

Abstract
We have used cell lines deficient in p120 Ras GTPase activating protein (Ras-GAP) to investigate the roles of Ras-GAP and the associated p190 Rho-GAP (p190) in cell polarity and cell migration. Cell wounding assays showed that Ras-GAP–deficient cells were incapable of establishing complete cell polarity and migration into the wound. Stimulation of mutant cells with growth factor rescued defects in cell spreading, Golgi apparatus fragmentation, and polarized vesicular transport and partially rescued migration in a Ras-dependent manner. However, for directional movement, the turnover of stress fibers and focal adhesions to produce an elongate morphology was dependent on the constitutive association between Ras-GAP and p190, independent of Ras regulation. Disruption of the phosphotyrosine-mediated Ras-GAP/p190 complex by microinjecting synthetic peptides derived from p190 sequences in wild-type cells caused a suppression of actin filament reorientation and migration. From these observations we suggest that although Ras-GAP is not directly required for motility per se, it is important for cell polarization by regulating actin stress fiber and focal adhesion reorientation when complexed with 190. This observation suggests a specific function for Ras-GAP separate from Ras regulation in cell motility.
Keywords: cytoskeleton, cell polarity, migration, actin, p190 Rho-GAP
Introduction
Within the Ras superfamily of small GTPases, the Rho subfamily of proteins has been shown to remodel the actin cytoskeleton. The Rho subfamily members, Cdc42, Rac and Rho, function as molecular switches through interconversion between an active GTP-bound state and an inactive GDP-bound configuration. Activation is regulated by guanine nucleotide exchange factors that favor GTP loading while deactivation is mediated by GTPase-activating proteins (GAPs) that catalyze the hydrolysis of GTP to GDP (Boguski and McCormick 1993). In mammalian cells, growth factor-induced activation of Rho proteins causes actin polymerization and reorganization, which leads to changes in cell shape. Specifically, activation of Cdc42 by bradykinin induces formation of filopodia, which are finger-like extensions of the cytoplasm that contain dense actin filaments (Kozma et al. 1995). Activation of Rac by PDGF leads to the formation of membrane ruffles and lamellipodia (Ridley et al. 1992). It has also been shown that activated Ras stimulates membrane ruffling in a Rac-dependent manner (Bar-Sagi and Feramisco 1986). Activation of Rho by lysophosphatidic acid, a major component of serum, results in actin stress fiber (SF) and focal adhesion (FA) formation (Ridley and Hall 1992) and the formation of stable microtubules (MTs; Cook et al. 1998). The use of dominant-negative mutants of the Rho proteins has further demonstrated that activation of Cdc42 is sufficient to activate Rac, which in turn activates Rho in a linear cascade in Swiss 3T3 cells (Nobes and Hall 1995). However, in some cell types such as neurons, Rho antagonizes the effects of Cdc42 and Rac (Kozma et al. 1997). GAPs can potentially negatively regulate actin dynamics. The p120 Ras-GTPase activating protein (Ras-GAP) and p190 Rho-GAP (p190) have both been implicated in remodeling the actin cytoskeleton.
Ras-GAP is composed of an NH2-terminal region containing two SH2 domains flanking an SH3 domain, a central region containing CalB and PH domains and a COOH-terminal catalytic domain that drives the conversion of Ras-GTP to Ras-GDP (Gawler et al. 1995; Pawson 1995). The SH2 domains of Ras-GAP bind to phosphotyrosine (pTyr) residues of p190 (Hu and Settleman 1997). P190, a protein initially identified as being tyrosine phosphorylated in v-Src–transformed cells (Ellis et al. 1990), is comprised of an NH2-terminal GTP binding domain and a COOH-terminal GAP domain (Settleman et al. 1992). This Ras-GAP/p190 complex may function together to regulate actin cytoskeletal dynamics. Over-expression of the NH2 terminus of Ras-GAP in RAT-2 cells induced abnormal actin SFs and decreased cell substratum adhesion and this correlated with increased in vitro Rho GAP activity compared with untransfected cells (McGlade et al. 1993). Other studies have suggested that while microinjection of the GAP domain of p190 into Swiss 3T3 cells inhibits SF formation (Ridley et al. 1993), the NH2-terminal SH3 domain of Ras-GAP controls Rho activation and SF formation (Leblanc et al. 1998). Such changes in the organization of actin filaments are known to affect cell morphology in a manner critical for directed cell movement. We have previously found that mouse embryos homozygous for a null mutation in the Ras-GAP gene show a defect in angiogenesis within the yolk sacs and aberrant sprouting of the intersomitic arteries during early development (Henkemeyer et al. 1995). We also demonstrated that Ras-GAP mutant cells exhibit elevated Ras-GTP levels after growth factor stimulation and are tyrosine hypophosphorylated on p190 (van der Geer et al. 1997). These defects in cellular organization and Ras regulation may result in impaired actin cytoskeletal dynamics.
Here, we have examined the role of the Ras-GAP complex in directed cell migration. Using a cellular wounding assay, we found that cells cultured from Ras-GAP mutant embryos exhibited a cell migration defect. To analyze the cellular basis for this phenotype, we examined cytoskeletal-based cell polarity markers that occur in a specific order before migration. We compared these markers between wild-type and Ras-GAP–deficient cell lines and determined the point at which these mutant cells became compromised during the establishment of cell polarity. Our results indicate that Ras-GAP–deficient fibroblasts can establish only partial polarity and show little movement upon wounding in culture. They have a fragmented Golgi apparatus (GA) that correlated with nonpolarized insertion of membrane mass by vesicular transport, and are incapable of organizing actin SFs and FAs in an orientation parallel to the direction of migration. We suggest that this failure is partially due to the absence of the Ras-GAP/p190 complex.
Materials and Methods
Cell Culture, DNA Transfections, and Wounding Assays
Mice, heterozygous for the Ras-GAP null mutation (Henkemeyer et al. 1995) were mated and d9.5(pc) embryos and yolk sacs were collected for tissue culture and genotyping, respectively. Early passage cells were maintained in DME + 10% FBS media and allowed to undergo crisis. From this, a wild-type, a heterozygous and two mutant cell lines were established. For wounding experiments, cells were plated either in tissue culture plastic dishes or on glass coverslips and allowed to grow to a confluency of 70–80%. After a 24-h period of serum deprivation, the cultures reached 100% confluency without crowding. Experimental wounds were made by dragging a Gilson plastic yellow pipette tip across the cell culture. Cultures were then rinsed with PBS and replaced with fresh serum-free media. Cell motility was then monitored at selected time intervals for a period of 24 h. In some experiments, wounded cultures were treated with 5 μM nocodazole (Sigma) for 30 min to promote depolymerization of dynamic MTs, before fixation. For other experiments, PDGF (30 ng/ml) stimulation was used increase cellular levels of Ras-GTP (van der Geer et al. 1997).
To quantify the extent of migration, cells were first plated onto tissue culture plates marked with grids. After making wounds across the grids, the cultures were photographed immediately (t = 0) and 24 h later. In total, nine wounds were sampled for each cell type. Total areas devoid of cells within the wound at these two time points were calculated using morphometric procedures and the difference was calculated. Hence, the extent of migration was expressed as a function of area reduction.
A human Ras-GAP cDNA (gift from Frank McCormick, UCSF, San Francisco, CA) was subcloned into either the pMSCV viral expression vector (Hawley et al. 1994) resulting in pMSCV-GAP or the pCDNA3 vector (Clontech). In addition, a truncated human Ras-GAP cDNA encoding only the entire NH2-terminal hydrophobic region and including the SH2-SH3-SH2 domains (GAP-N) as described by McGlade et al. 1993 was subcloned into the pMSCV vector. The pMSCV-GAP and pMSCV-GAP-N constructs, encoding a puromycin resistant gene, were transfected into the viral producer cell line GP+E (Markowitz et al. 1988). Puromycin-resistant colonies were picked, and expanded. Supernatants from the clones were used to infect Ras-GAP mutant cells and selected with puromycin. Such positive colonies were picked and screened for Ras-GAP expression by immunoblotting (see method below). Full-length Ras-GAP and GAP-N positive clones were then maintained in culture in puromycin-free media and cell wounding experiments were carried out. The pCDNA3-GAP vector was used for some microinjection experiments (see method below).
Microinjection
Cells used in microinjection experiments were seeded onto etched coverslips to facilitate location of injected cells. Injections were performed essentially as described by Bar-Sagi 1995. In brief, cell culture media was supplemented with 20 mM Hepes to buffer any pH changes. Microinjections were performed using a Leica DMIRB inverted microscope equipped with a heating stage and the Eppendorf 5242 microinjector. For GA-rescue experiments, the pCDNA3-GAP construct was microinjected into the nuclei of Ras-GAP mutant cells. For some experiments, p190 inhibitory peptides (peptide synthesis described below) were coinjected with 3 mg/ml rhodamine-conjugated goat IgG (Sigma) to identify injected wild-type cells.
VSV-G Vesicular Transport Assays
A cDNA encoding an EGFP-fusion of the temperature-sensitive vesicular stomatitis virus (VSV) coat protein G (Presley et al. 1997; kind gift from Dr. Jennifer Lippincott-Schwartz, NIH, Bethesda, MD) was microinjected into wild-type and Ras-GAP–deficient cells, immediately after wounding. After microinjection, the cells were then cultured in an incubator at 40°C for 4 h to allow sufficient time for VSV-G-protein expression. Then samples were transferred to a 32°C water bath to allow transport of VSV-G-protein to the plasma membrane. Samples were then fixed in 4% paraformaldehyde at select time intervals after the temperature shift, and then processed for immunofluorescence microscopy (see below).
Immunofluorescence Microscopy
Wounded cell cultures grown on coverslips were processed for microscopy by first rinsing briefly in PBS, and then fixing for 15 min with 4% paraformaldehyde in PBS. After three washes in PBS, the samples were permeabilized with 0.2% Triton X-100 in PBS for 5 min. For nocodazole-treated cultures, samples were first pre-extracted with 0.5% Triton X-100 in PHEM buffer (80 mM Pipes, 20 mM Hepes, 1 mM EGTA, and 2 mM MgCl2, pH 6.8) for 90 s to remove soluble proteins. Samples were then simultaneously fixed and extracted with 0.5% Triton X-100, 3.7% formalin, and 0.25% glutaraldehyde in PHEM buffer for 10 min followed by PBS rinses.
For VSV-G-protein transport assays, formaldehyde-fixed samples were initially stained with mouse anti–VSV-G antibodies (clone I14; gift from Dr. Doug Lyles; Wake Forest University, Winston-Salem, NC; see Lefrancios and Lyles 1982) followed by FITC-conjugated goat anti–mouse antibodies (Sigma). Samples were then permeabilized with 0.1% emulphogene (Sigma) for 5 min as described by Bergmann et al. 1983 before further double-labeling with antibodies to detect the GA.
To detect active Ras within wounded cultures, we made use of the minimal Ras-binding domain of Raf 1 fused to GST (gift from Dr. Johannes Bos, Utrecht University, The Netherlands; Rooij and Bos 1997). Using cell culture overlay technique described by Meriläinen et al. 1993 fixed wounded cultures were incubated with GST-Raf 1 (100 μg/ml) for 1 h and detected using an affinity-purified anti-GST antibody produced in rabbit in our lab. These samples were also double-labeled for activated MAP kinase as an indirect marker for activated Ras. Dual phosphorylation of ERK1 and ERK2 were detected using the monoclonal antibody to activated MAP kinase (Sigma; clone M8159) as described by Nobes and Hall 1999.
Single- or double-label immunofluorescence staining procedures were used. FAs were detected with mouse anti-vinculin antibodies (Sigma). MTs were stained with mouse anti-tubulin antibodies (clone DM1A; Amersham). Rabbit anti–mannosidase II antibodies (gift from Dr. Kelly Moremen, University of Georgia, Athens, GA) were used to detect the GA and either FITC- or rhodamine-conjugated phalloidin (Molecular Probes) was used to visualize actin filaments. Ras-GAP was stained with mouse anti–ras-GAP antibodies (Santa Cruz). The appropriate Cy5-, FITC-, or Texas red–conjugated goat anti–mouse or goat anti–rabbit antibodies (Sigma and Molecular Probes) were used to detect the primary antibodies. Hoechst 33258 (Molecular Probes) was used to stain nuclei. All samples were mounted in a 50% glycerol and PBS solution supplemented with p-phenylenediamine to retard photobleaching. Microscopy was carried out using the Leica DMRX microscope and the inverted Leica DMIRB microscope, both equipped with fluorescence and transmitted light optics. Samples were photographed on either KODAK EPH 1600 film for fluorescent images or KODAK 64T film for phase contrast images. Images in Fig. 7 and Fig. 9G–I were obtained using the Olympus 1X-70 inverted microscope equipped with fluorescence optics and Deltavision Deconvolution Microscopy software (Applied Precision). Remaining figure images were scanned using a Nikon Cool Laser Scan Jet and constructed in Adobe Photoshop.
Figure 7.
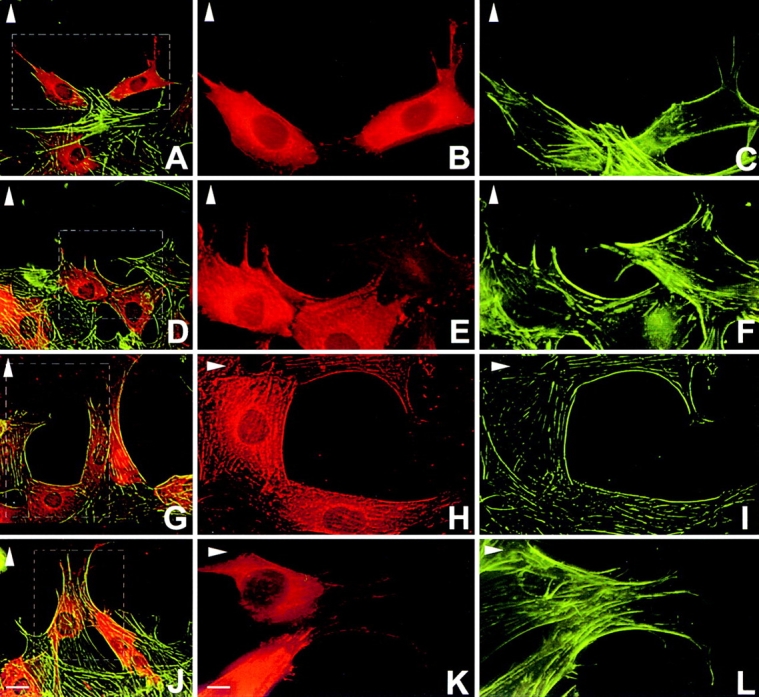
Inhibition of SF polarization and migration by microinjection of p190 inhibitory peptides into wild-type cells. (A–C, diphosphotyrosine peptide; D–F, Peptide 1 phosphonopeptide; G–I, Peptide 2 phosphonopeptide; J–L, rhodamine–goat-IgG alone). Actin filaments were visualized with FITC-phalloidin and peptides were coinjected with rhodamine-IgG. Injected cells within hatched boxes in panels at a lower magnification (A, D, G, and J) illustrating rhodamine-labeling and actin SFs, are shown separately at higher magnification in adjacent panels. Note that cells injected with the diphosphotyrosine peptide (A–C) and Peptide 1 (D–F) are largely confined to the wound edge and have no polarized SFs. Cells injected with Peptide 2 (G–I) exhibit some inhibition of polarity while those injected with IgG alone (J–L) result in no inhibition of polarized SF formation and motility. Bars: (A, D, G, and J) 50 μm; (B–C, E–F, H–I, and K–L) 50 μm.
Figure 9.

Effect of PDGF-induced Ras activation and Rho inhibition on rescuing cell polarity in Ras-GAP mutant cells. Wild-type cells (A and D) and Ras-GAP mutant cells (B–C and E–F) were stimulated with PDGF. Golgi morphology was assessed using anti-mannosidase antibodies (Texas red–labeled in A–C and Alexa 350–labeled in G). Nuclei were visualized by blue Hoechst staining (A–C). Actin filaments were visualized using FITC-phalloidin (D, E, and H). VSV-G-protein transport (FITC-labeled in C) in GAP mutant cells after PDGF stimulation was assessed (C and F). Phase-contrast image of two GAP mutant cells (F) is merged with image in C to show localization of VSV-G-protein to leading edge (arrows in C and F). GAP mutant cells (G–I) were microinjected with C3 transferase (0.3 mg/ml) to inhibit cellular Rho-GTP. Coinjection with rhodamine-IgG served to identify injected cells (I). Injected cells exhibit fewer SFs (H) and those at the wound edge (small arrow) show a polarized, elongate shape in the direction of migration. Only a partial rescue of Golgi morphology (G) was noted in the injected cell population. Arrowheads located at the left-hand corner of each panel indicate direction of migration. Bars: (A, B, D, E, and G–I) 50 μm; (C and F) 20 μm.
Cell Polarity Assays
Cell spreading is characterized by the formation of lamellipodia and membrane ruffling. Since these protrusive activities at the leading edge are a highly dynamic event, the degree of cell spreading was analyzed by a semiquantitative procedure. When the nucleus acquired a posterior position within the cell after wounding, that cell was scored as having spread (Euteneuer and Schliwa 1992). Orientation of stable MTs and GA around the nucleues in wound edge cells were scored as described by Gundersen and Bulinski 1988 and Kupfer et al. 1982, respectively. Orientation of actin SFs was scored as either being parallel, orthogonal or random with respect to the direction of migration. Greater than 500 cells were enumerated for each time point in all cell polarity assays. Also, experiments were repeated for reproducibility, and results for one experiment are shown to illustrate trends.
Immunoblotting
Cell lysates were prepared in PLC lysis buffer (30 mM Hepes, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 10 mM NaPPi, and 100 mM NaF). Protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM vanadate, and 1 mM PMSF) were added to lysis buffer before use. Protein was quantified using the Bio-Rad Protein Assay Kit. In some experiments, lysates were subject to immunoprecipitation using rabbit anti–Ras-GAP antibodies (McGlade et al. 1993) or mouse anti-p190 antibodies (gift from S.J. Parsons, University of Virginia, Charlottesville, VA). Samples were analyzed by 7.5% SDS-PAGE and proteins were transferred to nitrocellulose membranes. Filters were blocked with 5% milk powder in TBST and probed with either rabbit anti–Ras-GAP or mouse anti-p190 antibodies, followed by addition of appropriate HRP-conjugated secondary antibody (Biorad). Detection of bands was done using the enhanced chemiluminescence detection system (Amersham).
Synthesis of Inhibitory p190 Peptides
A phosphatase-resistant peptide containing a tyrosine at position 1105, based upon the p190 sequence EEENIpYSVPHDST-NH2, was synthesized using a racemic mixture of Nα-fluorenylmethoxycarbonyl-o-(4-di-t-butylphosphono-methyl)-d,l-phenylalanine (Colour Your Enzyme Inc.) as a nonhydrolyzable pTyr analogue. As a result, two monophosphonopeptide enantiomers were resolved by reverse-phase HPLC and designated as Peptide 1 and Peptide 2 based upon their elution profile. In addition, a longer phosphatase-sensitive peptide with the sequence, DpYAEPMDAVVKPRN-EEENIpYSVPHD-NH2 was synthesized using l-phosphotyrosine. This longer peptide contains pTyr residues at positions 1087 and 1105 of p190 and will be referred to as the diphosphotyrosine peptide from hereafter. Similar peptides have been shown to inhibit the interaction between Ras-GAP and p190 in vitro (Hu and Settleman 1997). All peptides were synthesized using the ABI 431 peptide synthesizer, Fmoc standard chemistry, and were further characterized by mass spectrometry and amino acid analysis. Peptides were kept in 40mM Hepes buffer at –20°C.
GST-mixing Experiments
Amino acid (aa) sequences corresponding to the NH2-terminal SH2 domain (aa 181–274), COOH-terminal SH2 domain (aa 351–442) and a hydrophobic region of Ras-GAP (GAP-H; aa 1–180), were subcloned into pGEX-2T vectors to yield GST-fusion constructs (gift from Dr. Louise LaRose, McGill University, Montrèal). To test the specificity of the inhibitory p190 phosphopeptides, competition experiments were carried out using cellular lysates derived from the src-overexpressor cell line, S7A (Koch et al. 1992) and the various GST-Ras-GAP fusion proteins. For the competition experiments, cellular lysates from the Src-transformed S7A cell line were used because they have increased levels of tyrosine-phosphorylated p190. These lysates were harvested by initially treating with 0.1% SDS to denature endogenous protein–protein complexes, boiled and then diluted with four parts of PLC lysis buffer before use. All peptides were used at a concentration of 50 μM and incubated with 10 μg of GST-Ras-GAP fusion proteins and 200 μg total S7A cellular lysate. The resulting mixture was centrifuged and beads were rinsed with PLC lysis buffer. Bound proteins were solubilized in Laemmli's sample buffer and then subject to SDS-PAGE and immunoblotting with anti-p190 antibodies.
Results
Ras-GAP–deficient Cells Are Migration Incompetent
During early development, mouse embryos with a null mutation in the Ras-GAP gene exhibit a defect in angiogenesis within the yolk sac suggesting a defect in cell movement (Henkemeyer et al. 1995). We tested the cellular basis for this migration defect using a classical in vitro wounding assay. In all cell motility assays, cultures were serum deprived for a minimum of 24 h to establish quiescence, ensuring that any presence of cells in the wounded area would be due to motility rather than cell proliferation. Fibroblasts derived from wild-type embryos exhibited a sixfold greater increase in their movement into a wound (Fig. 1A and Fig. C) compared with those derived from Ras-GAP mutant embryos (Fig. 1B and Fig. C). Cells heterozygous for the Ras-GAP mutation, showed movement that was intermediate between wild-type and mutant cells (Fig. 1 C).
Figure 1.
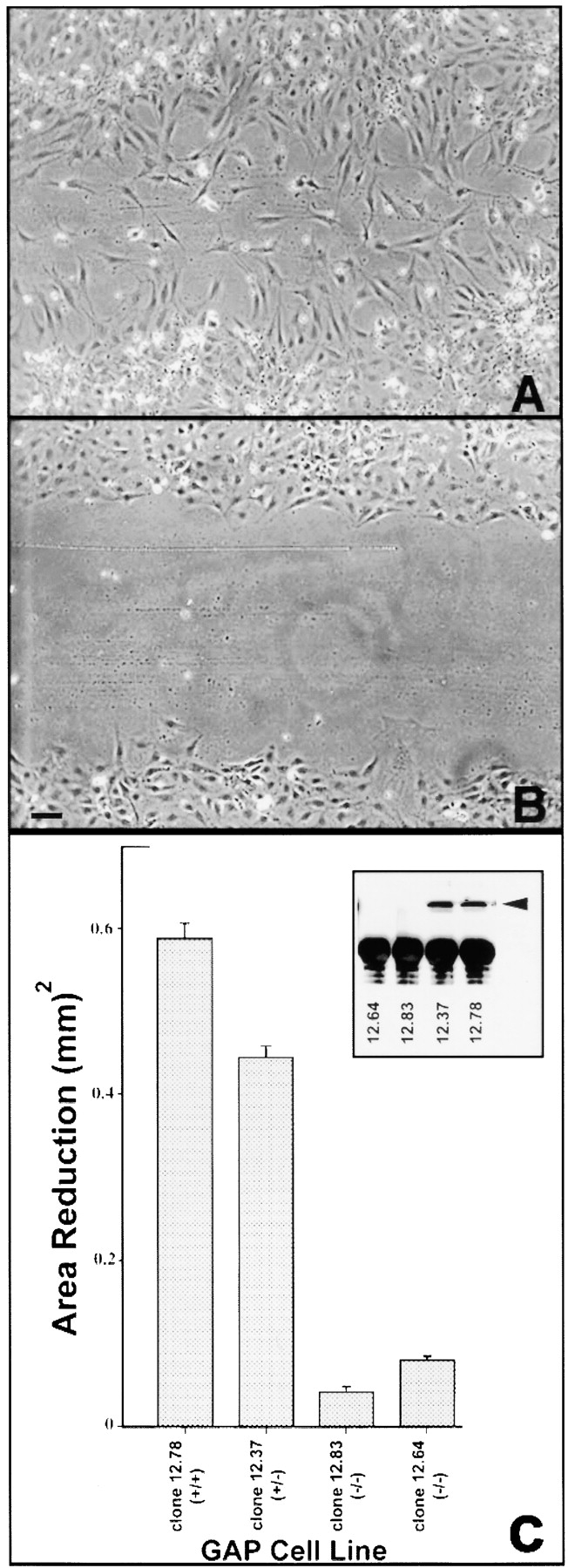
Wound-induced cell migration. (A and B, t = 24 h) Wild-type cells migrate into the wound after 24 h (A), while Ras-GAP mutant cells show little movement (B). (C) Extent of migration is expressed as a function of area reduction. Wounds in nine grids were analyzed for each cell line. Error bars represent standard error. Western blot of Ras-GAP expression for each cell line is shown in inset. Bar, 200 μm.
To determine if this inhibition of motility was due to clonal differences or lack of Ras-GAP, we performed rescue experiments by infecting mutant cells with a retrovirus containing full-length human Ras-GAP cDNA (see Materials and Methods). Ras-GAP–expressing clones were then analyzed for expression levels by Western blotting (Fig. 2 A) and movement using the wounding assay (Fig. 2, B–F). 75 puromycin-resistant clones were screened and all but two expressed Ras-GAP (data not shown). Some clones exhibited evidence of recombination events resulting in slightly truncated forms of Ras-GAP protein and were discarded. As shown in Fig. 2, clones expressing the lowest levels of full-length Ras-GAP (Fig. 2 C) or vector alone (Fig. 2 B) did not exhibit any significant movement. Those clones expressing intermediate levels (Fig. 2, D–E) showed some migration capabilities while cells exhibiting higher Ras-GAP expression levels (Fig. 2 F) displayed extreme sensitivity to serum deprivation and frequently rounded up and detached from the tissue culture dish. These data are consistent with the view that Ras-GAP is important for migration, and argue that the level of Ras-GAP expression may be an important factor for migration to proceed.
Figure 2.
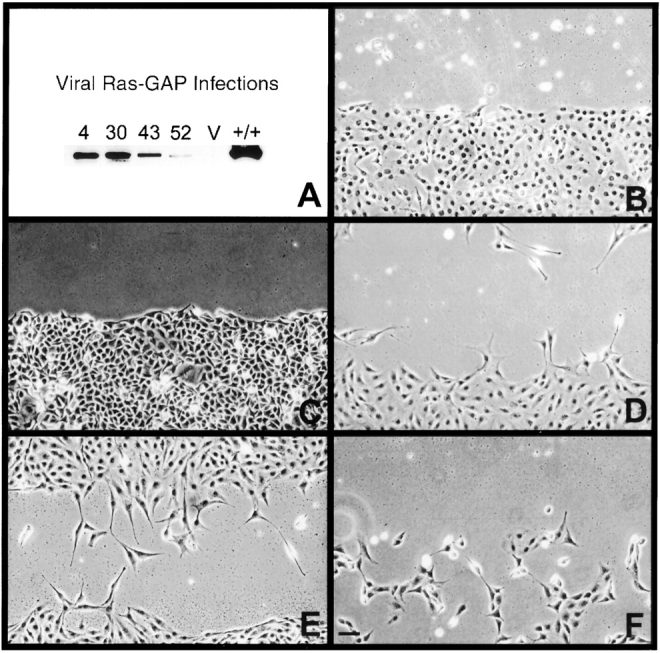
Rescue of migration in mutant cells infected with retroviral Ras-GAP. (A) Western blot of Ras-GAP–expressing clones using anti–Ras-GAP antibodies. Migration into denuded zones 24 h after wounding (B–F). B, vector alone; C, lowest Ras-GAP expressor clone 52; D and E, intermediate Ras-GAP expressors clone 4 and 43, respectively; F, highest Ras-GAP expressor clone 30. Bar, 100 μm.
Establishment of Cell Polarity as Measured by Cell Spreading
To analyze the migration defect in detail we examined early cytoskeletal-related cell polarity events that precede directed movement in a chronological order. We hypothesized that the migration defect might be due to a defect in the establishment of cell polarity. The first cell polarity marker examined was cell spreading, a process mediated by the cortical actin cytoskeleton. This early protrusive event is characterized by the formation of lamellipodia and membrane ruffling. Wild-type cells (Fig. 3 A) formed lamellipodia and exhibited membrane ruffling. A small percentage of mutant cells (Fig. 3B and Fig. C) did undergo some protrusive activity, but the majority of the population did not appear to spread as extensively as wild-type cells. In a 4-h time course experiment, >90% of wild-type cells had a posterior-positioned nucleus and <20% of Ras-GAP mutant cells exhibited cell spreading.
Figure 3.
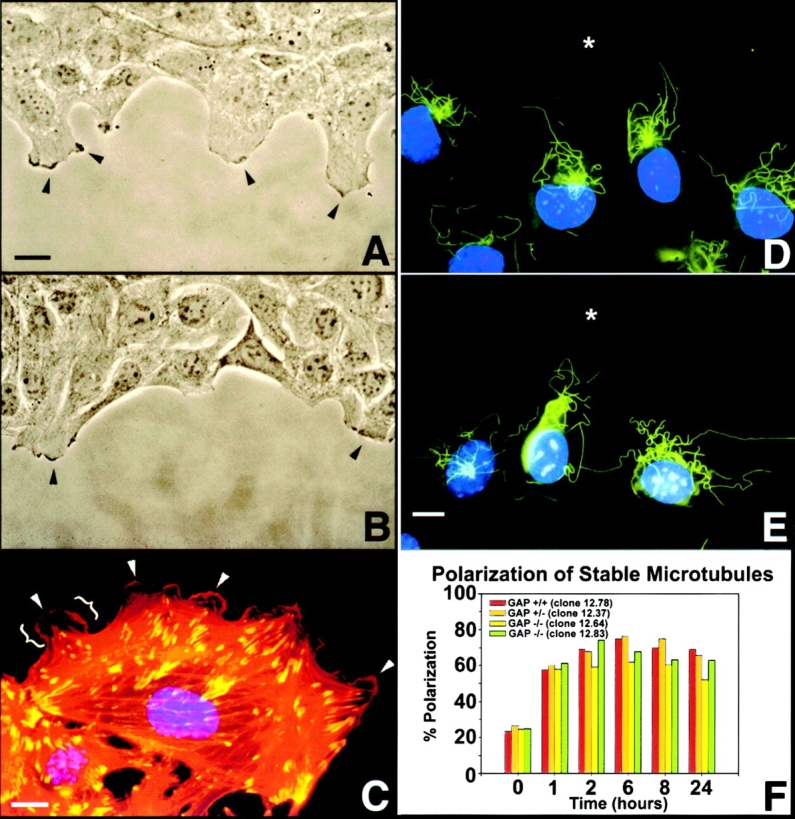
Cell spreading and establishment of a stable, polarized MT array. (A and B, t = 1 h; D and E, t = 2 h). Both wild-type cells (A) and Ras-GAP mutant cells (B) are capable of cell spreading by lamellar extension and membrane-ruffling (arrowheads). Gross-morphological examination revealed that wild-type cells spread more than Ras-GAP mutant cells. Note individual lamellipodia (brackets) and membrane ruffles (arrowheads) at the leading edge of a Ras-GAP mutant cell (C), as shown by rhodamine-phalloidin staining of actin filaments (red) and vinculin (FITC-labeled) staining. Wounded cultures revealed the presence of a nocodazole-stable population of MTs (FITC-labeled) preferentially oriented towards the direction of migration in both wild-type (D) and Ras-GAP mutant cells (E). Nuclei are visualized by Hoechst staining (blue). Asterisk (*) indicates wound edge. Persistence of a polarized stable MT array existed up to 24 h after wounding (F) in all cell types. Bars, 50 μm.
Ras-GAP Mutant Cells form Normal Stable MTs
We next analyzed the formation of a stable MT array oriented towards the leading edge of the cell in the direction of migration. One proposed function for this stable MT array is to provide a track for the polarized intracellular transport of secretory proteins, lipids, and other membrane components to the leading edge of migrating cells (Gundersen and Bulinski 1988). At selected time intervals after wounding, 5 μM nocodazole was added to cell cultures to promote depolymerization of dynamic MTs, leaving the stable population intact. At the time of wounding, nocodazole-stable MTs are predominantly randomly oriented about the nucleus (data not shown). As early as 1–2 h after wounding, this stable MT population was seen to radiate from the MT-organizing center toward the leading edge in both wild-type (Fig. 3 D) and Ras-GAP mutant cells (Fig. 3 E) and remained polarized up to 24 h (Fig. 3 F). This indicated that the foundation of polarized vesicular/membrane transport was intact, and that the migration defect was further downstream of this cell polarity marker.
Golgi Polarization and Vesicular Transport Is Impaired in Ras-GAP Mutant Cells
The GA has been suggested to play a role in polarized vesicular transport and membrane recycling in directed cell movement (Kupfer et al. 1987; Singer and Kupfer 1986). Although not likely responsible for force generation for forward movement, the GA functions in a passive context. In response to experimental wounds, this organelle has been shown to polarize to the leading edge face of the nucleus (Kupfer et al. 1982). Wounding assays showed that wild-type and heterozygous cells do have a polarized GA but no perinuclear orientation could be assigned to the Ras-GAP mutant cells (data not shown). Fig. 4 illustrates the morphology of this organelle by staining with anti–mannosidase II antibodies. Approximately 90% of wild-type (Fig. 4 A) and heterozygous cells (Fig. 4 B) show a normal perinuclear distribution of the GA (Fig. 4A and Fig. B). In contrast, 70–80% of the Ras-GAP mutant cell population contain a fragmented GA (Fig. 4C and Fig. D). The localization of this fragmentation was perinuclear, which is in sharp contrast to the complete cytoplasmic distribution normally seen in mitotic cells. Microinjection of full-length Ras-GAP cDNA into mutant cells rescued the fragmentation of the GA. 80% of the injected Ras-GAP mutant cells displayed a normal GA while the remaining 20% still contained a fragmented GA (Fig. 4E and Fig. F). A possible consequence of this fragmentation might be a nonvectorial transport of membrane components such as lipids and secretory proteins to the leading edge during migration.
Figure 4.
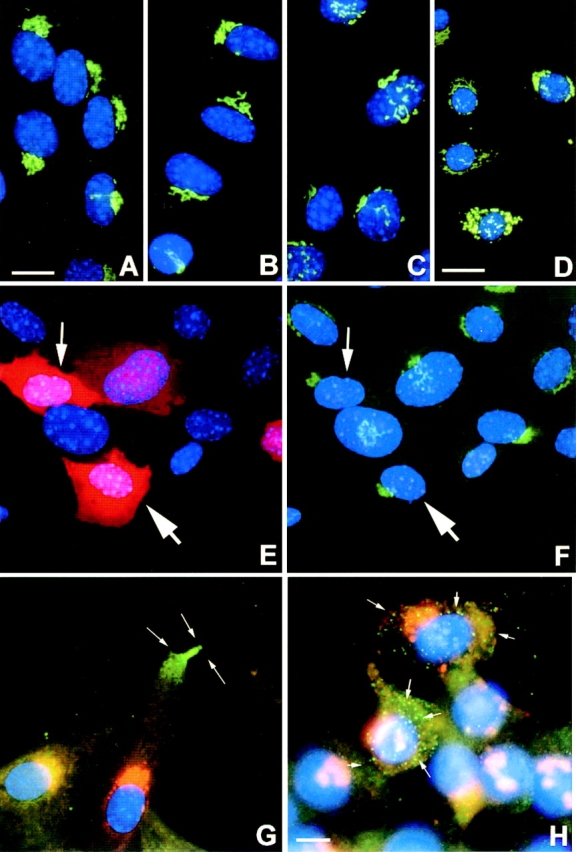
Golgi morphology and Ts-VSV-G-protein transport assay. (A and G, GAP +/+ cells clone 12.78; B, GAP (+/−) cells clone 12.37; C,H, GAP (−/−) clone 12.64; D–F, GAP (−/−) clone 12.83). Golgi morphology was visualized using anti–mannosidase II antibodies (FITC-labeled in A–F and Texas red–labeled in G–H). Golgi orientation about the nucleus (blue Hoechst staining) was assessed in all cell types. Golgi morphology was assessed (F) after microinjection of pCDNA3-GAP construct into GAP (−/−) cells (E and F). Ras-GAP expression was visualized with anti–ras-GAP antibodies followed by Texas red–labeled secondary antibody (E). Arrows in E and F identify injected cells. VSV-G-protein visualized with anti-I14 antibody (FITC-labeled in G-H). VSV-G-protein was delivered to leading edge of wild-type cells within 30 min after heat-shock (arrows in G) but was randomly inserted over the plasma membrane in Ras-GAP mutant cells in as early as 15 min (H) after the temperature shift. Bars: (A–C and E–H) 50 μm; (D) 125 μm.
To test this hypothesis, we microinjected a cDNA encoding an EGFP fusion of glycoprotein G from a temperature-sensitive strain of the VSV (Presley et al. 1997) into wild-type and Ras-GAP mutant cells. This G-protein has been used extensively to characterize the secretory pathway in mammalian cells (Bergmann et al. 1983, Bergmann et al. 1981). At nonpermissive temperatures of 40°C, the G-protein is unable to fold correctly and is trapped within the rough endoplasmic reticulum. At the permissive temperature of 32°C, the G-protein folds correctly, exits the endoplasmic reticulum and is transported to the GA. After modification, the G-protein bound within Golgi vesicles is transported to the plasma membrane of the leading edge in a polarized manner.
Our analysis of intracellular vesicular transport in wild-type cells indicated that the VSV-G-protein first appears at the leading edge of the migration front, 30 min after the temperature shift to 32°C (Fig. 4 G). The GA in these cells was intact, and polarized. No transport of VSV-G-protein was detected during earlier time points. In Ras-GAP mutant cells, the first appearance of the VSV-G-protein was over the entire surface of the cell with no asymmetric distribution (Fig. 4 H). The GA in these cells was not polarized and was fragmented. The polarized delivery of VSV-G to the leading edge of wild-type cells could not be examined during earlier time points because it took a minimum of 4 h for the VSV-G cDNA construct to express the VSV-G-protein. During the incubation of cells at the nonpermissive temperature after microinjection, cell spreading had already been extensive in wild-type cells, but not in mutant cells. Thus, we could not examine the polarized delivery of VSV-G to the leading edge soon after wounding. These observations suggest that random insertion of membrane mass may account for the reduction of cell spreading seen in these mutant cells. This observation also identifies a phenotypic difference between wild-type cells and Ras-GAP mutant cells as it relates to the establishment of cell polarity preceding migration.
Ras-GAP Mutant Cells Do Not Remodel Actin SFs and FAs
Since the cytoskeleton plays a major role for the production of traction forces necessary for forward movement and establishment of polarity, we examined the biased reorientation of actin SFs and FAs seen in several cell wounding systems (Ettenson and Gotlieb 1992). This dramatic reorganization presumably occurs to generate tension and contraction within the cortex of the cell in a polarized orientation for forward movement. In all cell lines analyzed, SFs and FAs were randomly distributed at the ventral surface of the cell at the time of wounding (Fig. 5A, Fig. D, and Fig. G). Only 20–25% of the population had these structures in an orientation parallel to the direction of cell movement. At later times after wounding, SF and FA polarization was only seen in Ras-GAP–expressing cells (Fig. 5B, Fig. C and Fig. G), concomitant with the acquisition of an elongated cell shape (Fig. 5B and Fig. H). Before the onset of motility, ∼8–16 h after wounding, there was a reduction in the number of SFs and FAs within wound edge Ras-GAP–expressing cells (Fig. 5 B). Cells further removed from the wound edge, however, had no such reduction (data not shown). Migrating cells detected within the denuded zone had reduced SFs and FAs (Fig. 5 C), were no longer polarized, and extended lamellipodia in many different directions.
Figure 5.
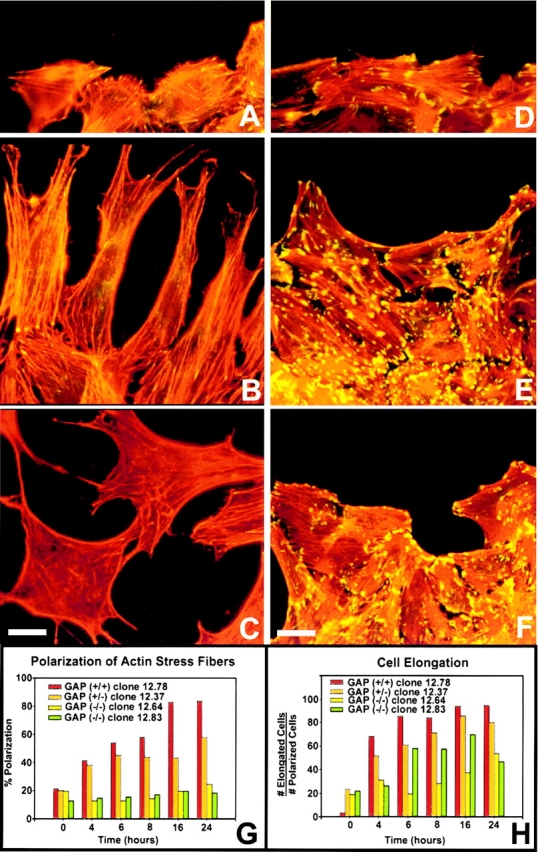
Actin SF and FA remodeling in wild-type and Ras-GAP mutant cells after wounding. Wild-type cells (A–C) and mutant cells (D–F) were wounded, fixed and stained for actin filaments (visualized with rhodamine-phalloidin) and vinculin-containing FAs (stained with anti-vinculin antibodies and FITC-labeled secondary antibodies). (A and D, 0 h; B and E, 16 h; C and F, 24 h) Polarized cells were scored as those having SFs parallel to the direction of migration (G). Percent cell elongation was expressed as the total number of elongated cells divided by total number of cells with polarized SFs and FAs. At all time points, >500 cells were sampled for each cell type (H). Bars: (A–D) 50 μm; (E–F) 125 μm.
During the time course experiment, there was no preferential increase in the number of polarized Ras-GAP mutant cells that had reorganized SFs and FAs (Fig. 5, D–F and G). However, 20–25% of the population did exhibit an elongated phenotype (Fig. 5 H). This elongation was only detected in cells that already contained polarized SFs and FAs. These results suggested that wound-edge cells containing polarized SFs and FAs at the time of wounding have the capacity to undergo elongation but not migration. At later stages, mutant cells at the wound edge do not display a reduction of SFs and FAs. Since SFs and FAs are formed in response to activated Rho (Ridley and Hall 1992), the failure of these cytoskeletal elements to reorient after wounding suggests that Ras-GAP mutant cells may be abnormal in their ability to regulate Rho. Since Ras-GAP and p190 are known to form a complex, these results raise the possibility that the interaction between Ras-GAP and p190 is potentially required for proper Rho-GAP function and that in the absence of Ras-GAP, p190 may be impaired in its ability to regulate Rho.
Ras-GAP and p190 Function to Remodel Actin SFs
To test the function of p190 in SF reorientation, we sought to disrupt the in vivo pTyr-mediated Ras-GAP/p190 protein complex in wild-type cells by using inhibitory peptides derived from p190 sequences. The specificity of these peptides was tested by an in vitro competition assay using detergent-treated lysates of RAT-2 cells transformed by v-Src (Koch et al. 1992). The NH2-terminal hydrophobic region of Ras-GAP (GAP-H; Fig. 6, lane 2) did not bind to p190 as expected. Positive control reactions demonstrated that both the GST-Ras-GAP-SH2 (NH2- and COOH-terminal) fusion proteins were able to bind to p190 (Fig. 6, lanes 3 and 4). In competition experiments, the longer diphosphotyrosine peptide that contained amino acid residues 1086–1110 inclusive of p190, was able to inhibit the interaction between p190 and both GST-Ras-GAP SH2 (NH2- and COOH-terminal) fusion proteins (Fig. 6, lanes 5 and 6). The shorter, phosphatase-resistant peptide-spanning amino acid residues 1100–1112 of p190, containing a single nonhydrolyzable pTyr analogue at position 1105, was eluted from the HPLC column in two peaks as enantiomers. The faster elution peak, designated as Peptide 1 inhibited the interaction with the NH2-terminal Ras-GAP SH2 domain (lane 7). However, the slower migrating fraction, designated as Peptide 2 inhibited the Ras-GAP/p190 interaction in the mixing experiments to a lesser extent (lane 8) than Peptide 1. From these results, we infer that Peptide 1 and Peptide 2 are in the L- and D-configurations, respectively. Both Peptide 1 and Peptide 2 decreased the interaction of the GST-Ras-GAP COOH-terminal SH2 domain with p190 (lanes 9 and 10). Presumably in this case the SH2 domain is recognizing the phenyl-phosphate side chain of each phosphatase-resistant phosphonopeptide and not the peptide backbone and the inhibition is due to the use of excess peptide.
Figure 6.

Disruption of p120 Ras-GAP/p190 interaction in S7A cellular lysates by inhibitory p190 synthetic peptides. Lanes 1–4, no peptide; lanes 5 and 6, diphosphotyrosine peptide; lanes 7 and 9, Peptide 1 phosphonopeptide; lanes 8 and 10, Peptide 2 phosphonopeptide. 50 μM peptide, 200 μg of total S7A cellular lysate, and 10 μg of GST-Ras-GAP fusion proteins were used for all mixing experiments. Samples were subject to blotting with anti-p190 antibodies.
To test whether the association between Ras-GAP and p190 influences SF and FA orientation, p190 inhibitory peptides were then microinjected into wounded wild-type cells. Coinjection with rhodamine-conjugated IgG antibodies facilitated identification of injected cells. The diphosphotyrosine peptide (Fig. 7, A–C) and Peptide 1 (Fig. 7, D–F) significantly reduced the extent of SF reorientation and migration (see also Table ). Injected rhodamine-IgG–labeled cells were primarily restricted to the wound edge. In contrast, microinjection of Peptide 2 (Fig. 7, G–I) and rhodamine-IgG antibody alone (Fig. 7, J–L) did not affect significantly SF reorientation and migration (see also Table ). FA reorientation was similarly affected by these p190 inhibitory peptides (data not shown). These results suggest that the pTyr-mediated interaction between Ras-GAP and p190 is required for SF and FA reorientation during directed movement, possibly through an effect on Rho-GAP function. However, we cannot exclude the possibility that a binding partner for the Ras-GAP SH2 domains other than p190 is responsible for communicating the Ras-GAP signal to the cytoskeleton.
Table 1.
Inhibition of Actin SF Reorientation in Wild-type Cells by p190 Inhibitory Peptides
| Peptide (100 μM) | Total no. injected cells at wound edge | Total no. cells with polarized SFs | Total no. cells in wound | N |
|---|---|---|---|---|
| Peptide 1 phosphonopeptide | 79 | 20 | 10 | 89 |
| Peptide 2 phosphonopeptide | 63 | 58 | 51 | 114 |
| Diphosphotyrosine peptide | 131 | 19 | 8 | 139 |
| Rhodamine-IgG | 103 | 56 | 30 | 133 |
Ras and Rho Function in Cell Polarity and Movement
We next examined the role of the Ras GTPase in directed movement. We have previously shown that growth factor stimulation by PDGF leads to a greater increase in cellular Ras-GTP levels in Ras-GAP mutant cells than in wild-type cells (van der Geer et al. 1997). As illustrated, PDGF-stimulated wild-type cells migrated rapidly into the wound, which frequently resulted in wound closure (Fig. 8 A′). Wild-type cells maintained an elongate morphology, whose longitudinal axis was pointed towards the wound (Fig. 8 A′). In contrast to this ordered migration of wild-type cells, the longitudinal axis of Ras-GAP mutant cells was randomly orientated with respect to the wound edge (Fig. 8 B′). Therefore, although the migration defect of Ras-GAP mutant cells is partially rescued by growth factor stimulation, correlating with increased Ras-GTP levels, the orientation of migration remained impaired. Higher magnification also revealed a loss of cell–cell contact in the mutant population (data not shown). These results suggest that the orientation defect was not due to aberrant Ras regulation and may be due to a Ras-independent function of Ras-GAP.
Figure 8.
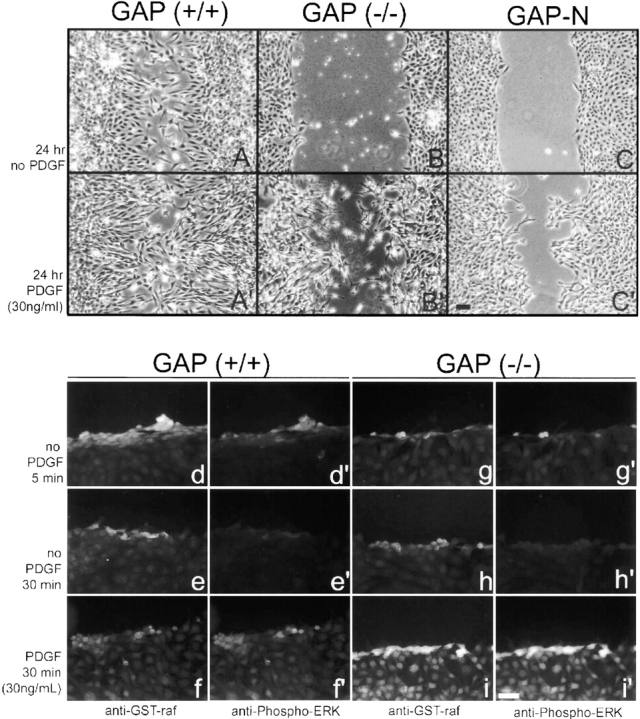
PDGF-induced Ras activation stimulates motility of Ras-GAP mutant cells. Wild-type cells (A and A′), Ras-GAP mutant cells (B and B′) and Ras-GAP mutant cells stably expressing GAP-N (C and C′), migrate into the denuded zone (A′–C′) more efficiently after PDGF stimulation than unstimulated cells (A–C). Ras activation after wounding and PDGF stimulation at the indicated times was visualized in wild-type cells (d–f and d′–f′) and in Ras-GAP mutant cells (g–i and g′–i′) by labeling with GST-Raf-RBD fusion protein followed by anti-GST antibodies (d–f and g–i). Cultures were also double-labeled for activated MAP kinase using antibodies to phosphorylated ERK1 and 2. Bars: (A–C′) 100 μm; (d–i′) 100 μm.
To test whether orientation of migration reflected a Ras-independent role of Ras-GAP, we constructed Ras-GAP mutant cells stably expressing the NH2-terminal region of Ras-GAP but lacking the catalytic domain (GAP-N; see Materials and Methods and McGlade et al., 1993). We then tested these cells for directed cell movement. Expression levels of GAP-N were analyzed by immunoblotting and subsequent immunoprecipitation experiments using anti–Ras-GAP antibodies showed that GAP-N associated with p190 (data not shown). As expected, unstimulated GAP-N cells did not exhibit movement (Fig. 8 C). However, in contrast to Ras-GAP mutant cells, GAP-N–expressing cells exhibited migration that was similar to wild-type cells after growth factor stimulation. The movement of GAP-N cells was ordered and the polarity was fully restored with elongate-shaped cells oriented uniformly to the direction of migration (Fig. 8 C′). Thus, these results suggest that Ras-GTP is required for cell motility, but the association between Ras-GAP and p190 is necessary to restore ordered cell polarization preceding movement.
To determine if these results were due to a change in the activation of Ras in wounded cells, we immunostained for active Ras using a GST-Raf-RBD fusion protein. The minimal Ras-binding domain (RBD) of Raf has been shown to be specific for activated Ras (Rooij and Bos 1997). Wounded cell cultures were fixed and stained for active Ras and activated MAP kinase (as a downstream marker for Ras activation). As shown in Fig. 8d and Fig. d′, wild-type cells showed more activation of Ras at the wound edge than did mutant cells (Fig. 8g and Fig. g′) 5 min after wounding. Ras activation tapered to basal levels 30 min after wounding (Fig. 8 e, e′, h, and h′). However, after growth factor stimulation, mutant cells exhibited more binding of the GST-Raf protein (Fig. 8 i) than did wild-type cells (Fig. 8 f), consistent with our previous finding of elevated Ras-GTP levels after PDGF stimulation (van der Geer et al. 1997). Activation of MAP kinase followed similar patterns of expression (Fig. 8d′–f′, g′–i′).
After PDGF stimulation, all mutant cells at the wound edge showed normal membrane ruffling and lamellipodial extensions (Fig. 9 F) to the same degree as wild-type cells. Phalloidin staining of stimulated mutant cells revealed elongate shaped cells with SFs oriented along the longitudinal axis but with the leading edges of cells pointed in a random manner (Fig. 9 E) in contrast to wild-type cells (Fig. 9 D). Stimulated GAP-N cells also produced elongate cell morphologies with polarized SFs but the leading edges of cells were oriented uniformly to the direction of migration, similar to wild-type cells (data not shown). PDGF-induced Ras activation also resulted in the rescue of Golgi morphology in eighty percent of actively migrating Ras-GAP mutant cells (Fig. 9 B) and rescued polarized vesicular transport of the VSV-G-protein to the leading edge of the cell (Fig. 9C and Fig. F). Interestingly, stimulated mutant cells that were further removed from the wound edge, still possessed a fragmented GA (data not shown).
We next examined the possibility that the failure of SFs and FAs to reorient in Ras-GAP mutant cells was due to the absence of the Ras-GAP/p190 complex, and a consequent effect on Rho regulation. This was accomplished by using the Rho inhibitor, C3 transferase, which ADP-ribosylates Rho, thus preventing guanine-nucleotide exchange activity (Aktories and Hall 1989). Microinjection of C3 transferase (visualized by rhodamine staining in Fig. 9 I), effectively reduced the number of SFs in Ras-GAP mutant cells. These cells also acquired an elongate shape and had SFs oriented towards the direction of movement (Fig. 9 H). However, the cells did not migrate further from the wound edge 24 h after C3 transferase treatment. Interestingly, treatment of Ras-GAP mutant cells with C3 transferase resulted in only a partial rescue of the Golgi morphology (Fig. 9 G). Only 40% of the C3 injected population had a normal GA and microinjection of a putative dominant-negative Rho (T19N Rho) cDNA resulted in only 50% rescue (data not shown), suggesting that Rho plays only a minor role in Golgi integrity. Although impaired Rho regulation appeared to affect polarized SF and FA turnover in Ras-GAP mutant cells, it did not inhibit movement.
Discussion
Cell motility is a complex process involving (a) the coordinated extension of lamellipodia and subsequent formation of new focal contacts to the underlying substratum at the leading edge, (b) the generation of traction and tension within the cell cortex, and (c) the release of focal contacts at the rear of the cell. The actin and MT cytoskeletons play an integral role in modulating cell shape changes in a manner critical for directed cell movement (for reviews, see Lee et al. 1993; Stossel 1993; Huttenlocher et al. 1995; Heidemann and Buxbaum 1998; Waterman-Storer and Salmon 1999). This control of actin cytoskeletal dynamics is regulated in part by GAPs. We have used a cell-wounding assay to analyze a migration defect in Ras-GAP–deficient cells. One advantage of a cell-wounding assay is that cell migration is unidirectional towards the wound face, providing a unique opportunity to examine cytoskeletal-based changes that lead to the generation of cellular asymmetry for polarity preceding movement.
In this study, we show that Ras-GAP serves two important functions in cell motility. First, Ras-GAP regulates Ras in a manner that is essential for movement. Second, Ras-GAP complexed to p190 affects SF and FA remodeling to direct the orientation of migration. Our data indicate that the role of Ras-GAP in the regulation of Ras is complex. A simplistic model would predict elevated levels of Ras-GTP in Ras-GAP mutant cells. However, we have found that Ras-GTP levels are, if anything, decreased in unstimulated wounded cells and it is only after stimulation of Ras-GAP mutant cells with growth factor are elevated Ras-GTP levels observed. This correlates with earlier data showing that Ras-GTP levels are only aberrantly elevated in mutant cells upon growth factor stimulation. Furthermore, the stimulation of mutant cells by growth factor rescued cell spreading and migration defects, suggesting these events are likely dependent upon Ras activation levels. This is consistent with previously described roles of Ras in generating membrane ruffling and lamellipodia formation (Bar-Sagi and Feramisco 1986; Ridley et al. 1992). Suppression of Ras activity by the use of a Ras-neutralizing antibody, a dominant-negative Ras (N17Ras), has been shown to cause a suppression of cell spreading (Fox et al. 1994; Ridley et al. 1995; Nobes and Hall 1999), similar to the defect observed in Ras-GAP mutant cells. Thus, low basal levels of activated Ras in Ras-GAP mutant cells may partially account for the migration defect observed.
Ras-GAP mutant cells are defective in polarized vesicular transport and display a fragmented Golgi phenotype. These data suggest a second causative role for the migration defects observed in these cells. However, our analysis of this defect indicates that this phenotype may also be dependent upon Ras activation levels. We observed that Ras-GAP mutant cells have normal ability to form stable MTs and to orient towards the wound face. However, nonpolarized vesicular transport was affected in Ras-GAP mutant cells as demonstrated by a fragmented GA and abnormal vesicular transport of VSV-G-protein. Recently, local MT dynamics at the leading edge have been shown to affect Ras/Rac activation (Waterman-Storer et al. 1999). In MT regrowth experiments, the restricted assembly of MTs at the leading edge has been shown to activate Rac1 resulting in lamellar protrusions. However, we found no detectable differences in the ability of Ras-GAP mutant cells to form stable MT complexes. The assembly of stable MTs has also been suggested to play a critical role for polarized vesicular transport (Gundersen and Bulinski 1988). The process of polarized vesicular transport of secretory proteins and membrane mass to the leading edge is required for initiation and continuation of lamellar extensions (Singer and Kupfer 1986). Our observations suggest that Ras-GAP may play a role in this process as evidenced by the growth factor dependence of the vesicular transport and fragmented Golgi phenotypes. A second explanation stems from recent observations by Liu and Li 1998, demonstrating an in vitro interaction between the catalytic domain of Ras-GAP and Rab5, a protein involved in vesicular transport. Thus, we cannot exclude the possibility that the lack of association between Ras-GAP and Rab5 may also contribute to this process.
We have shown that Ras-GAP also functions in a Ras-independent manner through a pTyr-dependent p190 interaction. This interaction apparently promotes SF and FA turnover, Rho-GAP regulation, and reorientation of SFs and FAs for directed movement. The involvement of the Ras-GAP/p190 complex in SF and FA remodeling has been suggested previously. Overexpression of GAP-N led to increased complex formation with p190 and resulted in a loss of SFs and FAs (McGlade et al. 1993). Conversely, we have demonstrated that disruption of the Ras-GAP/p190 interaction by phospho-specific peptides leads to a failure of SF and FA remodeling. Thus, this loss of turnover observed in Ras-GAP mutant cells is dependent upon Ras-GAP/p190 complex formation.
The association with Ras-GAP may also affect the Rho-GAP function of p190. We have previously shown that increased complex formation between GAP-N and p190 in RAT-2 cells enhances Rho-GAP activity (McGlade et al. 1993). Ridley et al. 1993 has shown that microinjection of the Rho-GAP catalytic domain of p190 into lysophospatidic acid–stimulated Swiss 3T3 cells, caused the dissolution of SFs and FAs. This suggests that the Rho-GAP function of native p190 is tethered in some way. Thus, although p190 is present in Ras-GAP mutant cells, interaction with Ras-GAP may be necessary for proper p190 regulation of Rho. Consistent with this hypothesis, the failure of SF and FA turnover and reorientation might be due to high levels of active Rho in Ras-GAP mutant cells. We tested this hypothesis by blocking Rho function with the Rho inhibitor, C3 transferase. In C3 transferase–treated mutant cells, SFs were reoriented towards the direction of migration and cells acquired an elongate morphology, suggesting the defects in SF reorientation and turnover can be rescued by inhibiting Rho. Ras-GAP mutant cells did not migrate further from the wound edge after C3 transferase treatment, due to lower levels of activated Ras as demonstrated. Taken together, these results indicate the formation of the Ras-GAP/p190 complex is required for the correct temporal Rho function of actin SFs and FAs and that this interaction is pTyr mediated. Roof et al. 1998 report major pTyr-dependent and minor pTyr-independent interactions between Ras-GAP and p190. It is attractive to visualize a mechanism whereby an active pTyr-dependent Ras-GAP/p190 complex regulates SF and FA turnover, and an inactive pTyr-independent interaction is functionally silent. Thus, when cells require such cytoskeletal remodeling for movement, the inactive complex converts to an active protein complex. The biological implications for these interactions in regulating the cytoskeleton require further investigation.
Our data show that when Ras-GAP is complexed to p190 it propagates uniform cell polarity at the wound edge and directs the orientation of migration. Ras-GAP mutant cells, induced to migrate after growth factor–induced Ras activation, had an appropriate elongate morphology for migration. However, these cells were oriented in a random manner with respect to the wound edge and the pattern of migration was similar to endothelial cells that were injected with the oncogenic form of Ha-ras (Fox et al. 1994). In contrast, expression of GAP-N in Ras-GAP–deficient cells not only rescued migration in a Ras-dependent manner, the orientation of migration was also ordered and uniform at the wound edge, similar to wild-type cells. These observations are consistent with Nobes and Hall 1999 who report Ras is not only essential for movement but may be responsible for SF and FA turnover.
In summary, our study shows that Ras-GAP has two important functions: (a) Ras-GAP negatively regulates Ras, which is a requirement for cell movement, and (b) Ras-GAP forms a complex with p190 in a pTyr-dependent manner that is required for the turnover and reorientation of SFs and FAs. Our data suggest that it is the absence of these functions that accounts for the early embryonic cell migration defects detected in the mouse embryo deficient for Ras-GAP (Henkemeyer et al. 1995). Further targeted mutational studies will be required to address the requirements of specific Ras-GAP domains for directed cell movement in vivo.
Acknowledgments
We thank Dr. Perry Howard for numerous useful discussions. We also thank Dr. Sacha Holland and Dr. Howard for critically reading the manuscript.
This work was supported by grants awarded to Tony Pawson from the Medical Research Council of Canada and the National Cancer Institute of Canada.
Footnotes
Dr. van der Geer's present address is University of California at San Diego, Department of Chemistry and Biochemistry, 9500 Gilman Dr., La Jolla, CA 92093-0601.
Dr. Henkemeyer's present address is University of Texas, South Western Medical Center, 6000 Harry Hines Blvd., Dallas, TX 75235-9133.
Abbreviations used in this paper: aa, amino acid; FA, focal adhesion; GA, Golgi apparatus; GAPs, GTPase-activating proteins; MT, microtubule; pTyr, phosphotyrosine; SF, stress fiber; VSV, vesicular stomatitis virus.
References
- Aktories K., Hall A. Botulinum ADP-ridosyltransferase C3a new tool to study low molecular weight GTP-binding proteins. Trends Pharmacol. Sci. 1989;10:415–418. doi: 10.1016/0165-6147(89)90191-0. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D. Mammalian cell microinjection assay. Methods Enzymol. 1995;255:436–442. doi: 10.1016/s0076-6879(95)55045-3. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D., Feramisco J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–1068. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- Bergmann J.E., Tokuyasu K.T., Singer S.J. Passage of an integral membrane protein, the vesicular stomatitis virus glycoprotein, through the Golgi apparatus en route to the plasma membrane. Proc. Natl. Acad. Sci. USA. 1981;78:1746–1750. doi: 10.1073/pnas.78.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann J.E., Kupfer A., Singer S.J. Membrane insertion at the leading edge of motile fibroblasts. Proc. Natl. Acad. Sci. USA. 1983;80:1367–1371. doi: 10.1073/pnas.80.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski M.S., McCormick F. Proteins regulating ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Cook T.A., Nagasaki T., Gunderson G.G. Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J. Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C., Moran M., McCormick F., Pawson T. Phosphorylation of GAP and GAP-associated proteins by transforming and mitogenic tyrosine kinases. Nature. 1990;343:377–381. doi: 10.1038/343377a0. [DOI] [PubMed] [Google Scholar]
- Ettenson D.S., Gotlieb A.I. Centrosomes, microtubules, and microfilaments in the reendothelialization and remodeling of double-sided in vitro wounds. Lab. Invest. 1992;66:722–733. [PubMed] [Google Scholar]
- Euteneuer U., Schliwa M. Mechanism of centrosome positioning during the wound response in BSC-1 cells. J. Cell Biol. 1992;116:1157–1166. doi: 10.1083/jcb.116.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox P.L., Sa G., Dobrowolski S.F., Stacey D.W. The regulation of endothelial cell motility by p21ras. Oncogene. 1994;9:3519–3526. [PubMed] [Google Scholar]
- Gawler D.J., Zhang L.J., Reedijk M., Tung P.S., Moran M.F. CaLBa 43 amino acid calcium-dependent membrane/phospholipid binding domain in p120 Ras GTPase-activating protein. Oncogene. 1995;10:817–825. [PubMed] [Google Scholar]
- Gundersen G.G., Bulinski J.C. Selective stabilization of microtubules oriented toward the direction of cell migration. Proc. Nat. Acad. Sci. USA. 1988;85:5946–5950. doi: 10.1073/pnas.85.16.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley R.G., Lieu F.H.L., Fong A.Z.C., Hawley T.S. Versatile retroviral vectors for potential use in gene therapy. Gene Therapy. 1994;1:136–138. [PubMed] [Google Scholar]
- Heidemann S.R., Buxbaum R.E. Cell crawlingfirst the motor, now the transmission. J. Cell Biol. 1998;141:1–4. doi: 10.1083/jcb.141.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkemeyer M., Rossi D.J., Holmyard D.P., Puri M.C., Mbamalu G., Harpal K., Shih T.S., Jacks T., Pawson T. Vascular system defects and neuronal apoptosis in mice lacking Ras-GTPase-activating protein. Nature. 1995;377:695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- Hu K.W., Settleman J. Tandem SH2 binding sites mediate the RasGAP-RhoGAP interactiona conformational mechanism for SH3 domain regulation. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:473–483. doi: 10.1093/emboj/16.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A., Sandborg R.R., Horwitz A.F. Adhesion in cell migration. Curr. Opinion Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Koch C.A., Moran M.F., Anderson D., Liu X., Mbamalu G., Pawson T. Multiple SH2-mediated interactions in v-Src-transformed cells. Mol. Cell. Biol. 1992;12:1366–1374. doi: 10.1128/mcb.12.3.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R., Ahmed S., Best A., Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 1995;15:1942–1952. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R., Sarner S., Ahmed S., Lim L. Rho family GTPases and neuronal growth cone remodellingRelationship between increased complexity induced by cdc42Hs, rac1, and acetylcholine and collapse induced by rhoA and lysophosphatidic acid. Mol. Cell. Biol. 1997;17:1201–1211. doi: 10.1128/mcb.17.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer A., Louvard D., Singer S.J. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc. Natl. Acad. Sci. USA. 1982;79:2603–2607. doi: 10.1073/pnas.79.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer A., Kronebusch P.J., Rose J.K., Singer S.J. A critical role for the polarization of membrane recycling in cell motility. Cell Mot. Cytoskelet. 1987;8:182–189. doi: 10.1002/cm.970080210. [DOI] [PubMed] [Google Scholar]
- Leblanc V., Tocque B., Delumeau I. Ras-GAP controls Rho-mediated cytoskeletal reorganization through its SH3 domain. Mol. Cell. Biol. 1998;18:5567–5578. doi: 10.1128/mcb.18.9.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Jacobson K. How do cells move along surfaces? Trends Cell Biol. 1993;3:366–370. doi: 10.1016/0962-8924(93)90084-e. [DOI] [PubMed] [Google Scholar]
- Lefrancios L., Lyles D.S. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virol. 1982;121:157–167. [PubMed] [Google Scholar]
- Liu K., Li G. Catalytic domain of the p120 ras-GAP binds to Rab5 and stimulates its GTPase activity. J. Biol. Chem. 1998;273:10087–10090. doi: 10.1074/jbc.273.17.10087. [DOI] [PubMed] [Google Scholar]
- Markowitz D., Goff S., Bank A.J. A safe packaging line for gene transferseparating viral genes on two different plasmids. J. Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlade J., Brunkhorst B., Anderson D., Mbamalu G., Settleman J., Dedhar S., Rozakis-Adcock M., Chen L.B., Pawson T. The NH2-terminal region of GAP regulates cytoskeletal structure and cell adhesion. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:3073–3081. doi: 10.1002/j.1460-2075.1993.tb05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriläinen J., Palovuori R., Sormunen R., Wasenius V.-M., Lehto V.-P. Binding of the α-fodrin SH3 domain to the leading lamellae of locomoting chicken fibroblasts. J. Cell. Sci. 1993;105:647–654. doi: 10.1242/jcs.105.3.647. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, rac and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999;114:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Presley J.F., Cole N.B., Schroer T.A., Hirschberg K., Zaal K.J.M., Lippincott-Schwartz J. ER-to Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Self A.J., Kasmi F., Paterson H.F., Hall A., Marshall C.J., Ellis C. Rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:5151–5160. doi: 10.1002/j.1460-2075.1993.tb06210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Comoglio P.M., Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol. Cell. Biol. 1995;15:1110–1122. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof R.W., Haskell M.D., Dukes B.D., Sherman N., Kinter M., Parsons S.J. Phosphotyrosine (pTyr)-dependent and –independent mechanisms of p190 rhoGAP-p120 rasGAP interactionTyr 1105 of p190, a substrate for c-src, is the sole p-Tyr mediator of complex formation. Mol. Cell. Biol. 1998;18:7052–7063. doi: 10.1128/mcb.18.12.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooij J., Bos J.L. Minimal ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene. 1997;14:623–625. doi: 10.1038/sj.onc.1201005. [DOI] [PubMed] [Google Scholar]
- Settleman J., Albright C.F., Foster L.C., Weinberg R.A. Association between GTPase activators for rho and ras families. Nature. 1992;359:153–154. doi: 10.1038/359153a0. [DOI] [PubMed] [Google Scholar]
- Singer S.J., Kupfer A. The directed migration of eukaryotic cells. Annu. Rev. Cell. Biol. 1986;2:337–365. doi: 10.1146/annurev.cb.02.110186.002005. [DOI] [PubMed] [Google Scholar]
- Stossel T.P. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- van der Geer P., Henkemeyer M., Pawson T. Aberrant ras regulation and reduced p190 tyrosine phosphorylation in cells lacking p120-GAP. Mol. Cell. Biol. 1997;17:1840–1847. doi: 10.1128/mcb.17.4.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer C.M., Salmon E.D. Positive feedback interactions between microtubule and actin dynamics during cell motility. Curr. Opin. Cell Biol. 1999;11:61–67. doi: 10.1016/s0955-0674(99)80008-8. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer C.M., Worthylake R.A., Liu B.P., Burridge K., Salmon E.D. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nature Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]