

Abstract
Fibroblast growth factors (FGF) play a critical role in bone growth and development affecting both chondrogenesis and osteogenesis. During the process of intramembranous ossification, which leads to the formation of the flat bones of the skull, unregulated FGF signaling can produce premature suture closure or craniosynostosis and other craniofacial deformities. Indeed, many human craniosynostosis disorders have been linked to activating mutations in FGF receptors (FGFR) 1 and 2, but the precise effects of FGF on the proliferation, maturation and differentiation of the target osteoblastic cells are still unclear. In this report, we studied the effects of FGF treatment on primary murine calvarial osteoblast, and on OB1, a newly established osteoblastic cell line. We show that FGF signaling has a dual effect on osteoblast proliferation and differentiation. FGFs activate the endogenous FGFRs leading to the formation of a Grb2/FRS2/Shp2 complex and activation of MAP kinase. However, immature osteoblasts respond to FGF treatment with increased proliferation, whereas in differentiating cells FGF does not induce DNA synthesis but causes apoptosis. When either primary or OB1 osteoblasts are induced to differentiate, FGF signaling inhibits expression of alkaline phosphatase, and blocks mineralization. To study the effect of craniosynostosis-linked mutations in osteoblasts, we introduced FGFR2 carrying either the C342Y (Crouzon syndrome) or the S252W (Apert syndrome) mutation in OB1 cells. Both mutations inhibited differentiation, while dramatically inducing apoptosis. Furthermore, we could also show that overexpression of FGF2 in transgenic mice leads to increased apoptosis in their calvaria. These data provide the first biochemical analysis of FGF signaling in osteoblasts, and show that FGF can act as a cell death inducer with distinct effects in proliferating and differentiating osteoblasts.
Keywords: craniosynostosis, apoptosis, fibroblast growth factors, fibroblast growth factor receptors, osteoblast
Introduction
Mutations in fibroblast growth factor receptors (FGFR) have been linked to a number of human autosomal dominant skeletal disorders, including dwarfism and craniosynostosis syndromes, thus defining an important role for FGF signaling in skeletogenesis (reviewed in Muenke and Schell 1995; Burke et al. 1998; Naski and Ornitz 1998). Missense mutations in FGFR1 and 2 have been found in a variety of human craniosynostosis syndromes, which are characterized by premature fusion of the calvarial sutures and can lead to a variety of abnormalities including malformed skull shape, proptosis and mental retardation. However, studies on null mice have not been able to define a specific role for these receptors in osteogenesis due to early embryonic lethality (Yamaguchi et al. 1994; Deng et al. 1994; Arman et al. 1998).
Craniosynostosis is a result of improper bone formation in the developing skull, which occurs by a complex and poorly understood process termed intramembranous ossification. Mesenchymal cells enter the osteoblastic lineage and undergo a spatial and temporal progression; early preosteoblasts proliferate and then mature to osteoblasts that can differentiate and produce a collagenous matrix, a stage which is correlated with a decrease in proliferative capacity (Stein et al. 1996). Subsequent mineralization of the matrix results in the formation of the calvarial flat bones. Ossification occurs outwards from bone centers with the advancing bone margins separated by the sutures that are comprised of mostly immature osteogenic stem cells. The entire process is coordinated by the interplay of several signaling molecules such as the FGFs, BMPs, hedgehog, and TGFβ (Wilkie 1997; Kim et al. 1998). Although clearly excessive or unregulated FGF signaling disrupts normal intramembranous ossification, the effects of FGFs on osteoblasts at different stages of maturation have not been studied in detail.
Signaling from the FGF receptors can lead to proliferation, growth inhibition, or differentiation in different cell types (Goldfarb 1996), but it is not clear how these different biological outcomes are achieved. Whereas in fibroblasts and myoblasts, FGF treatment leads to the activation of the RAS/MAP kinase pathway and to cell proliferation (Kouhara et al. 1997; Saxton et al. 1997), in chondrocytes, FGF signaling also leads to the activation of the STAT-1 pathway that is required for growth arrest (Sahni et al. 1999). Several lines of evidence indicate that FGFs regulate both proliferation and differentiation in osteoblasts, but apparently conflicting reports on the exact nature of these effects exist in the literature, probably as a result of differential effects of FGF depending on the stage of osteoblast maturation (Debiais et al. 1998; Pitaru et al. 1993). Furthermore, most studies on the effect of FGFR mutations in osteoblasts, thus far, have been restricted to analysis of osteoblasts derived from craniosynostosis patients, and have been limited by the varying age of the patients and controls, and by the unknown effects of the FGFR mutations on the previous in vivo progression of these cells through maturation and differentiation (Lomri et al. 1998; Fragale et al. 1999).
In this report we address these issues by studying the effect of FGF on immature osteoblasts and cells progressing to differentiation both in primary mouse calvarial cells and in a newly established murine osteoblast cell line, OB1. Furthermore, to study the effect of known craniosynostosis-linked mutations on osteoblast proliferation and differentiation, we generated point mutations in the FGFR2 cDNA and stably expressed them in OB1 osteoblasts. The genetic mutations that have been identified in FGFR2 generally lie in the extracellular domain of the receptor (Burke et al. 1998). The mutation S252W in the linker region between the IgII and IgIII domains accounts for 65% of the cases of Apert syndrome (Slaney et al. 1996; Wilkie 1997), and the substitution of cysteine 342 in IgIII is the most common mutation in FGFR2 associated with Crouzon, Jackson-Weiss, and Pfeiffer syndromes (Wilkie 1997). Biochemical analyses show that many of these mutations disrupt the intra Ig domain disulfide bond and the unpaired cysteines cause receptor-dimerization which leads to constitutive receptor-activation (Galvin et al. 1996; Mangasarian et al. 1997; Robertson et al. 1998).
Our results show that FGF signaling has distinct effects on immature and maturing osteoblasts. Whereas immature cells are induced to proliferate, more differentiated cells are not. Furthermore, treatment of differentiating osteoblasts with FGF leads to an increased rate of programmed cell death. In all cases FGF treatment results in inhibition of differentiation, characterized by decreased expression of alkaline phosphatase and by the absence of mineralized nodules. In comparing the growth and differentiation properties of OB1 osteoblasts expressing either the wild-type FGFR2 or mutant receptors, FGFR2/S252W (Apert) or FGFR2/C342Y (Crouzon), we find that cells bearing the mutant receptor have a higher basal rate of DNA synthesis. When the mutant receptor-containing osteoblasts are induced to differentiate the expression of alkaline phosphatase is strongly repressed and mineralized bone nodules fail to form. Furthermore, apoptosis is dramatically increased. We also show that transgenic mice overexpressing FGF2 have a greater proportion of cells undergoing apoptosis in the developing calvarial osteogenic fronts compared with wild-type littermates. Thus, FGF has multiple effects on osteogenesis which include the induction of programmed cell death in differentiating osteoblasts.
Materials and Methods
Cell Culture and Preparation of Primary Osteoblasts
Calvaria from newborn mice were dissected free of surrounding muscles and soft tissues and washed in PBS containing penicillin and streptomycin. Isolated calvaria were sequentially digested in αMEM (GIBCO BRL), containing 0.1% collagenase and 0.2% dispase at 37°C. Digested fractions were collected every 10 min and fractions 2–5 were pooled. Cells were collected by centrifugation and resuspended in αMEM supplemented with 10% FCS. ROS 17/2.8 cells, a rat osteosarcoma cell line, was obtained from Dr. D. Mellon (University of Wisconsin, Madison) and were grown in DME containing 10% FCS. To study the differentiation of osteoblasts, cells were cultured for up to 21 d in growth media containing ascorbic acid (AA; 100 μg/ml) and β-glycerophosphate (BGP; 4 mM) and medium was changed every 3 d.
Immortalization of Osteoblasts
Primary osteoblasts were infected with a retrovirus (pBabe-puro) expressing the Polyoma large T-Antigen (Su et al. 1999). The cells were infected for 1 h in the presence of 8 μg/ml of polybrene. The virus stock was produced in 293 cells as described previously (Bellosta et al. 1997). Clones were selected using 4 μg/ml of puromycin for 2 wk. Immortalized clones OB1-4 were characterized according to morphology, histochemical staining for alkaline phosphatase and for their ability to express osteocalcin upon differentiation. Expression of Polyoma large T-Ag was determined by immunofluorescence using an anti-Polyoma large T-Ag rat serum.
DNA Synthesis Assays and Alkaline Phosphatase Staining
1 × 104 cells were seeded on coverslips in 24-well plates in αMEM medium containing 10% FCS. After 24 h the cells were either serum starved for 48 h in the presence of 0.4% FCS, or were induced to differentiate with AA and BGP, in the presence or absence of FGF1 (10 ng/ml) and heparin (10 μg/ml). Cells were labeled with 4 μg/ml of bromodeoxyuridine (BrdU; Sigma-Aldrich) for 6 h at 37°C, and histochemical alkaline phosphatase staining was performed according to manufacturer's instructions (85L-2; Sigma-Aldrich). Stained cells were then treated for 5 min with 0.5% Triton in PBS and for 15 min with 1.5 M HCl. After washing, analysis of BrdU incorporation was performed using an anti-BrdU monoclonal antibody (Amersham) followed by anti–mouse secondary antibody conjugated with Texas Red (Molecular Probes 1:200). Nuclei were stained with a solution of 1 μg/ml of Hoechst 33342 dye in PBS for 5 min. The fluorescence was visualized using a Zeiss Axiophot II microscope. The frequency of S phase cells was calculated as a ratio of BrdU positive nuclei to the total Hoechst stained nuclei.
DNA Fragmentation and Apoptosis Assay
Cytosolic DNA from OB1 clones was isolated for fragmentation analysis as previously described (Bellosta et al. 1997). DNA was isolated from cells after 10 d of differentiation. Apoptotic nuclei of differentiating primary osteoblasts were visualized using Hoechst staining. Paraffin embedded sections of 10-d-old post-frontal mouse sutures were processed for the TUNEL assay (In situ cell death detection kit; Boehringer). Fluorescence was visualized using a Zeiss Axiophot II microscope and TUNEL staining was visualized by DAB.
Immunoprecipitation and Western Blot Analysis
Cells were lysed in RIPA buffer (10 mM, Tris-HCl, pH 7.2, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% Na deoxycholate, 1% Triton X-100) containing protease and phosphatase inhibitors. 500 μg protein extracts was immunoprecipitated with specific antibodies overnight at 4°C, after which time, 50 μl of protein A–Sepharose (CL-4B, Amersham Pharmacia Biotech) was added and incubated for 1 h at 4°C. The protein A antibody–antigen complexes were washed extensively with the RIPA buffer and separated on SDS-PAGE. Proteins were electrotransferred to nitrocellulose membrane and incubated with the specific antibodies. Proteins were visualized by ECL (Amersham Pharmacia Biotech). Antibodies against FGFR1, 2, and 3 were purchased from Santa Cruz. Other antibodies used included anti-FRS2 (provided by Dr. I. Lax, NYU Medical Center), anti-SHP2 (Transduction Laboratories), anti-phosphotyrosine 4G10 (Oncogene Science), anti-phospho MAP kinase (NEB), anti-Erk2, and anti-Myc antibodies (Santa Cruz).
Cloning and Expression of FGFR2 Mutants into OB1 Clone
The stop codon was removed from a murine FGFR2 cDNA (Mansukhani et al. 1992) by site-directed mutagenesis, replaced with a KpnI site and cloned in frame with the myc epitope in pcDNA3.1/Myc-His vector (Invitrogen). A 3-kb PmeI fragment encoding the myc-tagged receptor was subcloned in the HpaI site of the pLXSN retroviral vector under the mouse Moloney leukemia virus LTR. Myc-tagged FGFR2/C342Y (Crouzon) and FGFR2/S252W (Apert) mutations were also constructed as the FGFR2 above. The Crouzon mutation-containing cDNA (Mangasarian et al. 1997) was myc tagged and subcloned into pLXSN as described above. The Apert mutation, S252W, was generated by using site-directed mutagenesis and was confirmed by DNA sequencing. The pLXSN vector carries the neomycin resistance gene under the SV-40 promoter, allowing selection in G418. pLXSN/FGFR2 constructs were cotransfected with an ecotropic packaging vector in 293 cells in order to produce a replication-defective retrovirus (Muller et al. 1991). OB1 cells were then infected with the virus and pools of clones were selected using 400 μg/ml of G418 (GIBCO). The expression of the receptors in OB1 clones were analyzed by Western blot using an anti-Myc polyclonal antibody (Santa Cruz).
Von Kossa Staining
To detect mineralized nodules formed in vitro, cultures were fixed in 4% paraformaldehyde for 10 min and rinsed with water. Cells were stained with 1% silver nitrate, placed under a UV lamp for 30 min and rinsed with water before treatment with 5% sodium thiosulfate for 2 min. Cells were then washed twice with water and counterstained with 1% Safranin-O to visualize the matrix.
Results
FGF Stimulates the Proliferation of Primary Osteoblasts and Newly Established Cell Lines
Earlier studies indicated that FGFs have a modest proliferative effect on primary rat and human osteoblasts (Hurley and Florkiewicz 1996; Stein et al. 1996). To study the biological effect of FGF on primary murine calvarial osteoblasts, we determined the percentage of cells undergoing DNA synthesis by BrdU incorporation in the presence and absence of FGF1, a ligand which binds all the FGFR isoforms (Ornitz et al. 1996). Primary osteoblasts were obtained by serial digestion of dissected calvaria from 1-d-old newborn mice and only first or second passage cells were used for the assays. In primary murine osteoblasts, FGF1 modestly increases the fraction of BrdU-positive cells from 8% (untreated cells) to 21.5% (Fig. 1). This weak proliferative effect is in contrast to that observed using primary embryonic fibroblasts, where FGF1 routinely induces a >10-fold increase in DNA synthesis (Bellosta et al. 1997).
Figure 1.

Effect of FGF1 on BrdU incorporation: primary osteoblasts and OB1 cells. The cells were starved for 48 h in 0.4% FCS then either untreated (−) or treated with FGF1 (10 ng/ml) and heparin (10 ng/ ml) for 24 h, and BrdU (4 μg/ ml) was added from 18–24 h. 10% FCS was included as a positive control. The data show the percentage of cells incorporating BrdU. Data shown are mean ± SD from a representative experiment.
Primary osteoblast cultures contain a mixture of spindle-shaped (immature) and cuboidal cells (mature) that are at various stages of maturation. Upon initial plating ∼1–10% of primary osteoblasts are positive for alkaline phosphatase (ALP). Whereas spindle-shaped cells are negative for ALP staining, cuboidal cells range from negative to strongly ALP positive. When cells were simultaneously stained for BrdU and ALP, we observed that strongly ALP-positive cells had not incorporated BrdU, indicating that these mature cells have a low proliferative capacity (data not shown). To determine the effect of FGF1 on osteoblasts at different stages of maturation, we performed BrdU incorporation assays on clonal cell lines of the osteoblastic lineage that we derived by immortalizing primary osteoblasts with polyoma large T antigen (see Materials and Methods). Four of these clones, OB1–OB4, were characterized for their ability to differentiate and express the markers ALP and osteocalcin (OC). As summarized in Table , clones OB1 and 2 have a spindle-shaped morphology and are negative for ALP and OC but steadily increase ALP and OC expression upon differentiation. They acquire a cuboidal morphology and form mineralized nodules after 3 wk in differentiation medium. Clone OB3 and 4 have a more cuboidal morphology and express ALP and OC but ALP expression is not appreciably increased upon differentiation. Upon treatment with FGF1, clones OB1 and OB2 displayed a strong proliferative response, while OB3 and OB4 have lower levels of stimulation, more similar to that of primary cultures. As seen in Fig. 1, clone OB1 cells show an eightfold increase in BrdU incorporation after FGF treatment.
Table 1.
Characterization of Osteoblast Clones
| Cells | Stimulation of BrdUincorporation by FGF1 | Initial Morphology | ALP* | OC‡ | ||
|---|---|---|---|---|---|---|
| Day 0 | Day 5–8 | Day 0 | Day 5–8 | |||
| OB1 | 8-fold | Spindle shaped | ± | ++ | − | + |
| OB2 | 6-fold | Spindle shaped | ± | ++ | − | + |
| OB3 | 1.5-fold | Cuboidal | ++ | ++ | ++ | ++ |
| OB4 | 2-fold | Cuboidal | + | + | ++ | +++ |
| Primary OB | 3-fold | Mixed | + | +++ | + | +++ |
*Alkaline phosphatase (ALP) expression was determined by Northern analysis and by histochemical staining upon initial plating (day 0) and after 5–8 d of differentiation.
‡Osteocalcin (OC) levels were assessed by Northern analysis upon initial plating (day 0) and after 5–8 d of differentiation.
Clones OB1 and OB2 have the characteristics of immature osteoblasts, whereas clones OB3 and 4 represent more mature stages in the differentiation pathway. Thus, the weak proliferation seen in primary cultures is likely to be an average of the responses of osteoblasts at different stages of maturation. Clone OB1, which appears to be the least differentiated and which has the highest proliferative response to FGF was used for all further experiments.
FGF Signal Transduction in Osteoblasts
The strong proliferative response of fibroblasts to FGF is mediated through FGF receptors 1 and 2 (Li et al. 1994), whereas chondrocytes that are growth inhibited in their response to FGF treatment express primarily FGFR3 (Sahni et al. 1999). To determine which FGF receptors are expressed in primary calvarial osteoblasts and clone OB1, we performed Northern analysis, using probes specific for the extracellular domains of the different FGF receptors. Primary murine osteoblasts, OB1, and ROS cells were found to express the mRNAs for FGFR1, 2, and 3 (data not shown).
We then tested whether the FGFR mRNAs detected were expressed as functional FGF receptors that could be activated by FGF1. Protein lysates were collected from cells that had been untreated or treated with FGF1 for 10 min. Immunoprecipitation with FGFR-specific antibodies followed by Western blotting with an anti-phosphotyrosine antibody showed that in ROS cells all three receptor species are phosphorylated by FGF treatment. In primary osteoblasts and OB1 cells, however, only FGFR1 and FGFR2 are visibly phosphorylated (Fig. 2 A). Similar receptor activation experiments performed on clone OB2 cells also detect phosphorylation of only FGFR1 and FGFR2 upon FGF treatment (data not shown).
Figure 2.
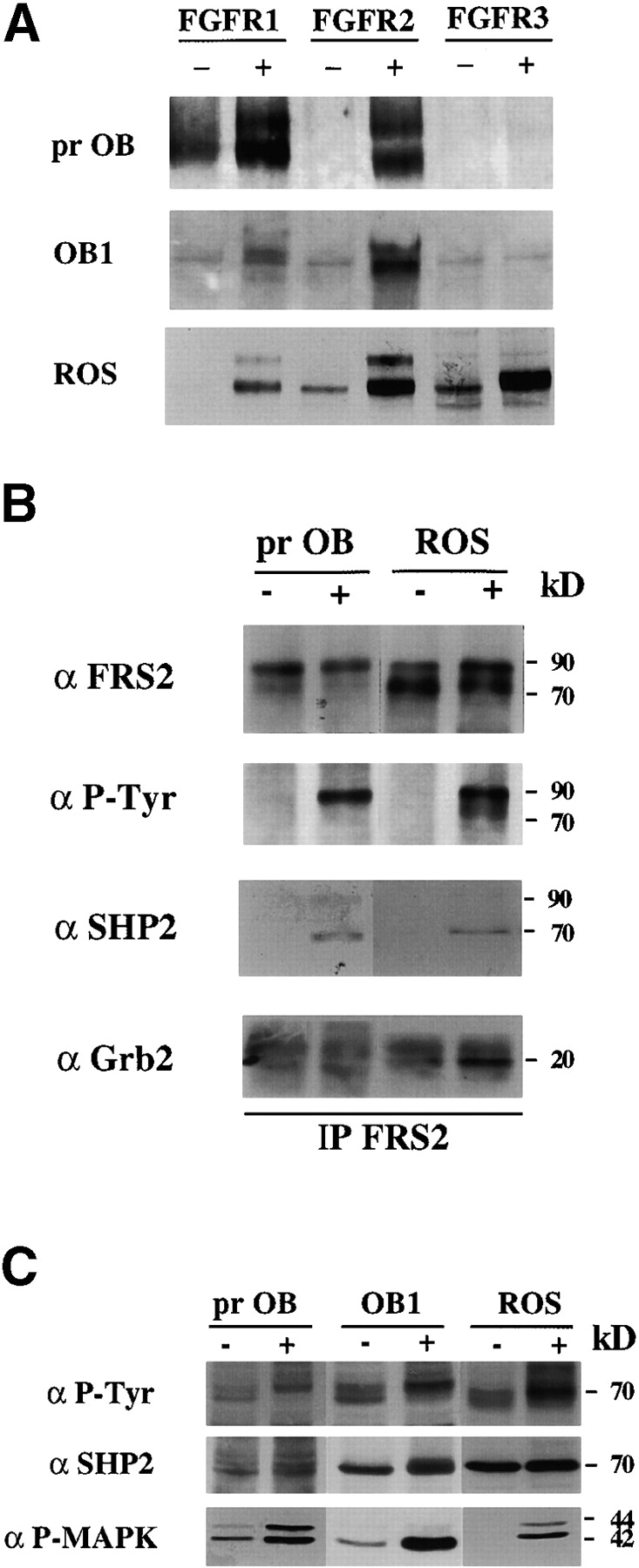
FGF Signaling in osteoblasts. (A) Activation of FGFRs in primary osteoblast, OB1 (prOB), and ROS cells. Cells were serum starved overnight in 0.4% FCS and were untreated (−), or FGF1-treated (+), at 100 ng/ml for 10 min and lysed. Total cell extract was subjected to immunoprecipitation with the anti-FGFR antibody indicated and blotted with anti-phosphotyrosine antibody 4G10. (B) Complex of FRS2, Shp2 and Grb2 in osteoblasts. Total cell extract from untreated (−) or FGF1-treated (+) primary osteoblasts and ROS cells was subjected to immunoprecipitation (IP) with anti-FRS2 antibodies. Immunoprecipitates were Western blotted (WB) with anti-phosphotyrosine (αP-Tyr), or with the indicated antibodies. 90-kD FRS2, 70-kD Shp2, and 20-kD Grb-2 bands are marked. (C) Phosphorylation of Shp2 and MAP kinase. 40 μg of total lysates was blotted with anti–P-Tyr, anti-Shp2, or anti-phospho MAP kinase (P-MAPK) antibodies.
When activated by ligand, receptor tyrosine kinases are rapidly autophosphorylated on several tyrosine residues, which are either essential for the catalytic kinase activity of the receptor or serve as docking sites for target proteins. The proliferative response to FGF in fibroblasts, for example, requires activation of the MAP kinase pathway and the phosphatase Shp2 (Saxton et al. 1997). Using protein lysates from unstimulated and FGF-stimulated primary osteoblasts and ROS cells, we studied the expression and activation of many previously identified FGF signaling targets. The docking protein FRS2 links the FGF receptors to the MAP kinase pathway by forming a complex with Shp2 and the adaptor protein Grb2 (Kouhara et al. 1997). Samples were immunoprecipitated with anti-FRS2 antibodies and blotted with anti-phosphotyrosine, anti-Shp2, and anti-Grb2 antibodies. Fig. 2 B shows that in both cell types, FRS2 is tyrosine phosphorylated in response to FGF (90-kD band). Shp2 (70 kD) and the adaptor protein Grb2 (20 kD) coimmunoprecipitate with FRS2 in FGF1-stimulated cells. Activation of Shp2 is detected by a slight shift in migration due to hyperphosphorylation and broadening of the 70-kD band in whole cell extracts which corresponds to the 70-kD tyrosine phosphorylated band seen in FGF-treated whole cell extracts (Fig. 2 C). Antibodies specific for the phosphorylated form of MAP kinase detect the activated form in FGF-stimulated primary, OB1 and ROS cells.
Recent findings on FGF signaling in chondrocytes (Sahni et al. 1999) show that activation of the STAT1 pathway is required for growth arrest. To determine whether the modest proliferation induced by FGF in primary osteoblasts may have been the result of the simultaneous activation of the STAT1 growth inhibitory pathway, we verified whether STAT1 is activated in response to FGF. However, FGF stimulation did not cause any detectable activation of STAT1 in ROS, OB1 or primary calvarial cells (data not shown). In conclusion, although osteoblasts coexpress FGFR1, 2 and 3, the signaling pathways activated by FGF treatment in these cells appear to be similar to those activated in fibroblasts.
Effect of FGF1 on Differentiating Osteoblasts
Long-term treatment of osteoblasts with ascorbic acid and β-glycerophosphate induces cell differentiation, characterized by increased expression of ALP and OC, cuboidal cellular morphology, and increased matrix production, followed by the mineralization of the extracellular matrix. To determine the effect of continuous FGF1 treatment on osteoblast differentiation, primary murine calvarial osteoblasts and OB1 cells were induced to differentiate in the presence or absence of FGF1 and heparin. After 3 wk, the cells were stained for ALP expression and by the Von Kossa method to visualize mineralized nodules. As seen in Fig. 3 A, differentiated primary osteoblasts stained strongly positive for ALP and mineralized areas were clearly visible. Continuous treatment with FGF1 markedly suppressed ALP expression and the formation of mineralized nodules. A similar pattern was also observed in clone OB1 cells (Fig. 6). The inhibition of ALP expression by FGF is reversible and the number of ALP-positive cells increased upon removal of FGF (data not shown).
Figure 3.
Effect of FGF on osteoblast differentiation. (A) Inhibition of alkaline phosphatase (ALP) and mineralization. Primary calvarial osteoblasts were maintained in differentiation medium for three weeks. FGF1 (10 ng/ml) and heparin were added from the start of the experiments and the differentiation medium was replaced every 3 d. Duplicate plates were fixed and stained for alkaline phosphatase enzymatic activity using Fast blue RR (Sigma) and for mineralization by Von Kossa staining. Positive cells stain purple for ALP expression and black areas indicate areas of mineralization. (B) Effect of FGF on osteoblast differentiation. 10,000 cells/well of primary osteoblasts were plated on coverslips in 10% FCS and differentiation medium was added to the wells at time 0. FGF1 was added at 10 ng/ml. BrdU (4 μg/ml) was added during the last 6 h of each time point. Cells were stained for alkaline phosphatase expression and for BrdU incorporation. Percentage of cells incorporating BrdU are shown. The percentage of cells that were positive for alkaline phosphatase were counted for each time point and are indicated on top of the graph. Data represent the mean of duplicate experiments. Bar, 800 μm.
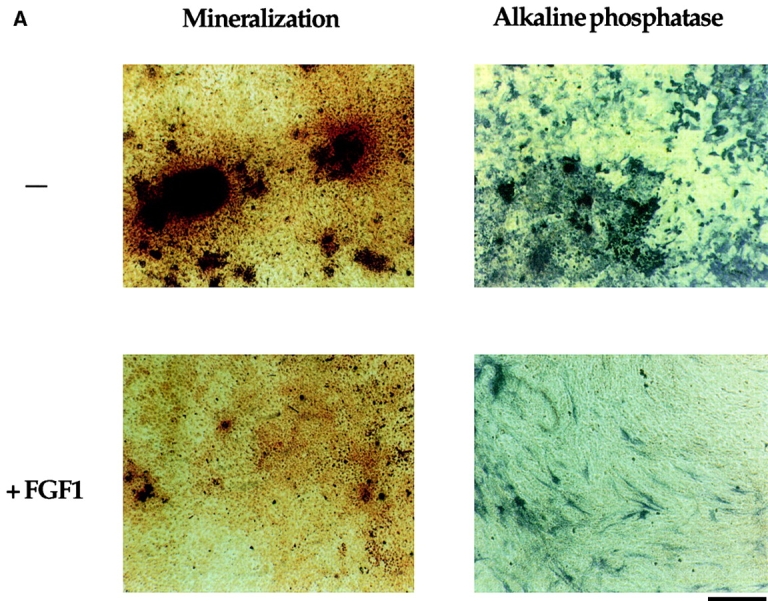

Figure 6.
Alkaline phosphatase and Von Kossa staining in OB1 cells expressing mutated FGFR2. Confluent cells were maintained in differentiation medium for three weeks (100 μg/ml ascorbic acid and 4 mM β-glycerophosphate). FGF1 (10 ng/ml) and heparin (10 μg/ml) were added where indicated after the first week. Medium was replaced every 3 d. (A) Plates were fixed and stained for alkaline phosphatase enzymatic activity using Fast blue RR (Sigma). Positive cells stain purple. (B) Plates were fixed and stained with silver nitrate for mineralization by the Von Kossa method. Bone nodules stain black and the matrix stains red when counterstained with Safranin O. Bars, 800 μm.


We then examined whether the effect of FGF on proliferation is altered as cells progress towards differentiation. We measured BrdU incorporation and expression of ALP in primary cells induced to differentiate either in the presence or absence of FGF1 after addition of differentiation medium. Fig. 3 B shows that as the cells differentiate, the rate of BrdU incorporation decreases steadily while ALP expression increases. Before addition of differentiation medium, 34% of the cells were incorporating BrdU. At day 1 after differentiation, only 22% of cells were positive for BrdU and this rate was reduced to 9.7% by day 3 and to 3.7 and 2.8% at day 5 and 9, respectively. During this time the percentage of ALP-positive cells increased from 1.6 to 38% at day 9. BrdU incorporation was only seen in cells that were either negative or weakly positive for ALP, i.e., cells that were strongly positive for ALP did not incorporate BrdU.
The effect of FGF1 on the level of DNA synthesis in the differentiating osteoblasts is quite different from the proliferative effect seen in the immature osteoblasts. At day 1, there was no significant change in the level of BrdU incorporation, but at all subsequent time points FGF1 further inhibited the rate of BrdU incorporation (Fig. 3 B). Thus, as the proliferative capacity of differentiating osteoblasts is progressively lost, FGF treatment no longer stimulates DNA synthesis and in fact inhibits it further at the later time points. ALP expression is drastically reduced by FGF treatment at all time points. A similar pattern was seen in differentiating OB1 osteoblasts (data not shown).
To determine if the lack of a mitogenic effect of FGF on differentiating osteoblasts was related to an attenuation of the MAP kinase pathway and/or alteration of FGFR activation, we studied the pattern of FGF receptor activation in OB1 and OB2 cells that had been differentiated for two weeks. Activation of FGFR1 and FGFR2, but not FGFR3, was evident by IP-Western analysis similar to what was found in immature osteoblasts that are growth stimulated by FGF. The pattern of activation of Shp2 and FRS2 was also unchanged and MAP kinase was still strongly activated as in immature osteoblasts (data not shown).
Expression of Activated FGFR2 in the OB1 Osteoblast Cell Line
Apert and Crouzon syndromes are predominantly associated with mutations in FGFR2. We created the most common Apert mutation in FGFR2, a serine 252 to tryptophan substitution in the linker region between Ig loops II and III in FGFR2 (Wilkie 1997). Crouzon syndrome mutations often involve the cysteine residues in Ig loop III and we altered the most commonly mutated cysteine 342 to tyrosine (Wilkie 1997). A wild-type FGFR2 cDNA and the mutated FGFR2 cDNAs (FGFR2/S252W or FGFR2/C342Y) were COOH-terminally tagged with a myc tag and cloned in the pLXSN retroviral vector. We had previously shown that the Crouzon receptor is constitutively activated, ligand unresponsive, and capable of transforming NIH3T3 cells (Mangasarian et al. 1997). The Apert receptor was reported to have higher affinity for FGF in solution assays (Anderson et al. 1998). We find that the Apert mutation-bearing receptor can also transform NIH3T3 cells although with a slightly lower efficiency (∼20%) than the Crouzon mutation-bearing FGFR2 (data not shown). Thus, the Apert mutation also causes a degree of ligand-independent receptor activation.
To study the effects of the mutant receptors on osteoblast proliferation and differentiation, we introduced the receptors into clone OB1 osteoblasts. Retroviral vectors expressing myc-tagged wild-type or mutant FGFR2 cDNA were used for infection and G418 resistant clones were selected and checked for the expression of the integrated constructs by Western blot analysis using anti-myc antibodies. Pooled clones were used for further analysis and tested for their ability to respond to FGF1 by receptor phosphorylation. Cell lysates were subjected to immunoprecipitation using anti-myc antibodies followed by Western blotting analysis with anti-phosphotyrosine antibodies. As seen in Fig. 4, cells expressing the wild-type receptor (FGFR2) show phosphorylation of the receptor upon stimulation with FGF1. The Apert mutation-bearing cells, FGFR2/S252W exhibit some ligand-independent receptor phosphorylation which is strongly enhanced by FGF1 treatment. As we had observed in fibroblasts, the FGFR2/C342Y (Crouzon) receptor in osteoblasts is also detected in an unglycosylated lower molecular mass form and its phosphorylation in the Crouzon cells is constitutive and is not further enhanced by FGF1 (Fig. 4 A). Western blotting with anti-myc antibodies shows that similar amounts of the myc-tagged receptor were immunoprecipitated in the FGF1-treated and untreated samples of each clone. The two bands recognized by the antibody are the unglycosylated (lower) and the glycosylated (upper) forms of FGFR2 (Fig. 4 B).
Figure 4.

Expression and activation of FGFR2 and FGFR2 mutant receptors in clone OB1. Parental OB1 cells as well as cells stably expressing the FGFR2 wild-type, Apert, or Crouzon mutants were serum starved for 24 h and left untreated (−) or treated (+) with 100 ng/ml FGF1 for 10 min. Expression and activation of the myc-tagged receptors was tested by immunoprecipitation with anti-myc antibodies followed by Western blotting as indicated. (A) Western blot with anti-phosphotyrosine antibodies (P-Tyr); (B) Western blot of with anti-myc antibodies; (C) Whole cell extracts blotted with anti-phospho–MAP kinase antibodies.
It has been shown that constitutive expression of activated tyrosine kinase receptors causes no detectable change in MAP kinase phosphorylation, most likely due to the rapid turnover of phosphorylated proteins (Su et al. 1997). Upon FGF treatment, inducible phosphorylation of MAP kinase is apparent in the Apert and Crouzon receptor-expressing cells (Fig. 4 C), which may indicate that the endogenous FGF receptors can still be activated by the addition of ligand.
Effect of Activated FGFRs on Osteoblast Proliferation and Differentiation
We examined the rate of proliferation of the Apert and Crouzon receptor-bearing osteoblasts in the presence or absence of FGF. Fig. 5 shows that compared with control cells or cells expressing the wild-type FGFR2, FGFR2/S252W (Apert) and FGFR2/C342Y (Crouzon) mutant expressing cells exhibit a twofold increase in the basal level of DNA synthesis. In Crouzon cells, this basal rate is weakly increased by FGF1 treatment, while Apert receptor–expressing cells are stimulated from 22 to 54% BrdU positive cells. Although the Crouzon mutation causes constitutive receptor activation, it prevents ligand binding in other cell types (Mangasarian et al. 1997). The stimulation of proliferation by FGF1 in OB1 cells expressing the Crouzon mutation is therefore probably due to activation of the endogenous wild-type FGFRs. The Apert mutation also causes an increase in osteoblast proliferation in the absence of exogenous FGF indicating a degree of ligand independence. However, the stronger proliferative response by these cells upon FGF treatment is likely to be due to the increased affinity of the Apert receptor for the ligand (Anderson et al. 1998).
Figure 5.

BrdU incorporation of OB1 cells, or cells expressing FGFR2, Apert, or Crouzon mutations. Cells were serum starved for 24 h in 0.4% FCS, then treated for 24 h with FGF (10 ng/ml) + heparin, and with 10% FCS as a positive control. BrdU was added from 13–24 h. The data show the percentage of cells incorporating BrdU.
We then tested whether expression of the Apert or Crouzon mutant FGFR2 affected the differentiation capacity of the OB1 cells. Cells were induced to differentiate upon confluence with the addition of ascorbic acid and beta glycerophosphate. They were analyzed for ALP expression and mineralized nodule formation after three weeks in differentiation medium in the absence or presence of FGF1. Fig. 6 A shows that control OB1 cells were well differentiated in the absence of FGF1 and stained strongly positive for ALP. Addition of FGF1 inhibited the expression of ALP both in control OB1 or in OB1 cells expressing wild-type FGFR2. In contrast, ALP expression in OB1-Apert or Crouzon cells was undetectable even in the absence of FGF1. Bone nodule formation was visualized by Von Kossa staining where nodules stain black. Von Kossa staining (Fig. 6 B) of parallel cultures showed that in clone OB1 osteoblasts, mineralization is repressed by addition of FGF1. In the mutant FGFR2-expressing cells, mineralized nodules are absent even in the absence of ligand indicating that the mutant receptors are constitutively signaling. Large areas of red staining are visible (matrix) indicating that some maturation does occur in the mutant cells. Thus, constitutive expression of activated receptors in osteoblasts blocks ALP expression and mineralization.
Activation of FGF Signaling during Osteoblast Differentiation Leads to Apoptosis
FGF treatment of immature osteoblasts increases proliferation. In contrast, we observed that continuous FGF treatment of differentiating osteoblasts caused a clear increase in cell death that became more apparent as differentiation progressed. Similarly, cultures of OB1 cells expressing the Crouzon or Apert FGFR2 mutations showed a large increase in cell death upon differentiating conditions. We thus investigated whether FGF treatment of osteoblast cultures induced to differentiate caused apoptosis.
Upon differentiation, apoptosis was clearly visible in Hoechst-stained nuclei of FGF-treated primary osteoblasts, as well as in the OB1 clone with large numbers of cells displaying the characteristic apoptotic nuclear blebbing and chromatin condensation. When induced to differentiate, OB1 cells expressing the Crouzon or Apert mutation undergo apoptosis even in the absence of added FGF. A quantitation of the percent of apoptotic nuclei during differentiation of primary osteoblasts is shown in Fig. 7 A. In untreated differentiating cultures, there is no appreciable increase in apoptosis over 14 d, while in cultures continuously treated with FGF1, the number of apoptotic nuclei is increased to 38%. We analyzed DNA from untreated and FGF-treated samples of clone OB1, FGFR2/S252W (Apert) and FGFR2/C342Y (Crouzon) cells that had been differentiated for 10 d, and checked for the DNA-laddering pattern typical of apoptotic cells. As seen in Fig. 7 B, DNA laddering is clearly visible in FGF-treated control OB1 cells indicating that activation of FGF signaling during differentiation induces programmed cell death. The same high degree of DNA laddering is observed in the OB1 cells expressing the mutated receptors, showing that constitutive FGFR2 activation is sufficient to induce apoptosis even in the absence of ligand stimulation (Fig. 7 B).
Figure 7.

Apoptosis in differentiating osteoblasts treated with FGF. (A) Cells were plated on coverslips in 24-well plates and cultured in differentiation medium for 14 d in the absence (−) or presence (+) of FGF1 (10 ng/ml + heparin). Cells were fixed on the indicated days. Hoechst dye–stained nuclei from at least seven different microscope fields were counted and apoptotic nuclei calculated as a percentage of total cells. Data shown are means ± SD from a representative experiment. (B) Cells were cultured for 10 d in differentiation medium. FGF1 was added at 10 ng/ml in the (+) lanes and the medium was changed every 3 d. DNA was extracted, electrophoresed on 0.8% agarose gel and visualized with ethidium bromide. Control, OB1 cells with vector alone.
Apoptosis is not generally observed in response to FGF, and in fact, FGF protects fibroblasts from apoptosis induced by serum-starvation (Bellosta et al. 1997). We therefore examined the signaling molecules activated by FGF that may be implicated in the cell death response. Since activation of AKT by PI-3 kinase is responsible for the protective effect of growth factors on apoptosis (Kennedy et al. 1997), we examined whether this pathway was impaired in differentiating osteoblasts. FGF treatment leads to the activation of AKT by serine phosphorylation (Miho et al. 1999). Using antibodies specific for phosphorylated AKT, we tested whether AKT was activated in response to FGF in OB1 osteoblasts. Fig. 8 A shows that while AKT phosphorylation is strongly increased in NIH3T3 fibroblasts after 15 min of treatment with FGF1, OB1 cells show no increase of AKT phosphorylation in proliferating or differentiating cells. MAP kinase was equally activated in NIH3T3 and OB1 cells. We then examined the steady state levels of phospho-AKT in OB1 cells induced to differentiate for 5 or 9 d. No increase was seen in phospho-AKT during differentiation both in the presence or absence of FGF1. We also found that the pro-apoptotic factor Bax is induced during differentiation and is further increased in the presence of FGF1. On the other hand, the anti-apoptotic factor Bcl2 is also induced during differentiation but is somewhat delayed in cells treated with FGF (Fig. 8 B). Thus, lack of phospho-AKT induction together with the increase in Bax level and the delay in Bcl2 accumulation may all contribute to the apoptotic effect of FGF seen in differentiating osteoblasts.
Figure 8.
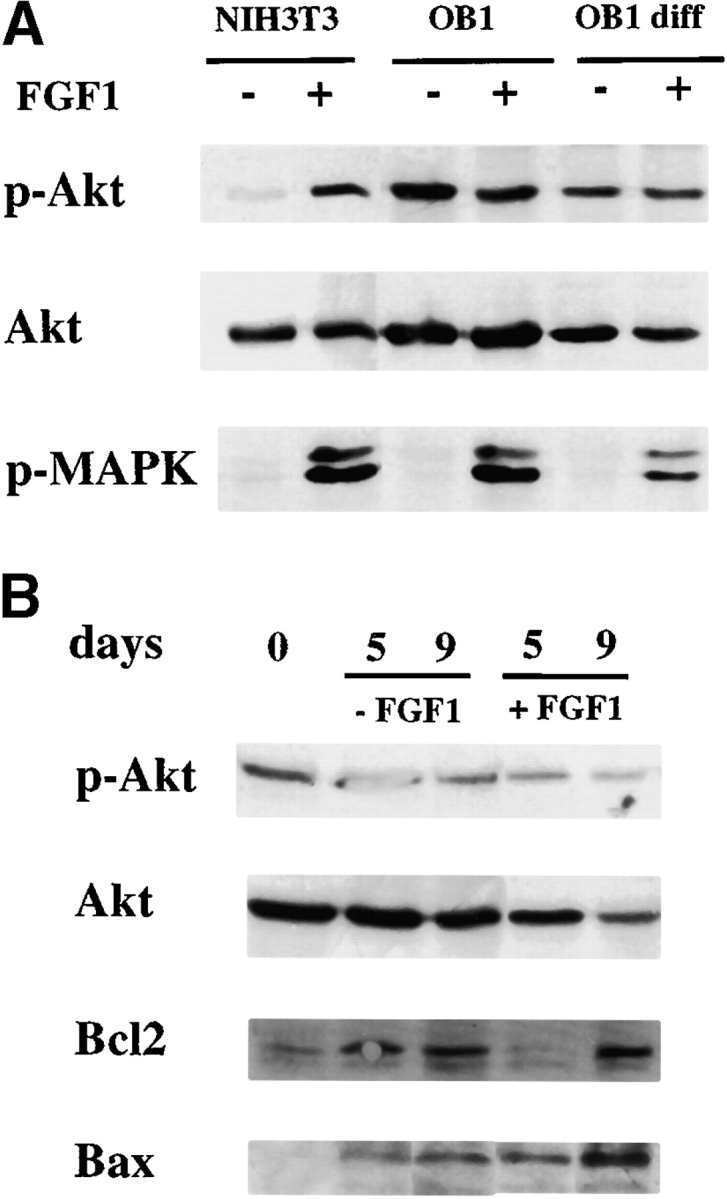
Expression activation of AKT, bax and bcl2 in osteoblasts. (A) Effect of FGF on AKT activation in osteoblasts. Total cell extracts were prepared from cells that were serum starved overnight in 0.4% FCS and were either left untreated (−) or treated with FGF1 (+) at 100 ng/ml for 15 min. OB1 diff, cells were differentiated for 7 d. Antibodies used are indicated. Anti-phospho AKT (anti–P-AKT) or anti-phospho MAPK (P-MAPK). (B) Effect of FGF on expression of AKT, bax, and bcl2 in differentiating osteoblasts. Whole cell extracts of cells that had been maintained in differentiation medium with 10% FCS for the indicated days in the absence (−FGF1) or presence (+FGF1) at 10 ng/ml. Antibodies used are indicated.
To determine whether FGF induced apoptosis also occurred in vivo, we examined a mouse model of unregulated FGF signaling. These animals overexpress a transgenic FGF2 under the control of the PGK promoter, and exhibit chondrodysplasia and abnormal skull formation (Coffin et al. 1995). We therefore examined calvarial sections from these mice and compared them to wild-type littermates. Serial sections of the post-frontal suture of 10-d-old mice were stained by the TUNEL assay to visualize apoptotic nuclei. Fig. 9 shows that compared with wild-type littermates, calvarial section of transgenic animals show a clear increase in apoptotic nuclei in the post frontal suture region. The positively stained cells are concentrated at the osteogenic front representing the position of differentiating cells. The immature cells in the middle of the suture are unstained. Although the relevance of this observation to the cranial malformations observed in these animals will require further investigation, these data show that unregulated FGF signaling can induce apoptosis in differentiating osteoblasts in vivo.
Figure 9.

Apoptosis in mouse calvarial sections. Sections of 10-d-old post-frontal suture of FGF2 transgenic (Tg) and wild-type (wt) littermates. Tissues were starved with Toluidine blue (upper panel) and for apoptosis using the TUNEL assay (lower panel). Cells in apoptosis are stained brown. Upper panel stained with Toluidine blue. OF, osteogenic front. The arrows point to the endocranial surface of the sections. Bar, 200 μm.
Discussion
The process of osteogenesis occurs in the developing calvarial bones, in the long bones, and continues in the adult to maintain the balance between bone formation and resorption. Although some progress has been made in understanding osteogenesis and the biology of premature suture closure, much, including the role of FGF, remains to be clarified. Whereas there has been general agreement that FGF stimulates the proliferation of osteoblasts in vitro, FGF appears to inhibit expression of differentiation markers ALP, OC in human and rat calvarial cells (Stein et al. 1996; Hurley and Florkiewicz 1996), but increases the levels of ALP, OC, and differentiation of osteogenic precursors in the bone marrow (Pitaru et al. 1993). Reports on osteoblasts analyzed from patients with FGFR2-linked craniosynostosis are also difficult to reconcile. Lomri et al. 1998 report that osteoblasts established from an Apert patient with an S252W mutation in FGFR2 have no alteration in their proliferation rate when compared with age-matched control cells, but express higher levels of differentiation markers. Fragale et al. 1999 studied osteoblasts from a patient with Apert syndrome with FGFR2/P253R and reported that the cells had decreased proliferation, higher ALP, and increased mineralization compared with age-matched control cells. Additionally, osteoblasts from a Pfeiffer's syndrome patient with FGFR2/C342R, an activating mutation also found in Crouzon's syndrome, showed decreased proliferation, higher ALP, but absence of mineralization. The effect of these mutations on osteoblast proliferation and differentiation cannot be easily compared because of varying ages of the patients and heterogeneity in the maturation stage of the initially isolated osteoblasts. Additionally, neither of these reports ascertained the expression of FGFRs, thus leaving the possibility that the mutant receptor may be downregulated in some cases (see below).
In this report, we studied the effect of FGF on osteoblasts before and during differentiation. We isolated clonal populations of osteoblasts at different stages of maturation and compared their responses to FGF. The clone OB1, an immature osteoblastic cell line that we generated, can undergo differentiation, upregulate expression of ALP and OC, and form mineralized nodules, thus providing a good model for osteoblastic differentiation. We found that FGF-induced stimulation of proliferation correlates with an immature phenotype whereas differentiating cells that are producing matrix and steadily upregulating ALP lose their proliferative response to FGF and respond with increased apoptosis. FGF treatment represses ALP expression and further inhibits the rate of DNA synthesis at later times during differentiation. We show that FGF signaling activates FRS2, Shp2, and the MAP kinase in osteoblasts and that there is no activation of STAT1. Although we find no detectable differences in activation of FGF receptors or MAP kinase in proliferating osteoblasts compared with osteoblasts that have been differentiated for two weeks, there may be differences at later times during differentiation or in other pathways.
We introduced two FGFR2 mutants in OB1 osteoblasts. Both mutations, FGFR2/S252W (Apert) and FGFR2/C342Y (Crouzon) increase the proliferative rate of OB1 cells and block the appearance of ALP and mineralized nodules upon differentiation. As in wild-type osteoblasts, the mutant receptor-expressing cells lose their proliferative capacity upon differentiation and display a striking increase in apoptosis. Thus, these constitutively activated receptors mimic the effect of treatment with exogenous FGF. Since the inhibition of proliferation during differentiation correlates with an increase in the rate of apoptosis, it suggests that increased programmed cell death may account for the reduced rate of DNA synthesis and the block in mineralization seen in these cultures. Furthermore, in vivo, we see that mice overexpressing FGF2 have increased apoptosis in the calvarial suture compared with wild-type littermates. These data indicate that our findings on FGF-induced apoptosis have relevance in vivo. Although it is unclear presently how this might affect the rate of osteogenesis and suture closure, we propose that FGF-induced apoptosis may represent a homeostatic mechanism controlling the rate of osteoblast differentiation and osteogenesis.
Apoptosis during osteogenesis has not been well studied but there are clear indications that it must play an important role in normal bone formation and remodeling. A major fraction of osteoblasts at bone remodeling sites cannot be accounted for and probably undergo apoptosis (Jilka et al. 1998) and the anabolic effect of PTH is due to its anti-apoptotic effect on osteoblasts (Jilka et al. 1999). A recent report by Rice et al. 1999 shows that apoptotic osteoblasts are present in the developing mouse calvarial bones. The FGFs have not previously been implicated in causing programmed cell death and generally protect from apoptotic effects induced by serum starvation (Hill et al. 1997; Bellosta et al. 1997). The fact that FGF induces apoptosis in differentiating osteoblasts in the presence of serum suggests a novel signaling mechanism by which FGF may regulate the rate of osteoblast proliferation and differentiation.
The mechanism by which FGF signaling increases proliferation in immature osteoblasts, and paradoxically inhibits DNA synthesis and increases apoptosis in differentiating cells is not clear at the moment. Differentiating osteoblasts could be expressing adaptors or substrates which are different from those expressed in immature cells and which could direct FGF signaling to novel pathways. Our data show that FGF signaling does not activate the anti-apoptotic AKT pathway in osteoblasts, which is in contrast to other cell types (Miho et al. 1999). Although we found no differences in AKT activation in immature versus mature osteoblasts, the lack of this response may contribute to the increased apoptosis in differentiating osteoblasts perhaps due to conflicting signals of proliferation and growth arrest similar to what has been observed for c-myc–induced apoptosis (Evan et al. 1992). However, in our experiments we find no increase in c-myc levels during differentiation (data not shown). Osteoblasts provide a good system in which to investigate the signaling pathways leading to divergent cellular response.
Our experiments suggest that the role of FGF signaling in intramembranous ossification could be dual. Initially FGF would stimulate the proliferation of immature osteoblasts, thus increasing the pool of osteoblast progenitors, and then act as a “brake” controlling the number of osteoblasts that undergo terminal differentiation. Alternatively, the concentration of apoptotic cells at the osteogenic front, seen in FGF2 transgenic mice, may imply that apoptosis enhances the progress of the bone fronts by clearing cells which must die in order for mineralization to occur. The increase of proliferation of immature cells by FGF and then an increase in apoptosis at the front may eventually result in accelerated suture closure. This latter hypothesis could be reconciled with the in vivo effects of FGF in enhancing bone formation (Mundy et al. 1999) and the craniosynostosis due to activated FGFRs. A detailed examination of apoptosis during calvarial osteogenesis and the effect of FGF should provide more insight into this process. The creation of mouse models of craniosynostosis, which is in progress in several laboratories, including ours, will help in answering these questions.
FGF2 and FGF9 are the ligands that are likely to be involved in FGF signaling during calvarial osteogenesis (Rice et al. 2000). The presence and concentration of ligand at different stages of osteoblast maturation will determine where and when FGF signaling is activated. Although it is unlikely, other FGFs could have different effects on differentiating osteoblasts. The timing of FGF signaling determined by regulation of receptor expression is also likely to be an important consideration in understanding cranial suture closure. FGF4 beads applied to the embryonic suture mesenchyme in the developing skull increases proliferation of undifferentiated cells but actually delays suture closure. However, FGF4 beads applied on the osteogenic front at the same stage of embryonic development accelerate suture closure (Kim et al. 1998). If FGF signaling is somehow downregulated or limited as cells move towards the front, it would allow differentiation to proceed. FGFR2 is expressed primarily in the preosteoblasts of the osteogenic front (Iseki et al. 1997, Iseki et al. 1999). If FGFR2 mRNA is downregulated, then its proliferative effect will be self limiting and cells may be able to progress to a later stage of differentiation. Furthermore, mutant FGFR2 genes that lead to craniosynostosis should be transcriptionally regulated in the same way as the normal gene. Thus, the regulation of FGFR2 expression at different stages of osteoblast maturation will be a critical factor in understanding the biology of craniosynostosis.
Acknowledgments
We wish to thank Dr. Lisa Dailey for the critical reading of this manuscript and Nina Mirsakov for technical assistance. We would especially like to thank Dr. J. Douglas Coffin (Department of Pharmaceutical Sciences, University of Montana) for providing us with the FGF2 Tg mice and Dr. Regina Raz (NYU School of Medicine) for genotyping the mice.
This investigation was supported by PHS grant CA42568 from the National Cancer Institute.
Footnotes
Alka Mansukhani and Paola Bellosta contributed equally to this work.
Abbreviations used in this paper: ALP, alkaline phosphatase; BrdU, bromodeoxyuridine; FGFR, fibroblast growth factor receptors; OC, osteocalcin.
References
- Anderson J., Burns H.D., Enriquez-Harris P., Wilkie A.O.M., Heath J.K. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Human Mol. Genet. 1998;7:1475–1483. doi: 10.1093/hmg/7.9.1475. [DOI] [PubMed] [Google Scholar]
- Arman E., Haffner-Krausz R., Chen Y., Heath J.K., Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl. Acad. Sci. USA. 1998;95:5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta P., Zhang Q., Goff S.P., Basilico C. Signaling through the ARK tyrosine kinase receptor protects from apoptosis in the absence of growth stimulation. Oncogene. 1997;15:2387–2397. doi: 10.1038/sj.onc.1201419. [DOI] [PubMed] [Google Scholar]
- Burke D., Wilkes D., Blundell T.L., Malcolm S. Fibroblast growth factor receptorslessons from the genes. Trends Biochem. Sci. 1998;23:259–262. doi: 10.1016/s0968-0004(97)01170-5. [DOI] [PubMed] [Google Scholar]
- Coffin J.D., Florkiewicz R.Z., Neumann J., Mort-Hopkins T., Dorn G.W., II, Lightfoot P., German R., Howles P.N., Kier A., O'Toole B.A. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol. Biol. Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiais F., Hott M., Graulet A.M., Marie P.J. The effects of fibroblast growth factor-2 on human neonatal calvaria osteoblastic cells are differentiation stage specific. J. Bone Miner. Res. 1998;13:645–654. doi: 10.1359/jbmr.1998.13.4.645. [DOI] [PubMed] [Google Scholar]
- Deng C.-X., Wynshaw-Boris A., Shen M.M., Daugherty C., Ornitz D.M., Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Deng C., Wynshaw-Boris A., Zhou F., Kuo A., Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Wyllie A.H., Gilbert C.S., Littlewood T.D., Land H., Brooks M., Waters C.M., Penn L.Z., Hancock D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fragale A., Tartaglia M., Bernardini S., Di Stasi A.M.M., Di Rocco C., Velardi F., Teti A., Battaglia P.A., Migliaccio S. Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. Am. J. Pathol. 1999;154:1465–1477. doi: 10.1016/S0002-9440(10)65401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin B.D., Hart K.C., Meyer A.N., Webster M.K., Donoghue D.J. Constitutive receptor activation by Crouzon syndrome mutations in fibroblast growth factor receptor (FGFR)2 and FGFR2/Neu chimeras. Proc. Natl. Acad. Sci. USA. 1996;93:7894–7899. doi: 10.1073/pnas.93.15.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M. Functions of fibroblast growth factors in vertebrate development. Cytokine Growth Factor Rev. 1996;7:311–325. doi: 10.1016/s1359-6101(96)00039-1. [DOI] [PubMed] [Google Scholar]
- Hill P.A., Tumber A., Meikle M.C. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology. 1997;138:3849–3858. doi: 10.1210/endo.138.9.5370. [DOI] [PubMed] [Google Scholar]
- Hurley M.M., Florkiewicz R.Z. Fibroblast growth factor and vascular endothelial cell growth factor families. In: Bilezikian J.P., Raisz L.G., Rodan G.A., editors. Principles of Bone Biology. Academic Press; New York: 1996. pp. 69–86. [Google Scholar]
- Iseki S., Wilkie A.O.M., Heath J.K., Ishimaru T., Eto K., Morriss-Kay G.M. Fgfr2 and osteopontin domains in the developing skull vault are mutually exclusive and can be altered by locally applied FGF2. Development. 1997;124:3375–3384. doi: 10.1242/dev.124.17.3375. [DOI] [PubMed] [Google Scholar]
- Iseki S., Wilkie A.O.M., Morriss-Kay G.M. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–5620. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- Jilka R.L., Weinstein R.S., Bellido T., Parfitt A.M., Manolagas S.C. Osteoblast programmed cell death (apoptosis)modulation by growth factors and cytokines. J. Bone Miner. Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- Jilka R.L., Weinsein R.S., Bellido T., Roberson P., Parfitt A.M., Manolagas S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 1999;104:439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S.G., Wagner A.J., Conzen S.D., Jordan J., Bellacosa A., Tsichlis P.N., Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Kim H.-J., Rice D.P.C., Kettunen P.J., Thesleff I. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125:1241–1251. doi: 10.1242/dev.125.7.1241. [DOI] [PubMed] [Google Scholar]
- Kouhara H., Hadari Y.R., Spivak-Kroizman T., Schilling J., Bar-Sagi D., Lax I., Schlessinger J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. 1997;89:693–702. doi: 10.1016/s0092-8674(00)80252-4. [DOI] [PubMed] [Google Scholar]
- Li Y., Basilico C., Mansukhani A. Cell transformation by fibroblast growth factors can be suppressed by truncated fibroblast growth factor receptors. Mol. Cell. Biol. 1994;14:7660–7669. doi: 10.1128/mcb.14.11.7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomri A., Lemonnier J., Hott M., de Parseval N., Lajeunie E., Munnich A., Renier D., Marie P.J. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J. Clin. Invest. 1998;101:1310–1317. [PMC free article] [PubMed] [Google Scholar]
- Mangasarian K., Li Y., Mansukhani A., Basilico C. Mutation associated with Crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. J. Cell. Physiol. 1997;172:117–125. doi: 10.1002/(SICI)1097-4652(199707)172:1<117::AID-JCP13>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Mansukhani A., Dell'Era P., Moscatelli D., Kornbluth S., Hanafusa H., Basilico C. Characterization of the murine BEK fibroblast growth factor (FGF) receptoractivation by three members of the FGF family and requirement for heparin. Proc. Natl. Acad. Sci. USA. 1992;89:3305–3309. doi: 10.1073/pnas.89.8.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miho Y., Kouroku Y., Fujita E., Mukasa T., Urase K., Kasahara T., Isoai A., Momoi M.Y., Momoi T. bFGF inhibits the activation of caspase-3 and apoptosis of P19 embryonal carcinoma cells during neuronal differentiation. Cell Death Differ. 1999;6:463–470. doi: 10.1038/sj.cdd.4400506. [DOI] [PubMed] [Google Scholar]
- Muenke M., Schell U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995;11:308–313. doi: 10.1016/s0168-9525(00)89088-5. [DOI] [PubMed] [Google Scholar]
- Muller A.J., Young J.C., Pendergast A.M., Pondel M., Landau N.R., Littman D.R., Witte O.N. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol. Cell. Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy G., Garrett R., Harris S., Chan J., Chen D., Rossini G., Boyce B., Zhao M., Gutierrez G. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–1949. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- Naski M.C., Ornitz D.M. FGF signaling in skeletal development. Front. Biosci. 1998;3:781–794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- Ornitz D.M., Xu J., Colvin J.S., McEwen D.G., MacArthur C.A., Coulier F., Gao G., Goldfarb M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Pitaru S., Kotev-Emeth S., Noff D., Kaffuler S., Savion N. Effect of basic fibroblast growth factor on the growth and differentiation of adult stromal bone marrow cellsenhanced development of mineralized bone-like tissue in culture. J. Bone Miner. Res. 1993;8:919–929. doi: 10.1002/jbmr.5650080804. [DOI] [PubMed] [Google Scholar]
- Rice D., Kim H., Thesleff I. Apoptosis in murine calvarial bone and suture development. Eur. J. Oral Sci. 1999;107:265–275. doi: 10.1046/j.0909-8836.1999.eos107406.x. [DOI] [PubMed] [Google Scholar]
- Rice D.P.C., Aberg T., Chan Y.S., Tang Z., Kettunen P.J., Pakarinen L., Maxson R.E., Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Robertson S.C., Meyer A.N., Hart K.C., Galvin B.D., Webster M.K., Donoghue D.J. Activating mutations in he extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proc. Natl. Acad. Sci. USA. 1998;95:4567–4572. doi: 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni M., Ambrosetti D.-C., Mansukhani A., Gertner R., Levy D., Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1261–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton T.M., Henkemeyer M., Gasca S., Shen R., Rossi D.J., Shalaby F., Feng G.S., Pawson T. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2352–2364. doi: 10.1093/emboj/16.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaney S.F., Oldridge M., Hurst J.A., Morris-Kay G.M., Hall C.M., Poole M.D., Wilkie A.O.M. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- Stein G.S., Lian J.B., Stein J.L., van Wijnen A.J., Frenkel B., Montecino M. Mechanisms regulating osteoblast proliferation and differentiation. In: Bilezikian J.P., Raisz L.G., Rodan G.A., editors. Principles of Bone Biology. Academic Press; New York: 1996. pp. 69–86. [Google Scholar]
- Su W.-C.S., Kitagawa M., Xue N., Xie B., Garofalo S., Cho J., Deng C., Horton W.A., Fu X.-Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 1997;386:288–292. doi: 10.1038/386288a0. [DOI] [PubMed] [Google Scholar]
- Su J., Muranjan M., Sap J. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr. Biol. 1999;9:505–511. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- Wilkie A.O.M. Craniosynostosisgenes and mechanisms. Human Mol. Genet. 1997;6:1647–1656. doi: 10.1093/hmg/6.10.1647. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T.P., Harpal K., Henkemeyer M., Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;15:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]