

Abstract
Rit is a novel member of the Ras superfamily of small GTP-binding proteins that regulates signaling pathways controlling cellular fate determination. Constitutively activated mutants of Rit induce terminal differentiation of pheochromocytoma (PC6) cells resulting in a sympathetic neuron-like phenotype characterized by the development of highly-branched neurites. Rit signaling has been found to activate several downstream pathways including MEK/ERK, p38 MAPK, Ral-specific guanine nucleotide exchange factors (GEFs), and Rit associates with the Par6 cell polarity machinery. In this study, a series of Rit effector loop mutants was generated to test the importance of these cellular targets to Rit-mediated neuronal differentiation. We find that Rit-mediated neuritogenesis is dependent upon MEK/ERK MAP kinase signaling but independent of RalGEF activation. In addition, in vivo binding studies identified a novel mechanism of Par6 interaction, suggesting that the cell polarity machinery may serve to spatially restrict Rit signaling.
Keywords: Neuronal differentiation, PC12 cell, Rit, GTPase, Ras, ERK MAP kinase, Par6
Introduction
The Ras superfamily of small GTPases is composed of more than 200 members that act as molecular switches at key regulatory nodes to direct a variety of cellular functions [1]. Within this varied superfamily, the Ras subfamily is particularly important for the regulation of cellular proliferation, differentiation, and survival [1–3]. The exchange of bound GDP for GTP, stimulated by external cues that activate guanine nucleotide exchange factors (GEFs), results in a conformational rearrangement of the G-protein exposing previously buried residues including a region known as the effector domain, which is primarily responsible for the interaction with, and activation of, downstream signaling partners [4–6]. Determining the identity of effector protein targets and understanding how their activation leads to alterations in cellular function are important goals in Ras-mediated signal transduction [7–10].
One recently identified branch of the Ras subfamily is composed of Rit, Rin and Drosophila Ric proteins [11, 12]. These GTPases contain a unique effector binding domain and lack the usual C-terminal signal for lipid addition, suggesting that they may function to regulate signaling pathways distinct from Ras [11, 13]. Initially investigated for their abilities to promote tumor formation in NIH 3T3 fibroblasts, only a constitutively activated version of Rit, RitL79, was found to support tumorigenic growth in a manner distinct from that for oncogenic Ras [14, 15]. However, like Ras, Rit binds and activates the Ral GEF, RGL3, to activate Ral GTPase signaling [16].
While only Rit was found to promote NIH 3T3 cell transformation, all members of this subfamily potently stimulate differentiation in rat pheochromocytoma (PC12) cells [17–21]. PC12 cells are a well-defined model system that has been used extensively to examine the extracellular stimuli and intracellular signaling cascades that regulate neuronal differentiation [22–28]. Nerve Growth Factor (NGF) stimulation promotes neurite outgrowth in PC12 cells, induces differentiation and survival of neurons in vivo, and leads to the activation of both Rit and Rin GTPases [24, 29–33]. Selective knock-down of either Rit or Rin in PC6 cells, a sub-line of PC12, using small interfering RNA (siRNA) methods significantly decreases neurite outgrowth and activation of p38 MAP kinase signaling following NGF stimulation [29, 30]. RNAi studies have also demonstrated a role for Rit in NGF-mediated ERK MAP kinase activation in PC6 cells [29].
More recently, Rit has been shown to interact with the cellular polarity machinery by directly binding to Par6 [34]. The Par3-Par6-atypical Protein Kinase C (aPKC) complex is a known scaffold for directing apical-basal polarity in epithelial cells and for axon-specification in neurons, at least in part through control of Rho family-mediated actin remodeling [35–37]. GTP-bound Rac1 and Cdc42 bind to the semi-Cdc42/Rac1 interactive binding (CRIB) domain of Par6 [38]. Interestingly, GDP-bound Rac1 also indirectly interacts with the Par3/6 scaffold, associating with the Rac activator Tiam 1, which is a component of the larger Par complex [39]. This association is necessary for tight junction formation in keratinocytes as well as directing neuronal polarity in hippocampal neurons [39, 40]. Hoshino and colleagues have shown that Rit directly associates with the PDZ domain of Par6 in a GTP-dependent fashion, even though Rit does not contain a consensus PDZ recognition site at its C-terminus [34]. While Par6 association has been found to promote Rit-mediated NIH 3T3 cell transformation, the importance of Par6 association to Rit-directed neuronal differentiation has not been examined [34]. Since Rit-mediated neurite outgrowth has been proposed to require Rac/Cdc42 activation, association with the Par6 complex may play a critical role in both Rit-mediated Rho-family activation and neuronal differentiation [19].
While it is clear that Rit family GTPase signaling is important for appropriate PC6 cell and neuronal signaling, the contribution of individual Rit-mediated effector signaling pathways to this process remains unknown. To begin to understand the consequences of Rit-dependent activation of these distinct cellular signaling pathways, a series of alanine mutations was made to the effector domain of constitutively activated Rit (residues 48 to 59). These Rit effector mutant proteins were tested for their ability to induce PC6 cell differentiation and to associate with, and activate, known Rit effector targets.
Materials and Methods
Plasmids and reagents
Flag-tagged wild type, constitutively activated, and dominant negative Rit in p3xFLAG-CMV-10 vector, and Glutathione-S-Transferase-tagged activated Rit in the pEBG vector and wild-type Rit in the pGEX-KG vector have been described previously [29, 41]. Hemagglutinin-tagged pKH3 Par6A and Par6B were the kind gift of Dr. Ian Macara, University of Virginia, Charlottesville, VA. Par6C (or Par6α) was PCR isolated from Homo sapiens cDNA clone IMAGE: 4844008. ΔPDZ (residues 1-139) and ΔN (residues 154-345) mutants lacking the PDZ domain or the amino terminus, respectively, were isolated by PCR. Full length, ΔPDZ, and ΔN constructs were verified by sequence analysis by Agencourt Bioscience Corp. (Beverly, MA). The following commercially available antibodies were used: Flag (Sigma, Saint Louis, MO), GST (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), HA (12CA5), phospho-specific-ERK1/2 mouse monoclonal antibody and phospho-specific-p38 mitogen-activated protein kinase (MAPK) mouse monoclonal antibody (Cell Signaling, Beverly, MA). The GST-RGL3-RBD glutathione-agarose beads were prepared from bacterially-expressed GST-RGL3-RBD as described previously [16, 17, 29]. The full-length biotinylated human Par6C (or Par6α) cDNA-PinPoint Xa-3 and GST–tagged PKCι PB1 domain [PKCι-(1-113)] −pGEX-6P plasmids were constructed previously [42].
Expression and purification of proteins
Expression and purification of biotinylated human Par6 protein has been described previously [43]. Cmr plasmids carrying αA crystallin gene and GST-PKCι-(1-113) pGEX-6P were co-transformed into E. coli BL21(DE3) and transformants were grown in LB containing 35 μg/ml chloramphenicol and 50 μg/ml ampicillin at 22 °C (220 rpm). At OD600 of 0.8, IPTG was added to a final concentration of 0.1 mM and cells were harvested after 6 hours of induction. Soluble GST-tagged PKCι (1-113) protein was isolated using B-PER GST Fusion Protein Purification Kit (Pierce, Rockford, IL) and dialyzed against Tris buffer (50 mM Tris (pH 8.0), 135 mM NaCl, 10% glycerol). Expression and purification of GST-Rit, as well as guanine nucleotide exchange, has been described previously [13]. GDP- and GTPγS-bound GST-Rit were aliquoted into 50 μg samples and frozen at −80°C prior to use.
Cell lines, cell culture, and transfection
PC6 is a sub-line of PC12 that produce neurites in response to NGF, but grow as well isolated cells in culture (the generous gift of Dr. T. C. Vanaman, University of Kentucky, Lexington, KY). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT), 5% (v/v) heat-inactivated horse donor serum (HS; Life Technologies), 100 μg/ml streptomycin and 100 U/ml penicillin at 37°C in a humidified atmosphere of 5% CO2. PC6 cells were transfected using Effectene (Qiagen, Valencia, CA) as described previously [30, 41]. COS cells were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% (v/v) FBS, 100 μg/ml streptomycin and 100 U/ml penicillin at 37 °C in a humidified atmosphere of 5% CO2. Transfection of COS cells was performed using Superfect (Qiagen) as described previously [29].
Generation of Rit effector loop mutants
Nine effector loop mutants, each with one residue changed to alanine, were produced in the constitutively active RitL79 background using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Successful mutation was verified by DNA sequence analysis. The generation of RitL79S53 was described previously [13].
Neurite Induction
PC6 cells were exposed to the indicated Effectene-DNA complexes for 12–16 hours [41]. The cells were then diluted (1:4), re-plated, and subjected to G418 selection (Life Technologies, San Diego, CA). At day 3 after neurite induction, cells were examined with a Zeiss Axiovert25 inverted phase-contrast microscope using a 20x objective. The percentage of cells bearing neurites greater than one cell body diameter was determined in three separate experiments. At least 150 cells in 10–15 random fields were counted for each condition. For representative images of the mutants, cells were fixed at day 3 after neurite induction for 15 minutes in fresh 4% paraformaldehyde, rinsed three times with phosphate buffered saline (PBS), and permeabilized for 5 minutes in 0.1% Triton X-100 [44]. Cells were then incubated for 1 hour at room temperature with shaking in 1:1000 FITC-phalloidin (Sigma, Saint Louis, MO) in 5% bovine serum albumin, washed three times in PBS, and images captured with an Orca ER camera attached to a Zeiss Axiovert 200M fluorescence microscope using a 32x objective.
MAP kinase immunoblotting
To examine ERK and p38 MAP kinase activation, PC6 cells seeded in 6-well plates were transfected with p3xFlag-RitL79, a p3xFlag-tagged effector loop mutant, or empty p3xFlag vector as control. 48 h after transfection, cells were starved for serum 5 h prior to the preparation of whole cell lysates. The phosphorylation status of ERK1/2 and p38 MAP kinases was determined by immunoblotting with phospho-specific antibodies as described previously [29].
Immunoprecipitation, Glutathione-S-Transferase (GST) pull-down analysis, and Par6 binding assay
To examine the ability of the Rit effector loop mutants to bind to a known downstream target of Rit, the Ral GEF, RGL3, GST fusion proteins containing the Rit binding domain of RGL3 (residues 610-709) were expressed and purified as described previously [16, 18, 29, 41]. PC6 cells seeded in 6-well plates were transiently transfected with p3xFlag-RitWT, p3xFlag-RitL79, p3xFlag-tagged Rit effector loop mutant, or empty p3xFlag vector and incubated for 36 h to allow maximal expression. Cell monolayers were lysed in GST pull-down assay buffer (20 mM HEPES [pH 7.4], 250 mM NaCl, 50 mM KF, 50 mM β-glycerol phosphate, 1% Triton X-100, 10% glycerol, and 1x protease inhibitor cocktail) with sonication. Insoluble material was pelleted by centrifugation. GST resin (20 μgGST-RGL3-RBD/20 μL glutathione beads) was added to 200 μg lysate, incubated for 1 h with end-over-end rotation at 4°C, and the resin collected by brief centrifugation. The pelleted beads were washed once with ice-cold GST pull-down buffer, twice with ice-cold GST pull-down buffer supplemented with 500 mM NaCl, and twice with ice-cold GST pull-down buffer. Bound Rit proteins were released by boiling in Laemmli sample buffer, separated by sodium dodecylsulfate (SDS)-10% polyacrylamide gels, transferred to nitrocellulose (Protran; Schleicher & Schuell Bioscience Dassel, Germany), and detected by immunoblotting with Flag antibody.
To examine the Par6 isoform Rit binds, COS cells were transiently transfected with pKH3-Par6A, pKH3-Par6B, or p3xFlag-Par6C and empty GST vector or GST-RitL79. Cells were allowed to recover for 36 h after transfection. Cells were harvested by scraping in ice-cold GST pull-down assay buffer and lysed with sonication on ice. Insoluble material was pelleted by centrifugation. Pre-washed glutathione-sepharose resin (GE Healthcare Bio-sciences Corp., Piscataway, NJ) was added to 500 μg lysate in a total volume of 1 ml and incubated with end-over-end rotation for 2 h at 4 °C. The resin was collected by brief centrifugation and the supernatant discarded. The resin was extensively washed, twice with ice-cold GST pull-down buffer, once with ice-cold GST pull-down buffer supplemented with 1 M NaCl, and twice with ice-cold GST pull-down buffer. Bound proteins were released by boiling in Laemmli sample loading buffer. Bound proteins and 10 μg total cell lysate from each sample were resolved using SDS-10% polyacrylamide gels, transferred to nitrocellulose, and detected by immunoblotting with hemagglutinin (HA), Flag, or GST antibody.
To examine the ability of the Rit effector loop mutants, wild type, activated, and dominant negative Rit to bind Par6C, cell lysates were generated from COS cells transiently transfected with empty p3xFlag vector, p3xFlag-RitWT, p3xFlag-RitL79, p3xFlag-RitN35, or p3xFlag-tagged effector loop mutant along with empty GST vector, GST-Par6C, GST-Par6CΔPDZ, or GST-Par6CΔN. GST pulldowns were performed with whole-cell lysates as described above.
To assess Rit binding to Par6 in vitro, recombinant biotinylated human Par6 protein was covalently bound to streptavidin-coated 96-well microtiter plates (Nunc, Roskilde, Denmark) as described previously [42]. Purified GST-tagged PKCι (1-113), GST-tagged GTPγS-bound Rit, GST-tagged GDP-bound Rit or active unfused GST (Biovision, Mountain View, CA) was added to the wells at the concentrations indicated and allowed to bind to Par6 overnight at 4°C. Wells were then washed twice with PBS-Tween 20 to remove unbound protein and incubated with rabbit anti-GST polyclonal antibody (1:2500) (Chemicon International) at room temperature for 2 h. After two washes with PBS-Tween 20, Alexa Fluor® 532 goat anti-rabbit IgG (1:250, Invitrogen) was added to each well and incubated at room temperature for 3 h. The wells were washed twice with PBS-Tween 20, once with PBS, and binding determined by measuring fluorescence intensity using a Typhoon 9410 imager (GE Healthcare Bio-sciences Corp., Piscataway, NJ) and ImageQuant software (Amersham). Specific binding was calculated as total binding minus binding of GST at each concentration. Data were analyzed using SigmaPlot (Systat Software, Inc).
Results and Discussion
Mutations within the Rit effector loop alter biological activity
Effector domain mutants have proven to be useful reagents for defining the contribution of specific effector pathways to the actions of Ras and Rho family proteins, including their actions in PC12 cell differentiation [7, 45, 46]. To examine the contribution of known signaling pathways to Rit-mediated neurite outgrowth, single amino acids within the effector domain (residues 48-59) were mutated to alanine in the context of constitutively active RitL79 (Fig. 1A). These double mutants were tested for their ability to induce PC6 cell differentiation as measured by the extension of neurites. Another Rit mutant, RitL79S53, previously shown to allow preferential activation of Raf/MEK/ERK signaling but not of RalGEF, was also examined [13]. Expression of activated RitL79 in PC6 cells induced 10-fold higher neurite formation than cells transfected with empty vector alone and caused 1.5 times as many cells to exhibit neurites as wild-type Rit (Fig. 1B, C). The neurite induction of the various effector loop mutations could be classified into those with activity equal to that of RitL79 (RitL79A48, RitL79A49), those with a significantly reduced ability to induce neurite formation compared to wild-type Rit (RitL79A54, RitL79A55, and RitL79A56), and those with an intermediate phenotype with induction slightly greater than wild-type Rit (RitL79A50, RitL79A51, RitL79S53, RitL79A58, and RitL79A59). Since no single effector domain mutation was found to completely disrupt Rit-dependent neuritogenesis, these data suggest that activation of more than one effector pathway is likely necessary for full neurite induction.
Figure 1. Mutation of the Rit effector loop alters neurite outgrowth ability.

(A) Sequence alignment of the effector domains from several Ras family GTPases. (B) PC6 cells were transiently transfected, subjected to G418 (400 μg/ml) selection for 3 days, fixed and stained with FITC-phalloidin to label actin filaments. Representative fluorescent micrographs from random fields are presented. (C) The percentage of neurite-bearing cells was calculated at day 3 and is presented relative to RitL79 (100%) as means ± standard error from three independent experiments.
ERK activation correlates with neurite formation
Several pathways are known to be required for NGF-mediated PC12 cell differentiation including the Raf/MEK/ERK and p38 MAPK cascades [23, 27, 47, 48]. Constitutively activated mutants of Ras, Raf, and MEK are all capable of inducing neuronal differentiation in PC12 cells highlighting the importance of this MAP kinase signaling cascade in cell fate [17, 27, 49, 50]. Expression of RitL79, but not the empty Flag vector, increased the cellular levels of both phospho-ERK1/2 and phospho-p38 (Fig. 2) in PC6 cells, consistent with earlier studies [29, 30, 51]. All of the Rit effector loop mutants appear to be expressed to approximately equal levels (see Figs. 2 and 3) suggesting that the alanine substitutions did not result in global destabilization. Surprisingly, each of the Rit mutant proteins was found to promote p38 MAP kinase activation to levels seen here with RitL79 (Fig. 2, top panel) and with previous studies using wild-type Rit [29]. These data indicate that none of these substitutions disrupt the interaction(s) required for activation of the p38 MAP kinase signaling cascade. The organization of molecular complexes by protein scaffolds is one mechanism known to confer specificity to MAP kinase cascades and may contribute to Rit-dependent p38 signaling [52]. Thus, the nature of the molecular machinery that links Rit and p38 remains to be characterized and is a goal of ongoing studies.
Figure 2. Mutation of the Rit effector loop alters activation of downstream MAPK pathways.
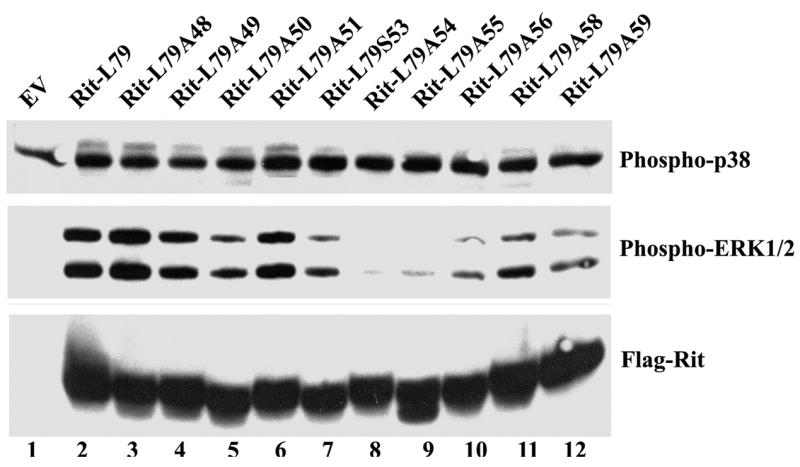
PC6 cells were transiently transfected with expression vectors for the indicated Rit mutant, subjected to serum starvation, and harvested as described under “Materials and Methods”. 50 μg of pre-cleared lysate was resolved on SDS 10%-polyacrylamide gels and the levels of activated ERK1/2 and p38 MAPK were determined by immunoblotting with the appropriate phospho-specific antibodies. Blots are representative of three independent trials.
Figure 3. Rit effector loop mutants exhibit differential binding to RGL3-RBD.

PC6 cells were transiently transfected with the indicated expression vectors, and the cells starved with serum-free DMEM for 5 h prior to the preparation of whole cell lysates. Cell lysates (200 μg) were incubated at 4 °C for 1 h with GST-RGL3-RBD pre-bound to glutathione beads. The RGL3-RBD complexed affinity beads were pelleted and washed extensively, and the amount of bound Rit protein in the pellet fraction was determined by anti-Flag immunoblotting. The expression of Rit present in each lysate was also determined by immunoblot analysis. Blots are representative of three independent trials.
Although each of the Rit mutants were expressed at comparable levels in PC6 cells (Fig. 2, lower panel), their ability to stimulate ERK MAP kinase activity could be divided into three groups: mutants that induce phospho-ERK1/2 to the same level as RitL79 (RitL79A48, RitL79A49), mutations that impair Rit-mediated ERK activation similar to the level of RitWT in previous studies (RitL79A54, RitL79A55, RitL79A56), and those with a slight reduction in the ability to activate ERK (RitL79A50, RitL79A51, RitL79S53, RitL79A58, RitL79A59) [29]. Importantly, ERK activation mirrored the ability of individual effector loop mutants to induce neuronal differentiation, consistent with earlier studies suggesting a critical role for MEK-ERK signaling in Rit-mediated neuritogenesis [17, 29].
RGL3 binding is not required for Rit-mediated neurite induction
Ral GEF proteins are downstream targets of both Ras and Rit signaling [16, 53]. The importance of Ral GTPase function to neurite induction is currently unclear, with some studies suggesting that Ral-mediated activation of the exocyst complex is necessary for membrane fusion to drive protrusion of neurites, while others suggest that Ral signaling impedes neuritogenesis by inhibiting Rho family GTPase-regulated cytoskeletal reorganization [54, 55]. Since the Ras/Rit Binding Domain (RBD) of RGL3 is both necessary and sufficient for GTP-dependent Rit binding, we examined the ability of Rit effector loop mutants to associate with GST-RGL3-RBD by glutathione bead precipitation [16]. As seen previously, both wild-type Rit and RitL79 were found to potently bind RGL3-RBD [16]. This is explained by the observation that overexpressed wild-type Rit is predominantly GTP-bound and is thus able to both bind RGL3-RBD and promote neurite outgrowth [17]. Unlike p38 activation, RGL3 association was particularly sensitive to effector domain mutation. Only RitL79A49 and RitL79A55 displayed robust RGL3-RBD association (see Fig. 3 lanes 4 and 9), while both RitL79A48 and RitL79A59 exhibited weak but detectable binding (Fig. 3 lanes 3 and 12). Interestingly, both RitL79A48 and RitL79A49 induce neurite formation to the same extent (Fig 1C), which is significantly higher than that for RitL79A55, suggesting that interaction with RalGEFs and activation of Ral signaling are not required for Rit-mediated neurite induction.
Interaction of effector mutants with Par6 correlates with neurite induction
Directing polarization of the actin cytoskeleton is critical for initiating neurite formation and is controlled, in part, by the Par3-Par6-aPKC complex, which localizes to the tips of growing neurites [36, 56–58]. Hoshino and colleagues first described the interaction between GTP-Rit and the PDZ domain of Par6, and found that Par6 synergistically enhanced RitL79-dependent NIH 3T3 cell transformation [34]. Because of the critical role for the Par complex in axonal growth, we next examined whether Par6 contributes to Rit-mediated neuritogenesis [34, 36]. There are three mammalian members of the Par6 family (Par6A-C), and while Hoshino and colleagues described a Rit-Par6 interaction, it was unclear which isoform was used in their studies [34, 59]. To address this issue, we co-expressed GST or GST-RitL79 and the three Par6 isoforms in COS cells and assessed Par6 binding following glutathione resin precipitation. GST-RitL79, but not unfused GST, interacted with all three Par6 isoforms (Fig. 4A). Because Par6C is preferentially expressed in the brain, this isoform was used in the remaining GST pull-down assays [59].
Figure 4. Rit interacts with Par6C in an effector loop dependent manner.
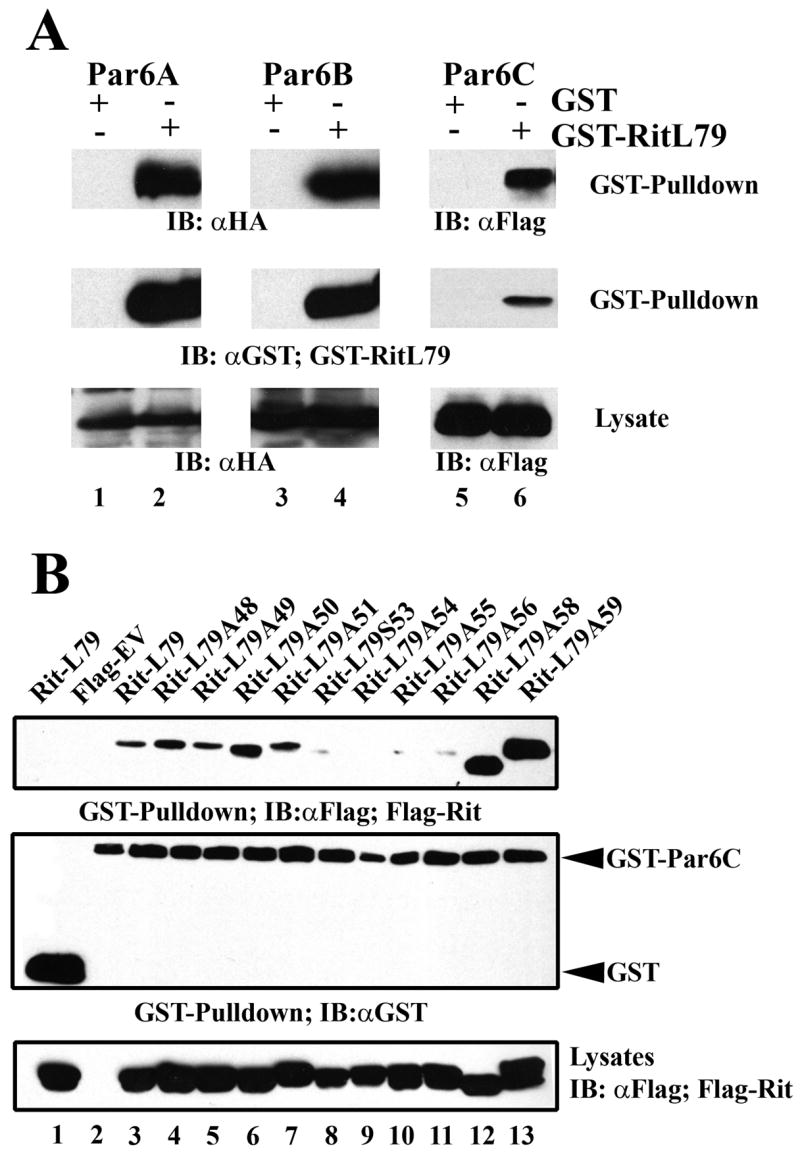
(A) COS cells were transfected with GST or GST-RitL79 and 3xHA-Par6A, 3xHA-Par6B, or 3xFlag-Par6C, whole-cell lysates were generated, and GST pull-down assays performed as described under “Materials and Methods”. Precipitated Par6 proteins were detected by immunoblot analysis using anti-HA, or anti-Flag antibodies. Equal expression of Par6 proteins was detected from total lysates by immunobloting. Blots are representative of three independent trials. (B) COS cells were co-transfected with either unfused GST or GST-Par6C expression vectors and the indicated Flag-tagged Rit mutants. Whole-cell lysates were generated and GST pull-down assays performed as described under “Materials and Methods”. Precipitated Rit and Par6 protein was detected by immunoblot analysis using anti-Flag and anti-GST antibodies, respectively. Expression of Rit protein was examined by anti-Flag immunoblotting of total lysate. Blots are representative of three independent trials.
To examine the effector-domain dependence of Par6C binding, cell lysates co-expressing either RitL79 or Rit effector loop mutants and GST-Par6C or unfused GST were generated and subjected to glutathione-sepharose bead pull-down analysis. As expected, Flag-RitL79 bound to full length Par6C, but not unfused GST (Fig. 4B). Unlike the strong correlation found between ERK1/2 activation and PC6 cell differentiation, binding of the Rit effector mutants to Par6C did not clearly mirror neurite induction. Those Rit effector mutants that promoted robust neurite induction, RitL79A48 and RitL79A49, bound Par6C, while mutants with significantly impaired neurite initiation (alanine substitutions at residues 54-56) failed to bind Par6C, suggesting that interaction with Par6C might contribute to Rit-mediated neurite induction. However, Par6 association is not essential since RitL79S53 does not display appreciable Par6 binding (Fig. 4B) but promotes neurite formation (Fig. 1C). In addition, except for RitL79S53, all the Rit effector mutants that fail to bind Par6C also fail to activate ERK (Fig. 2). Thus, it not possible to separate the relative contributions of these two effector pathways to Rit function. Additional studies will be necessary to more fully characterize the role of Par6 in Rit-mediated neuronal signaling.
Characterization of an indirect Rit-GDP-Par6 interaction
Because this is the first reported interaction of Rit and Par6C, we next examined the nucleotide dependence of binding. Lysates expressing Flag-tagged wild-type Rit, RitL79, or RitN35 with GST-Par6C or unfused GST were generated and GST pull-down assays performed. As expected, none of the Rit constructs interacted with unfused GST while both wild-type Rit and RitL79 displayed modest Par6C binding (Fig. 5A). Surprisingly, dominant negative Rit, RitN35, a mutant which is predominantly GDP-bound, does not promote neurite outgrowth, and fails to interact with all previously identified Rit effectors, displayed robust association with Par6C (Fig. 5A) [13, 17, 29]. Thus, in addition to the previously characterized GTP-dependent interaction of Rit with the PDZ domain of Par6, these data suggest that Rit associates with Par6C in a GTP-independent manner [34].
Figure 5. Dominant negative Rit strongly interacts with Par6 independent of its PDZ domain.
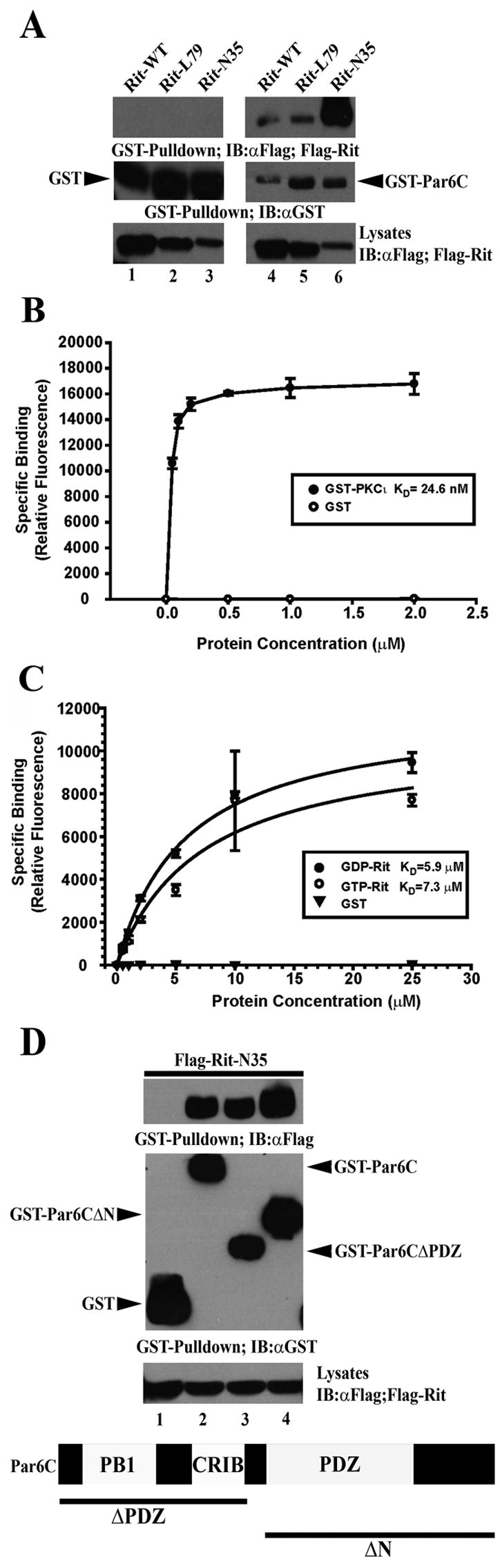
(A) COS cells were transfected with GST or GST-Par6C and Flag-tagged RitWT, RitL79 or RitN35. GST precipitations were performed as in Figure 4B. Note that RitN35 displays stronger association with Par6 than either RitWT or RitL79. Blots are representative of three independent trials. (B,C) Biotinylated Par6C was bound to streptavidin-coated plates and incubated with the indicated concentrations of recombinant GST, GST-PCKι, or GST-Rit pre-loaded with non-hydrolyzable GTPγS (GTP) or GDP. Binding isotherms for PKCι (B) and GTPγS-Rit and GDP-Rit (C) to Par6 indicate that PKCι binds Par6 with much higher apparent KD than Rit and that GTP- and GDP-Rit bind to Par6 with similar affinities. (D) COS cells were co-transfected with GST, GST-Par6C full length, GST-Par6CΔPDZ, or GST-Par6CΔN and Flag-RitN35and whole cell lysates were generated and subjected to GST pull-down analysis as described under “Materials and Methods”. Blots are representative of three independent trials.
To further characterize the GTP-independent interaction of Rit with Par6, recombinant wild-type GST-Rit protein was pre-loaded with either GDP or the non-hydrolyzable GTP analog, GTPγS [13]. We took advantage of a well established in vitro human Par6C binding assay [42]. As seen in Fig. 5B, binding studies were first performed with unfused GST or GST-PKCι, an established binding partner of Par6 [43]. As expected, no association between GST and Par6 was observed whereas GST-PKCι demonstrated strong binding with an apparent KD of 24.6 nM. Interestingly, both GDP- and GTPγS-Rit bound Par6 with approximately equal affinity (KD=5.9 μM and KD=7.3 μM, respectively) (Fig. 5C). These data support the observations of Hoshino and colleagues that Rit can directly bind Par6, albeit with a much lower binding affinity when compared with GST-PKCι, a known component of the Par3-Par6 complex [34, 35]. Additionally, these data indicate that the direct in vitro interaction between Rit and Par6 is GTP-independent, since GDP- and GTPγS-bound Rit display approximately equal binding (Fig. 5C). Moreover, they suggest that the direct binding of GDP-Rit to Par6 does not account for the robust binding of RitN35 to Par6 seen in whole cell lysates. Indeed, the very low affinity observed for the in vitro binding of Rit to Par6 suggests that the observed intracellular association is likely not due to direct interaction, but instead is mediated by Rit association to the larger Par6 scaffolding complex.
To begin to explore the nature of the RitN35-Par6C interaction, we examined which region of Par6 was bound by RitN35. Lysates expressing Flag-tagged RitN35 and either GST-Par6C (full-length: residues 1-345), GST-Par6CΔPDZ (residues 1-139), or GST-Par6CΔN (Residues 154-345) were generated and subjected to GST pull-down analysis. Surprisingly, RitN35 was found to bind all three Par6C constructs tested (Fig. 4D), suggesting that the GTP-independent association of Rit with Par6C does not require an intact PDZ domain [34].
Recently, the Rac1 GTPase has been shown to interact in a complex fashion with the Par3/6 polarity complex. In this case, active GTP-bound Rac1 binds to the semi-CRIB domain of Par6 while GDP-Rac1 interacts with the Rho family GEF, Tiam1, which associates with components of the larger Par3-Par6-aPKC complex [39, 40]. The Par/Tiam1 complex allows for rapid and localized Rac1 activation following chemokine stimulated-induction of Rap1 in T lymphocytes, providing a mechanism by which Rap1 activation can control chemotaxis [60]. Thus, it is tempting to speculate that dominant negative Rit (RitN35) can associate with the Par3/6 complex in a similar fashion, involving a scaffolded RitGEF protein. This interaction would not have been detected in the study by Hoshino and colleagues, since they examined the nucleotide-dependence of the Rit-Par6 interaction with recombinant proteins and additional cellular co-factor(s) would be absent in their assays [34]. The possibility that the Par6 complex contains a RitGEF is supported by the robust association seen with RitN35, which bound more tightly than either wild-type or activated Rit (Fig. 5A). Dominant-negative Ras GTPases are preferentially bound by cellular GEFs but not by other effectors which favor a GTP-bound conformational state [4]. Taken together, these data suggest that Rit-Par6 association is unexpectedly complex, and may provide a novel mechanism for regulating Rit activity. Indeed, we have recently found that Rit is controlled by cross-talk between the NGF and BMP signaling pathways and suggest a role for Rit in regulating axonal and dendritic growth via activation of the ERK MAP kinase signaling pathway [44]. Since the Par3/6 complex is involved in axon-dendrite specification in hippocampal neurons [36], studies are currently underway to further investigate the role of Par3/6 binding in Rit-mediated control of axonal and dendritic growth.
In summary, using a series of Rit effector loop mutants we have found that Rit-mediated neuronal differentiation depends upon activation of the ERK MAP kinase cascade, but does not require activation of RalGEF/Ral signaling. In addition, our data demonstrates an unexpectedly complex association of Rit with Par6 and suggests that the larger Par3/6/aPKC polarity complex may contain a scaffolded RitGEF. Since Rit has recently been shown to regulate axonal-dendritic growth via activation of ERK MAP kinase signaling, it will be important to examine the contribution of Par3/6 complex to the regulation of this process.
Acknowledgments
This work was supported by Public Health Service grant NS045103 (to D.A.A) from the National Institute of Neurological Disorders and Stroke, by Grant P20RR20171 from the COBRE program of the National Center for Research Resources, a Predoctoral Fellowship from the Ohio Valley Affiliate of the American Heart Association (to J.L.R.), the National Institutes of Health (CA81436 to A.P.F), The James and Esther King Biomedical Research Program and the American Lung Assocation/LUNGevity Foundation (to A.P.F.) We thank Dr. Ian Macara for the generous gift of reagents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004:RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–32. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 3.Reuther GW, Der CJ. The Ras branch of small GTPases: Ras family members don't fall far from the tree. Curr Opin Cell Biol. 2000;12:157–65. doi: 10.1016/s0955-0674(99)00071-x. [DOI] [PubMed] [Google Scholar]
- 4.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–27. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 5.Satoh T, Nakafuku M, Kaziro Y. Function of Ras as a molecular switch in signal transduction. J Biol Chem. 1992;267:24149–52. [PubMed] [Google Scholar]
- 6.Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc Natl Acad Sci U S A. 2001;98:4944–9. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–41. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 8.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 9.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–8. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–54. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CH, Della NG, Chew CE, Zack DJ. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci. 1996;16:6784–94. doi: 10.1523/JNEUROSCI.16-21-06784.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wes PD, Yu M, Montell C. RIC, a calmodulin-binding Ras-like GTPase. Embo J. 1996;15:5839–48. [PMC free article] [PubMed] [Google Scholar]
- 13.Shao H, Kadono-Okuda K, Finlin BS, Andres DA. Biochemical characterization of the Ras-related GTPases Rit and Rin. Arch Biochem Biophys. 1999;371:207–19. doi: 10.1006/abbi.1999.1448. [DOI] [PubMed] [Google Scholar]
- 14.Rusyn EV, Reynolds ER, Shao H, Grana TM, Chan TO, Andres DA, Cox AD. Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene. 2000;19:4685–94. doi: 10.1038/sj.onc.1203836. [DOI] [PubMed] [Google Scholar]
- 15.Sakabe K, Teramoto H, Zohar M, Behbahani B, Miyazaki H, Chikumi H, Gutkind JS. Potent transforming activity of the small GTP-binding protein Rit in NIH 3T3 cells: evidence for a role of a p38gamma-dependent signaling pathway. FEBS Lett. 2002;511:15–20. doi: 10.1016/s0014-5793(01)03264-1. [DOI] [PubMed] [Google Scholar]
- 16.Shao H, Andres DA. A novel RalGEF-like protein, RGL3, as a candidate effector for rit and Ras. J Biol Chem. 2000;275:26914–24. doi: 10.1074/jbc.M002241200. [DOI] [PubMed] [Google Scholar]
- 17.Spencer ML, Shao H, Andres DA. Induction of neurite extension and survival in pheochromocytoma cells by the Rit GTPase. J Biol Chem. 2002;277:20160–8. doi: 10.1074/jbc.M201092200. [DOI] [PubMed] [Google Scholar]
- 18.Spencer ML, Shao H, Tucker HM, Andres DA. Nerve growth factor-dependent activation of the small GTPase Rin. J Biol Chem. 2002;277:17605–15. doi: 10.1074/jbc.M111400200. [DOI] [PubMed] [Google Scholar]
- 19.Hoshino M, Nakamura S. Small GTPase Rin induces neurite outgrowth through Rac/Cdc42 and calmodulin in PC12 cells. J Cell Biol. 2003;163:1067–76. doi: 10.1083/jcb.200308070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi GX, Rehmann H, Andres DA. A Novel cAMP-dependent Epac-Rit Signaling Pathway Contributes to PACAP38-mediated Neuronal Differentiation. Mol Cell Biol. 2006 doi: 10.1128/MCB.00332-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison SM, Rudolph JL, Spencer ML, Wes PD, Montell C, Andres DA, Harrison DA. Activated RIC, a small GTPase, genetically interacts with the Ras pathway and calmodulin during Drosophila development. Dev Dyn. 2005;232:817–26. doi: 10.1002/dvdy.20346. [DOI] [PubMed] [Google Scholar]
- 22.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–8. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–17. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- 24.Klesse LJ, Meyers KA, Marshall CJ, Parada LF. Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene. 1999;18:2055–68. doi: 10.1038/sj.onc.1202524. [DOI] [PubMed] [Google Scholar]
- 25.Burry RW. p21(ras) stimulates pathways in addition to ERK, p38, and Akt to induce elongation of neurites in PC12 cells. J Neurosci Res. 2001;63:45–53. doi: 10.1002/1097-4547(20010101)63:1<45::AID-JNR6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 26.Kao S, Jaiswal RK, Kolch W, Landreth GE. Identification of the mechanisms regulating the differential activation of the mapk cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem. 2001;276:18169–77. doi: 10.1074/jbc.M008870200. [DOI] [PubMed] [Google Scholar]
- 27.Morooka T, Nishida E. Requirement of p38 mitogen-activated protein kinase for neuronal differentiation in PC12 cells. J Biol Chem. 1998;273:24285–8. doi: 10.1074/jbc.273.38.24285. [DOI] [PubMed] [Google Scholar]
- 28.Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–9. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 29.Shi GX, Andres DA. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol Cell Biol. 2005;25:830–46. doi: 10.1128/MCB.25.2.830-846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi GX, Han J, Andres DA. Rin GTPase couples nerve growth factor signaling to p38 and b-Raf/ERK pathways to promote neuronal differentiation. J Biol Chem. 2005;280:37599–609. doi: 10.1074/jbc.M507364200. [DOI] [PubMed] [Google Scholar]
- 31.Klesse LJ, Parada LF. Trks: signal transduction and intracellular pathways. Microsc Res Tech. 1999;45:210–6. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<210::AID-JEMT4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 32.Levi-Montalcini R, Angeletti PU. Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol. 1963;7:653–9. doi: 10.1016/0012-1606(63)90149-0. [DOI] [PubMed] [Google Scholar]
- 33.Misko TP, Radeke MJ, Shooter EM. Nerve growth factor in neuronal development and maintenance. J Exp Biol. 1987;132:177–90. doi: 10.1242/jeb.132.1.177. [DOI] [PubMed] [Google Scholar]
- 34.Hoshino M, Yoshimori T, Nakamura S. Small GTPase proteins Rin and Rit Bind to PAR6 GTP-dependently and regulate cell transformation. J Biol Chem. 2005;280:22868–74. doi: 10.1074/jbc.M411592200. [DOI] [PubMed] [Google Scholar]
- 35.Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–9. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 36.Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimura T, Arimura N, Kaibuchi K. Signaling networks in neuronal polarization. J Neurosci. 2006;26:10626–30. doi: 10.1523/JNEUROSCI.3824-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–7. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- 39.Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–7. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- 40.Mertens AE, Rygiel TP, Olivo C, van der Kammen R, Collard JG. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J Cell Biol. 2005;170:1029–37. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andres DA, Rudolph JL, Sengoku T, Shi GX. Analysis of rit signaling and biological activity. Methods Enzymol. 2005;407:499–512. doi: 10.1016/S0076-6879(05)07040-0. [DOI] [PubMed] [Google Scholar]
- 42.Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–74. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- 43.Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–9. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- 44.Lein PJ, Guo X, Shi GX, Moholt-Siebert M, Bruun D, Andres DA. The novel GTPase Rit differentially regulates axonal and dendritic growth. J Neurosci. 2007;27:4725–36. doi: 10.1523/JNEUROSCI.5633-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuguchi T, Kraft AS. Regulation of myeloid cell growth by distinct effectors of Ras. Oncogene. 1998;17:2701–9. doi: 10.1038/sj.onc.1202201. [DOI] [PubMed] [Google Scholar]
- 46.Shirouzu M, Koide H, Fujita-Yoshigaki J, Oshio H, Toyama Y, Yamasaki K, Fuhrman SA, Villafranca E, Kaziro Y, Yokoyama S. Mutations that abolish the ability of Ha-Ras to associate with Raf-1. Oncogene. 1994;9:2153–7. [PubMed] [Google Scholar]
- 47.Jaiswal RK, Moodie SA, Wolfman A, Landreth GE. The mitogen-activated protein kinase cascade is activated by B-Raf in response to nerve growth factor through interaction with p21ras. Mol Cell Biol. 1994;14:6944–53. doi: 10.1128/mcb.14.10.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwasaki S, Iguchi M, Watanabe K, Hoshino R, Tsujimoto M, Kohno M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J Biol Chem. 1999;274:26503–10. doi: 10.1074/jbc.274.37.26503. [DOI] [PubMed] [Google Scholar]
- 49.Wood KW, Qi H, D'Arcangelo G, Armstrong RC, Roberts TM, Halegoua S. The cytoplasmic raf oncogene induces a neuronal phenotype in PC12 cells: a potential role for cellular raf kinases in neuronal growth factor signal transduction. Proc Natl Acad Sci U S A. 1993;90:5016–20. doi: 10.1073/pnas.90.11.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–52. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 51.Hynds DL, Spencer ML, Andres DA, Snow DM. Rit promotes MEK-independent neurite branching in human neuroblastoma cells. J Cell Sci. 2003;116:1925–35. doi: 10.1242/jcs.00401. [DOI] [PubMed] [Google Scholar]
- 52.Dard N, Peter M. Scaffold proteins in MAP kinase signaling: more than simple passive activating platforms. Bioessays. 2006;28:146–56. doi: 10.1002/bies.20351. [DOI] [PubMed] [Google Scholar]
- 53.White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem. 1996;271:16439–42. doi: 10.1074/jbc.271.28.16439. [DOI] [PubMed] [Google Scholar]
- 54.van Dam EM, Robinson PJ. Ral: mediator of membrane trafficking. Int J Biochem Cell Biol. 2006;38:1841–7. doi: 10.1016/j.biocel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Goi T, Rusanescu G, Urano T, Feig LA. Ral-specific guanine nucleotide exchange factor activity opposes other Ras effectors in PC12 cells by inhibiting neurite outgrowth. Mol Cell Biol. 1999;19:1731–41. doi: 10.1128/mcb.19.3.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bose R, Wrana JL. Regulation of Par6 by extracellular signals. Curr Opin Cell Biol. 2006;18:206–12. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Arimura N, Kaibuchi K. Key regulators in neuronal polarity. Neuron. 2005;48:881–4. doi: 10.1016/j.neuron.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 58.Schwamborn JC, Puschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–9. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- 59.Gao L, Macara IG. Isoforms of the polarity protein par6 have distinct functions. J Biol Chem. 2004;279:41557–62. doi: 10.1074/jbc.M403723200. [DOI] [PubMed] [Google Scholar]
- 60.Gerard A, Mertens AE, van der Kammen RA, Collard JG. The Par polarity complex regulates Rap1- and chemokine-induced T cell polarization. J Cell Biol. 2007;176:863–75. doi: 10.1083/jcb.200608161. [DOI] [PMC free article] [PubMed] [Google Scholar]