

Abstract
The globus pallidus (GP) plays a central integrative role in the basal ganglia circuitry. It receives strong GABAergic inputs from the striatum and significant glutamatergic afferents from the subthalamic nucleus (STN). The change in firing rate and pattern of GP neurons is a cardinal feature of Parkinson’s disease pathophysiology. Kainate receptor GluR6/7 subunits immunoreactivity is expressed presynaptically in GABAergic striatopallidal terminals (Kane-Jackson and Smith 2003; Jin et al., 2006), which provides a substrate for regulation of GABAergic transmission in GP. To test this hypothesis, we recorded GABAA-mediated inhibitory postsynaptic currents (IPSCs) in the GP following electrical stimulation of the striatum. Following blockade of AMPA and NMDA receptors with selective antagonists, bath application of kainate (KA) (0.3–3 μM) reduced significantly the amplitude of evoked IPSCs. This inhibition was associated with a significant increase in paired-pulse facilitation ratio and a reduction of the frequency, but not amplitude, of miniature IPSCs (mIPSCs), suggesting a presynaptic site of KA action. The KA effects on striatopallidal GABAergic transmission were blocked by the G-protein inhibitor, N-ethylmaleimide (NEM), or protein kinase C (PKC) inhibitor calphostin C. Our results demonstrate that KAR activation inhibits GABAergic transmission through a presynaptic G protein-coupled, PKC-dependent metabotropic mechanism in the rat GP. These findings open up the possibility for the development of kainate-mediated pharmacotherapies aim at decreasing the excessive and abnormally regulated inhibition of GP neurons in Parkinson’s disease.
Keywords: IPSC, patch-clamp, striatum, PKC, paired-pulse facilitation
Kainate receptors (KARs) are one of the three subtypes of ionotropic glutamate receptors in the CNS. The family of KARs is composed of five different genes that encode for the subunits GluR5, GluR6, GluR7, KA1 and KA2 subunits (Hollmann and Heinemann, 1994; Bettler and Mulle, 1995). These receptors are widely expressed pre-and post-synaptically throughout the brain (Charara et al., 1999; Lerma et al., 2001; Kieval et al., 2001; Kullmann, 2001; Huettner, 2003; Lerma, 2003; Jin et al., 2006), but their role remains poorly understood compared to other ionotropic glutamate receptors. However, recent years have witnessed significant developments in the understanding of KARs physiology, largely based on data gathered from the hippocampus (Huettner, 2003; Lerma, 2003). These studies have highlighted the unique properties of KARs as pre-synaptic auto- or hetero-receptors that mediate their effects through ion channel or G protein-coupled mechanisms (Rodríguez-Moreno and Lerma, 1998; Rodríguez-Moreno et al., 2000; Cunha et al., 2000; Jiang et al., 2001; Kang et al., 2004; Jin et al., 2006).
Despite the widespread distribution of KARs subunits through the basal ganglia, very little is known about the physiology and mechanisms of action through which these receptors regulate synaptic transmission in these nuclei. KARs are expressed in striatal glutamatergic, but not GABAergic, axons terminals in the monkey striatum (Charara et al., 1996; Kieval et al., 2001). KAR activation regulates inhibitory post-synaptic currents (IPSCs) evoked locally in the rat striatum (Chergui et al., 2000) and substantia nigra pars compacta (SNc) (Nakamura et al., 2003). Recent findings from our laboratory showed that KARs also play a significant role in regulating glutamatergic transmission in the rat GP (Jin et al., 2006). Although it has long been considered as a mere relay center, it is now well established that the globus pallidus (GP) (external globus pallidus, GPe, in primate) plays a central integrative role in the basal ganglia circuitry (Plenz and Kitai, 1999; Bevan et al., 2002). It receives massive GABAergic inputs from the striatum (Str) and a significant glutamatergic innervation from the subthalamic nucleus (STN). In turn, it sends GABAergic projections back to the STN and other basal ganglia nuclei. In contrast to most neurons in the CNS, more than 80% of synaptic inputs to GP neurons originate from GABAergic striatal projections (Shink and Smith, 1995; Smith et al., 1998). The loss of striatal dopamine in Parkinson’s disease (PD) has opposite effects on the so-called “direct and indirect” striatofugal systems of the basal ganglia circuitry (DeLong, 1990). Increased GABAergic activity from the striatum and abnormal bursting pattern of the pallido-subthalamopallidal loop are cardinal features of Parkinson’s disease pathophysiology (DeLong, 1990; Wichmann and Delong, 1998). The introduction of novel drug therapy that may help regulate the abnormally increased striatal GABAergic influences towards GP neurons, therefore, represents a potential avenue for future pharmacotherapeutic development in Parkinson’s disease. Recent findings from our laboratory have demonstrated GluR6/7 immunoreactivity in putative striatal GABAergic terminals in the monkey and rat GP (Kane-Jackson and Smith, 2003; Jin et al., 2006). Activation of these presynaptic KARs may, therefore, modulate striatopallidal GABAergic synaptic transmission. To further address this important issue, we employed whole cell patch-clamp recording techniques to examine the presynaptic function of KAR on striato-pallidal synapses in the rat GP.
Findings of this study have been presented on abstract form (Jin and Smith 2005, 2006).
EXPERIMENTAL PROCEDURES
Slice preparation
All electrophysiological experiments were performed on slices from 13 to 17-d old Sprague Dawley rats (Charles River Laboratories, Wilmington, MA). After decapitation, brains were removed and quickly submerged in the ice-cold oxygenated sucrose buffer containing (in mM): 233.4 sucrose, 20 glucose, 47.3 NaHCO3, 3 KCl, 1.9 MgSO4, 1.2 KH2PO4, 2 CaCl2, (Poisik et al. 2003; Jin et al. 2006). Parasagittal slices (300μm in thickness) were made on a Vibratome 3000 (The Vibratome Company, St. Louis, MO) in ice-cold oxygenated sucrose buffer. This plane of section was chosen based on previous anatomical data showing that striatal axons to the globus pallidus travel in the parasagittal plane from their origin to their pallidal targets in both rats and monkeys (Kawaguchi et al., 1990; Parent et al., 1995). Slices were stored at room temperature in a chamber containing artificial cerebrospinal fluid (ACSF) (in mM): 124 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 2.0 CaCl2, 20 glucose, 26 NaHCO3, at pH 7.3–7.4 with 95% O2, 5% CO2 bubbling through it. The osmolarity of the ACSF was ~ 310 mOsm.
Whole cell patch-clamp recordings
Whole cell patch-clamp recordings were performed as described previously (Poisik et al., 2003; Jin et al., 2006). During the recording, the slice was maintained fully submerged in the recording chamber and perfused with oxygenated ACSF (~3ml/min). GP neurons were visualized by IR-differential interference contrast microscopy (BX51Wl) using a 40X water immersion objective (Olympus, Pittsburgh, PA). Whole cell patch electrodes were pulled from borosilicate glass on a vertical patch pipette puller (Narishige, Tokyo, Japan) to have resistance in the range of 3–5 M′Ω when filled with an intracellular patch solution. All experiments were performed at room temperature. Tight-seal (>1GΩ) whole-cell recording was obtained from the cell body of GP neurons. Series resistance was regularly monitored during recording, and cells were rejected if the series resistance changed by > 20%.
For whole cell voltage- clamp experiments, patch pipettes were filled with the internal solution containing (in mM): 124 Cs-methanesulfonate, 11 KCl, 2 MgCl2, 10 HEPES, 2 Na2ATP, 0.3 GTP, 5 N-(2,6-Dimethylphenylcarbamoylmethyl) triethylammonium bromide (QX314) and 0.5% biocytin (pH 7.4 (and 300–310 mOsm). Neurons were voltage-clamped at a holding potential of −10 mV and whole-cell membrane currents were recorded with a Patch-Clamp PC-501A (Warner Instruments, Hamden, CT). To isolate GABAA receptor mediated-IPSCs, 100 μM of the AMPA receptor antagonist, 4-(8-methyl-9H-1,3-dioxolo[4,5 h] {2,3} benzodiazepine-5-yl)-benzenamine hydrochloride (GYKI 52466) and 50 μM of the NMDA receptor antagonist, D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5), were added to the ACSF. Three IPSCs were averaged and their peak amplitude was measured before, during and after KA application.
To record miniature IPSCs (mIPSCs), pipettes contained the following (in mM): 125 KCl, 10 NaCl, 1 CaCl2, 2 MgCl2, 10BAPTA, 10 HEPES, 2 Na2-ATP, 0.3 GTP and 0.5% biocytin (pH 7.4 and 300–310 mOsm). The mIPSCs were recorded at a holding potential of −60 mV in the presence of 1μM tetrodotoxin (TTX), 100 μM GYKI and 50 μM D-AP5. Data were collected over continuous 10 – 20 min periods.
In some experiments, N-ethylmaleimide (NEM) (200 μM), a pertussis toxin-sensitive G-proteins inhibitor, calphostin C (1 μM), a selective protein kinase C (PKC) inhibitor, staurosporine (0.5 μM), a broad spectrum inhibitor for protein kinases, or (H-89) (0.5 μM), a selective protein kinase A inhibitor (PKA) were applied. Kainate, AMPA, D-AP5, CNQX, GYKI 52466, TTX and staurosporine were purchased from Tocris Cookson (Ellisville MO). Calphostin C and H-89 were purchased from Sigma (St Louis MO). All compounds were made in a 1000x and diluted into the ACSF immediately before use. Compounds were aliquoted and stored at −20°C. To assess their exact location, recorded GP neurons were filled with biocytin and their location within the GP was confirmed at the light microscopic level.
Electrical stimulation
Bipolar matrix stimulating electrodes (FHC, Bowdoinham, ME) were placed in the striatum close to the GP. Evoked inhibitory postsynaptic currents (IPSCs) in GP neurons were recorded by stimulation of striatum with single pulses that ranged from 3 to 10 V, 150–200 μsec, delivered once every 20 sec. The paired-pulse facilitation (PPF) of evoked IPSCs was performed as follows: two stimuli of the striatum were paired with an interstimulus interval of 40–50ms. The ratio of peak 2/peak1 was calculated.
Biocytin histochemistry
Following overnight fixation in 10% neutral formalin, the slices were rinsed in PBS for 30 min and incubated in PBS containing 1% sodium borohydride for 20 min. They were then incubated in an ABC solution overnight at room temperature. After two 10 min washes in PBS and one 10 min wash in TRIS buffer (0.05 M, pH 7.6), the immunostaining was revealed by incubation for 10 min in a solution containing 0.025% 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma, St. Louis, MO), 0.01 M imidazole (Fisher Scientific, Atlanta, GA), and 0.006% hydrogen peroxide (H2O2). The reaction was stopped by repeated washes in PBS. The biocytin-labeled neurons were then viewed with a Leica DMRB microscope, equipped with Leica digital Imaging system software.
Data analysis
Signals were filtered at 5 kHz and digitized with a Digidata 1200 analog-to-digital converter (Axon Instruments, Foster City, CA). Data were analyzed off-line using pClamp 6 (Axon Instruments). The mIPSCs were detected and analyzed using Mini Analysis software (Synaptosotf, Fort Lee, NJ). The cumulative probability distributions were compared by the Kolmogorov-Smirnov test. All group data were expressed as means ± SEM. Statistical significance was assessed by Student’s t-test.
RESULTS
Activation of KAR depresses GABAergic synaptic transmission in GP
Previous studies have demonstrated that KARs modulate GABAergic transmission in the striatum and substantia nigra pars compacta (SNc) (Chergui et al., 2000; Nakamura et al., 2003). Our recent findings have shown GluR6/7 immunoreactivity in putative striatal GABAergic terminals in the monkey and rat GP (Jin et al., 2006; Kane-Jackson and Smith, 2003). We, therefore, examined whether bath application of KA modulates GABAergic transmission in slices of rat GP. GABAA receptor- mediated inhibitory post synaptic currents (IPSCs) were evoked every 20 seconds in GP neurons at holding potential of −10 mV in the presence of 50 μM D-AP5 and 100 μM GYKI 52466. Bath application of KA (0.3–3 μM) reversibly decreased IPSC amplitude in all cells tested (Fig. 1, A and B). On average, the IPSC amplitude was 76.5 ± 6.4% (n = 8, p < 0.001), 45.8 ± 4.76% (n = 13, p < 0.001) and 29.1 ± 3 (n = 7, p < 0.001) of control when perfused with 0.3, 1 and 3 μM KA, respectively (Fig. 1B). We did not detect any effect of KA on IPSC amplitude with KA concentrations below 0.3 μM (Fig. 1B). To confirm that the KA-induced IPSC inhibition was due to KAR activation, we tested the effect of 1 μM KA on IPSC amplitude in the presence of 50 μM CNQX, an AMPA/KA receptor antagonist. As demonstrated in figure 1B, we found no significant effect of KA on IPSC amplitude under this condition (91.2 ± 4.6% of control, n = 5, p > 0.05). The evoked inhibitory synaptic currents by striatal stimulation were GABAA receptor-mediated because they could be completely and reversibly blocked by bicuculline (20 μM) (n = 9) (Fig. 1, C and D), a selective GABAA receptor antagonist. All biocytin-filled recorded neurons were confined to the GP and displayed morphological features consistent with those described in our previous study (Jin et al., 2006).
Fig. 1.

Kainate receptor (KAR) activation depressed GABAergic synaptic transmission in the GP. A: time course of the effect of 1 μM KA on GABAA-mediated inhibitory postsynaptic current (IPSC) amplitude (pA) in the presence of 100 μM GYKI 52466 and 50 μM D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5). Three IPSCs are averaged in each trace at the time indicated by the corresponding letters in the graph. B: a summary bar graph showing that KA (0.3–3 μM) significantly reduced the IPSC amplitude and this effect was blocked when 1 μM KA was applied together with 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX) (50 μM). C: time course of the effect of bicuculline (20 μM) on IPSC amplitude in the presence of D-AP5 and CNQX. Three IPSCs are averaged in each trace at the time indicated by corresponding letters in the graph. D: a summary bar graph shows that 20 μM bicuculline completely blocked IPSC amplitude recorded from nine neurons. There was a significant difference from control: * P < 0.001. In this and subsequent figures, “NS” indicates non-significant differences and ‘n’ indicates the number of cells tested under each condition. All averaged data except figure 2, are presented as percent of control ± SEM.
KAR-mediated depression of GABAergic transmission in GP involves presynaptic mechanisms
Although 1 μM KA GluR6/7 induces small postsynaptic currents in GP neurons, these effects are small and almost assuredly do not account for a majority of the effect on glutamatergic transmission in rat GP (Jin et al. 2006). Therefore, one would predict that the effects KA (0.3–1 μM) on IPSCs evoked from the striatum are likely mediated by activation of presynaptic KARs. To test this hypothesis, we examined the effect of KA on PPF of evoked IPSCs. To record paired IPSCs, two stimuli of the striatum close to the GP were paired with an interstimulus interval of 40–50 ms. We then calculated ratio of peak2/peak1 in the presence or absence of KA (1 μM). The ratio of peak2/peak1 was significantly increased in the presence of KA compared to control (1.91 ± 0.1 and 1.25 ± 0.15, respectively. n = 6, p < 0.01) (Fig. 2A, B and C), indicating a presynaptic effect.
Fig. 2.
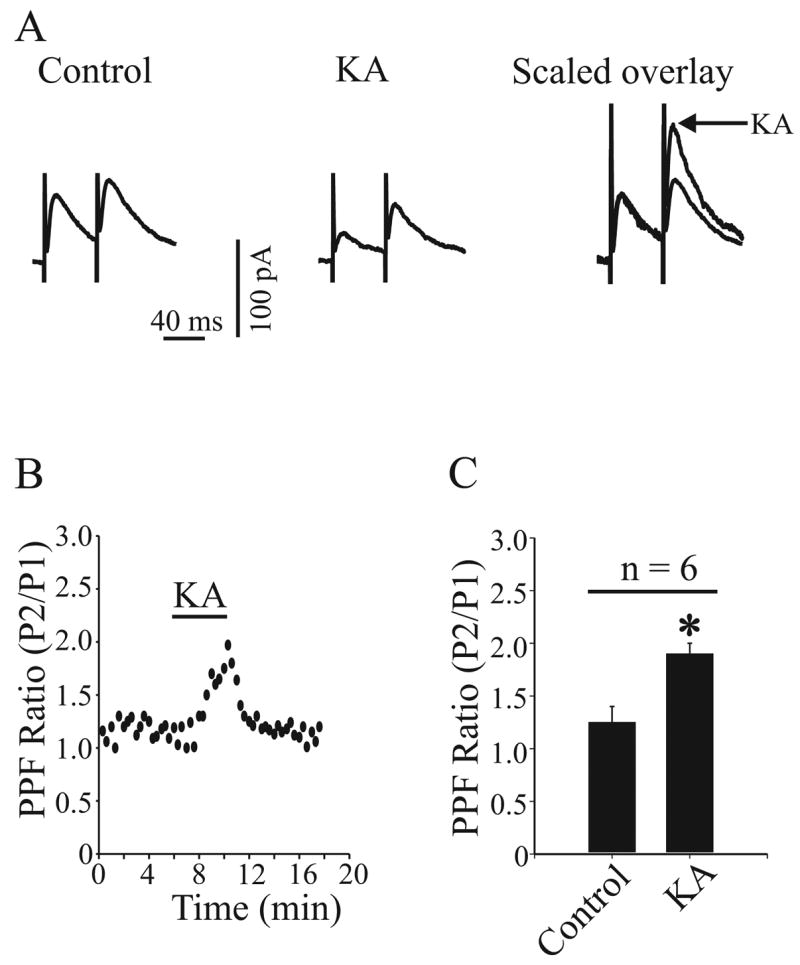
Kainate receptor (KAR) activation increased paired-pulse facilitation (PPF) at GABAergic synapse in the GP. A: paired IPSCs were recorded before (left trace) and during (middle) 1μM KA application. The right trace shows the KA-induced effect after scaling to the peak of the first IPSC. B: the same neuron presented in (A) shows the time course of increased paired-pulse facilitation ratio (PPFR) of IPSCs in response to 1 μM KA application. C: a bar graph shows that the PPFR expressed as a mean ratio of P2/P1 ± SEM was significantly increased in the presence of 1 μM KA (* P < 0.01).
To provide further evidence, we recorded mIPSCs from GP neurons using a high-Cl internal solution at holding potential −60 mV in the presence of 1 μM TTX, 100 μM GYKI 52466 and 50 μM D-AP5. Figure 3A shows that 1 μM KA application induced a significant reduction of the frequency of mIPSC. On average, the IPSC frequency was 79.4 ± 4% (n = 8, p < 0.005) and 63 ± 5.2% (n = 7, p < 0.005) of controls when perfused with 0.3 or 1μM KA respectively (Fig. 3E). This inhibitory effect of KA on mIPSCs frequency was blocked by 50 μM CNQX (101 ± 7.4%, p > 0.5, n = 5) (Fig. 3E). The inter-mIPSC intervals were significantly increased following 1 μM KA application (P < 0.01, Kolmogorov-Smirnov test, Fig. 3C). In contrast, KA (0.3 and 1 μM) had no significant effect on their mean amplitude (96 ± 5%, p > 0.5, n = 8; 99.8 ± 11.75%, p > 0.05, n = 7, Fig. 3F) or the amplitude distribution (Fig. 3D) of mIPSCs. We confirmed that mIPSCs were GABAA receptor-mediated events since they were completely blocked by 20 μM bicuculline (Fig. 3B, E and F). Taken together, these data strongly support the hypothesis that KA-induced inhibition of GABAergic transmission at striatopallidal synapses is mediated by presynaptic mechanisms.
Fig. 3.

Kainate receptor (KAR) activation reduced the frequency but not the amplitude of miniature IPSCs (mIPSCs). A: Sample traces show mIPSCs before, during and after 1 μM KA application. These mIPSCs were recorded in the presence of 100 μM GYKI 52466, 50 μM D-AP5 and 1μM TTX. B: Sample traces show mIPSCs before, during and after 20 μM bicuculline application. After three mins application of bicuculline, mIPSCs were completely abolished (bottom trace). C and D: The cumulative distributions of the inter-mIPSC intervals and cumulative amplitude distributions of mIPSCs obtained from the same neuron as in panel A. KA (1 μM) significantly shifted the inter event interval distribution curve to the right (left, P < 0.05), but had no significant effect on the distribution of mIPSCs amplitude (right, P > 0.5). E: A summary bar graph shows KA (0.3–1 μM) significantly reduced the frequency of mIPSCs, which was blocked in the presence of 50 μM CNQX. F: A summary bar graph shows that neither KA nor KA together with CNQX affected the amplitude of mIPSCs. Both the frequency and amplitude of mIPSCs were significantly blocked by 20 μM bicuculline (* P < 0.005).
KA- induced modulation of GABAergic transmission in GP requires activation of NEM toxin-sensitive G-protein
Data obtained so far indicate that KARs mediate their effects through metabotropic and/or iontropic mechanisms (Rodriguez-Moreno and Lerma, 1998). We have recently shown that KAR-mediated inhibitory effect on glutamatergic transmission in the rat GP involves a G-protein-dependent signal transduction pathway (Jin et al., 2006). Therefore, we tested whether the KA-induced depression of GABAergic transmission was also due to a G-protein-coupled transduction cascade. First, we studied the effect of the G-protein inhibitor NEM (200 μM) on the KA induced presynaptic inhibition of evoked IPSCs. As reported in several brain regions and spinal cord, bath application of NEM alone increased synaptic transmission (Frerking et al., 2001; Kubota et al., 2003; Rozas et al., 2003 Jin et al., 2006). The amplitude of IPSC was 132 ± 5.3% of control in the presence of NEM (n = 7, P < 0.001) (Fig. 4A and B). After 15 min of NEM perfusion, application of KA had no effect on IPSC amplitude. The IPSC amplitude in the presence of KA together with NEM was 122.6 ± 4% of control, which was not significantly different from the amplitude of IPSC recorded with NEM alone (132 ± 5.3%, P > 0.5, n = 7) (Fig. 4B).
Fig. 4.

Application of G-protein inhibitor, N-ethylmaleimide (NEM) but not G protein coupled receptors antagonist blocked the KAR activation-induced inhibition on IPSCs. A: The time course of the effect of 1 μM on IPSC amplitude in the presence of 200 μM NEM. Three IPSCs are averaged in each trace at the time indicated by corresponding letters in the graph. B: A summary bar graph shows that the inhibitory effect of KA on IPSC amplitude was blocked in the presence of NEM. C: Bar graph shows that 1 μM KA significantly reduced the IPSCs amplitude in the presence of mGluR antagonist (1 mM MCPG and 100 μM CPPG), the GABAB antagonist (20 um SCH 50911), the adenosine receptor antagonist (10μM CGS 15943), or dopamine receptor antagonists (10 μM SCH 23390 and 10 μM sulpiride).
Since several studies have reported that KA-induced inhibition of GABAergic transmission may involve G-protein-coupled GABAB and adenosine receptors (Frerking et al., 1999; Chergui et al., 2000; Nakamura et al., 2003), we performed experiments to test whether the KA-induced inhibition of evoked IPSCs in the GP was due to the secondary activation of G-protein-coupled receptors. Slices were pretreated with a metabotropic glutamate receptor antagonist cocktail (1 mM MCPG and 100μM CPPG), a GABAB receptor antagonist (20 μM SCH 50911), a dopamine receptor antagonist cocktail (10 μM sulpiride and 10 μM SCH 23390) or a non-selective adenosine receptor antagonist (10 μM CGS 15943) for 10 min prior to bath application of KA. Pretreatment with any of these antagonists had no significant effect on KA-induced inhibition of evoked IPSCs (Fig. 4C).
To test whether activation of G-protein was also required for KA-induced regulation of spontaneous GABA release, we examined the effect of NEM (200 μM) on KA-induced inhibition of mIPSCs. As expected, NEM increased the mIPSC frequency (Fig. 5A, D). In the cumulative distributions of the inter-mIPSC intervals, 200 μM significantly shift the curve towards shorter inter-event intervals (P < 0.01, Kolmogorov-Smirnov test, Fig. 5B). On average, the frequency of mIPSCs was 279 ± 44% (P < 0.05, n = 6) of control after NEM application (Fig. 5D). However, NEM did not affect either the amplitude distribution (Fig. 5C) or mean amplitude (111 ± 7%, P > 0.5, n = 6, Fig. 5D) of mIPSCs. After 15 min of NEM perfusion, KA had no significant effect on mIPSC frequency and amplitude. The mIPSC frequency and amplitude in the presence of KA together with NEM was 269 ± 40% and 102 ± 7.1% of control respectively, which was not significantly different from the frequency and amplitude of mIPSC recorded with NEM alone (279 ± 44%, P > 0.5, 111 ± 7%, P > 0.5, n = 6) (Fig. 5D and E). Taken together, these results suggest that the inhibitory effect of KA on evoked IPSC and mIPSC is modulated through NEM-sensitive G protein.
Fig. 5.

Application of G-protein inhibitor (NEM) blocked the KAR activation-induced inhibition on mIPSCs frequency. A: Sample traces show mIPSCs in control condition (left trace), in the presence of 200 μM NEM (middle trace) or after the application of NEM together with 1 μM KA (right trace). B and C: The cumulative distributions of the inter-mIPSC intervals and cumulative amplitude distributions of mIPSCs obtained from the same neuron as in A. KA (1 μM) did not show any significant effect on the either the inter event interval (B) or the amplitude distribution (C) curves of mIPSCs in the presence of NEM (P > 0.5). D and E: A summary bar graph shows that KA had no effect on either mIPSCs frequency or amplitude in the presence of NEM.
KA- induced inhibition of GABAergic transmission requires activation of PKC
To examine whether protein kinase activation downstream of the G protein activity is needed for the effect of KA on GABAergic transmission in the rat GP, we performed the following experiments. First, we tested the effect of KA on evoked IPSC amplitude after pretreatment of slices with staurosporine (0.5 μM, 2–4 h), a broad-spectrum inhibitor of protein kinase. As shown in figure 6B, staurosporine significantly prevented the inhibitory effect of KA on evoked IPSC amplitude (100.3 ± 3% of control, n = 6, P > 0.5). We then performed further experiments to address which protein kinase might be responsible for the effect of staurosporine on KA-induced inhibition on IPSC amplitude. Pretreatment of slices with H-89 (0.5 μM, 2–4 h), a selective PKA inhibitor, did not prevent the effect of KA on IPSC amplitude (49.2 ± 2.7% of control, n = 5, P < 0.005) (Fig. 6B). However, the effect of KA on evoked IPSC amplitude was significant blocked (98 ± 3.1% of control, n = 6, P > 0.05) (Fig. 6A and B) after slices were incubated with calphostin C (1 μM, 2–4 h), a specific PKC inhibitor. We also found that pretreatment of slices with staurosporine or calphostin C but not H-89 prevented the effect of KA on mIPSC frequency (Fig. 7A–D). The mean mIPSC frequency was 95 ± 5%, 91.4 ± 4.7% and 62.4 ± 6.5% of control in the presence of staurosporine, calphostin and H-89 respectively (n = 6, P > 0.05, n = 7, P > 0.05 and n = 7, P < 0.05 respectively, Fig. 7E). These results demonstrate that activation of PKC, but not PKA, is involved in KA-induced inhibition of GABAergic transmission in rat GP.
Fig. 6.
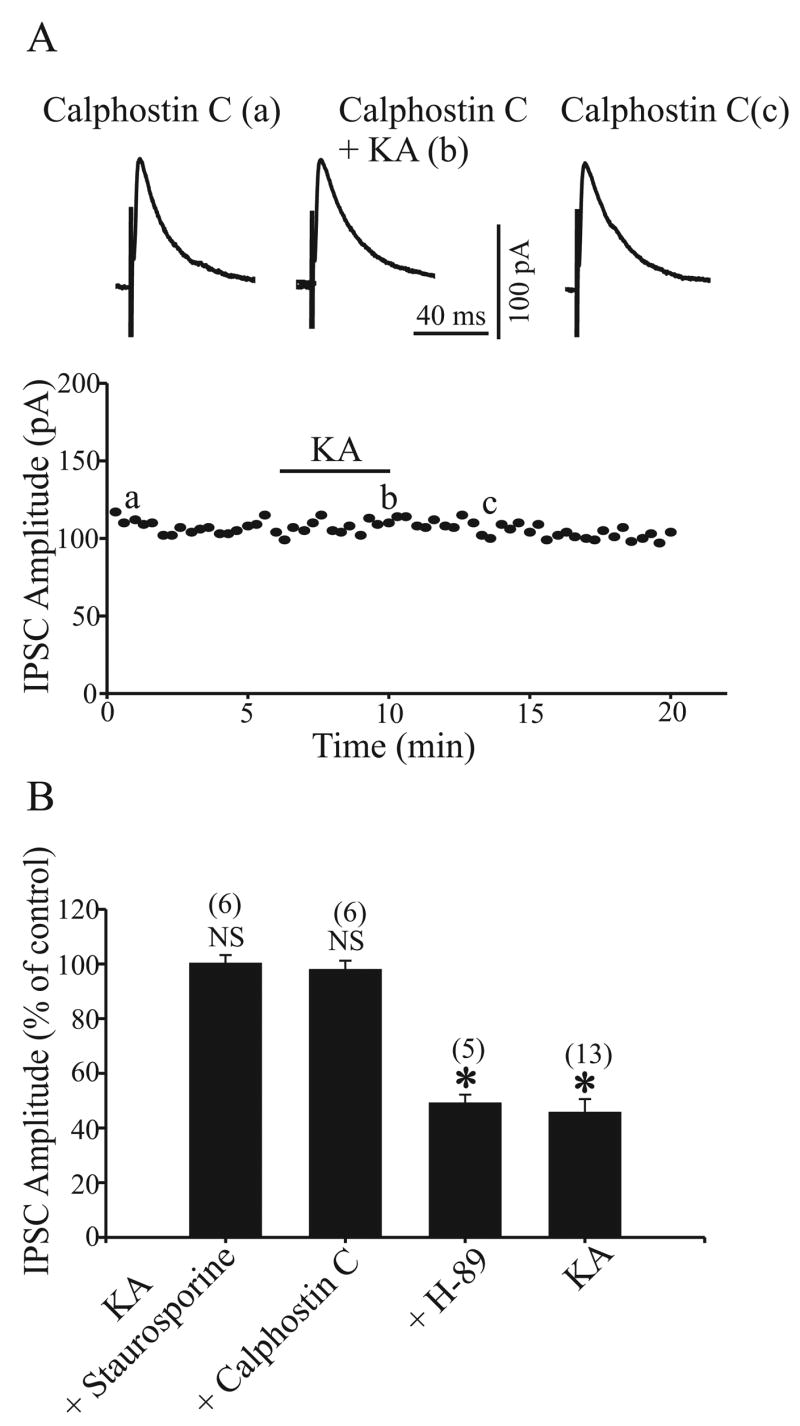
Pretreatment with protein kinase C inhibitor (calphostin C) but not protein kinase A inhibitor (H-89) prevents KAR activation-induced inhibition on IPSCs. A: The time course of the effect 1 μM KA on IPSC amplitude in the presence of 1 μM calphostin C. Three IPSCs are averaged in each trace at the time indicated by corresponding letters in the graph. B: A bar graph shows that the KAR activation-induced inhibition on IPSCs was blocked by 0.5 μM staurosporine, a broad-spectrum inhibitor for protein kinase and calphostin C but not H-89. For comparison, the effect of 1 μM KA on IPSCs in control conditions from Fig. 1B is shown. There was a significant difference from control, * P < 0.01.
Fig. 7.

Pretreatment with protein kinase C inhibitor (calphostin C) but not protein kinase A inhibitor (H-89) prevents KAR activation-induced inhibition of mIPSCs. A: mIPSCs were recorded in the presence of 1 μM calphostin C and calphostin C together with 1 μM KA. B: mIPSCs were recorded in the presence of 0.5 μM H-89 and H-89 together with 1 μM KA. C and D: the cumulative distributions of the inter-mIPSC intervals and amplitude distributions of mIPSCs obtained from the same neuron as in A. KA had no effect on inter-event interval (left) and mIPSCs amplitude (right) distribution curves in the presence of calphostin C (P > 0.5). E: summary bar graph shows that KA had no effect on mIPSCs frequency in the presence of 0.5 μM staurosporine and calphostin C but not in the presence of H-89. F: summary bar graph shows that KA had no effect on mIPSCs amplitude in the presence of staurosporine, calphostin C and H-89. * P < 0.05.
DISCUSSION
The findings presented in this study demonstrate that activation of presynaptic KARs suppresses GABAergic transmission in the GP through a G-protein-dependent mechanism that involves PKC. Together with our recent data showing that KA also regulates glutamatergic transmission (Jin et al., 2006), these findings indicate that KARs are located to subserve subtle, but widespread, regulatory influences towards excitatory and inhibitory synaptic transmission in the GP.
Presynaptic KAR modulation of GABAergic transmission
It has been reported that inhibition of GABAergic synaptic transmission by KA is mediated, at least in part, by activation of presynaptic KARs in the hippocampus (Clarke et al., 1997; Rodriguez-Moreno et al., 1997; Rodriguez-Moreno et al., 1998; Maingret et al., 2005) and SNc (Nakamura et al., 2003). Similarly, our results show that KA (03–1 μM)-induced inhibition of GABAergic transmission is associated with a significant increase in PPF ratio of evoked IPSC and decrease in the frequency, but not amplitude of mIPSCs in the rat GP. These results are consistent with our previous electron microscopic immunocytochemical data showing that GluR6/7 immunoreactivity is expressed in GABAergic axon terminals in rat GP (Jin et al., 2006). This effect is unlikely due to changes in the membrane properties of GP neuron since 1 μM KA does not induce significant whole-cell membrane current or depolarization (Jin et al., 2006). However, 1 μM KA could induce dendritic conductance changes that are unlikely to be monitored by somatic recordings. Thus, we cannot rule out that such dendritic effects may contribute to some of the KA-induced inhibition of evoked and miniature IPSCs.
Since some of the GP neurons project back to the striatum and have local axon collaterals in the GP (Kita and Kitai, 1991; Bevan et al., 1998; Smith et al., 1998; Kita et al., 1999; Kita and Kita, 2001; Sadek et al., 2007), electrical stimulation of the striatum could activate both striatopallidal and pallidostriatal fibers. Because it is technically not possible to clearly discriminate striatopallidal from pallidostriatal IPSCs in our preparation, we cannot rule out that both sets of afferents could potentially contribute to the evoked IPSCs recorded in the present study. However, our electron microscopic immunocytochemical data showing that GluR6/7 immunoreactivity is expressed in GABAergic terminals morphologically similar to striatal, but not pallidal boutons (Smith et al., 1998; Jin et al., 2006), strongly suggest that striatopallidal axon terminals are likely to be the main targets of KAR-mediated effect on IPSCs in the GP.
KAR activation can modulate GABA release in a bidirectional manner in the hippocampus (Jiang et al., 2001) and basolateral amygdala (Braga et al., 2003). For instance, bath application of a low dose of KA (0.3 μM) and ATPA (0.3 μM) facilitates GABA release, whereas a high dose of KA (5 μM) or ATPA (10 μM) inhibits GABAergic transmission (Jiang et al., 2001; Braga et al., 2003). In the present study, the amplitude of evoked IPSCs or the frequency of miniature IPSCs were constantly reduced by bath application of different concentrations (0.3–1 μM) of KA. We did not find any effect of KA on either evoked or mIPSC when concentrations lower than 0.3 μM KA were applied, indicating that presynaptic KAR activation does not modulate GABA release in a bidirectional manner in the rat GP. It is not clear why we could not find the facilitatory effects of low KA concentration (<300nM) on GABAergic transmission in the GP. One possibility is that the GluR5 subunit is not part of KARs in GP, while it is a constitutive subunit of KARs in the amygdala and hippocampus (Bischoff et al., 1997; Braga et al., 2003). It is also possible that activation of axonal KARs mediates this facilitatory effect in the hippocampus and amygdala, while in the GP the KARs-induced regulation of GABAergic transmission may be largely mediated by pre-synaptic KARs in GABAergic terminals.
Taking into consideration that axo-axonic synapse are rare in the GP, extrasynaptic glutamate spillover and /or glial release of glutamate are the two most likely sources of transmitter to activate these presynaptic heteroreceptors. The GP receives its main glutamatergic innervation from STN though modest inputs have also been described from the thalamus and brainstem (Kincaid et at., 1991; Naito and Kita, 1994; Shink and Smith, 1995; Mouroux et al., 1997). In contrast to many brain regions, glutamate transporter 1(GLT-1)-immunoreactive astrocytic processes do not reach the synaptic cleft of glutamatergic synapse in the GP (Charara et al., 2002). They rather tightly ensheat axo-dendritic complexes (Charara et al., 2002), which suggests that synaptically released glutamate could spill over from glutamatergic synapse to activate KARs in neighboring GABAergic terminals. The close spatial arrangement of glutamatergic and GABAergic terminals onto individual GP dendrites favors this possibility (Shink and Smith, 1995; Smith et al., 1998). A similar mechanism has been described for mGluR-mediated inhibition of GABAergic transmission in other brain regions (Mitchell and Silver, 2000; Semyanov and Kullmann, 2000; Valenti et al., 2003). In addition to synaptic glutamate spillover, non-synaptic glial release of glutamate should also be considered as a potential source of activating transmitter for these receptors (Bezzi et al., 2004; Volterra and Steinhauser, 2004; Volterra and Meldolesi, 2005; Jourdain et al., 2007). Future studies are needed to determine the physiological or pathological conditions necessary for synaptically released glutamate to activate presynaptic KARs on GABAergic terminals in the GP.
Mechanism(s) underlying KAR-mediated presynaptic regulation of GABAergic transmission
Several hypotheses have been proposed to explain the possible mechanism (s) underlying the presynaptic effects of KARs on GABAergic synaptic transmission (for reviews, see Huettner, 2003; Lerma, 2003). For example, a direct depolarization of GABAergic axon terminals through Ca2+ permeable GluR5-containing KARs (Braga et al., 2003) or activation of axonal KARs to bring the axon close to firing threshold, could account for KA-mediated increase in GABA release (Semyanov and Kullmann, 2001). In the present study we show that presynaptic activation of KARs reduces the amplitude of evoked IPSCs and decreases the frequency of mIPSCs. As mentioned above, differences in subunit composition may account for this differential degree of kainate sensitivity between GP and hippocampal KARs (see above).
Another alternative is that KA activates somatodendritic KARs on local neurons to cause the release of neurotransmitters, such as adenosine or GABA, which in turn, could activate GABAB and A2A receptors heterosynaptically on neighboring boutons to modulate the release of GABA (Frerking et al., 1999; Chergui et al., 2000; Nakamura et al., 2003). This is unlikely to be the case in GP, since blockade of GABAB, mGluRs, adenosine and dopamine receptors did not have any significant effects on KAR-mediated inhibition of GABAergic transmission, which is consistent with data from other brain regions (Rodríguez-Moreno and Lerma, 1998; Rodríguez-Moreno et al., 2000; Braga et al., 2003; Maingret et al., 2005).
There is a growing body of evidence suggesting that presynaptic KAR activation could modulate GABAergic and glutamatergic transmission via a direct, presynaptic metabotropic action (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Rodríguez-Moreno et al., 2000; Frerking et al., 2001; Lauri et al., 2005; Jin et al., 2006). In these studies, the presynaptic inhibitory effects of KARs were abolished by G-protein inhibitors (NEM) or pertussis toxin (PTx) (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Frerking et al., 2001; Jin et al., 2006), but not by KA-mediated depolarization (Rodríguez-Moreno and Lerma, 1998; Rodríguez-Moreno et al., 2000). Furthermore, recent studies indicated that PKC activation downstream of G-protein activity is essential for KAR-mediated pre and post-synaptic effects. For instance, KA-induced presynaptic inhibition of GABAergic or glutamatergic transmission and KA-mediated postsynaptic inhibition of slow afterhyperpolarization currents (IsAHP) were blocked by PKC inhibitor (Calphostin C) but not by PKA inhibitor (H-89) (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Rodríguez-Moreno et al., 2000; Frerking et al., 2001; Melyan et al., 2002, 2004; Lauri et al., 2005; Jin et al., 2006). In line with these findings, we showed that presynaptic KAR-mediated effects on evoked and mIPSCs were blocked by NEM and Calphostin but not by H-89 in the rat GP. These results therefore, demonstrate that KAR-induced depression of GABAergic synaptic transmission in the rat GP requires G-protein and PKC activation but does not rely on the secondary activation of G protein-coupled receptors. Thus, together with our recent study showing KAR-mediated regulation of glutamatergic transmission (Jin et al., 2006), these findings demonstrate that KARs mediate their presynaptic effects on both GABAergic and glutamatergic transmission in the GP through a metabotropic mode of action.
Functional implications
The KAR-induced presynaptic inhibition of evoked and mIPSCs suggests KARs can function as heteroreceptors to modulate GABAergic transmission at the striatopallidal synapse. Knowing that increased GABAergic transmission from striatum to GP is one of the cardinal features of Parkinson’s disease (PD) pathophysiology (DeLong, 1990), KAR activation may have beneficial effects in PD through attenuation of the overactive “indirect” GABAergic outflow from the striatum. It is noteworthy that other ionotropic or metabotropic glutamate receptor antagonists have proven to be successful in alleviating parkinsonian symptoms in both rat and primate models of PD (Brotchie et al., 1991; Klockgether et al. 1991; Greenamyre et al., 1994; Conn et al., 2005). However, the use of AMPA/NMDA receptor antagonists in humans is severely hampered by the widespread distribution, fast excitatory effects and need of these receptors for normal brain functioning. In contrast, KARs mediate slower modulatory effects that resemble those induced by mGluRs activation. It is noteworthy that blockade of mGluR5 receptors or activation of group III mGluRs in the GP alleviates akinesia by normalizing activity of selective basal ganglia structures in parkinsonian rats (Breysse et al., 2003; Marino et al., 2003; Valenti et al., 2003; Lopez et al., 2007). Through the possible regulation of overactive striatopallidal GABAergic synapses and glutamatergic subthalamopallidal transmission (Jin et al., 2006), KARs represent an additional target of interest for the development of novel pharmacotherapeutic approaches in Parkinson’s disease.
Acknowledgments
We thank Jean-Francois Paré and Susan Maxson for technical assistance. We also thank Dr Don Rainnie for a critical reading of the manuscript. This research was supported by a grant from the US Army, the Yerkes Primate Center NIH base Grant and an Award from Merck/Center for Neurodegenerative Disease at Emory University.
Abbreviations
- ABC
avidin-biotin-peroxidase complex
- ACSF
artificial cerebrospinal fluid
- AMPA
α-amino-3-hydroxyl-5-methyl-4-isoxazole propionic acid
- ATPA
(RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid
- CNQX
6-cynao-7-nitroquinoxaline-2, 3-dione
- CPPG
(RS)-α-cyclopropyl-4-phosphonophenylglycine
- DAB
3,3-diaminobenzidine tetrahydrochloride
- D-AP5
D-(-)-2-amino-5-phosphonopentanoic acid
- IPSCs
inhibitory post synaptic currents
- GLT-1
glutamate transporter 1
- GP
globus pallidus
- GYKI 52466
4-(8-Methyl-9H-1,3-dioxolo[4,5 h]{2,3}benzodiazepine-5-yl)-benzenamine hydrochloride
- KARs
kainate receptors
- MCPG
α-Methyl-4-carboxyphenylglycine
- NEM
N-ethylmaleimide
- NMDA
N-methyl-D-aspartate
- PD
Parkinson’s disease
- PBS
phosphate-buffered saline
- PKA
protein kinase A
- PKC
protein kinase C
- PPF
paired-pulse facilitation
- QX314
N-(2,6-Dimethylphenylcarbamoylmethyl) triethylammonium bromide
- SNc
substantia nigra parts
- STN
subthalamic nucleus
- TTX
tetrodotoxin
Footnotes
I have read and have abided by the Authorship Responsibility, Financial Disclosure, and Acknowledgment Form.
Section Editor: Dr. Menahem Segal, Weizmann Institute of Science, Department of Neurobiology, Hertzl Street, Rehovot 76100, Israel.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhäuser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nature Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- Bettler B, Mulle C. Neurotransmitter receptors II. AMPA and kainate receptors. Neuropharmacology. 1995;34:123–139. doi: 10.1016/0028-3908(94)00141-e. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: Oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 2002;25:525. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Booth PA, Eaton SA, Bolam JP. Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci. 1998;18:9438–9452. doi: 10.1523/JNEUROSCI.18-22-09438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff S, Barhanin J, Bettler B, Mulle C, Heinemann S. Spatial distribution of kainate receptor subunit mRNA in the mouse basal ganglia and ventral mesencephalon. J Comp Neurol. 1997;379:541–562. doi: 10.1002/(sici)1096-9861(19970324)379:4<541::aid-cne6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Braga MFM, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23 (2):442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breysse N, Amalric M, Salin P. Metabotropic glutamate 5 receptor blockade alleviates akinesia by normalizing activity of selective basal-ganglia structures in parkinsonian rats. J Neurosci. 2003;23:8302–8309. doi: 10.1523/JNEUROSCI.23-23-08302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotchie JM, Mitchell IL, Sambrook MA, Crossman AR. Alleviation of parkinsonism by antagonism of excitatory amino acid transmission in the medial segment of the globus pallidus in rat and primate. Mov Disord. 1991;6:133–138. doi: 10.1002/mds.870060208. [DOI] [PubMed] [Google Scholar]
- Charara A, Blankstein E, Smith Y. Presynaptic kainate receptors in the monkey striatum. Neuroscience. 1999;91(4):1195–200. doi: 10.1016/s0306-4522(99)00099-8. [DOI] [PubMed] [Google Scholar]
- Charara A, Paquet M, Rothstein JD, Smith Y. Subcellular and subsynaptic localization of glutamate transporters in the monkey basal ganglia. The Basal Ganglia. 2002;VI:599–613. [Google Scholar]
- Chergui K, Bouron A, Normand E, Mulle C. Functional GluR6 kainate receptors in the striatum: indirect downregulation of synaptic transmission. J Neurosci. 2000;20:2175–2182. doi: 10.1523/JNEUROSCI.20-06-02175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nature Reviews Neuroscience. 2005;6(10):787–98. doi: 10.1038/nrn1763. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Stanford IM. Electrophysiological and morphological characteristics of three subtypes of rat globus pallidus neurons in vitro. J Physiol. 2000;527:291–304. doi: 10.1111/j.1469-7793.2000.t01-1-00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder TL, Weiner JL. Functional characterization of kainate receptors in the rat nucleus accumbens core region. J Neurophysiol. 2002;88:41–48. doi: 10.1152/jn.2002.88.1.41. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Malva JO, Ribeiro JA. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [3H] GABA release. FEBS Letters. 2000;469(2–3):159–62. doi: 10.1016/s0014-5793(00)01272-2. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Frerking M, Petersent CC, Nicoll RA. Mechanisms underlying kainate receptor-mediated disinhibition in the hippocampus. Proc Natl Acad Sci USA. 1999;96:12917–12922. doi: 10.1073/pnas.96.22.12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Nicoll RA. Synaptic kainate receptors. Curr Opin Neurobiol. 2000;10:342–351. doi: 10.1016/s0959-4388(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3—CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21(9):2958–66. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, Eller RV, Zhang Z, Ovadia A, Kurlan R, Gash DM. Antiparkinsonian effects of remacemide hydrochloride, a glutamate antagonist, in rodent and primate models of Parkinson’s disease. Ann Neurol. 1994;35(6):655–661. doi: 10.1002/ana.410350605. [DOI] [PubMed] [Google Scholar]
- Hollman M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Kainate receptors and synaptic transmission. Prog in Neurobiol. 2003;70(5):387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- Jiang Li, Xu J, Nedergaard M, Kang J. A kainate receptor increases the efficacy of GABAergic synapses. Neuron. 2001;30:503–513. doi: 10.1016/s0896-6273(01)00298-7. [DOI] [PubMed] [Google Scholar]
- Jin X-T, Smith Y. Pre-synaptic kainate receptor modulates GABAergic and glutamatergic transmission in the rat globus pallidus. Soc Neurosci Abstr. 2005;631:8. [Google Scholar]
- Jin X-T, Smith Y. Pre-synaptic kainate receptor activation modulates GABAergic transmission in the rat globus pallidus. Soc Neurosci Abstr. 2006:254.3. doi: 10.1016/j.neuroscience.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X-T, Paré JF, Raju DV, Smith Y. Localization and function of pre-and postsynaptic kainate receptors in the rat globus pallidus. Eur J Neurosci. 2006;23:374–386. doi: 10.1111/j.1460-9568.2005.04574.x. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nature Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kane-Jackson R, Smith Y. Pre-synaptic kainate receptors in GABAergic and Glutamatergic axon terminals in the monkey globus pallidus. Neuroscience. 2003;120:285–289. doi: 10.1016/s0306-4522(03)00596-7. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 1990;10:3421–3438. doi: 10.1523/JNEUROSCI.10-10-03421.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieval JZ, Hubert GW, Charara A, Pare JF, Smith Y. Subcellular and subsynaptic localization of presynaptic and postsynaptic kainate receptor subunits in the monkey striatum. J Neurosci. 2001;21:8746–8757. doi: 10.1523/JNEUROSCI.21-22-08746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic and nigral stimulation. Brain Res. 1991;564:296–305. doi: 10.1016/0006-8993(91)91466-e. [DOI] [PubMed] [Google Scholar]
- Kita H, Tokuno H, Nambu A. Monkey globus pallidus external segment neurons projecting to the neostriatum. Neuro Report. 1999;10:1467–1472. doi: 10.1097/00001756-199905140-00014. [DOI] [PubMed] [Google Scholar]
- Kita H, Kita T. Number, origins, and chemical types of rat pallidostriatal projection neurons. J Comp Neurol. 2001;437:438–448. doi: 10.1002/cne.1294. [DOI] [PubMed] [Google Scholar]
- Klockgether T, Turski L, Honore T, Zhang ZM, Gash DM, Kurlan R, Greenamyre JT. The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine-depleted and MPTP-treated monkeys. Ann Neurol. 1991;30:717–723. doi: 10.1002/ana.410300513. [DOI] [PubMed] [Google Scholar]
- Kubota H, Katsurabayashi S, Moorhouse AJ, Murakami N, Koga H, Akaike N. GABAB receptor transduction mechanisms, and cross-talk between protein kinase A and C, in GABAergic terminals synapsing onto neurons of the nucleus basalis of Meynert. J Physiol. 2003;551:263–276. doi: 10.1113/jphysiol.2003.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Presynaptic kainate receptors in the hippocampus: slowly emerging from obscurity. Neuron. 2001;32:561–564. doi: 10.1016/s0896-6273(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Segerstrale M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. J Neurosci. Vol. 25. 2005. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus; pp. 4473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodriguez-Moreno A, Lopez-Garcia JC. Molecular physiology of kainate receptors. Physiol Rev. 2001;81:971–998. doi: 10.1152/physrev.2001.81.3.971. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nature Reviews Neuroscience. 2003;4(6):481–95. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Acher F, Leonibus ED, Mele A, Amalric M. Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson’s disease. J Neurosci. 2007;27(25):6701–6711. doi: 10.1523/JNEUROSCI.0299-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Lauri SE, Taira T, Isaac JT. Profound regulation of neonatal CA1 rat hippocampal GABAergic transmission by functionally distinct kainate receptor populations. J Physiol. 2005;567:131–42. doi: 10.1113/jphysiol.2005.089474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, Conn PJ. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc NatI Acad Sci U S A. 2003;23:13668–73. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Lancaster B, Wheal HV. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci. 2004;24:4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34(1):107–14. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Mouroux M, Hassani OK, Féger J. Electrophysiological and Fos immunohistochemical evidence for the excitatory nature of the parafascicular projection to the globus pallidus. Neuroscience. 1997;81:387–397. doi: 10.1016/s0306-4522(97)00110-3. [DOI] [PubMed] [Google Scholar]
- Naito A, Kita H. The cortical-pallidal projection in the rat: an anterograde tracing study with biotinylated dextran amine. Brain Res. 1994;653:251–257. doi: 10.1016/0006-8993(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Jang I1-Sung, Ishibaski H, Watanabe S, Akaike N. Possible roles of kainate receptors on GABAergic presynaptic nerve terminals projecting to rat substantia nigra dopaminergic neurons. J Neurophysiol. 2003;90:1662–1670. doi: 10.1152/jn.01165.2002. [DOI] [PubMed] [Google Scholar]
- Parent A, Charara A, Pinault D. Single striatofugal axons arborizing in both pallidal segments and in the substantia nigra in primates. Brain Res. 1995;698:280–284. doi: 10.1016/0006-8993(95)01017-p. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kitai ST. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400 (6745):677–82. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- Poisik OV, Mannaioni S, Traynelis S, Smith Y, Conn PJ. Distinct functional roles of the metabotropic glutamate receptors 1 and 5 in the rat globus pallidus. J Neurosci. 2003;23:122–120. doi: 10.1523/JNEUROSCI.23-01-00122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-I/EAAT2 subtype. Neurochem Int. 1999;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Lopez-Garcia JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci USA. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- Rozas TL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Sadek AR, Magill PJ, Bolam JP. A single-cell analysis of intrinsic connectivity in the rat globus pallidus. J Neurosci. 2007;27(24):6352–6362. doi: 10.1523/JNEUROSCI.0953-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Modulation of GABAergic signaling among interneurons by metabotropic glutamate receptors. Neuron. 2000;25:663–672. doi: 10.1016/s0896-6273(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Kainate receptor-dependent axonal depolarization and action potential initiation in interneurons. Nature Neurosci. 2001;4:718–723. doi: 10.1038/89506. [DOI] [PubMed] [Google Scholar]
- Shink E, Smith Y. Differential synaptic innervation of neurons in the internal and external segments of the globus pallidus by the GABA- and glutamate-containing terminals in the squirrel monkey. J Comp Neurol. 1995;358:119–141. doi: 10.1002/cne.903580108. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86(2):353–87. doi: 10.1016/s0306-4522(98)00004-9. [DOI] [PubMed] [Google Scholar]
- Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, Conn PJ. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci. 2003;23(18):7218–26. doi: 10.1523/JNEUROSCI.23-18-07218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nature Reviews Neuroscience. 2005;6(8):626–40. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Volterra A, Steinhauser C. Glial modulation of synaptic transmission in the hippocampus. GLIA. 2004;47(3):249–57. doi: 10.1002/glia.20080. [DOI] [PubMed] [Google Scholar]
- Wichmann T, DeLong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurgery Clinics of North America. 1998;9(2):223–36. [PubMed] [Google Scholar]