addition, the data were analyzed by the computer program Sedfit. This software analyses analytical ultracentrifuge velocity data files by direct fitting with numerical solutions of the Lamm Equation75.
Sedimentation equilibrium experiments were performed at 20°C according to procedures described76. Typically, three 120-μl samples of 10 μM SOD were centrifuged to equilibrium at 3 different speeds from 15,000–22,000 rpm. Scans were collected with absorbance optics at wavelengths between 230 and 280 nm. The radial step size was 0.001 cm, and each c versus r data point was the average of 15 independent measurements. Wavelengths were chosen so that no points exceeded an absorbance of 1.0. Using UltraScan software, equilibrium data were fit to multiple models using global fitting. The most appropriate model was chosen based on the statistics and on visual inspection of the residual patterns.
Coordinates
The coordinates and structure factors have been deposited in the Protein Data Bank (http://www.pdb.org/) as entry XXXX.
Acknowledgments
This publication was made possible in part by the Environmental Health Sciences Center supported by grant number P30 ES00210 from the National Institute of Environmental Health Sciences, NIH. This research was supported by grants ES00040 and AT002034-02 from the National Institute of Environmental Health Sciences to JSB and NIH GM39345 (JAT) and NIH GM37684 (EDG).
Abbreviations
- ALS
amyotrophic lateral sclerosis
- SOD
Cu,Zn superoxide dismutase
- DTT
dithiothreitol
- rmsd
root-mean squared deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–51. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nature Structural Biology. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 5.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 6.Parge HE, Hallewell RA, Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci U S A. 1992;89:6109–13. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Getzoff ED, Tainer JA, Stempien MM, Bell GI, Hallewell RA. Evolution of CuZn superoxide dismutase and the Greek key beta-barrel structural motif. Proteins. 1989;5:322–36. doi: 10.1002/prot.340050408. [DOI] [PubMed] [Google Scholar]
- 8.Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–90. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 9.Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–7. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- 10.Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, Rodriguez JA, Whitson LJ, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A. 2004;101:5976–81. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–15. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- 12.Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 13.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 14.Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci U S A. 2002;99:9010–4. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardoso RM, Thayer MM, DiDonato M, Lo TP, Bruns CK, Getzoff ED, Tainer JA. Insights into Lou Gehrig’s disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J Mol Biol. 2002;324:247–56. doi: 10.1016/s0022-2836(02)01090-2. [DOI] [PubMed] [Google Scholar]
- 16.Hart PJ, Liu H, Pellegrini M, Nersissian AM, Gralla EB, Valentine JS, Eisenberg D. Subunit asymmetry in the three-dimensional structure of a human CuZnSOD mutant found in familial amyotrophic lateral sclerosis. Protein Sci. 1998;7:545–55. doi: 10.1002/pro.5560070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–7. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 18.Elam JS, Malek K, Rodriguez JA, Doucette PA, Taylor AB, Hayward LJ, Cabelli DE, Valentine JS, Hart PJ. An alternative mechanism of bicarbonate-mediated peroxidation by copper-zinc superoxide dismutase: rates enhanced via proposed enzyme-associated peroxycarbonate intermediate. J Biol Chem. 2003;278:21032–9. doi: 10.1074/jbc.M300484200. [DOI] [PubMed] [Google Scholar]
- 19.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 20.Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci U S A. 2004;101:15893–8. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khare SD, Caplow M, Dokholyan NV. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2004;101:15094–9. doi: 10.1073/pnas.0406650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doucette PA, Whitson LJ, Cao X, Schirf V, Demeler B, Valentine JS, Hansen JC, Hart PJ. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J Biol Chem. 2004;279:54558–66. doi: 10.1074/jbc.M409744200. [DOI] [PubMed] [Google Scholar]
- 23.Hornberg A, Logan DT, Marklund SL, Oliveberg M. The Coupling between Disulphide Status, Metallation and Dimer Interface Strength in Cu/Zn Superoxide Dismutase. J Mol Biol. 2007;365:333–42. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 24.Banci L, Bertini I, Cantini F, D’Amelio N, Gaggelli E. Human SOD1 before harboring the catalytic metal: solution structure of copper-depleted, disulfide-reduced form. J Biol Chem. 2006;281:2333–7. doi: 10.1074/jbc.M506497200. [DOI] [PubMed] [Google Scholar]
- 25.Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–30. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–38. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–64. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- 28.Ge WW, Wen W, Strong W, Leystra-Lantz C, Strong MJ. Mutant copper-zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. J Biol Chem. 2005;280:118–24. doi: 10.1074/jbc.M405065200. [DOI] [PubMed] [Google Scholar]
- 29.Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- 30.Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:29771–9. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- 31.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 32.Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci U S A. 2005;102:10516–21. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–44. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 36.Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown RH, Jr, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci U S A. 1994;91:8292–6. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, Hart PJ, Hayward LJ, Valentine JS, Hasnain SS. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–91. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- 38.Strange RW, Antonyuk SV, Hough MA, Doucette PA, Valentine JS, Hasnain SS. Variable metallation of human superoxide dismutase: atomic resolution crystal structures of Cu-Zn, Zn-Zn and as-isolated wild-type enzymes. J Mol Biol. 2006;356:1152–62. doi: 10.1016/j.jmb.2005.11.081. [DOI] [PubMed] [Google Scholar]
- 39.Ferraroni M, Rypniewski W, Wilson KS, Viezzoli MS, Banci L, Bertini I, Mangani S. The crystal structure of the monomeric human SOD mutant F50E/G51E/E133Q at atomic resolution. The enzyme mechanism revisited. J Mol Biol. 1999;288:413–26. doi: 10.1006/jmbi.1999.2681. [DOI] [PubMed] [Google Scholar]
- 40.Ogihara NL, Parge HE, Hart PJ, Weiss MS, Goto JJ, Crane BR, Tsang J, Slater K, Roe JA, Valentine JS, Eisenberg D, Tainer JA. Unusual trigonal-planar copper configuration revealed in the atomic structure of yeast copper-zinc superoxide dismutase. Biochemistry. 1996;35:2316–21. doi: 10.1021/bi951930b. [DOI] [PubMed] [Google Scholar]
- 41.Burmeister WP. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr D Biol Crystallogr. 2000;56:328–41. doi: 10.1107/s0907444999016261. [DOI] [PubMed] [Google Scholar]
- 42.Weik M, Ravelli RB, Kryger G, McSweeney S, Raves ML, Harel M, Gros P, Silman I, Kroon J, Sussman JL. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc Natl Acad Sci U S A. 2000;97:623–8. doi: 10.1073/pnas.97.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart PJ, Balbirnie MM, Ogihara NL, Nersissian AM, Weiss MS, Valentine JS, Eisenberg D. A structure-based mechanism for copper-zinc superoxide dismutase. Biochemistry. 1999;38:2167–78. doi: 10.1021/bi982284u. [DOI] [PubMed] [Google Scholar]
- 44.Carugo O, Carugo KD. When X-rays modify the protein structure: radiation damage at work. Trends Biochem Sci. 2005;30:213–9. doi: 10.1016/j.tibs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Stroppolo ME, Nuzzo S, Pesce A, Rosano C, Battistoni A, Bolognesi M, Mobilio S, Desideri A. On the coordination and oxidation states of the active-site copper ion in prokaryotic Cu,Zn superoxide dismutases. Biochem Biophys Res Commun. 1998;249:579–82. doi: 10.1006/bbrc.1998.9199. [DOI] [PubMed] [Google Scholar]
- 46.Cardoso RM, Silva CH, Ulian de Araujo AP, Tanaka T, Tanaka M, Garratt RC. Structure of the cytosolic Cu,Zn superoxide dismutase from Schistosoma mansoni. Acta Crystallogr D Biol Crystallogr. 2004;60:1569–78. doi: 10.1107/S0907444904016798. [DOI] [PubMed] [Google Scholar]
- 47.Strange RW, Yong CW, Smith W, Hasnain SS. Molecular dynamics using atomic-resolution structure reveal structural fluctuations that may lead to polymerization of human Cu-Zn superoxide dismutase. Proc Natl Acad Sci U S A. 2007;104:10040–4. doi: 10.1073/pnas.0703857104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banci L, Bertini I, Cantini F, D’Onofrio M, Viezzoli MS. Structure and dynamics of copper-free SOD: The protein before binding copper. Protein Sci. 2002;11:2479–92. doi: 10.1110/ps.0210802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS. Solution structure of Apo Cu,Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry. 2003;42:9543–53. doi: 10.1021/bi034324m. [DOI] [PubMed] [Google Scholar]
- 50.Lisa M, Ellerby DEC, Graden Janet A, Valentine JS. Copper-Zinc Superoxide Dismutase: Why Not pH-Dependent? Journal of American Chemical Society. 1996;118:6556–6561. [Google Scholar]
- 51.Pantoliano MW, Valentine JS, Burger AR, Lippard SJ. A pH-dependent superoxide dismutase activity for zinc-free bovine erythrocuprein. Reexamination of the role of zinc in the holoprotein. J Inorg Biochem. 1982;17:325–41. doi: 10.1016/s0162-0134(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 52.Burk RF, Christensen JM, Maguire MJ, Austin LM, Whetsell WO, Jr, May JM, Hill KE, Ebner FF. A combined deficiency of vitamins E and C causes severe central nervous system damage in guinea pigs. J Nutr. 2006;136:1576–81. doi: 10.1093/jn/136.6.1576. [DOI] [PubMed] [Google Scholar]
- 53.Spagnolo L, Toro I, D’Orazio M, O’Neill P, Pedersen JZ, Carugo O, Rotilio G, Battistoni A, Djinovic-Carugo K. Unique features of the sodC-encoded superoxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking zinc in the active site. J Biol Chem. 2004;279:33447–55. doi: 10.1074/jbc.M404699200. [DOI] [PubMed] [Google Scholar]
- 54.D’Orazio M, Folcarelli S, Mariani F, Colizzi V, Rotilio G, Battistoni A. Lipid modification of the Cu,Zn superoxide dismutase from Mycobacterium tuberculosis. Biochem J. 2001;359:17–22. doi: 10.1042/0264-6021:3590017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blencowe DK, Morby AP. Zn(II) metabolism in prokaryotes. FEMS Microbiol Rev. 2003;27:291–311. doi: 10.1016/S0168-6445(03)00041-X. [DOI] [PubMed] [Google Scholar]
- 56.Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O’Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–8003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- 57.Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr, Walz T, Lansbury PT., Jr An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43:4899–905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 58.Fukada K, Nagano S, Satoh M, Tohyama C, Nakanishi T, Shimizu A, Yanagihara T, Sakoda S. Stabilization of mutant Cu/Zn superoxide dismutase (SOD1) protein by coexpressed wild SOD1 protein accelerates the disease progression in familial amyotrophic lateral sclerosis mice. Eur J Neurosci. 2001;14:2032–6. doi: 10.1046/j.0953-816x.2001.01828.x. [DOI] [PubMed] [Google Scholar]
- 59.Borgstahl GE, Parge HE, Hickey MJ, Johnson MJ, Boissinot M, Hallewell RA, Lepock JR, Cabelli DE, Tainer JA. Human mitochondrial manganese superoxide dismutase polymorphic variant Ile58Thr reduces activity by destabilizing the tetrameric interface. Biochemistry. 1996;35:4287–97. doi: 10.1021/bi951892w. [DOI] [PubMed] [Google Scholar]
- 60.Leinweber B, Barofsky E, Barofsky DF, Ermilov V, Nylin K, Beckman JS. Aggregation of ALS mutant superoxide dismutase expressed in Escherichia coli. Free Radic Biol Med. 2004;36:911–8. doi: 10.1016/j.freeradbiomed.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 61.Banci L, Bertini I, Cabelli DE, Hallewell RA, Tung JW, Viezzoli MS. A characterization of copper/zinc superoxide dismutase mutants at position 124. Zinc-deficient proteins. Eur J Biochem. 1991;196:123–8. doi: 10.1111/j.1432-1033.1991.tb15794.x. [DOI] [PubMed] [Google Scholar]
- 62.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–26. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 63.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methosd of Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 64.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 65.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 ( Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 66.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, Schirmer RH. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J Biol Chem. 2002;277:2779–84. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 67.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 68.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–93. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 69.DeLano WL. The PyMOL Molecular Graphics System 2002 [Google Scholar]
- 70.Olsen PH, Esmon NL, Esmon CT, Laue TM. Ca2+ dependence of the interactions between protein C, thrombin, and the elastase fragment of thrombomodulin. Analysis by ultracentrifugation. Biochemistry. 1992;31:746–54. doi: 10.1021/bi00118a016. [DOI] [PubMed] [Google Scholar]
- 71.Perkins SJ. Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem. 1986;157:169–80. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 72.Weischet WO, Tatchell K, Van Holde KE, Klump H. Thermal denaturation of nucleosomal core particles. Nucleic Acids Res. 1978;5:139–60. doi: 10.1093/nar/5.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Anal Biochem. 2004;335:279–88. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 74.Cao W, Demeler B. Modeling analytical ultracentrifugation experiments with an adaptive space-time finite element solution of the Lamm equation. Biophys J. 2005;89:1589–602. doi: 10.1529/biophysj.105.061135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ausio J, Malencik DA, Anderson SR. Analytical sedimentation studies of turkey gizzard myosin light chain kinase and telokin. Biophys J. 1992;61:1656–63. doi: 10.1016/S0006-3495(92)81969-0. [DOI] [PMC free article] [PubMed] [Google Scholar]