

Abstract
Regulation of cell migration/invasion is important for embryonic development, immune function, and angiogenesis. However, migratory cells must also coordinately activate survival mechanisms to invade the extracellular matrix and colonize foreign sites in the body. Although invasive cells activate protective programs to survive under diverse and sometimes hostile conditions, the molecular signals that regulate these processes are poorly understood. Evidence is provided that signals that induce cell invasion also promote cell survival by suppressing apoptosis of migratory cells. Extracellular-regulated kinase (ERK) activation and molecular coupling of the adaptor proteins p130 Crk-associated substrate (CAS) and c-CrkII (Crk) represent two distinct pathways that induce cell invasion and protect cells from apoptosis in a three-dimensional collagen matrix. CAS/Crk-mediated cell invasion and survival requires activation of the small GTPase Rac, whereas ERK-induced cell invasion, but not survival requires myosin light chain kinase activation and myosin light chain phosphorylation. Uncoupling CAS from Crk or inhibition of ERK activity prevents migration and induces apoptosis of invasive cells. These findings provide molecular evidence that during invasion of the extracellular matrix, cells coordinately regulate migration and survival mechanisms through ERK activation and CAS/Crk coupling.
Keywords: cell survival, mitogen-activated protein kinases, signal transduction, three-dimensional collagen, integrins
Introduction
In the adult organism, the capacity for cell migration is tightly controlled, as most cells in the body do not move. However, it is generally believed that aberrant cell migration events occur in normal tissue, and that apoptotic programming is one mechanism used to eliminate these misguided cells. This process is important for maintaining tissue integrity and homeostasis of the body (Raff 1992; Steller 1995). In fact, abnormal regulation of cell survival and migratory mechanisms is thought to contribute to developmental abnormalities, tumor cell metastasis, and autoimmune disease (Raff 1992; Steller 1995; Lauffenburger and Horwitz 1996). On the other hand, scheduled migration events are part of normal development, wound healing, and immune function. In this case, migratory cells suppress or inactivate their apoptotic program as they invade and colonize distinct sites in the body. For example, neural crest cells migrate on guided pathways, leading to different derivative cell types in unique embryonic sites. Cells that deviate from their specific path or do not adapt to their new environment are eliminated by apoptosis (Wakamatsu et al. 1998; Poelmann and Gittenberger-de Groot 1999). Immune cells infiltrating during an inflammation response and metastatic tumor cells are also believed to suppress their apoptotic program in order to invade the extracellular matrix (ECM) (Glinsky and Glinsky 1996; Takaoka et al. 1997; Keely et al. 1998; Brezinschek et al. 1999).
Although the mechanisms responsible for regulating survival of migratory cells are not known, adhesive proteins present in the ECM are likely to play a central role (Meredith et al. 1993; Aplin et al. 1998). In fact, the collagen-rich environment of the ECM is one of the first barriers that a migratory cell encounters during invasion (Keely et al. 1998). Recent evidence indicates that the ECM is necessary for cell migration, and cells deprived of ECM components do not progress through the cell cycle and readily undergo apoptosis (Meredith et al. 1993; Brooks et al. 1994; Frisch and Francis 1994; Ruoslahti and Reed 1994; Lauffenburger and Horwitz 1996). This suggests that inappropriate remodelling or recognition of ECM components by invasive cells may lead to loss of ECM contacts and activation of an apoptotic program and termination of cell movement. Therefore, cells that utilize ECM components for both migration and survival are likely to be successful at invading foreign tissues. In contrast, cells that stray out of their normal environment and migrate down the wrong pathway would be destined to commit suicide.
Recent evidence indicates that growth factor and integrin adhesion receptors and their downstream signals play a central role in facilitating cell movement and survival (Aplin et al. 1998). Regulation of these cellular processes is complex, and involves several potential signaling cascades including focal adhesion kinase (FAK), phosphatidylinositol 3-kinase (PI3K), Akt kinase, nuclear factor (NF)-κB, and mitogen-activated protein (MAP) kinases (Frisch et al. 1996; Khwaja et al. 1997; Parrizas et al. 1997; Berra et al. 1998; Ilic et al. 1998; Scatena et al. 1998). However, it is not yet clear how these signals facilitate survival or whether they operate during cell invasion and remodelling of the ECM. In this report, we demonstrate that extracellular-regulated kinase (ERK) 1 and ERK2 activation, and molecular coupling of p130 Crk-associated substrate (CAS) and c-CrkII (Crk) adaptor proteins are two signaling pathways that facilitate cell invasion and protect cells from apoptosis. Both signaling pathways lie downstream of integrin and cytokine receptors, and are necessary for cell survival and migration during invasion of a three-dimensional (3-D) collagen matrix.
Materials and Methods
Expression Vectors and Reagents
The expression plasmid pUCCAGGS containing either full-length myc-tagged Crk, Crk cDNA with a mutated NH2-terminal Src-homology (SH) 3 domain (tryptophan 109 to cysteine), or Crk with a mutated SH2 domain (arginine 38 to valine) have been described previously (Matsuda et al. 1993; Tanaka et al. 1993, Kiyokawa et al. 1998). The pSSRα and pEBG expression plasmid containing glutathione S-transferase–tagged or untagged wild-type CAS or CAS with an in-frame deletion of its substrate domain (CAS-SD, amino acids 213–514) have been described previously (Mayer et al. 1995; Klemke et al. 1998). Green fluorescent protein (GFP) in pEGFP-C1 was from Clontech and pCMV5 encoding β-galactosidase (β-gal) has been described (Klemke et al. 1998). Bcl-2 in pcDNA3.1 was provided by Dr. S. Huang (The Scripps Research Institute, La Jolla, CA). Mutationally activated MEK1 in pcDNA3 has been described (Klemke et al. 1997). PD98059 (2-[2′-amino-3′methoxyphenyl]-oxanaphthalen-4-one) was from Calbiochem. Hemagglutinin (HA)-tagged myosin light chain (MLC) kinase (MLCK) and myc-tagged Rac1 constructs have been described previously (Klemke et al. 1997, Klemke et al. 1998). The phosphoERK antibody was from Promega. Bcl-2, MEK, ERK, and CAS specific antibodies were from Santa Cruz Biotechnology, and anti-Crk was from Transduction Laboratories. Antibodies to myosin IIB isotype and MLC have been described (Klemke et al. 1997). HA antibodies and human recombinant insulin were from Boehringer Mannheim. Epidermal growth factor (EGF) and insulin-like growth factor (IGF-1) were from Genzyme. Bromodeoxyuridine (BrdU) and anti-BrdU antibodies were from Sigma Chemical Co. Z-VAD-fmk (halomethyl ketone benzyloxycarbonyl-VAD fluoromethyl ketone) was from Calbiochem. Apoptosis detection kit was from Oncogene Sciences. Collagen (Invitrogen 100) for 3-D cultures was from Biomaterials. FG-M cells are pancreatic carcinoma cells that show increased migration and metastasis compared with the noninvasive parental FG cells (Klemke et al. 1998). COS-7 cells are low passage monkey kidney cells obtained from American Type Tissue Culture. COS-7 cells stably overexpressing full-length untagged Crk in pMEXneo and untagged wild-type CAS in pBABEpuro vector were selected in media containing G418 (0.5 mg/ml; Sigma Chemical Co.) and puromycin (1 μg/ml, Calbiochem). NIH3T3 cells were provided by Dr. Tony Hunter (Salk Institute, La Jolla, CA). Embryonic fibroblast cells isolated from wild-type and CAS-deficient mice were provided by Dr. H. Hirai (University of Tokyo, Tokyo, Japan).
Detection of Apoptosis in 3-D Collagen
3-D collagen gels were made from bovine dermal collagen (2.4–2.7 mg/ml) according to the manufacturer's recommendation (Biomaterials) and as described previously (Keely et al. 1995). The collagen preparation is 95–98% native collagen type I with the remainder being collagen type III. Cells were serum-starved for 24 h, removed from the dish with dilute trypsin, and added immediately to ice-cold collagen solution at 3 × 105 cells/ml. The cell suspension in 5–15-μl drops was added to wells of a 24-well culture dish, then immediately inverted and warmed to 37°C for 20–30 min to initiate polymerization of the collagen. Culture medium (500 μl of fibroblast basal medium [FBM] with 0.5% BSA) was added to each well containing various cytokines, FBS, the caspase inhibitor Z-VAD-fmk (100 μM), anti-integrin antibodies (25 μg/ml), or PD98059. Apoptotic cells were identified directly in the 3-D collagen gels at various times with an apoptosis detection kit and FITC-conjugated Annexin V/propidium iodide (PI) staining method according to the manufacturer's recommendation. The number of apoptotic cells staining with FITC-annexin V, but not PI was determined blindly by two observers using an inverted fluorescent microscope (Ziess axiovert 100), and is reported as the percentage of cells undergoing apoptosis (i.e., annexin V–positive cells/PI-negative/total number of cells per high-powered field). Apoptotic cells were also identified by morphological criteria. Apoptotic cells with substantial membrane blebbing, cytoplasmic and nuclear condensation, and cell fragmentation were scored as positive. A representative image is shown in Fig. 1 A.
Figure 1.

Migratory cells are protected from apoptosis during invasion of 3-D collagen. (A) Confocal images of COS-7 cells expressing GFP and suspended in 3-D collagen in the absence or presence of insulin (25 μg/ml) for the indicated times (hours). The arrowheads show membrane blebs and fragments of apoptotic cells. The time of cell culture is shown in the upper left hand corner of each photomicrograph. Time 0 represents cells immediately after polymerization of the collagen gel. Bar, 10 μm. (B) COS-7 cell apoptosis in 3-D collagen in the absence (Buffer) or presence of insulin (25 μg/ml), EGF (100 ng/ml), or FBS (10%). The percentage of apoptotic cells was determined at the indicated times by annexin V/PI staining as described in Materials and Methods. The arrow denotes cells incubated for 24 h with buffer containing Z-VAD-fmk (z-VAD; 100 μM), which served as a general inhibitor of apoptosis by blocking caspase proteases (Garcia-Calvo et al. 1998). Results are the mean ± SEM of cell counts from three individual cultures of three separate experiments. (C) COS-7 cell migration in 3-D collagen in the absence (Buffer) or presence of insulin (25 μg/ml), EGF (100 ng/ml), or FBS (10%). The number of cells migrating per field was determined as described in Materials and Methods. Results are the mean ± SEM of three independent experiments.
Cell Migration in 3-D Collagen
The membrane of a Transwell migration chamber was coated on both sides with collagen solution (100 μl top, 50 μL bottom) and allowed to polymerize as described above. Cells (1–2 × 106) placed in the upper chamber were allowed to invade to the underside of the porous membrane for various times in FBM 0.5% BSA containing various cytokines and growth factors, which were placed in the lower compartment as a chemoattractant. In some cases, the medium was supplemented with anti-integrin antibodies, PD98059, or Z-VAD-fmk as described above. Noninvasive cells were removed from the top with a cotton swab and the invasive cells present in the 3-D collagen on the underside of the membrane were fixed and stained with crystal violet as described previously (Klemke et al. 1997). The number of migratory/invasive cells were counted per field (40×) from three migration chambers with an inverted microscope as described previously (Klemke et al. 1997).
Transfection of Cells, Immunoprecipitation, and Immunoblotting
Transient tranfection of COS-7, FG-M, and fibroblast cells as well as Transwell migration assays were performed as described previously (Klemke et al. 1998; Cheresh et al. 1999). In brief, cells were cotransfected with Lipofectamine and expression vectors containing cDNAs encoding wild-type or mutant forms of either Bcl-2, MEK, Crk, CAS, Rac, or MLCK along with a reporter construct encoding β-gal or GFP. Control cells were mock-transfected with the appropriate amount of the empty expression vectors along with the β-gal or GFP reporter. Cells were allowed to incorporate the cDNA constructs for 6–8 h, washed, and incubated for 40 h, which provides optimal transient expression of proteins in most cells. Cells were then prepared for cell migration and survival in 3-D collagen as described above. The transfected cells incorporating the reporter constructs were detected as described above using X-gal as a substrate (Klemke et al. 1998), or with an inverted fluorescence microscope. Controls for transfection efficiency and cell adhesion of transfected cells to purified ECM proteins were monitored as described below. Preparation of detergent lysates of cells in 3-D collagen was performed as described (Lin and Grinnell 1993), and immunoprecipitation and immunoblotting of proteins performed as reported previously (Klemke et al. 1998). Metabolic labeling of cells with [32P]orthophosphate, isolation of MLC, and Western blotting were performed as described previously (Klemke et al. 1997).
Cell Adhesion and Transfection Efficiency
Controls for transfection efficiency and cell adhesion to ECM proteins were performed as described previously (Klemke et al. 1998). In brief, an aliquot of cells from the transfection experiments above were allowed to attach to culture dishes coated with purified collagen. The dishes were washed and adherent cells transfected with the β-gal reporter, or GFP gene detected using X-gal as a substrate or visualized with an inverted fluorescence microscope, respectively. In typical transfection experiments with these cells, we obtain 70–80% efficiency as determined by counting the number of β-gal or green positive cells relative to the total number of cells attached per field (200×). It is important to note that in an individual experiment, transfection efficiently varies <10%. The efficiency and adhesion control assures that changes observed in cell migration/survival is not simply the result of differences in transfection efficiency or expression of the reporter gene or differences in the ability of transfected cells to attach to the ECM protein.
BrdU Incorporation and Laser Confocal Imaging of Apoptotic Cells in 3-D Collagen
Serum-starved COS-7 cells were transfected with a construct encoding GFP, then cultured in 3-D collagen gels for various times as described above, fixed, and examined directly within the 3-D matrix for apoptotic morphology using a laser confocal microscope (BioRad 1024 and a Zeiss Axiovert microscope) and Adobe Photoshop software. In some cases, serum-starved cells were transfected with vectors encoding the appropriate cDNAs along with the GFP reporter construct, then cultured in 3-D collagen for 6 h as described above. BrdU (20 μg/ml) was added and the cells cultured for an additional 20 h at 37°C, then fixed, permeabilized, and stained with anti-BrdU and secondary antibodies conjugated with rhodamine. Similar experiments were performed with NIH3T3 fibroblasts using the procedures described above, except cells were cultured on glass coverslips and serum-starved in buffer with 0.2% FBS for 18 h before BrdU incorporation and immunostaining. The number of GFP-transfected cells stained with the anti-BrdU antibodies was recorded blindly by two investigators. Results are reported as percentage of total transfected cells (i.e., green) incorporating BrdU (i.e., red).
Results
Cells Migrating in a 3-D Collagen Matrix Are Protected from Apoptosis
Cell invasion of the ECM requires signals for both cell migration and survival. To begin to investigate whether signals that initiate cell movement also regulate cell survival, we used a 3-D collagen matrix and purified growth factors and cytokines, since this culture system closely simulates the collagen-rich ECM of normal tissue (Keely et al. 1995; Koyama et al. 1996; Alford et al. 1998). This model allows us to monitor apoptosis in cells continuously anchored to a natural 3-D matrix. Apoptosis in this system is unique from anoikis in which cell–ECM connections are purposely broken and the ensuing death monitored as a secondary event (Frisch and Francis 1994). Serum-starved COS-7 cells were placed into 3-D collagen gels supplemented with or without cytokines and examined for morphological characteristics of apoptotic cells, including membrane blebbing, nuclear condensation, and fragmentation (Cohen 1993). As shown in Fig. 1 A, cells readily attach to and spread along the collagen fibers, forming a dendritic-like morphology within 8–12 h of culture. However, although these cells initially attach and spread in response to the collagen matrix, at later times (∼12 h) the dendritic processes retract and cells begin to display membrane blebbing and nuclear condensation (Fig. 1 A). In fact, after 24 h of culture, ∼60–70% of the cells are apoptotic with extensive fragmentation, a characteristic of cells in the late phase of apoptosis (Fig. 1 A). Importantly, cells are undergoing apoptosis rather than necrosis, since an increasing proportion of cells stain only with annexin V and not the vital dye PI during the culture period (Fig. 1 B). However, by 24 h, 70–80% of the cells are in the late stages of apoptosis and have lost membrane integrity, and therefore, stain with both annexin V and PI (data not shown). In addition, overexpression of Bcl-2 or exposure of cells to an inhibitor of caspase proteases (Z-VAD-fmk), which blocks apoptosis in some cells (Garcia-Calvo et al. 1998), is sufficient to rescue these cells from apoptosis (Fig. 1 B and Fig. 2 B). Together these findings indicate that COS-7 cells readily undergo apoptosis when cultured in a 3-D collagen matrix in the absence of growth factors and cytokines.
Figure 2.

Bcl-2 expression or exposure to Z-VAD rescues cells from apoptosis, but does not induce cell migration. (A) COS-7 cell migration in 3-D collagen after mock transfection with the empty vector or the vector with Bcl-2, along with a β-gal reporter constructed. In some cases, mock cells were exposed to Z-VAD (100 μM) or insulin (25 μg/ml). The number of transfected cells migrating was determined at the indicated times using X-gal as a substrate as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. (B) COS-7 cells treated as for the migration experiment above and examined for apoptosis after 24 h of culture in 3-D collagen. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Bcl-2 expression was confirmed in these cells by immunoblotting with anti–Bcl-2 antibodies. (C) COS-7 cells treated with various concentrations of EGF or (D) insulin, then evaluated for migration for 6 h and apoptosis after 24 h in 3-D collagen. The number of apoptotic cells was determined by morphological criteria as described in Materials and Methods. The results shown are the percentage of surviving cells (i.e., cells without apoptotic morphology/total number of cells per high powered field), and are the mean ± SEM of three cultures from three independent experiments.
To investigate whether cytokines could rescue cells from apoptosis as well as induce cell movement in 3-D collagen, the kinetics of migration and survival were monitored in cells exposed to FBS, insulin, or EGF. The kinetics of these responses are critical, since cells in the late stages of apoptosis may stop migrating and not adhere to the ECM. However, as shown in Fig. 1A–C, the kinetics of migration and programmed cell death are significantly different. In fact, we can measure changes in cell migration within 4 h (Fig. 1 C), whereas apoptosis is not detected until 8–12 h of culture as measured by annexin V staining, an early indicator of apoptotic cells (Fig. 1 B) (Martin et al. 1995). Importantly, although the kinetics of cell migration and apoptosis are clearly different in 3-D collagen, both cell processes are regulated by similar levels of cytokine in a dose-response manner (Fig. 2C and Fig. D). This and the kinetic difference allow us to evaluate these processes as separate events during invasion of the ECM. Moreover, the morphological and cytoskeletal changes associated with apoptotic cells are not detected until 12 h of culture (Fig. 1 A). In contrast, by this time a significant number of cells have already migrated in 3-D collagen in response to cytokines (Fig. 1 C). Therefore, cytokine-induced cell migration is not the result of a simple increase in the number of surviving cells available to invade the ECM. In fact, overexpression of Bcl-2 in cells or exposure to Z-VAD promotes cell survival, but not migration (Fig. 2A and Fig. B). It appears then that activation of the migration machinery of cells promotes cell survival by suppressing apoptosis. However, simple prevention of apoptosis in cells by overexpression of Bcl-2 or inhibition of caspases does not induce cell migration.
Ligation of Integrin and Cytokine Receptors Are Required for Cell Migration and Survival in 3-D Collagen
Integrin and cytokine receptors cooperate to promote signals that control cellular responses to ECM components (Renshaw et al. 1997; Schneller et al. 1997; Aplin et al. 1998). To determine if integrin receptors were involved in mediating cytokine-induced survival and motility in 3-D collagen, COS-7 cells were cultured in the presence of FBS, EGF, or insulin along with function-blocking anti-integrin antibodies. Antibodies directed to the β1 integrin subunit, but not the αvβ5 vitronectin receptor present on these cells (data not shown), significantly blocked both cell survival and migration responses induced by these cytokines (Fig. 3A and Fig. B). Importantly, cells not attached to the ECM and suspended in agar showed significant apoptosis even in the presence of cytokines (Fig. 3 C). These findings indicate that ligation of both cytokine and integrin receptors promote signals necessary for survival and migration of cells in a 3-D collagen matrix.
Figure 3.
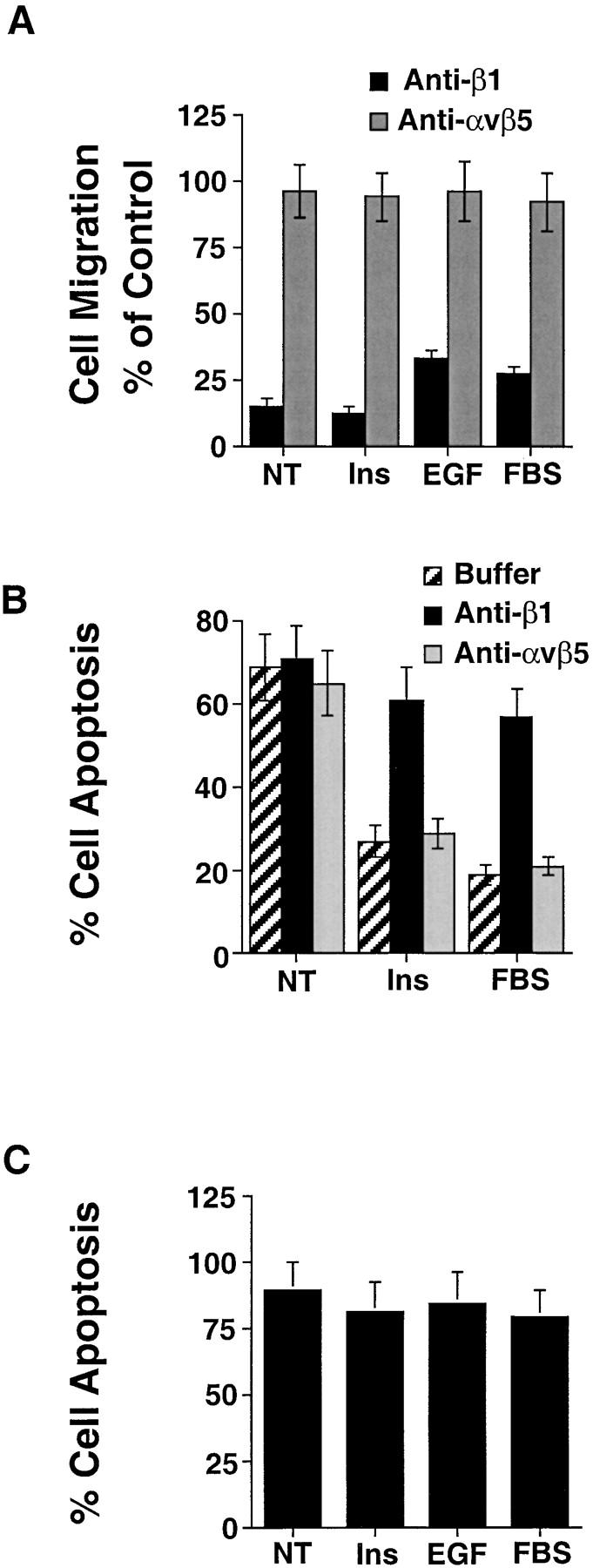
Ligation of both integrin and cytokine receptors are required for COS-7 cell survival in 3-D collagen. (A) COS-7 cell migration in 3-D collagen for 6 h in buffer (NT), buffer with insulin (25 μg/ml), EGF (100 ng/ml), or FBS (10%) together with function-blocking anti-integrin antibodies to the β1 subunit (25 μg/ml) or αvβ5 (25 μg/ml). Results are the mean ± SEM and are expressed as the percentage of cells migrating relative to cells not exposed to antibodies as described in Materials and Methods. (B) COS-7 cells cultured in 3-D collagen for 24 h in buffer (NT), buffer with insulin (25 μg/ml), or FBS (10%) together with function-blocking anti-integrin antibodies to β1 or αvβ5. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. (C) COS-7 cells cultured in soft agar for 24 h in buffer (NT), buffer with insulin, EGF, or FBS as described above. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments.
CAS/Crk and ERK Signaling Pathways Facilitate both Cell Invasion and Survival in 3-D Collagen
The activation of ERK and molecular coupling of the adaptor proteins CAS and Crk operate as distinct signaling pathways downstream of integrin and cytokine receptors capable of inducing two-dimensional haptotaxis cell migration (Cheresh et al. 1999; Nguyen et al. 1999). To determine whether these migratory signals could also promote cell invasion and survival in 3-D collagen, cells were transfected with mutationally activated MEK (ERK kinase) or CAS and Crk, then examined for apoptosis and migration in the absence of cytokines. Cells transiently transfected with either CAS and Crk or MEK showed increased migration and survival relative to mock cells expressing the empty vector (Fig. 4 A). In fact, the migration and survival responses initiated by these signals were comparable to cells exposed to insulin (Fig. 4 B) or EGF (data not shown). Importantly, the migratory and survival responses were associated with both increased CAS/Crk coupling and ERK activation in cells cultured in 3-D collagen (Fig. 4 C). In addition, exposure of cells in 3-D collagen to insulin or EGF (data not shown) promoted ERK activity and the endogenous association of CAS and Crk (Fig. 4 D). Stable expression of CAS and Crk in cells was also sufficient to promote increased migration and survival of cells in the absence of cytokines (Fig. 4E and Fig. F), and this event was blocked with antibodies to β1 integrins (data not shown). Together these findings indicate that CAS/Crk and ERK signals promote integrin-mediated cell invasion and protect migratory cells from apoptosis.
Figure 4.

CAS/Crk coupling and ERK signaling promote cell migration and protect cells from apoptosis in 3-D collagen. (A) COS-7 cells examined for apoptosis in 3-D collagen after transfection with the empty vector (Mock) or the vector with CAS and Crk (C+C) or mutationally activated MEK along with a β-gal reporter construct. The percentage of transfected apoptotic cells was determined by morphological criteria after 24 h of culture as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (B) Cells were transfected as described above and allowed to migrate in 3-D collagen for the indicated times. In some cases, mock-transfected cells were allowed to migrate in buffer or buffer with insulin (25 μg/ml). The number of transfected cells migrating was determined at the indicated times using X-gal as a substrate as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. (C) Detergent lysates of cells transfected as described above and Western blotted for CAS, Crk, MEK, ERK2, or the phosphorylated/activated form of ERK1 and ERK2. CAS and Crk are glutathione S-transferase– and myc-tagged, respectively, and show reduced mobility in SDS-PAGE compared with endogenous forms of these proteins. (D) CAS and ERK2 immunoprecipitates from detergent lysates of cells in 3-D collagen for 2 h in buffer (NT) or buffer containing insulin (25 μg/ml). CAS immunoprecipitates were examined for Crk binding by Western blotting with anti-Crk antibodies as described in Materials and Methods. ERK2-P represents the phosphorylated/activated form of ERK2, which migrates slower in SDS-PAGE. (E) COS-7 cells stably transfected with the empty vector (Mock) or the vector with CAS and Crk (C+C) and examined for cell migration for 6 h or (F) apoptosis in 3-D collagen for 24 h as described above. An aliquot of these cells was also lysed in detergent and examined for CAS and Crk expression by Western blotting with anti-CAS and Crk antibodies, respectively.
CAS/Crk and ERK Signaling Pathways Differentially Regulate Cell Proliferation
During embryonic development and certain pathological conditions associated with inflammation and tumor cell metastasis, some migratory cells not only show the ability to survive, but also proliferate as they invade the ECM. To investigate whether CAS/Crk coupling and ERK activation induced proliferation of migratory cells in 3-D collagen, cells were transfected with CAS and Crk or activated MEK, then examined for their ability to proliferate using BrdU incorporation into DNA and anti-BrdU immunostaining. As shown in Fig. 5, COS-7 cells expressing activated MEK showed significant BrdU incorporation that was comparable to cells exposed to FBS. Control cells treated with EGF and insulin also showed BrdU incorporation, although it was not as strong as cells exposed to FBS or cells expressing MEK. Surprisingly, <10% of cells expressing CAS/Crk showed BrdU staining (Fig. 5), even though they displayed significantly increased survival (Fig. 4A and Fig. F). We also examined the ability of CAS/Crk coupling and ERK activity to promote proliferation of cultured 3T3 fibroblast cells, since these cells are commonly used to measure DNA synthesis in response to ECM components (Meredith et al. 1993; Renshaw et al. 1997). As shown in Fig. 5 B, ERK activation, but not CAS/Crk coupling promoted cell proliferation. Together these findings indicate that CAS/Crk coupling supports cell survival and migration, but does not stimulate cell cycle progression leading to DNA synthesis and cell proliferation. In contrast, ERK signaling appears to support cell migration, survival, and proliferation under these conditions.
Figure 5.
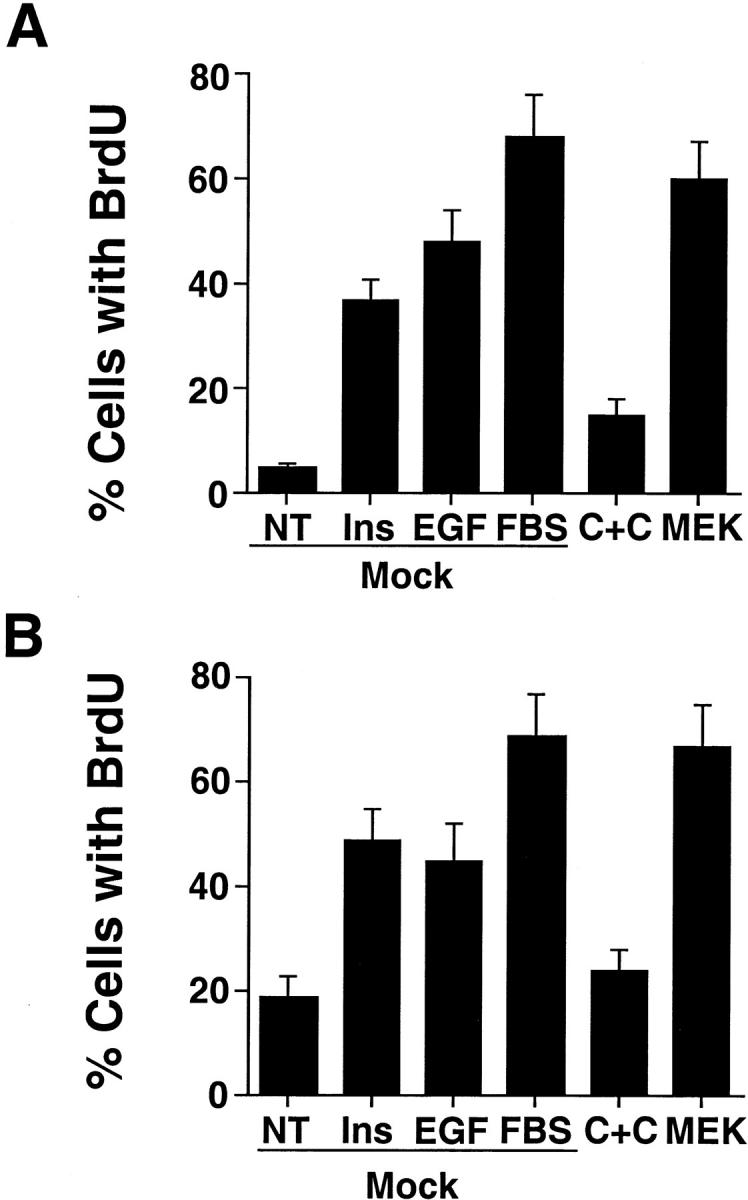
ERK activation, but not CAS/Crk coupling promotes cell proliferation. (A) COS-7 cells transfected with the empty vector (Mock) or the vector containing CAS and Crk (C+C) or mutationally activated MEK along with a GFP reporter construct. The percentage of transfected cells expressing GFP and incorporating BrdU into DNA was determined in cells cultured in 3-D collagen for 24 h in the presence or absence of insulin (25 μg/ml), EGF (100 ng/ml), or FBS (10%) as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (B) NIH3T3 fibroblasts transfected as described in A and examined for BrdU incorporation. The percentage of transfected cells expressing GFP and incorporating BrdU into DNA was determined in cells cultured on glass coverslips for 24 h in buffer (NT), buffer with insulin (25 μg/ml), EGF (100 ng/ml), or FBS (10%) as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments.
CAS/Crk Coupling and ERK Activation Are Necessary for Cell Migration and Survival
Crk with a mutated SH2 domain (Crk-SH2) and CAS without its substrate domain (CAS-SD) serve as dominant-negative proteins by preventing the coupling of these adaptor molecules, thereby blocking cell migration (Klemke et al. 1998). PD98059 is an inhibitor of MEK that prevents ERK activity and cell migration (Klemke et al. 1997; Cheresh et al. 1999). Expression of either CAS-SD or Crk-SH2 in cells significantly blocked insulin, EGF, and FBS-induced survival and migration in 3-D collagen (Fig. 6A and Fig. B). Similarly, inhibition of ERK activity by PD98059 prevented insulin and EGF-induced cell survival and migration (Fig. 6C and Fig. D). Interestingly, PD98059 did not significantly impact cell survival or migration induced by FBS, even though it blocked ERK activity in these cells (Fig. 6, C–F). These findings suggest that serum contains additional components (growth factors/cytokines) capable of promoting migration and survival independent of ERK signaling. However, although blocking ERK activity in cells did not induce cell death, it did prevent BrdU incorporation in response to FBS (Fig. 6 E). Therefore, ERK activity was required for FBS-induced cell proliferation, but not survival and migration. Together these findings indicate that CAS/Crk and ERK signals are required for cell migration and survival in response to insulin and EGF. However, additional signaling pathways exist to regulate migration and survival in response to serum components that are independent of ERK activity.
Figure 6.

CAS/Crk coupling and ERK activation are required for cell survival and migration in 3-D collagen. (A) COS-7 cell migration in 3-D collagen after transfection with the empty vector (Mock), the vector containing CAS without its substrate domain (CAS-SD), or Crk with a mutated SH2 domain (Crk-SH2), which prevents CAS/Crk coupling in cells (Klemke et al. 1998). These cells were transfected along with a β-gal reporter construct, and the number of transfected cells migrating for 6 h in buffer (NT), buffer with EGF (100 ng/ml), insulin (25 μg/ml), or FBS (10%) was determined using X-gal as a substrate as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. Note that detergent lysates were also prepared from these cells and examined for expression of CAS-SD and Crk-SH2 by Western blotting with CAS and Crk antibodies. The changes in CAS-SD and Crk-SH2 mobility on SDS-PAGE compared with endogenous forms of these proteins results from the truncation of the substrate domain of CAS and the myc-tag present on Crk, respectively. (B) COS-7 cells transfected as described for the migration experiment above in A and examined for apoptosis after 24 h of culture in 3-D collagen. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (C) COS-7 cell migration in 3-D collagen for 8 h in buffer (NT), buffer with insulin (25 μg/ml), EGF (100 ng/ml), FBS (10%), or PD98059 (25 μM) as indicated. The number of cells migrating was determined as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. (D) COS-7 cells treated as described in C above and monitored for apoptosis after 24 h in 3-D collagen. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (E) COS-7 cell proliferation in 3-D collagen. The percentage of cells incorporating BrdU into DNA was determined in cells cultured in 3-D collagen for 24 h in the presence or absence of FBS (10%) or PD98059 (25 μM) as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (F) ERK2 immunoprecipitated from detergent lysates from COS-7 cells cultured in 3-D collagen for 2 h in buffer (NT), buffer with insulin (25 μg/ml), EGF (100 ng/ml), FBS (10%), or PD98059 (25 μM) as indicated. ERK2 immunoprecipitates were Western blotted with ERK2 antibodies to detect the phosphorylated/activated form of this protein (ERK2-P), which shows reduced mobility by SDS-PAGE as the result of phosphorylation.
To further examine the requirement of CAS for cell migration and survival in 3-D collagen, we used primary fibroblasts isolated from CAS-deficient or wild-type embryos (Honda et al. 1998). In this case, basal as well as FBS-induced invasion was significantly impaired in CAS-deficient cells compared with wild-type cells (Fig. 7 A). Associated with the decreased invasiveness of these cells was decreased survival (Fig. 7 B). After 24 h of culture in FBS, ∼69% of CAS-deficient cells were apoptotic compared with only 20% of wild-type cells with CAS (Fig. 7 B). Importantly, reexpression of CAS in CAS-deficient cells restored their ability to migrate and rescued them from apoptosis (Fig. 7C and Fig. D). Therefore, CAS is required for both fibroblast cell migration and survival in 3-D collagen. Together these findings indicate that ERK and CAS/Crk signals regulate cell migration and suppress apoptosis during invasion of the ECM.
Figure 7.

Embryonic fibroblast cells deficient in CAS show reduced migration and survival in 3-D collagen. (A) Migration of embryonic fibroblast cells isolated from wild-type (CAS +/+) or CAS-deficient mice (CAS −/−) for 6 h in buffer (NT) or buffer with FBS (10%). The number of migrating cells invading into the lower chamber was counted as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. (B) Cells treated as for the migration experiment above and examined for apoptosis after 24 h of culture in 3-D collagen in the presence of various concentrations of FBS. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. (C) CAS −/− cells transiently transfected with the empty vector (Mock) or the vector containing wild-type CAS (CAS −/− plus CAS), along with a β-gal reporter construct. The number of migratory cells invading into the lower chamber was detected with X-gal and counted as described in Materials and Methods. Results are the mean ± SEM of three independent experiments. CAS immunoblotted from an aliquot of mock (lane 1) or CAS −/− plus CAS transfected cells (lane 2) is also shown. (D) Cells from the migration experiment in C are examined for apoptosis after 24 h of culture in 3-D collagen in the presence or absence of FBS (10%). The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three independent experiments.
Metastatic Carcinoma Cells Show Enhanced Survival in 3-D Collagen Relative to Nonmetastatic Cells
To investigate whether CAS/Crk and ERK migratory signals are associated with carcinoma cell survival and invasion, we used FG-M adenocarcinoma cells. These cells have a four- to fivefold increase in metastasis in animal models relative to the poorly metastatic FG cell line (Klemke et al. 1998). FG-M cells showed significantly increased ability to invade 3-D collagen compared with FG cells (Fig. 8 A). Associated with the increased invasiveness of FG-M cells was enhanced cell survival (Fig. 8 B). In fact, after 72 h in culture, 58% of FG cells were apoptotic compared with only 28% of FG-M cells. Importantly, FG-M cells showed increased CAS/Crk complexes and ERK activity compared with the noninvasive FG cells (Fig. 8 C). Furthermore, inhibition of ERK activity or expression of either CAS-SD or Crk-SH2 blocked FG-M cell invasion and induced apoptosis of these cells (Fig. 8, D–F). These findings provide additional evidence that invasive signals mediated by CAS/Crk and ERK coordinately protect cells from apoptosis.
Figure 8.

Carcinoma cells selected for increased invasive behavior in vitro and in vivo show increased survival in 3-D collagen. FG-M is a pancreatic carcinoma cell line that displays high migratory and metastatic properties in vivo. These cells were selected from a low invasive FG cell line as described previously (Klemke et al. 1998). (A) The number of FG and FG-M cells invading 3-D collagen was determined at the indicated times as described in the Materials and Methods. Results are the mean ± SEM of three independent experiments. (B) FG and FG-M cell apoptosis in 3-D collagen. The percentage of apoptotic cells was determined at the indicated times by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (C) CAS and ERK immunoprecipitates from detergent lysates of FG and FG-M cells in 3-D collagen for 2 h. CAS immunoprecipitates were examined for Crk binding by Western blotting with anti-Crk antibodies. ERK activity was evaluated by Western blotting with anti-phosphoERK antibodies that recognize the phosphorylated/activated form of ERK1 and ERK2 as described in Materials and Methods. (D) FG and FG-M cell migration in 3-D collagen after transfection with the empty vector (Mock) or the vector containing CAS without its substrate domain (CAS-SD) or Crk with a mutated SH2 domain (Crk-SH2). These cells were transfected along with a β-gal reporter constructed and the number of transfected cells migrating for 8 h determined using X-gal as a substrate as described in Materials and Methods. Mock-transfected cells were also allowed to migrate in 3-D collagen for 8 h in buffer (NT) or buffer with PD98059 (25 μM). Results are the mean ± SEM of three independent experiments. (E) Cells treated as described for the migration experiment above in D and examined for apoptosis after 48 h of culture in 3-D collagen. The percentage of apoptotic cells was determined by morphological criteria as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. (F) Detergent lysates were also prepared from cells treated as described above and examined for expression of CAS-SD and Crk-SH2 by Western blotting with CAS-, Crk-, and phosphoERK-specific antibodies as described in C. The changes in CAS-SD and Crk-SH2 mobility on SDS-PAGE compared with endogenous forms of these proteins result from the truncation of the substrate domain of CAS and the myc-tag present on Crk, respectively.
Rac and MLCK activation are downstream signals that mediate CAS/Crk and ERK-induced two-dimensional haptotaxis migration, respectively (Klemke et al. 1997, Klemke et al. 1998). To determine whether these signals were also necessary for cell invasion and survival in 3-D collagen, COS-7 cells were transfected with a dominant-negative form of either Rac (RacN17) or MLCK with its kinase domain mutated (MLCK-KD). These cells were then examined for apoptosis and invasion. As shown in Fig. 9 A, MEK-induced cell migration was readily blocked by MLCK-KD, but not the wild-type form of this protein. Interestingly, MLCK-KD did not impact MEK-induced cell survival, even though it prevented MLC phosphorylation in response to ERK activation (Fig. 9 A). Expression of MLCK-KD or wild-type MLCK alone in cells did not significantly impact migration or survival in the absence of ERK activation (Fig. 9 A). These findings indicate that MLCK activity is specifically required for ERK-mediated cell invasion, but not survival. In contrast, inhibition of Rac activity readily prevented both CAS/Crk-mediated cell invasion and survival (Fig. 9 B). Associated with the inhibition of migration and induction of apoptosis was reduced cell spreading and formation of dendritic-like processes compared with control and CAS/Crk-expressing cells (data not shown). Thus, Rac activity is a critical downstream component necessary for CAS/Crk-mediated invasion and survival. These events are likely linked to Rac's ability to organize the actin cytoskeleton and regulate cell shape changes necessary for migration and survival in response to ECM proteins. Interestingly, although cells expressing a constitutively activated form of Rac (RacQ61L) alone showed membrane ruffling (data not shown), there was no difference in migration or survival (Fig. 9 B). Moreover, exogenous expression of the Rac-activating protein DOCK180 in cells (Kiyokawa et al. 1998) did not support invasion or survival, even though it did facilitate significant Rac activity (data not shown). Similar findings were obtained with CAS-deficient fibroblast cells expressing activated Rac or DOCK180 (data not shown). Therefore, Rac activation alone in cells is not sufficient to support these processes. These findings also suggest that an additional effector molecule(s) and signaling pathway works in conjunction with Rac and DOCK180 to facilitate CAS/Crk-mediated invasion and survival.
Figure 9.

CAS/Crk-mediated cell invasion and suppression of apoptosis requires Rac activity, whereas ERK-induced invasion, but not survival, requires MLCK activity. (A) COS-7 cells examined for cell migration and apoptosis in 3-D collagen after transfection with the empty vector (Mock), mutationally activated MEK alone, HA-tagged wild-type (WT), or kinase-dead (KD) MLCK and a β-gal reporter construct. The number of transfected migratory cells after 6 h or the percentage of apoptotic cells was determined by morphological criteria after 24 h of culture in 3-D collagen as described in Materials and Methods. Results are the mean ± SEM of three cultures from three independent experiments. Cells transfected as described above were lysed in detergent and Western blotted for MEK and HA. In some cases, MLC was immunoprecipitated from cells metabolically labeled with [32P]orthophosphate and examined for changes in phosphorylation and total MLC protein as described in Materials and Methods. (B) COS-7 cells examined for cell migration and apoptosis in 3-D collagen after transfection with the empty vector (Mock), CAS and Crk alone, myc-tagged mutationally inactivated (RacN17), or activated Rac (RacQ61L). Cells transfected as described above were evaluated for migration and apoptosis as described above. Results are the mean ± SEM of three independent experiments. Cells transfected as described above were lysed in detergent and Western blotted with myc, Crk, and CAS antibodies as indicated. CAS and Crk are glutathione S-transferase– and myc-tagged, respectively, and show reduced mobility in SDS-PAGE compared with endogenous forms of these proteins.
Based on the findings above and the work of others, a model can be proposed showing how ERK activation and CAS/Crk coupling mediate cell migration and suppress the apoptotic machinery of cells during invasion of the ECM (Fig. 10). Cellular recognition of adhesive proteins and cytokines present in the ECM trigger ERK and CAS/Crk coupling, leading to MLCK and Rac activation, respectively. CAS/Crk coupling to DOCK180 facilitates Rac activation, actin organization, and cell shape changes necessary for remodelling and invasion of the ECM. In this model, Rac plays a central role in promoting CAS/Crk-mediated cell survival through its ability to regulate the actin cytoskeleton and cell architecture. ERK activation represents a second pathway that mediates cell invasion and suppresses apoptosis. In this study, MLCK is identified as a critical downstream component that specifically mediates cell contraction and invasion, but not survival in response to ERK activation. Although little is known regarding how ERK suppresses the apoptotic machinery of migratory cells, recent evidence indicates that ERK may directly regulate survival mechanisms by inactivating the death effector proteins BAD and hid (Fig. 10, and Discussion below).
Figure 10.

Model depicting the role of CAS/Crk and ERK signaling pathways in mediating cell migration and suppressing apoptosis during invasion of the ECM. Interaction of cells with adhesive proteins and growth factors/cytokines present in the ECM facilitate integrin and cytokine receptor activation on the cell surface. This initiates two distinct intracellular signaling pathways that control migration and suppress apoptosis as cells invade the ECM. Pathway One shows that ligation of integrin and cytokine receptors facilitate CAS tyrosine phosphorylation and the coupling of Crk to Cas via its SH2 domain (hatched box). FAK and src represent two potential upstream tyrosine kinases that mediate CAS/Crk coupling in migratory cells. DOCK180 is an effector molecule that specifically binds to the SH3 domain of Crk and regulates Rac activity. The CAS/Crk/DOCK180 protein module serves, in a Rac-dependent manner, as a critical signaling complex required for maintenance of the actin cytoskeleton and cell shape changes necessary for cellular invasion and survival. Additional signaling pathways downstream of Rac that may also operate to support survival during cell invasion are shown. These include inhibition of BAD and caspase 9 by Akt as well as gene transcriptional elements regulated by nicotinamide adenine dinucleotide phosphate (NADPH) burst oxidase and NF-κB. Interestingly, work in this study revealed that Rac activation alone in cells, in the absence of a CAS/Crk complex, is not sufficient to facilitate these cellular responses. Therefore, an additional effector molecule(s) and signaling pathway(s) must be associated with CAS/Crk that works in conjunction with Rac to induce cell migration and suppress apoptosis (black box). Integrin- and cytokine receptor–mediated Ras/ERK activation represents a separate pathway capable of mediating cell migration and suppressing apoptosis during invasion of the ECM. Once ERK is phosphorylated (ERK-P) and activated by MEK, it can directly phosphorylate MLCK (MLCK-P), which phosphorylates MLC (MLC-P). MLC-P can then associate with actin, leading to cell contraction and invasion. Work in this study indicates that ERK-mediated cell invasion, but not survival, requires MLCK activity. Although little is known regarding the specific signals that facilitate ERK-induced survival in migratory cells, recent work by others indicates that ERK activation in cells leads to phosphorylation and inactivation of BAD and its dissociation from Bcl family members. The uncoupling of these proteins increases the ratio of Bcl to Bax, which facilitates protection from apoptosis through mechanisms that are not yet understood. hid is a death-effector gene discovered in Drosophila that induces cell apoptosis by activating caspases. Interestingly, ERK can directly phosphorylate and inactivate hid and protect cells from apoptosis in this system. Although the mammalian homologue of this gene has not been identified, it is possible that a hid-like protein exists in mammalian cells which is inhibited during invasion of the ECM in an ERK-dependent manner. Alternatively, activated ERK is well known to translocate to the nucleus and regulate gene transcription processes important for cell cycle progression, leading to DNA synthesis and cell proliferation.
Discussion
Migratory cells must activate survival mechanisms as they move out of their normal environment and invade the collagen-rich matrix of the surrounding tissue. Ligation of integrin and cytokine receptors plays a central role in these processes, as they transmit signals that facilitate both migration and survival (Meredith et al. 1993; Aplin et al. 1998; Keely et al. 1998). In fact, when cells are denied contact with ECM components, they rapidly enter an apoptotic program and die in a processes referred to as anoikis (Meredith et al. 1993; Frisch and Francis 1994; Ruoslahti and Reed 1994). In this study, we used a 3-D collagen matrix that provides a physiological model to examine both survival and invasion of cells continuously anchored to the ECM. Collagen is one of the primary constituents of the extracellular environment that cells encounter during dissemination from their primary site. Therefore, signal transduction mechanisms that control migration and survival in 3-D collagen are more likely to reflect natural changes that occur in vivo as invasive cells interact with and remodel the collagen-rich ECM (Keely et al. 1995, Koyama et al. 1996; Alford et al. 1998; Haas et al. 1998; Satake et al. 1998; Farrelly et al. 1999). Our findings suggest that during cell invasion of a 3-D ECM, migration and survival mechanisms are coordinately regulated through activation of similar signaling pathways involving ERK activity and the molecular coupling of CAS and Crk. Several lines of evidence indicate that these signals operate downstream of integrin and cytokine receptors to coordinately regulate invasion and survival. First, the cytokines EGF, insulin, and insulin-like growth factor (IGF-1) are sufficient to induce both migration and survival of cells, and these events require ligation of β1 integrins. Second, ERK activation of MLCK and CAS/Crk coupling and Rac activation are two distinct signaling pathways that induce cell migration (Cheresh et al. 1999) and promote survival of cells during invasion of a collagen matrix. Third, coordinate activation of these signaling pathways is critical for survival of invasive cells, since uncoupling CAS/Crk complexes or inhibition of ERK activity blocks cell migration and induces apoptosis. Finally, carcinoma cells selected for increased cell motility and metastasis in vivo show increased survival compared with noninvasive cells that depend on ERK activity and CAS/Crk coupling. Interestingly, we found that prevention of apoptosis in cells with Z-VAD-fmk, a general inhibitor of caspases, or overexpression of Bcl-2, does not impact the general migration capacity of cells. Therefore, signaling components that regulate cell survival do not directly couple to and stimulate the migration system of invasive cells. In contrast, signals for cell migration directly regulate the survival machinery of cells by suppressing apoptotic mechanisms.
Although the specific mechanism responsible for suppression of apoptosis in migratory cells is not yet known, our findings indicate that dual signals from β1 integrins and cytokine receptors are required for both processes. Ligation of integrin and cytokine receptors facilitate CAS/Crk coupling and ERK activation leading to cell migration (Klemke et al. 1998; Cheresh et al. 1999). In this report, we show that these signals also suppress the apoptotic machinery of migratory cells as they invade a 3-D collagen matrix. However, additional signals are known to regulate cell survival in response to ECM components, including Akt, NF-κB, PI3K, and FAK (Frisch et al. 1996; Khwaja et al. 1997; Parrizas et al. 1997; Berra et al. 1998; Ilic et al. 1998; Scatena et al. 1998). Interestingly, FAK and PI3K have also been linked to integrin-mediated cell migration (Keely et al. 1997; Reiske et al. 1999). In fact, recent evidence indicates that PI3K and FAK can regulate CAS/Crk coupling as well as ERK activity in cells (Ojaniemi and Vuori 1997; Aplin et al. 1998). Therefore, PI3K and FAK may regulate survival and migration by facilitating CAS/Crk coupling and ERK activation. PI3K can also bind to the SH3 domain of Crk, suggesting that CAS/Crk coupling may directly regulate its activity or cellular localization (Sattler et al. 1997). However, it is not known if these survival signals are linked to CAS/Crk and ERK pathways that operate during cell invasion and remodelling of the ECM.
Although it is not yet clear how integrin and cytokine signals impact cell survival and migration, a common element of all of these signaling events is their ability to regulate the actin cytoskeleton and cell shape. Integrin and cytokine-mediated signals are known to regulate turnover of adhesive contacts, actin polymerization, and cell contraction (Aplin et al. 1998). CAS/Crk coupling facilitates Rac activation through its interaction with the small GTPase-activating protein DOCK180. Rac activation in turn is associated with actin polymerization and membrane ruffling (Ridely et al. 1992; Kiyokawa et al. 1998). Embryonic fibroblasts isolated from CAS-deficient animals show reduced actin-polymerization, delayed spreading and migration, as well as survival (Fig. 8) (Honda et al. 1998, Honda et al. 1999). In this study, we demonstrate that CAS/Crk coupling facilitates both cell invasion and survival in a Rac-dependent manner. We found that inhibition of Rac activity reduces cell spreading, migration, and induces apoptosis of invasive cells, suggesting that these events are linked to organization of the actin cytoskeleton. However, we also show that Rac activation alone in cells in the absence of a CAS/Crk complex is not sufficient to facilitate cell invasion or promote survival. This suggests an additional signaling pathway(s) and effector molecule(s) operate downstream of CAS/Crk that works in conjunction with Rac to induce cell invasion and survival. Interestingly, it has been reported that stable expression of wild-type or constitutively activated Rac in T47D breast adenocarcinoma cells is sufficient to induce cell invasion of collagen (Keely et al. 1997). This suggests that Rac activity is the limiting factor in the invasive process of these cells. These cells may express the necessary effector molecule(s) or may have constitutively activated the appropriate signaling pathway(s) that works along with CAS/Crk and Rac to induce cell invasion. Since PI3K activity is necessary for Rac-induced cell invasion, the ability of migratory cells to survive in the ECM may be directly linked to regulation of this protein (Keely et al. 1997). In fact, PI3K is known to activate Akt, which promotes cell survival through its ability to regulate BAD and caspase 9 activity, as well as gene transcriptional elements in the nucleus (Fig. 10) (Datta et al. 1997; Cardone et al. 1998; Brunet et al. 1999). Alternatively, Rac may regulate survival independent of PI3K through nicotinamide adenine dinucleotide phosphate (NADPH)/NF-κB and/or Jun NH2-terminal kinase (JNK) activation (Perona et al. 1997).
Previous work has shown that ERK activation can regulate myosin phosphorylation, leading to actin–myosin association and cell contraction of the ECM (Klemke et al. 1997; Cheresh et al. 1999; Nguyen et al. 1999). In this report, we show that ERK can facilitate cell invasion and protect cells from apoptosis. MLCK activity was found to be a critical component necessary for ERK-mediated cell invasion, but not survival. Recent evidence indicates that MLCK activity and MLC contraction may provide the force necessary for membrane extension and retraction processes associated with blebbing in apoptotic cells (Mills et al. 1998). Our findings suggest that ERK-mediated MLCK activation is not involved in the apoptotic process per se, but rather is specifically involved in contraction events that facilitate cell movement. In fact, we observed that inhibition of ERK or MLCK activity in apoptotic cells did not impact membrane blebbing (data not shown). In this case, RhoA may regulate MLC phosphorylation and membrane blebbing in apoptotic cells (Mills et al. 1998). Although the downstream mediator of ERK-induced cell survival is not yet known, it was shown recently that ERK can directly phosphorylate and inactivate the death effector gene hid in Drosophila (Bergmann et al. 1998). A hid-like gene may also exist in mammalian cells which is regulated through a similar mechanism. ERK activation has also been implicated in regulating phosphorylation of the Bcl-2 family protein BAD, which could lead to inactivation of the apoptotic machinery (Scheid et al. 1999). Alternatively, ERK is known to translocate to the nucleus and regulate gene transcription processes involved in cell cycle progression (Aplin et al. 1998).
Understanding the signaling networks that coordinately regulate migration and survival is crucial, as this serves to protect cells as they invade and remodel the foreign ECM. The ability of invasive cells to regulate the apoptotic machinery plays a central role during embryonic development, immune function, angiogenesis, and wound healing, as well as during certain pathological conditions associated with atherosclerosis, autoimmune disease, and tumor cell metastasis. Our findings that CAS/Crk coupling and ERK operate as common signaling pathways that induce cell migration and suppress apoptosis help provide a molecular understanding of how these processes are regulated as cells invade the ECM.
Acknowledgments
We thank Drs. R. Adelstein, H. Hirai, P. Gallagher, and M. Matsuda for providing reagents for this project. We also thank Drs. Kristiina Vuori and Dwayne Stupack for providing advice concerning this work, and Dr. S.R. Spencer for assistance with confocal microscopy.
R.L. Klemke was supported by National Institutes of Health grant CA 78493-01 and the American Cancer Society RPG-99-180-0. This manuscript is number 12819-IMM from The Scripps Research Institute.
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; BrdU, bromodeoxyuridine; CAS, p130 Crk-associated substrate; Crk, c-CrkII; ECM, extracellular matrix; ERK, extracellular-regulated kinase; FAK, focal adhesion kinase; GFP, green fluorescent protein; HA, hemagglutinin; MEK, ERK kinase; MLC, myosin light chain; MLCK, MLC kinase; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinase; SH, Src-homology; 3-D, three-dimensional.
References
- Alford D., Baeckstrom D., Geyp M., Pitha P., Taylor-Papadimitriou J. Integrin-matrix interactions affect the form of the structures developing from human mammary epithelial cells in collagen or fibrin gels. J. Cell Sci. 1998;111:521–532. doi: 10.1242/jcs.111.4.521. [DOI] [PubMed] [Google Scholar]
- Aplin A.E., Howe A., Alahari S.K., Juliano R.L. Signal transduction and signal modulation by cell adhesion receptorsthe role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharm. Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- Bergmann A., Agapite J., McCall K., Steller H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell. 1998;95:331–341. doi: 10.1016/s0092-8674(00)81765-1. [DOI] [PubMed] [Google Scholar]
- Berra E., Diaz-Meco M., Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J. Biol. Chem. 1998;273:10792–10797. doi: 10.1074/jbc.273.17.10792. [DOI] [PubMed] [Google Scholar]
- Brezinschek R.I., Marks-Oppenheimer N., Lipsky P.E. Activated T cells acquire endothelial cell surface determinants during transendothelial migration. J. Immunol. 1999;162:1677–1684. [PubMed] [Google Scholar]
- Brooks P.C., Montgomery A.M., Rosenfeld M., Reisfeld R.A., Hu T., Klier G., Cheresh D.A. Integrin alpha V beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Cheresh D.A., Leng J., Klemke R.L. Regulation of cell contraction and membrane ruffing by distinct signals in migratory cells. J. Cell Biol. 1999;146:1107–1116. doi: 10.1083/jcb.146.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J.J. Apoptosis. Immunol. Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Farrelly N., Lee Y., Oliver J., Dive C., Streuli C.H. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 1999;144:1337–1347. doi: 10.1083/jcb.144.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell. Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Vuori K., Ruoslahti E., Chan-Hui P. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Glinsky G.V., Glinsky V.V. Apoptosis and metastasisa superior resistance of metastatic cancer cells to programmed cell death. Cancer Lett. 1996;101:43–51. doi: 10.1016/0304-3835(96)04112-2. [DOI] [PubMed] [Google Scholar]
- Haas T.L., Davis S., Madri J.A. Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMMP-2 in microvascular endothelial cells. J. Biol. Chem. 1998;273:3604–3610. doi: 10.1074/jbc.273.6.3604. [DOI] [PubMed] [Google Scholar]
- Honda H., Oda H., Nakamoto T., Honda Z., Sakai R., Suzuki T., Saito T., Nakamura K., Nakao K., Ishikawa T. Cardiovascular anomaly, impaired actin bunding and resistance to src-induced transformation in mice lacking p130CAS. Nat. Genet. 1998;19:361–365. doi: 10.1038/1246. [DOI] [PubMed] [Google Scholar]
- Honda H., Nakamoto T., Sakai R., Hirai H. P130CAS, an assembling molecule of actin filaments, promotes cell movement, cell migration, and cell spreading in fibroblasts. Biochem. Biophys. Res. Commun. 1999;262:25–30. doi: 10.1006/bbrc.1999.1162. [DOI] [PubMed] [Google Scholar]
- Ilic D., Almeida E.A., Schlaepfer D.D., Dazin P., Aizawa S., Damsky C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P., Fong A.M., Zutter M., Santoro S.A. Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense α2 integrin mRNA in mammary cells. J. Cell Sci. 1995;108:595–607. doi: 10.1242/jcs.108.2.595. [DOI] [PubMed] [Google Scholar]
- Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness via PI 3-kinase. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Keely P., Parise L., Juliano R. Integrins and GTPases in tumor growth, motility and invasion. Trends Cell Biol. 1998;8:101–106. doi: 10.1016/s0962-8924(97)01219-1. [DOI] [PubMed] [Google Scholar]
- Khwaja A., Rodriguez-Viciana P., Wennstrom S., Warne P.H., Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa E., Hashimoto Y., Kobayashi S., Sugimura H., Kurata T., Matsuda M. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R.L., Cai S., Giannini A.L., Gallagher P.J., de Lanerolle P., Cheresh D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997;137:481–482. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R.L., Leng J., Molander R., Brooks P.C., Vuori K., Cheresh D.A. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama H., Raines E., Bornfeldt K.E., Roberts J., Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- Lauffenburger D.A., Horwitz A.F. Cell migrationa physically integrated process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lin Y., Grinnell F. Decreased level of PDGF-stimulated receptor autophosphorylation by fibroblasts in mechanically relaxed collagen matrices. J. Cell Biol. 1993;122:663–672. doi: 10.1083/jcb.122.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.J., Reutelingsperger C.P., McGahon A.J., Rader J.A., Van Schie R., LaFace D.M., Green D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulusinhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M., Nagata S., Tanaka S., Nagashima K., Kurata T. Structural requirement of Crk SH2 region for binding to phosphotyrosine-containing proteins. J. Biol. Chem. 1993;268:4441–4446. [PubMed] [Google Scholar]
- Mayer B.J., Hirai H., Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- Meredith J.E., Frazeli B., Schwartz M.A. The extracellular matrix as a survival factor. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J.C., Stone N.L., Erhardt J., Pittman R.N. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J. Cell Biol. 1998;140:627–636. doi: 10.1083/jcb.140.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.H., Catling A.D., Webb D.J., Sanlovic M., Walker L.A., Somlyo A.V., Weber M.J., Gonias S.L. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J. Cell Biol. 1999;146:149–164. doi: 10.1083/jcb.146.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojaniemi M., Vuori K. Epidermal growth factor modulates tyrosine phosphorylation of p130CAS. J. Biol. Chem. 1997;272:25993–25998. doi: 10.1074/jbc.272.41.25993. [DOI] [PubMed] [Google Scholar]
- Parrizas M., Saltiel A.R., LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- Perona R., Montaner S., Saniger L., Sanchez-Perez I., Bravo R., Lacal J.C. Activation of the nuclear factor-kappaB by Rho, cdc42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- Poelmann R.E., Gittenberger-de Groot A.C. A subpopulation of apoptosis-prone cardiac neural crest cells targets to the venous polemultiple functions in heart development. Dev. Biol. 1999;207:271–286. doi: 10.1006/dbio.1998.9166. [DOI] [PubMed] [Google Scholar]
- Raff M. Social controls on cell survival and death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Reiske H.R., Kao S.C., Cary L.A., Guan J.L., Lai J.F., Chen H.C. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J. Biol. Chem. 1999;274:12361–12366. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- Renshaw M., Ren X., Schwartz M.A. Growth factor activation of MAP kinase requires cell adhesion. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridely A.J., Paterson H.F., Johnstone C.L., Diekmann D., Hall A. The small GTP-binding protein Rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E., Reed J. Anchorage independence, integrins and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Satake S., Kuzuya M., Ramos M., Kanda S., Iguchi A. Angiogenic stimuli are essential for survival of vascular endothelial cells in three-dimensional collagen lattice. Biochem. Biophys. Res. Commun. 1998;244:642–646. doi: 10.1006/bbrc.1998.8313. [DOI] [PubMed] [Google Scholar]
- Sattler M., Salgia R., Shrikhande G., Verma S., Pisick E., Prasad K., Griffin J.D. Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-kit, phosphatidylinositol 3-kinase, and p120(CBL) J. Biol. Chem. 1997;272:10248–10253. doi: 10.1074/jbc.272.15.10248. [DOI] [PubMed] [Google Scholar]
- Scatena M., Almeida M., Chaisson M., Fausto N., Nicosia R.F., Giachelli C.M. NF-κB mediates αvβ3 integrin-induced endothelial cell survival. J. Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid M.P., Schubert K.M., Duronio V. Regulation of Bad phosphorylation and association with Bcl-xL by the MAPK/ERK kinase. J. Biol. Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- Schneller M., Vuori K., Ruoslahti E. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5600–5607. doi: 10.1093/emboj/16.18.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- Takaoka A., Adachi M., Okuda H., Sato S., Yawata A., Hinoda Y., Takayama S., Reed J., Imai K. Anti-cell death activity promotes pulmonary metastasis of melanoma cells. Oncogene. 1997;14:2971–2977. doi: 10.1038/sj.onc.1201147. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Hattori S., Kurata T., Nagashima K., Fukui Y., Nakamura S., Matsuda M. Both the SH2 and SH3 domains of human Crk protein are required for neuronal differentiation of PC12 cells. Mol. Cell. Biol. 1993;13:4409–4415. doi: 10.1128/mcb.13.7.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu Y., Mochii M., Vogel K.S., Weston J.A. Avian neural crest-derived neurogenic precursors undergo apoptosis on the lateral migration pathway. Development. 1998;125:4205–4213. doi: 10.1242/dev.125.21.4205. [DOI] [PubMed] [Google Scholar]