

Abstract
Monitoring the fusion of constitutive traffic with the plasma membrane has remained largely elusive. Ideally, fusion would be monitored with high spatial and temporal resolution. Recently, total internal reflection (TIR) microscopy was used to study regulated exocytosis of fluorescently labeled chromaffin granules. In this technique, only the bottom cellular surface is illuminated by an exponentially decaying evanescent wave of light. We have used a prism type TIR setup with a penetration depth of ∼50 nm to monitor constitutive fusion of vesicular stomatitis virus glycoprotein tagged with the yellow fluorescent protein. Fusion of single transport containers (TCs) was clearly observed and gave a distinct analytical signature. TCs approached the membrane, appeared to dock, and later rapidly fuse, releasing a bright fluorescent cloud into the membrane. Observation and analysis provided insight about their dynamics, kinetics, and position before and during fusion. Combining TIR and wide-field microscopy allowed us to follow constitutive cargo from the Golgi complex to the cell surface. Our observations include the following: (1) local restrained movement of TCs near the membrane before fusion; (2) apparent anchoring near the cell surface; (3) heterogeneously sized TCs fused either completely; or (4) occasionally larger tubular-vesicular TCs partially fused at their tips.
Keywords: exocytosis, membrane fusion, green fluorescent protein, total internal reflection, docking
Introduction
The dynamics of the late exocytic constitutive pathway are largely unexplored. For instance, how do post-Golgi carriers dock and fuse with the plasma membrane, and what influences the site of fusion? To study docking and fusion, diverse techniques have been employed (Rothman and Wieland 1996; Betz and Angleson 1997; Henry et al. 1998). Biochemical and genetic approaches have provided considerable insight into the molecular docking and fusion machinery and the possible molecular orders of events; however, they alone cannot address the dynamics. One classic technique, patch-clamping, relies on eliciting fusion with a specific stimulus and monitoring the changes in the membrane area or capacitance over time. While this technique has been championed for studying regulatory pathways, it is ill-suited for monitoring constitutive fusion. Moreover, it provides little information on the events before fusion or where fusion occurs.
Another approach is to microscopically monitor trafficking and fusion of the constitutive pathway using fluorescently labeled proteins. Wide-field or confocal time-lapse imaging of green fluorescent protein (GFP)–tagged proteins in living cells have challenged the view that small vesicles transport constitutive cargo from the TGN to the plasma membrane (Rothman and Wieland 1996). Rather, the transport containers (TCs) appeared as heterogeneously sized tubular-vesicular structures, often several microns in length (Hirschberg et al. 1998; Nakata et al. 1998; Toomre et al. 1999). The TCs emerged from the Golgi region and trafficked along microtubules (Toomre et al. 1999) to the cell periphery in a stop-and-go manner. Occasionally, a TC would remain static and vanish, sometimes in a small puff (Hirschberg et al. 1998; Toomre et al. 1999). However, the latter events were difficult to observe and, while they tentatively appeared to represent fusion, other interpretations were possible. It was also unclear if the large TCs represented clusters of vesicles in close proximity, or a single continuous tubular-vesicular structure. Postimaging fixation and electron microscopy of one axon favored the latter interpretation (Nakata et al. 1998). Whether large TCs remain intact until docking at the cell surface is also unknown. Thus, while these studies have suggested that large post-Golgi carriers exist, it has been difficult to definitively monitor fusion using such probes in standard epi-illuminated (EPI) wide-field or confocal microscopy systems.
Total internal reflection (TIR) fluorescence microscopy (also called evanescent wave microscopy) has been employed to monitor regulated exocytosis of chromaffin granules (Steyer et al. 1997; Oheim et al. 1999; Steyer and Almers 1999). Shortly after stimulation, fluorescently labeled secretory cargo was expulsed as a fluorescent puff into the medium. The theoretical aspects of this technique are well established (for review see Axelrod et al. 1992). Essentially, the bottom of the cell is externally illuminated with a laser beam at an angle that causes the light to be totally internally reflected at the fluid–coverslip interface (see Fig. 1 a). However, some of the light penetrates into the cell as an exponentially decaying evanescent wave, typically penetrating 35–200 nm. One advantage of TIR microscopy is that only a very thin layer at the bottom of the cell is illuminated; thus, an excellent fluorescent signal-to-noise ratio is obtained and photo damage is minimized. In addition, the light emitted by the fluorophore will exponentially increase as it approaches the coverslip (i.e., the bottom of the cell) and can facilitate detection of fusion.
Figure 1.

Live cell visualization of the late exocytic pathway by combined EPI and TIR. (a) Principle of a combined epifluorescence/evanescent wave microscope. See also accompanying QuickTime movies available at http://www.jcb.org/cgi/content/full/149/1/33/DC1. Organelles and TCs away from the plasma membrane (>100 nm) are visible by EPI (red) only. Once TCs approach the plasma membrane, they also become visible by TIR (green). In an overlay of the two channels, originally red, TCs turn yellow and subsequently green as they approach the plasma membrane and later fuse. (b) Two live cells imaged by EPI and TIR. Individual channels and a merge are shown. Boxes indicate areas enlarged in c and d. (c) A TC exits the Golgi complex and moves to the plasma membrane (small >), where it docks and partially fuses (large >). A running subtraction of successive frames tracks the movement of the TC from the Golgi complex to the apparent docking site (last frame). (d) Two TCs that undergo successive fusion in close proximity. Only the second (>) undergoes complete fusion. The time is indicated in seconds. (e) Plot of the relative fluorescence intensity of the second fusion event boxed in d. Note that the decrease in EPI fluorescence (red) is accompanied by a large, sharp increase in TIR fluorescence (green) when fusion occurs (>). The small TIR peak is due to an earlier (222 s) nearby fusion event. Bars: (b) 20 μm; (c) 4 μm; (d) 2 μm.
We have used a combination of EPI and TIR microscopy to follow constitutive membrane trafficking of vesicular stomatitis virus glycoprotein tagged with the yellow fluorescent protein (VSVG3-SP-YFP) from Golgi exit to plasma membrane fusion. We observed and analyzed the dynamics of single fusion events with good temporal and spatial resolution.
Materials and Methods
Cell Culture
PtK2 cells were grown in MEM, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 1× nonessential amino acids (GIBCO BRL), and 10% FCS (complete medium). Cells used for microscopy were grown on 1-mm-thick, 30-mm-diam sapphire slides (Rudolf Brügger AG) in complete medium without phenol red.
Infection with Recombinant Adenoviruses
VSVG3-SP-YFP is based on previously described temperature-sensitive VSVG3-GFP (Toomre et al. 1999), and differs from it by having a longer spacer between the last amino acid of VSVG and the start of YFP. Its construction, generation of recombinant adenoviruses, and characterization will be described elsewhere (Keller, P., D. Toomre. J. White, and K. Simons, manuscript submitted for publication). PtK2 cells were infected for 1 h at 37°C in 1 ml complete medium. After changing the medium, the cells were incubated for 6–20 h at 39.5°C to accumulate the protein in the ER, and then used for microscopy. Sapphire slides were transferred to a closed perfusion chamber (POC), which contained a glass coverslip (0.17 × 42 mm), a 25-μm Teflon spacer, and complete medium with 20 μg/ml cycloheximide. Samples were imaged 20–50 min after a shift to 32°C, conditions under which VSVG3-SP-YFP can exit the ER.
TIR Instrumentation
A prism type (Axelrod 1981) TIR fluorescent setup with an inverted microscope (Axiovert 135TV) and a hemicylindrical prism was used. The 514-nm laser line of an argon-krypton laser (Innova 70C Spectrum; Coherent) was selected by an acousto-optic tunable filter (model N48062; NEOS Technologies), which also acts as a fast shutter. The beam was coupled (model LA08-VIS; Spindler & Hoyer) into a single mode fiber (488 nm), which was connected to a focusable collimator (model MB 02; Spindler & Hoyer) mounted on x-y translation and a rotary stage was positioned 0.5 m to the side of the microscope. This allowed angular adjustments of the laser beam in a vertical plane, such that the emerging beam hit the sample area at an oblique angle. A hemicylindrical sapphire prism (radius, 6.5 mm to top, 7.5 mm to side; n = 1.77; Vision) was optically coupled to the sapphire sample slide chamber by a thin layer of diiodomethane immersion oil (n = 1.74; Merck) and fixed in position by an adjustable clamp. After mounting a sample, the final laser illumination spot position was adjusted with the x-y translator on the collimator. Fluorescence light was collected by a Zeiss water immersion objective (63×, 1.2 NA water; Zeiss). TIR- or EPI-illuminated light both passed through a YFP filter block (ex = 500/20, dichroic = 515, em = 535/30; Chroma Technology Corp.). The microscope was enclosed in a thermal insulation box and heated to 32 ± 0.5°C (Air-Therm; World Precision Instruments). At a measured incidence angle of 65° with n sapphire = 1.77 and n cytosol ∼1.38, a 514-nm beam had a calculated penetration depth of ∼45 nm (Axelrod et al. 1992).
Image Acquisition and Analysis
Images typically were recorded on a 12-bit–cooled CCD IMAGO digital camera (0.134 μm/pixel with 2 × 2 binning), controlled by TILLvisION v3.3 software (both TILL Photonics). Data were acquired at 0.5–20 Hz with TIR and EPI exposure times of 20–150 ms and 0.5–0.8 s, respectively, with acousto-optic switching to limit exposure between frames. Laser power levels were typically 30–100 mW per line.
Most postacquisition analysis, including tracking (single projection of difference images), area calculations and back-subtraction was done using TILLvisION macros. To analyze the data, fusion events were manually selected, a 10 × 10 pixel square (1.80 μm2) was centered around the fusion site, and the average pixel intensity over time was calculated. The frequency of fusion events and the fate of immobile TCs in cells were manually counted while looping 300–700 frame time-lapses. Sequences were exported as single TIFF files, and further processed using Adobe Photoshop 5.0 and Illustrator 7.0, or they were converted into QuickTime movies using IPLab v.3.2 (Scanalytics) or NIH Image v1.62.
Determination of the Diffusion Coefficient and Increase in Fluorescence upon Fusion
To determine the diffusion coefficient of VSVG3-SP-YFP in the plasma membrane, we analyzed the diffusional spread of the fluorescent cloud right after fusion. Three round concentric regions of interest, with radii 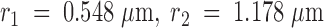 , and
, and 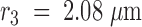 , were centered on a solitary fluorescent spot that expanded because of exocytosis during the recorded sequence (MetaMorph; Universal Imaging). Intensities (I
1 and I
2) are defined by the total fluorescence intensity within regions 1 and 2, divided by the area of the regions. I
annulus is the total fluorescence intensity within the annulus bordered by r
2 and r
3, divided by the area of this annulus. Its intensity is used as a measure of background fluorescence, and is subtracted in the model from both I
2 and I
1. We fitted the ratio of the background-subtracted intensities, I2 and I1, using the following formula:
, were centered on a solitary fluorescent spot that expanded because of exocytosis during the recorded sequence (MetaMorph; Universal Imaging). Intensities (I
1 and I
2) are defined by the total fluorescence intensity within regions 1 and 2, divided by the area of the regions. I
annulus is the total fluorescence intensity within the annulus bordered by r
2 and r
3, divided by the area of this annulus. Its intensity is used as a measure of background fluorescence, and is subtracted in the model from both I
2 and I
1. We fitted the ratio of the background-subtracted intensities, I2 and I1, using the following formula:
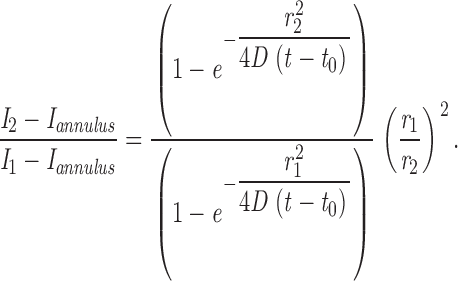 |
The model assumes instantaneous release of the dye from a point source at time t 0 and subsequent two-dimensional diffusion in the plasma membrane with the diffusion constant (D). The free parameters were D and t 0. The fit included seven data points beginning with the frame that precedes fusion. The fit was weighted by error bars that were calculated by taking the SEM of the last five intensity values before fusion over time and applying the error propagation formula. The background intensity (I annulus) after fusion was set to the average of the last five values directly preceding fusion to avoid overestimation of the background by spread of fluorescence into the annulus.
The fusion of a TC is accompanied by an increase in the total fluorescence. To calculate this increase when going from a vesicle or tubular structure near the plasma membrane (I vesicle) to the situation shortly after fusion when the dye is in the plasma membrane (I plasma membrane), we used the following formula, where a is the total length of the tube, L is the penetration depth of the evanescent wave, and d is the distance of the tube from the plasma membrane before fusion:
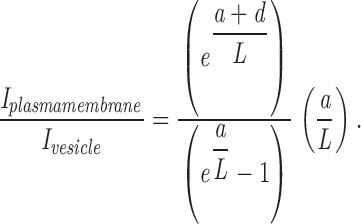 |
The model assumes that all of the dye is located in the membrane and that the axis of the tube is perpendicular to the plasma membrane.
Online Supplemental Material
We highly recommend that the interested reader consult the supplementary videos, which are available online with instructions for viewing them at http://www.jcb.org/cgi/content/full/149/1/33/DC1.
Results
Visualization of the Entire Late Secretory Pathway by Combined EPI and TIR Microscopy
As a novel approach to monitor cellular trafficking, we combined EPI and TIR fluorescent techniques (Fig. 1 a). Whereas EPI (red) illuminated the entire cell volume, TIR (green) only excited fluorophores near the coverslip, at a calculated penetration depth of ∼45 nm (see Materials and Methods). As a probe to follow exocytosis, we used a YFP-tagged temperature-sensitive mutant of VSVG, VSVG3-SP-YFP. Exocytosis of VSVG is considered a constitutive event in fibroblasts and related cell types. Around 30 min after release from the ER temperature block, VSVG3-SP-YFP was localized by EPI to the Golgi region and peripheral post-Golgi TCs (Fig. 1 b). Only the plasma membrane and a subset of TCs, but not the Golgi complex, was visible in the corresponding TIR image. Time-lapse video microscopy, with sequential EPI and TIR acquisition, allowed us to monitor cargo exit from the Golgi and approach the cell surface (see supplemental movies to Figure 1 at http://www.jcb.org/cgi/content/full/149/1/33/DC1). Fig. 1 c shows a red TC that exited the Golgi, turned yellow as it approached the surface, where it paused for ∼45 s before it partially fused. The projection of difference images depicted in the last frame shows the path of the movement. Another example is shown in Fig. 1 d, where a TC (indicated by >) entered the field, approached the surface, paused, and completely fused with the plasma membrane. At fusion, the signal observed by EPI dropped as VSVG3-SP-YFP diffused into the membrane (Fig. 1 e, red trace). In contrast, the corresponding TIR signal rapidly increased at fusion, as more cargo was excited near the coverslip (Fig. 1 e, green trace).
Fusion of Membrane Cargo with the Cell Surface Gives a Distinct Signature
Fusion of membrane cargo with the plasma membrane was apparent by visual inspection (see supplemental movies to Figure 1). Upon fusion (Fig. 2 a, top), VSVG3-SP-YFP diffused into the membrane like ripples in a pond. However, other means were needed to unambiguously identify fusion, quantitate these events, and eventually permit automated detection. When sequential frames of images were subtracted from one another and displayed as a sequence, fusion was identifiable by an expanding donut that was first white and then black. This pattern was readily distinguishable from the two other observed motions of TCs: rapid movement across the field or hovering near the plasma membrane (Fig. 2b and Fig. c, respectively; see also supplemental movie to Figure 2). The traces in Fig. 2 d show the relative fluorescence intensity in the corresponding fields over time.
Figure 2.
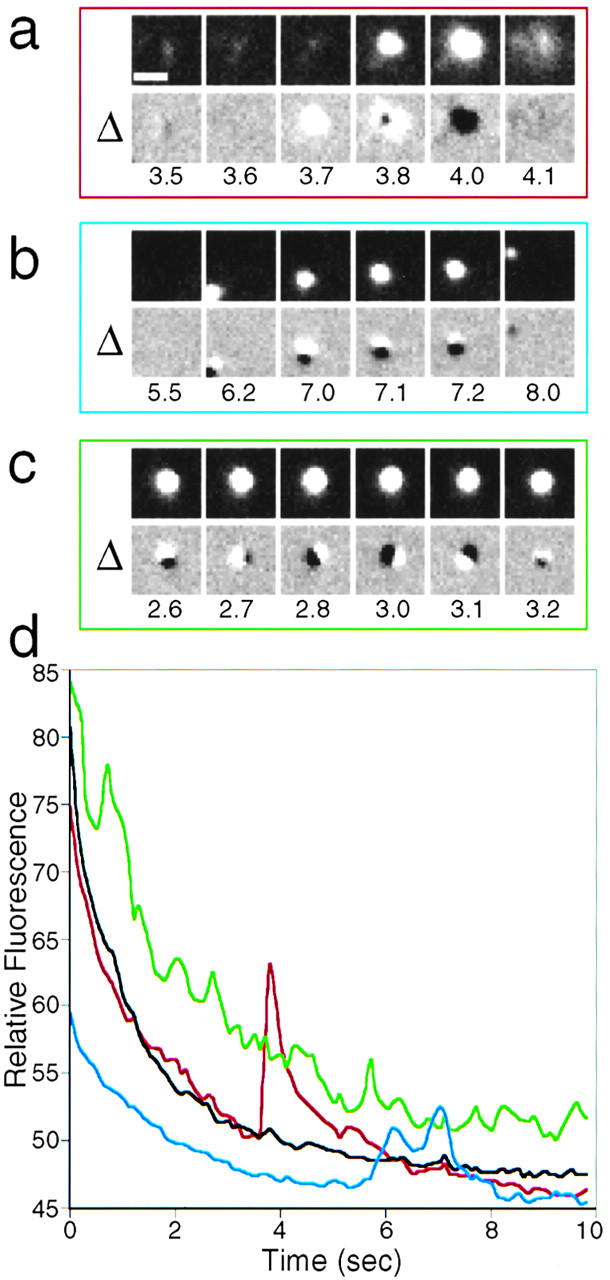
Fusion at the plasma membrane gives a unique signature. See also accompanying QuickTime movie available at http://www.jcb.org/cgi/content/full/149/1/33/DC1. In any given field (a–c, top) TCs were observed to either fuse (a), move across (b), or hover (tethered, but with motion in the z-axis) above the plasma membrane without undergoing fusion (c). A differential analysis (Δ, see Materials and Methods) allows one to distinguish these possibilities. A true fusion event results in sequential white and black clouds which expand as donutlike rings (a). TCs moving across the field result in black and white dots that keep their respective orientation (b). A TC hovering above the plasma membrane yields an asymmetric pattern in which the relative orientation of the dots changes over time (c). (d) Relative fluorescence intensity measured by TIR in fields (a–c) over time. Note that only a true fusion event (red trace) leads to a sharp increase in the fluorescence signal, generating an asymmetric peak with a long trailing edge as the protein diffuses into the membrane. The black curve shows the fluorescence intensity in an adjacent area without TCs; the initial decrease in fluorescence intensity is due to photobleaching at the plasma membrane which also facilitates detection of newly fusing TCs. Bar: 1 μm. Time is indicated in seconds.
We calculated the diffusion coefficient for VSVG3-SP-YFP upon fusion with the plasma membrane to be ∼1.3 × 10−9 cm2/s (see Materials and Methods). This value is in line with the diffusion constants of membrane proteins that are highly mobile in the plasma membrane (Sako and Kusumi 1995; Jacobson et al. 1997). As seen in the examples (Fig. 2 a, 3 b, and 4 c), ∼85% of the fluorescent profiles during and after fusion appeared circular in shape with width to height ratios of 1.06 ± 0.21, also suggesting free diffusion. The remaining noncircular shapes might represent fusion at microvilli (Polishchuk et al. 2000) or other mechanisms.
Local Tethering often Precedes Membrane Fusion
Most fusion events in any given cell (Fig. 3 a; see also supplemental movie to Figure 3 and Figure 4 at http://www.jcb.org/cgi/content/full/149/1/33/DC1) were preceded by a variable static. While TCs barely moved (<0.5 μm) in the x-y horizontal plane as if tethered, they oscillated slightly in the z-axis, as manifested by becoming alternately brighter and dimmer before fusion (Fig. 3 b, red and blue events). Intensity plots of the region, where fusion occurred, illustrate this rapid flux before fusion (Fig. 3 c). Although docking was common, some TCs rapidly approached the surface and fused after minimal docking (Fig. 3, black event). Occasionally, tubular-vesicular TCs approached at an angle and fused (Fig. 3, green event). On average the prefusion tethering phase (movement <2 μm) was 39 ± 33 s (n = 28; Fig. 3 d). There was no obvious correlation between the time of tethering and the size or approach angle of the TC. Interestingly, ∼64% of these TCs quickly approached the tethering site along curvilinear paths, suggesting the possible involvement of microtubules.
Figure 3.

Surface tethering often precedes membrane fusion. See also accompanying QuickTime movies available at http://www.jcb.org/cgi/content/full/149/1/33/DC1. (a) Cell surface visualized by TIR. (b) Sequential frames (1-s intervals) of the areas boxed in a show differential behavior of TCs. Many oscillate along the z-axis and then rapidly fuse (red), or stay tethered at the same position for up to a minute before fusion (blue). The intensity of the TIR signal changes over time but hardly moves within the xy-plane, suggesting the TC is tethered. Occasionally, TCs approach rapidly and only become visible at fusion (black). A trace of a tubular TC that slowly approaches the surface at an angle, and then slowly fuses is shown (green). The time of the first frame of the sequences is indicated in seconds. (c) The relative fluorescence intensity of the respective boxed regions is shown over time. Fusion events are indicated by arrows. (d) Histogram of the docking time (movement <2 μm) before fusion. Bars: (a) 10 μm; (b) 1 μm.
At any time, many TCs were close to the surface with an average density of 1.1 ± 0.3 TCs/μm2 (214 TCs analyzed in 5 cells). Many of those remained stationary for a considerable time, suggesting that they may represent a bound pool. As a comparison, TC fusion averaged 0.18 ± 0.02 events/min·μm2 (1,100 frames analyzed in 3 cells). To determine the fate of these tethered TCs, we monitored them to see whether they fused, stayed in place, or detached and moved back inside the cell. 2 TCs (10%) fused, 10 (52%) remained docked, and 7 (36%) detached in one cell within 5 min. In another cell monitored for 12 min, 4 TCs (24%) fused, 2 (12%) remained docked, and 11 (65%) detached. Thus, tethering or docking for extensive periods of time (>5 min) is common to many TCs, often precedes fusion, and is reversible.
Exclusion of Fusion in Peripheral Zones
Fusion of TCs appeared randomly distributed across the central region of the cell (Fig. 4 a; see also supplemental movie to Figure 3 and Figure 4 at http://www.jcb.org/cgi/content/full/149/1/33/DC1). However, at the edge of the cell, there was a wide belt where hardly any fusion was observed (Fig. 4 b, dark region). Manual tracking of fusion events also showed little events at the cell edge (data not shown). A lack of fusion could not be trivially explained by poor contact of such regions with the coverslip, since the VSVG3-SP-YFP present in the plasma membrane showed a relatively even fluorescence intensity (Fig. 4 a). These data are consistent with our earlier observations that few TCs appeared to move or tentatively fuse in the peripheral microtubule-poor, actin-rich lamella of PtK2 cells (Toomre et al. 1999). Instead, TCs appear to move off microtubules, after which the released TCs dock and fuse.
Figure 4.
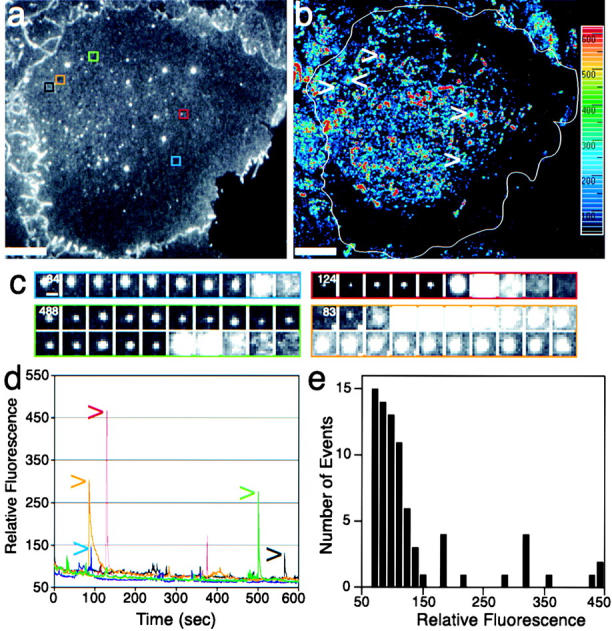
TCs fusing at the plasma membrane are heterogeneous in size and kinetics. See also accompanying Quick-Time movies available at http://www.jcb.org/cgi/content/full/149/1/33/DC1. (a) Cell surface visualized by TIR. (b) A time projection of difference images (see Materials and Methods) highlights TC fusion and movement. While there is substantial activity in the central region (red to light blue), only a few fusion events occur in the cell periphery (purple to black). Strong fusion events are seen as a red dot surrounded by a blue halo. Sequential frames (c) and the fluorescence intensity plot (d) of the areas marked in a and b show fusion events of different magnitude. Frames of the black square are shown in Fig. 3. Time of the first frame in the sequence is indicated in seconds. (e) Histogram of the relative intensity for all fusion events observed in this cell. Bars: (a and b) 10 μm; (c) 1 μm.
Heterogeneously Sized TCs Fuse with the Cell Surface
The widely variable intensity of flashes caused by fusion events (see also supplemental movie to Figure 3 and Figure 4) suggested that both small and large TCs fuse with the plasma membrane. Both small and large fusion peak times were very rapid (<100 ms; our maximal detection rate), as generally expected for vesicular fusion (Henry et al., 1988). By comparing the relative peak heights of various TCs (Fig. 4c and Fig. d), up to 10-fold differences in fluorescence release into the plasma membrane were observed. This indicates that either large peaks were due to large TCs, or that cargo was more concentrated in those containers. We favor the first interpretation. First, and most importantly the fluorescence intensity during high intensity fusion events often was much greater than at the docked state, sometimes by 20-fold. Such a dramatic change could not occur upon fusion of small vesicles 100 nm in diameter. However, it could arise from a 100-nm-wide tubule oriented perpendicular to the membrane, which we estimate would have to be ∼950 nm long to give a 20-fold increase in fluorescence upon fusion (see Materials and Methods). Second, TCs that appeared larger by EPI often gave larger TIR flashes (Fig. 1 and Fig. 5; supplemental movies to Fig. 1 and Fig. 5). Recent independent data using a similar marker and correlative light-electrion microscopy has confirmed the existence of large tubular-saccular TCs trafficking from the Golgi complex to the plasma membrane (Polishchuk et al. 2000). Last, we have plotted the relative intensity of all fusion events observed in a single cell as a histogram (Fig. 4 e). These data suggest that there exist at least two populations of carriers. While ∼80% of the TCs that fused were small, the larger TCs contained almost half of the total fluorescence and, thus, represent a significant entity.
Figure 5.
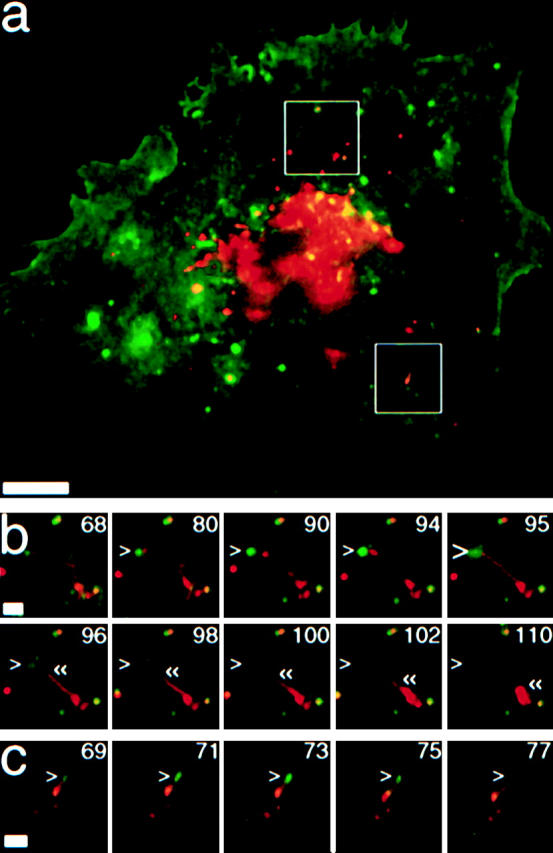
Large tubular-vesicular TCs can undergo partial fusion at the tip. See also accompanying QuickTime movies available at http://www.jcb.org/cgi/content/full/149/1/33/DC1. (a) Merge of a cell imaged by EPI (red) and TIR (green). Boxes indicate areas enlarged in (b) and (c). (b) A TC approaches the plasma membrane by a tubular extension, tethers with the tip of the tubule (small >), and fuses (large >) at that site. Fusion is transient and incomplete. The tubular extension detaches and collapses back into a globular structure («). (c) Another tubular-vesicular TC approaches the plasma membrane (small >), but retracts without undergoing fusion. Time is indicated in seconds. Bars: (a) 10 μm; (b and c) 2 μm.
Partial Fusion at the Tip of Tubular-vesicular Structures
As seen in Fig. 1, TCs appeared capable of undergoing partial fusion. A striking example of this is shown in Fig. 5, a and b (see also supplemental movies to Figure 5 at http://www.jcb.org/cgi/content/full/149/1/33/DC1), where a tubular-vesicular TC extended a tubule towards the plasma membrane. After partial fusion at the tip of this extension, the rest of the tubule immediately retracted, as if membrane tension was lost. Another example of local probing of the cell surface at the tips of tubules is shown in Fig. 5 c.
Discussion
We have applied a specialized microscopy technique, TIR, to detect and analyze fusion of individual constitutive post-Golgi TCs with the cell surface. TIR was previously applied to monitor fusion of regulated chromaffin granules (Steyer et al. 1997; Oheim et al. 1999; Steyer and Almers 1999), which are considerably larger than average TCs of constitutive transport. Unlike earlier studies (Hirschberg et al. 1998; Toomre et al. 1999), we now provide strong direct evidence that we can monitor fusion. Plasma membrane fusion was visually (see supplemental movies) and analytically (Fig. 1 e and 2) distinguishable from other trafficking phenomena. One advantage of following a fluorescent transmembrane protein, rather than a secreted protein or dye, is that it disperses two-dimensionally into the membrane plane and, thus, is easier to detect and quantify. Moreover, when combined TIR and EPI microscopy were used (Fig. 1 and Fig. 5) it permitted us to simultaneously monitor events at the cell surface and within the cell. Thus, we could monitor the entire late constitutive pathway: exit of TCs from the Golgi, trafficking towards the periphery, docking, and fusion with the plasma membrane.
TIR microscopy also allowed us to study the kinetics and dynamics of fusion. Parameters investigated included the relative size, fate of docked TCs, as well as the diffusion coefficient of VSVG3-SP-YFP immediately after fusion with the plasma membrane. Our studies indicate that large TCs do exist and can directly fuse with the plasma membrane. Evidence supporting the existence of large TCs included the following: (1) fusion of both small and large TCs; (2) a large rapid (<50 ms) increase of fluorescence after fusion as compared with the docked state; and (3) more intense and/or multiple fusion events commensurate with disappearance of larger TCs, as observed by dual EPI and TIR.
These studies also lead to several surprising observations. First, many TCs were associated near the membrane and were immobile for several minutes. While some eventually fused, the majority detached and moved back to the cell interior, suggesting that docking may be reversible. The finding of a large pool of apparently docked containers is novel and unanticipated for constitutive cargo. On the other hand, many secretory granules of the regulated pathway are docked near the cell surface (Steyer et al. 1997), suggesting that constitutive cargo may share similar mechanisms. Second, only a few fusion events occurred in the very outer cell periphery (Fig. 4), a region which often contains few microtubules in PtK2 cells (Toomre et al. 1999). Most movements before docking were fast, unidirectional, and long range. Together this would imply, albeit indirectly, that microtubules may play a role in directing cargo towards the final exit site and not just to the general periphery. Third, although most TCs fused completely, TCs also were capable of partial fusion (Fig. 1 and Fig. 5), in particular at the tips of larger tubular-vesicular TCs. Transient connections of large post-Golgi exocytic TCs with the plasma membrane were also recently observed by correlative light-electron microscopy (Polishchuk et al. 2000).
Overall, our studies reveal the complexity of constitutive docking and fusion that would be hard, if not impossible, to detect with other techniques. Schmoranzer et al. 2000 have reported similar results using TIR microscopy in this issue. In the future it should be possible to biochemically inhibit exocytic pathways in permeabilized cells so that, by TIR, the molecular components involved in docking and fusion can be discriminated at the single TC level.
Acknowledgments
We thank TILL Photonics for generously providing equipment and support; in particular Christian Heinemann has been very helpful. Without the support from the Max Planck Institut für medizinische Forschung (staff and equipment) this study would not have been possible; special thanks are due to Dietmar Manstein, Günter Giese, Michael U. Müller, and Jürgen Tritthard (all from Max Planck Institut). Alexander Rohrbach is acknowledged for the diffusion coefficient calculation program, Jamie White for his help with the supplemental online material, and Jan Ellenberg (all from European Molecular Biology Laboratory, Heidelberg, Germany) for critical comments on the manuscript.
D. Toomre was supported by a Marie Curie Research grant (no. ERBFMBICT961424), J.A. Steyer and P. Keller were supported by fellowships from the Max Planck Gesellschaft, and K. Simons was supported by an EU network grant and a grant from the Deutsche Forschungsgemeinschaft (SFB 352).
Footnotes
The online version of this article contains supplemental material.
Abbreviations used in this paper: EPI, epi-illuminated; GFP, green fluorescent protein; TCs, transport containers; TIR, total internal reflection; VSVG3-SP-YFP, YFP-tagged vesicular stomatitis virus glycoprotein; YFP, yellow fluorescent protein.
References
- Axelrod D. Cell–substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981;89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D., Hellen E.H., Fulbright R.M. Total internal reflection fluorescence. In: Lakovicz J. R., editor. Topics in Fluorescence Spectrometry. Plenum Press; New York: 1992. p. 289. [Google Scholar]
- Betz W.J., Angleson J.K. Cellular secretion. Now you see it, now you don't. Nature. 1997;388:423–424. doi: 10.1038/41213. [DOI] [PubMed] [Google Scholar]
- Henry J.P., Darchen F., Cribier S. Physical techniques for the study of exocytosis in isolated cells. Biochimie. 1998;80:371–377. doi: 10.1016/s0300-9084(00)80005-x. [DOI] [PubMed] [Google Scholar]
- Hirschberg K., Miller C.M., Ellenberg J., Presley J.F., Siggia E.D., Phair R.D., Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K.A., Moore S.E., Yang B., Doherty P., Gordon G.W., Walsh F.S. Cellular determinants of the lateral mobility of neural cell adhesion molecules. Biochim. Biophys. Acta. 1997;1330:138–144. doi: 10.1016/s0005-2736(97)00128-4. [DOI] [PubMed] [Google Scholar]
- Nakata T., Terada S., Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J. Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oheim M., Loerke D., Chow R.H., Stuhmer W. Evanescent-wave microscopya new tool to gain insight into the control of transmitter release. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1999;354:307–318. doi: 10.1098/rstb.1999.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk R.S., Polishchuk E.V., Marra P., Alberti S., Buccione R., Luini A., Mironov A.L. Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J. Cell Biol. 2000;148:45–58. doi: 10.1083/jcb.148.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J.E., Wieland F.T. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Sako Y., Kusumi A. Barriers for lateral diffusion of transferrin receptor in the plasma membrane as characterized by receptor dragging by laser tweezersfence versus tether. J. Cell Biol. 1995;129:1559–1574. doi: 10.1083/jcb.129.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoranzer J., Goulian M., Axelrod D., Simon S.M. Imaging constitutive exocytosis with total internal reflection fluorescence microscopy. J. Cell Biol. 2000;149:23–31. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer J.A., Almers W. Tracking single secretory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys. J. 1999;76:2262–2271. doi: 10.1016/S0006-3495(99)77382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer J.A., Horstmann H., Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Toomre D., Keller P., White J., Olivo J.C., Simons K. Dual-color visualization of trans-Golgi network to plasma membrane traffic along microtubules in living cells. J. Cell Sci. 1999;112:21–33. doi: 10.1242/jcs.112.1.21. [DOI] [PubMed] [Google Scholar]