

Abstract
In Saccharomyces cerevisiae, endocytic material is transported through different membrane-bound compartments before it reaches the vacuole. In a screen for mutants that affect membrane trafficking along the endocytic pathway, we have identified a novel mutant disrupted for the gene YJL204c that we have renamed RCY1 (recycling 1). Deletion of RCY1 leads to an early block in the endocytic pathway before the intersection with the vacuolar protein sorting pathway. Mutation of RCY1 leads to the accumulation of an enlarged compartment that contains the t-SNARE Tlg1p and lies close to areas of cell expansion. In addition, endocytic markers such as Ste2p and the fluorescent dyes, Lucifer yellow and FM4-64, were found in a similar enlarged compartment after their internalization. To determine whether rcy1Δ is defective for recycling, we have developed an assay that measures the recycling of previously internalized FM4-64. This method enables us to follow the recycling pathway in yeast in real time. Using this assay, it could be demonstrated that recycling of membranes is rapid in S. cerevisiae and that a major fraction of internalized FM4-64 is secreted back into the medium within a few minutes. The rcy1Δ mutant is strongly defective in recycling.
Keywords: endocytosis, recycling, FM4-64, ubiquitin, yeast
Introduction
The endocytic pathway in S. cerevisiae can be divided into several steps of membrane transport from the plasma membrane to the vacuole, the degradative compartment in yeast. Individual transport steps can be studied using the pheromone α-factor, which is internalized by receptor-mediated endocytosis (Chvatchko et al. 1986). Once inside the cell, the pheromone passes sequentially through two distinct compartments, called early and late endosomes, before it reaches the vacuole where it is degraded (Singer-Kruger et al. 1993). A recent morphological study in which endocytosis of positively charged nanogold was followed has allowed visualization of the endocytic compartments in yeast (Prescianotto-Baschong and Riezman 1998). Accordingly, early endosomes are vesicular-tubular structures with often horseshoe-like shape, whereas late endosomes are oval in shape, multivesicular, and are found close to the vacuole (Hicke et al. 1997; Prescianotto-Baschong and Riezman 1998).
To identify components that are involved in different trafficking steps along the endocytic pathway, several mutant screens have been performed (Chvatchko et al. 1986; Davis et al. 1993; Raths et al. 1993; Munn and Riezman 1994; Wendland et al. 1996). Most of these newly isolated endocytosis (end) mutants affect the internalization step of endocytosis, leading to a better understanding of the molecular mechanisms underlying internalization of plasma membrane proteins (Geli and Riezman 1998). These studies clearly identified roles for actin (Kubler and Riezman 1993) and ubiquitination (Galan et al. 1996; Hicke and Riezman 1996) in this step. A second group of mutants affecting endocytosis has been identified independently during screens for proper sorting of vacuolar proteins (Bryant and Stevens 1998). Vacuole protein sorting (vps) mutants of the class E type led to the enlargement of a compartment called the prevacuolar compartment (PVC) (Raymond et al. 1992; Piper et al. 1995), and inhibited traffic of vps and endocytic markers from the PVC to the vacuole. These results led to the conclusion that the endocytic pathway intersects with the vps pathway at the level of the PVC (Munn and Riezman 1994; Piper et al. 1995). In contrast, only a few mutants have been studied that potentially affect endocytosis at a postinternalization step before the intersection with the vps pathway (Holthuis et al. 1998a; Seron et al. 1998).
Endocytosis is a rapid process. In response to a stimulus, yeast receptors or transporters are degraded with half-lives of <20 min (Hicke and Riezman 1996; Springael and Andre 1998). In response to ligand binding, 50% of the α-factor receptor Ste2p is internalized in 6–8 min at 24°C (Munn and Riezman 1994; Geli and Riezman 1996). For comparison, the half-time of internalization of transferrin into mammalian cells has been determined to be 4 min at 37°C (Ghosh and Maxfield 1995). Early work with mammalian cells has shown that these high rates of internalization are achieved in part because major fractions of the plasma membrane are being internalized over a relatively short time. In fibroblasts, membranes corresponding to 50% of the entire cell surface can be internalized per hour (Steinman et al. 1976). To balance this enormous loss of plasma membranes due to the constitutive formation of endocytic vesicles, membranes are recycled back to the plasma membrane (Mayor et al. 1993; Mukherjee et al. 1999). The same is true for plasma membrane proteins. Only a fraction of the internalized proteins are targeted to the lysosome (Mayor et al. 1993; Ghosh et al. 1994). To protect membrane proteins from lysosomal degradation, they are sorted away from the linear pathway (early endosome, late endosome, lysosome) at the level of the early endosome (Ghosh and Maxfield 1995; Sheff et al. 1999). This sorting process allows proteins to be reused for further cycles of ligand binding and internalization. In yeast, little is known about recycling of plasma membrane but given the high rates of internalization observed, it is likely that this pathway also exists. The first proteins that were proposed to follow a recycling pathway in yeast were two chitin synthases (Chs1p and Chs3p) (Chuang and Schekman 1996; Ziman et al. 1996). These chitin synthases are present in the plasma membrane and in an intracellular pool called chitosomes (Chuang and Schekman 1996; Ziman et al. 1996). It was proposed that these two pools of chitin synthases are kept in balance through their continuous internalization and recycling. Because Chs1p and Chs3p exert their functions at specific times and locations during the cell cycle, sorting in the endosomal system and recycling could be a mechanism to target these enzymes to subregions of the plasma membrane (Chuang and Schekman 1996; Shaw et al. 1991). Recently, localization of Chs3p has been shown to be affected in cells disrupted for genes encoding the soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs) Tlg1p or Tlg2p (Holthuis et al. 1998b). The absence of one of these t-SNAREs may affect recycling of Chs3p by preventing transport through a compartment along the recycling pathway in yeast.
In this study, we identify and characterize a novel mutant defective both in endocytosis and for the recycling pathway in yeast. The mutant is a disruptant of the open reading frame (ORF) YJL204c named RCY1 (for recycling 1). The rcy1Δ mutant was identified in a screen for mutants defective for the accumulation of the fluorescent dye Lucifer yellow carbohydrazide (LY) in the vacuole. The European functional analysis network (EUROFAN) yeast mutant collection that was used here contains yeast strains disrupted for single nonessential ORFs that have been discovered during the sequencing of the yeast genome, and have only limited homologies to previously identified genes (Oliver 1996).
The rcy1Δ mutant strain was found to be defective in endocytosis at an early postinternalization step, before the intersection of the endocytic with the vps pathway. In addition to this rare endocytic phenotype, this mutant is also defective for recycling of internalized material to the plasma membrane. Rcy1p, which is required for these trafficking steps, encodes a protein with an F-box near its NH2 terminus. F-boxes mediate the interaction of target proteins with the SKP1/cullin/F-box (SCF)–ubiquitin ligase complex (Patton et al. 1998b). The presence of this motif in a protein that affects vesicular traffic and another potential role of ubiquitination in the endocytic pathway are discussed.
Materials and Methods
Yeast Strains, Media, and Reagents
Yeast strains used in this study are listed in Table . YPUAD and the minimal medium SD was prepared as described by Dulic et al. 1991. Yeast nitrogen base (YNB) medium contains 0.67% yeast nitrogen base (Difco Laboratories) and 2% glucose. SD medium containing 5 fluoroorotic acid (5FOA; PCR Inc.) was prepared according to Munn and Riezman 1994. To select for disruptant strains containing the kanMX-module (Wach et al. 1994), cells were grown on plates containing 200 μg/ml Geneticin (GIBCO BRL). The following nutrients (40 μg/ml) were added to the SD and yeast nitrogen base media when required: adenine, uracil, tryptophan, histidine, lysine, leucine, and tyrosine. Solid media contained 2% Bactoagar (Difco). Biosynthetically 35SO4-labeled α-factor was purified using the amberlite resin, CG-50 (Serva) followed by C18 reverse-phase HPLC as described in detail by Dulic et al. 1991. Preparative silica gel 60 (2.2-mm thick; 20 × 20 cm) TLC plates were supplied by MERCK. LY was obtained from Fluka (Buchs), FM4-64 from Molecular Probes. Glucose oxidase, HRP, and o-dianisidine were purchased from Fluka.
Table 1.
Yeast Strains Used in this Study
| Strain | Genotype | Source |
|---|---|---|
| RH4344 | Mata yjl204c::kanMX his4 leu2 ura3 lys2 bar1 | This study |
| RH4345 | Mata his4 leu2 ura3 lys2 bar1 | This study |
| RH4739 | Mata yjl204c::kanMX pma1::PMA1HA:LEU2 his3 ura3 ade2 trp1 | This study |
| RH4740 | Mata pma1::PMA1HA:LEU2 his3 ura3 ade2 trp1 | This study |
| RH2180 | Matα vps1-Δ2 pep4 his4 leu2 ura3 ade6 | T. Stevens |
| RH2517 | Mata ypt7::LEU2 his4 ura3 bar1 | H. Riezman |
| RH2906 | Mata end13/vps4::URA3 his4 leu2 lys2 bar1 | H. Riezman |
| RSY271 | Matα sec18-1 his4 ura3 | R. Schekman |
| RSY255 | Matα leu2 ura3 | R. Schekman |
| SEY6210 | Matα his3 leu2 ura3 lys2 trp1 suc2 | H. Pelham |
| JHY016 | Mata tlg1::TRP1 his3 leu2 ura3 lys2 suc2 | H. Pelham |
| TSYKO3 | Matα tlg2::HIS5pL leu2 ura3 lys2 trp1 suc2 | H. Pelham |
Polyclonal antiserum against carboxypeptidase Y (CPY) was used as described previously (Moreau et al. 1996). Antisera against Tlg1p and Sed5p were a kind gift of H.R.B. Pelham (Medical Research Council [MRC] Laboratory of Molecular Biology, Cambridge, UK).
Yeast Genetic Techniques
Yeast standard techniques were used for mating, sporulation, and tetrad analysis. For transformation of yeast cells, a lithium acetate method was used (Gietz et al. 1992).
DNA Constructs Used
The plasmids carrying the deletion cassette and the cognate gene of the ORF YJL204c (which we named RCY1) were obtained from European functional analysis network (EUROFAN) and were constructed by Wysocki et al. 1999. The deletion cassette containing a kanMX-module was cloned into the pUG7 vector (pYORCYJL204c). The wild-type gene was cloned into the pRS416 centromeric plasmid to yield the plasmid pYCGYJL204c. Details on the construction of these plasmids can be found elsewhere (Wysocki et al. 1999).
LY Accumulation in the Vacuole
Fluid-phase endocytosis of LY was performed as described by Dulic et al. 1991. Cultures of yeast cells were grown to logarithmic phase (OD600 = 0.2–0.5) in YPUAD at 24°C. Cells were concentrated to 2–5 OD600 U/ml in YPUAD and incubated for 1 h at 24°C in the presence of 4 mg/ml LY. After LY accumulation, the cells were washed three times in 1 ml of ice-cold 50 mM sodium phosphate, 10 mM sodium azide, 10 mM sodium fluoride, pH 7.5. Samples were viewed by fluorescence microscopy using FITC optics.
α-Factor Endocytosis Assays
Internalization of α-factor was followed using the pulse-chase protocol described by Dulic et al. 1991. 100 ml of mid-log phase cells (107 cells/ml) was harvested and resuspended in ice-cold YPUAD. [35S]α-factor was prebound to the cells for 50 min on ice. Uptake of α-factor was initiated by resuspending the cells in YPUAD prewarmed to 24°C. Internalization was calculated by dividing the internalized counts (remaining cell bound in a 50 mM citrate buffer, pH 1.1) by the total cell-associated counts (cells washed in 50 mM potassium phosphate buffer, pH 6).
Extraction of total bound or internalized [35S]α-factor from the yeast cells and subsequent separation of intact and degraded α-factor on TLC plates was performed as described (Dulic et al. 1991).
Uracil Permease Assays
Wild-type and rcy1Δ cells were transformed with a construct (p195gF) (2μURA3-Gal-FUR4) to overexpress uracil permease (Volland et al. 1994). These strains were grown in yeast nitrogen base medium plus 4% galactose and the required amino acids to an OD600 of 0.6. Cycloheximide (100 μg/ml) was then added to the medium. Uracil uptake was measured at different timepoints after the addition of cycloheximide. Aliquots of 1 ml of yeast culture were incubated with 5 μM [14C]uracil (NEN) for 20 s at 30°C and then quickly filtered through a Whatman GF/C filter, which was washed twice with ice-cold water and counted for the retained radioactivity. To examine permease degradation, 6 × 107 cells were harvested at the different timepoints and lysed for 10 min on ice with 0.185 N NaOH, 2% 2-mercaptoethanol. The protein samples were precipitated using 5% trichloroacetic acid. The pellets were resuspended in 45 μl sample buffer (4% sodium dodecyl sulfate, 0.1 M Tris-hydrochloride, pH 6.8, 4 mM EDTA, 20% glycerol, 2% mercaptoethanol, 0.02% bromophenol blue) plus 25 μl of 1 M Tris base. The samples were heated at 37°C for 10 min before separation of the proteins on 10% SDS-polyacrylamide tricine gels. The uracil permease was detected using a rabbit antiserum raised against the last 10 residues of the permease as described (Galan et al. 1996).
Staining with FM4-64
Endocytosis of FM4-64 was modified from the protocol described by Vida and Emr 1995. Yeast strains were grown in YPUAD to an optical density OD600 of 0.2–0.5. Cells were harvested and resuspended at 3–5 OD600 U/ml. FM4-64 (16 mM in DMSO) was diluted to 400 μM in H2O and added to the cells at a final concentration of 40 μM. Cells were then incubated in the presence of the dye for 10–12 min at 24°C. After this internalization step, the yeast cells were washed three times in ice-cold medium to remove surface-bound dye. The cells were resuspended in YPUAD and incubated for various times at 24°C. Stained cells were placed on slides and visualized using RITC optics.
FM4-64 Recycling Assay
Yeast cells were allowed to internalize FM4-64 for 12 min as described above. The cells were diluted in ice-cold SD medium and harvested by centrifugation (1 min at low speed in a benchtop microfuge). The cells were then washed three times in ice-cold SD medium (each wash step takes 2–5 min to completely remove surface-bound dye). After the last wash step, the cells are resuspended in 10 μl SD medium and kept on ice. Prewarmed SD medium was added to the cells to a final optical density OD600 of 0.25. The cell suspension (1.5 ml) was added to 10 × 10 mm acryl cuvettes (Sarstedt). The fluorescence was recorded at an angle of 90°. Excitation was at 515 nm with a bandwidth of 10 nm, and emission was measured at 680 nm with a bandwidth of 10 nm. Fluorescence was usually measured for 10 min. During the entire measurement, cells were stirred and kept at indicated temperature. The measurements were performed on a JASCO Spectrofluorometer FP-777 (Japan Spectroscopic Co., Ltd.).
To estimate the amount of FM4-64 in the medium, cells were incubated in the presence of FM4-64 for 12 min as above. The cells were then washed in ice-cold SD medium until no fluorescence could be detected in the washes after adding 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate (CHAPS) to 1% (from a 20% stock solution in SD). Cell pellets were resuspended in prewarmed SD medium as described above and allowed to recycle. Aliquots of cells corresponding to 0.375 OD600 units were taken at different timepoints and total fluorescence was measured for 30 s. CHAPS was added to 1% final concentration and mixed. Fluorescence was measured for an additional 60 s. The difference between the values after and before CHAPS addition was calculated. CHAPS had no effect on the fluorescence of cells that had not been treated with FM4-64.
Invertase Secretion
Yeast cells were grown at 24°C to an OD600 of 0.2–0.5 in YPUAD medium. Cell were harvested and washed in distilled water to remove glucose from the medium. The cells were resuspended in YPUAS medium (containing 0.05% glucose and 2% sucrose) to induce invertase expression at 24°C. Samples were taken every 15 min and membrane transport was blocked by the addition of sodium azide to 10 mM final concentration and incubation on ice. Equal amounts of cells were used to measure secreted invertase activity. To measure total invertase activity (internal and secreted invertase), 50 μl of 10% Triton X-100 (Sigma Chemical Co.) was added to 0.5 ml of yeast cell suspension OD600 of 0.5. Lysates were prepared by freezing the cells in liquid nitrogen and thawing them at room temperature. Invertase was assayed as described (Goldstein and Lampen 1975).
Pulse-Chase Analysis and Secretion of CPY
Cells were grown to a density of 2 × 107 cells/ml (OD600 = 1). The cells were collected and resuspended in fresh medium at OD600 5 and labeled for 4 min by adding 150 μCi/ml [35S]Translabel (NEN) per ml of culture. The metabolically labeled cells were then chased with cold 10 mM methionine and 10 mM cysteine. 0.3-ml aliquots of culture were removed at various times during the chase. Cell extracts were prepared and the proteins prepared for immunoprecipitation as described previously (Moreau et al. 1996). For immunoprecipitation, anti-CPY antibody was used at a dilution of 1:500. Immunoprecipitated proteins were separated by SDS-PAGE on 7.5% tricine gels.
To detect secreted CPY, cells were grown on YPUAD plates in contact with a 0.45-μm nitrocellulose filter (Schleicher and Schuell). After incubation for 48 h, cells were eluted from the filter with water. The filter was probed using anti-CPY antibodies as described (Munn and Riezman 1994).
Immunofluorescence
Staining of Ste2p was performed exactly as described by Hicke et al. 1997. For double immunofluorescence, strains were used that express a hemagglutinin-tagged version of Pma1p. After fixation and removal of the cell wall using recombinant lyticase, the cells were incubated with a polyclonal antibody to Ste2p (1:200) and a mAb recognizing the hemagglutinin epitope of the hemagglutinin protein (1:500; Boehringer Mannheim). Cy3- and Oregon green–conjugated secondary antibodies were used to detect the rabbit and mouse primary antibodies, respectively.
Immunoelectron Microscopy
Yeast strains were grown in YPUAD medium to early logarithmic phase and then fixed overnight at 4°C by direct addition of glutaraldehyde (final 0.2%) and formaldehyde (final 3%) to the culture medium. The cells were washed in 50 mM Hepes, pH 7.5, 3 mM KCl, incubated in 1% NaIO4, and free aldehyde groups were quenched with 50 mM NH4Cl as described (van Tuinen and Riezman 1987). Dehydration, infiltration, and polymerization in LR GOLD resin (London Resin) was done according to the supplier's instructions. Thin sections of ∼50 nm were cut and mounted on 200-mesh nickel grids. Antibodies against Sed5p and Tlg1p were raised in rabbits and kindly provided by H.R.B. Pelham (MRC Laboratory of Molecular Biology). The IgG fraction of each antiserum was purified on protein A–Sepharose columns and used for immunolabeling. As secondary antibodies, IgG-colloidal gold conjugates (BIO Cell) were used. Grids were placed upside down on 50-μl droplets of blocking solution (150 mM NaCl, 10 mM potassium phosphate, pH 7.5, 0.1% Tween 20, 2% fatty acid–free BSA [Sigma Chemical Co.]) for 20 min. The grids were then transferred to droplets containing appropriate dilutions of primary antibodies in blocking buffer and incubated for 4 h at room temperature. The grids were then washed five times for 5 min with PBS solution, pH 7.5. The grids were incubated for 10 min in blocking buffer before transferring to droplets with secondary antibodies and incubation for 2 h and washed as described above. After washing in water, the grids were stained in 6% uranyl acetate for 10 min and in Reynold's lead citrate for 30–60 s.
Results
Unusual Nonvacuolar Accumulation of LY in a Novel Endocytosis Mutant
During a systematic screen of the European functional analysis network (EUROFAN) (Oliver 1996) mutant collection for strains defective in the accumulation of LY in the vacuole (Chvatchko et al. 1986), several candidate endocytosis mutants were found. In this report, one of these mutants was further characterized. The strain, disrupted for the ORF YJL204c, showed decreased levels of LY in the vacuole consistent with a defect in fluid-phase endocytosis (compare Fig. 1 A and Fig. 1 B). Strikingly, small LY-positive structures were observed in the bud of small budded cells, close to sites of cell growth (Fig. 1A and Fig. C). At this level of resolution, one to three punctuate structures could be seen in the bud. When viewed by Nomarski optics, vacuolar morphology in the mutant cells was apparently normal, and the LY-labeled structures did not colocalize with vacuoles, even when the latter were visible in buds (Fig. 1 C). Accumulation of LY in punctate nonvacuolar membranes has not been observed previously.
Figure 1.
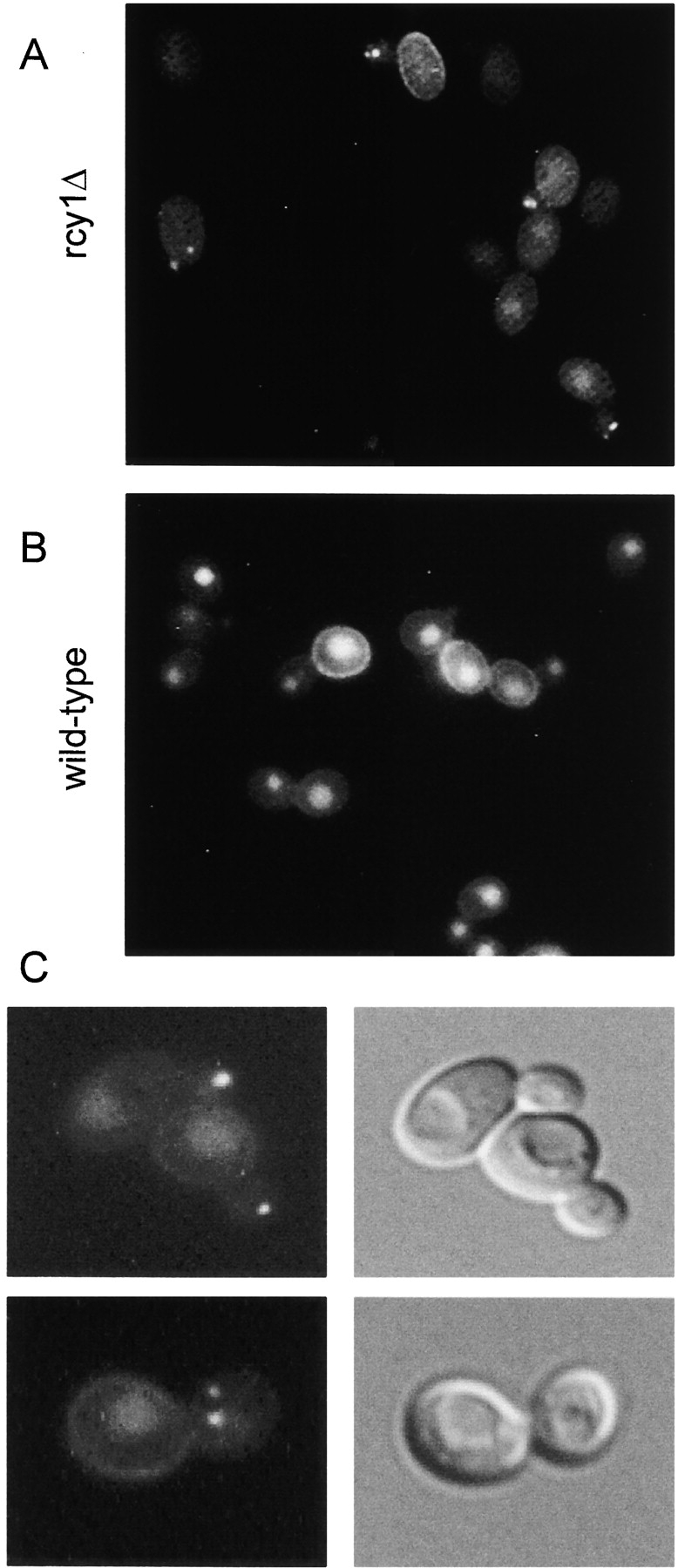
Accumulation of LY in punctuate nonvacuolar structures close to and in the bud of the rcy1-null mutant cells. rcy1 mutant (A and C) and wild-type cells (B) were incubated in YPUAD containing LY (4 mg/ml) for 1 h at 24°C. The cells were then washed and visualized using fluorescence optics. (C) rcy1 mutant cells visualized at higher magnification using either fluorescence (left panels) or Nomarski optics (right panels).
The mutant with this unusual endocytic phenotype contains a disruption of the gene YJL204c (Wysocki et al. 1999). To confirm the linkage of the phenotype to the disruption of YJL204c, a heterozygous diploid was created by replacing one copy of YJL204c with a kanMX cassette in our genetic background. After sporulation and dissection of this strain, each of the tetrads yielded two large and two small colonies. The two small colonies were the yjl204c::kanMX disruptants, confirmed by their ability to grow on geneticin-containing YPUAD plates and by PCR (data not shown). Dissection of the heterozygous diploid carrying the complementing gene on a centromere-containing vector led to the formation of equal-sized colonies. Furthermore, the complemented strains also showed wild-type accumulation of LY in the vacuole and no LY-positive structures in the bud (data not shown). The observed changes in LY distribution are therefore the result of the disruption of YJL204c.
The gene YJL204c, which we renamed RCY1 (recycling 1), encodes a protein of 840 amino acids in length (Swissprot P39531). The presence of a CAAX box at the very COOH terminus of the proteins indicates that Rcy1p may be prenylated (Omer and Gibbs 1994). In addition, Rcy1p contains a putative F-box (Patton et al. 1998b). This degenerate 40–amino acid motif is found in a number of proteins and has been shown to mediate interaction with Skp1p, a component of the SCF–ubiquitin ligase complex (Skowyra et al. 1997; Patton et al. 1998b). Interestingly, the COOH-terminal third of the protein (275AA) shows some sequence homology to Sec10p, a protein known to be involved in membrane trafficking (Wysocki et al. 1999; see Discussion).
Large Compartment Found in the rcy1Δ Mutant
EM was used to visualize and characterize the compartment that accumulated LY in the rcy1Δ mutant strain. When compared with wild-type cells, mutant cells contained unusually large compartments in the bud or when seen in the mother cell, localized near the bud neck (Fig. 2, A–C). Compartments of similar shape with a large lumenal compartment were not seen in wild-type cells (Fig. 2, D–F). The membrane compartments observed in the mutant cells have at least two different morphologies. There are straight elongated membrane structures and horseshoe-shaped cisternae-like structures (Fig. 2). Furthermore, in certain areas, these membranes have a large lumenal compartment up to 0.15 μm across. The large lumen of this compartment in rcy1Δ cells may explain why these structures could be visualized using LY.
Figure 2.
Tlg1p localizes to the enlarged compartment of the rcy1Δ mutant. Logarithmically growing cells were fixed, dehydrated, and embedded for EM as described in Materials and Methods. Cell sections were then incubated with purified anti-Tlg1p antibodies followed by anti-IgG secondary antibodies. The gold-coupled secondary antibodies can be seen on the sections as black dots with a diameter of 10 nm. Immunogold-labeled sections of rcy1Δ (A–C) and the corresponding wild-type (D–F) are shown. Bars, 200 nm. (D) Tlg1p-positive membranes are indicated by a star.
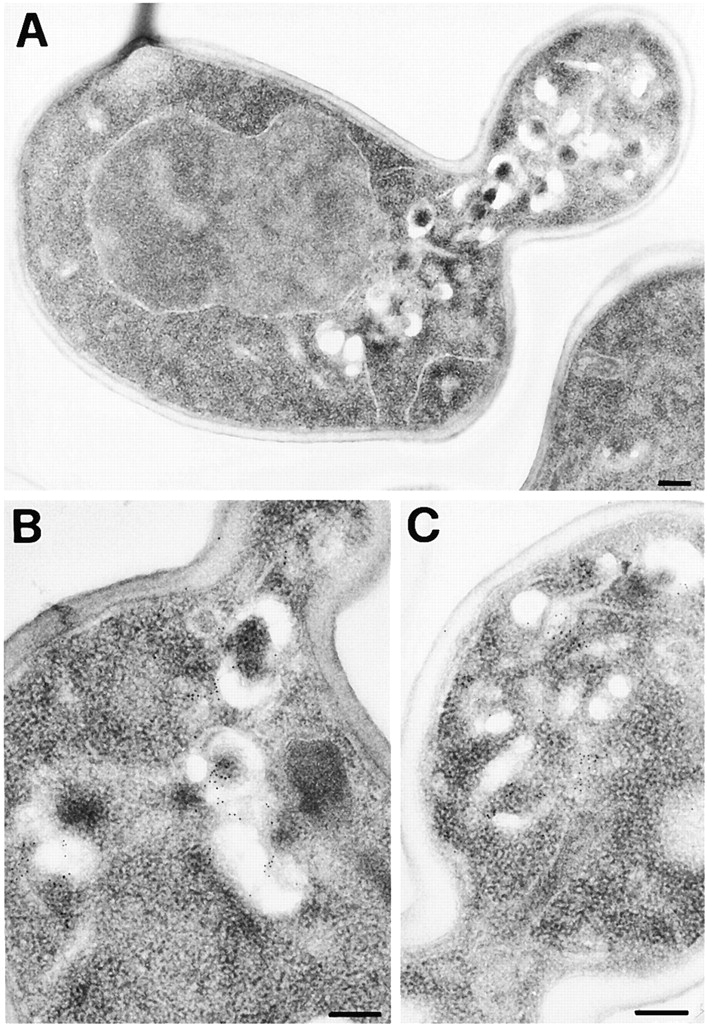
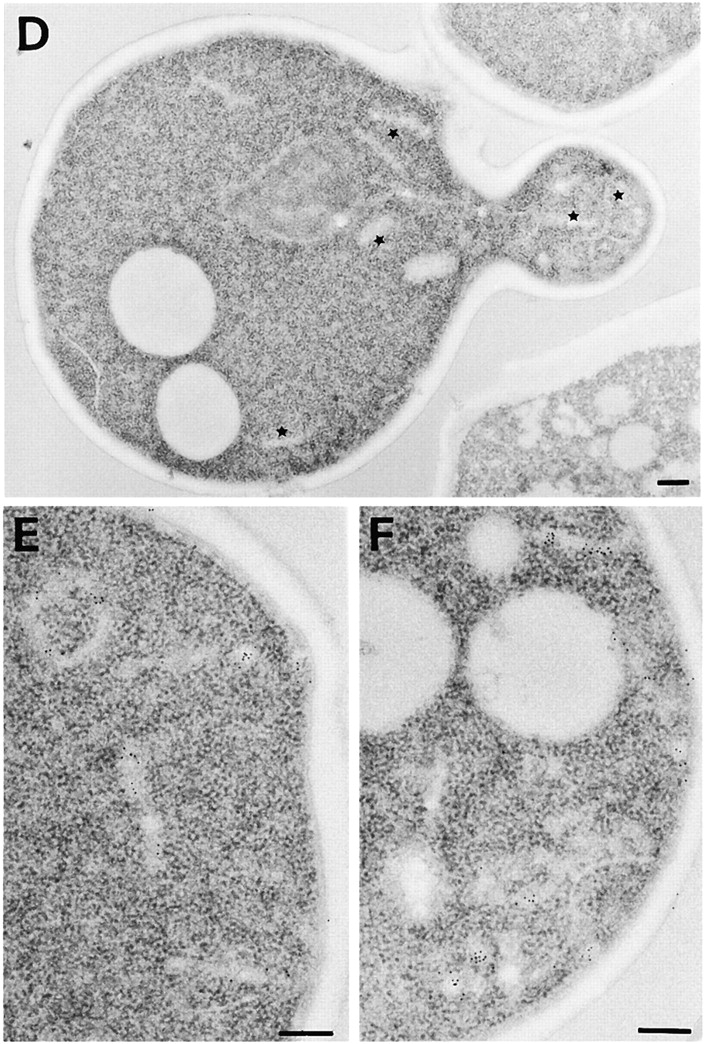
During the characterization of the enlarged compartment present in the mutant cells, an antibody raised against the t-SNARE Tlg1p (Holthuis et al. 1998a) was found to label the enlarged cisternae strongly (Fig. 2). Tlg1p labeling was specific for these compartments because no immunogold labeling was observed on other membranes such as ER, mitochondria, plasma membrane, or the vacuole. In wild-type cells, Tlg1p was present on small cisterna and vesicles. In the absence of Tlg1p labeling, these compartments are difficult to precisely identify in wild-type cells. In contrast to the compartment observed in mutant cells (Fig. 2, A–C), the Tlg1p-positive wild-type compartments lacked a large lumen (Fig. 2, D–F).
Tlg1p has been shown previously to localize to Golgi and/or endosomal membranes (Holthuis et al. 1998a). An antibody recognizing the cis-Golgi t-SNARE Sed5p (Banfield et al. 1994; Pelham 1999) revealed that this marker for early Golgi membranes was not present on the enlarged compartment of the rcy1Δ mutant cells (data not shown). Together, these data suggest that the Tlg1p-positive, aberrant compartment may be either of late Golgi complex or endosomal origin.
The rcy1Δ Mutant Has a Traffic Defect from an Endocytic Compartment to the Vacuole
We speculated that the enlargement of this compartment and the reduction in accumulation of LY in the vacuole in rcy1Δ cells is the result of a defect in endocytosis. To address which step in endocytosis is affected, the traffic of two well characterized endocytic markers, pheromone α-factor and the plasma membrane protein uracil permease (Fur4p), were followed along the endocytic pathway (Fig. 3). The uptake of radiolabeled α-factor and the disappearance of uracil uptake activity from the cell surface were used as measures for the internalization step of endocytosis. Both markers were internalized at similarly rapid rates in rcy1Δ and wild-type cells (Fig. 3A and Fig. B). It has been shown previously that once internalized, the two markers are further transported to the vacuole (Singer-Kruger et al. 1993; Galan et al. 1996). Transport from endosomal membranes to a proteolytically active compartment can be monitored by following degradation of α-factor or the uracil permease. When compared with wild-type cells, degradation was delayed for both markers in the rcy1Δ mutant cells (Fig. 3C and Fig. D). The disappearance of intact α-factor and the decrease of uracil permease signal was quantified (Fig. 3E and Fig. F, respectively). Based on these results, endocytosis is most likely inhibited at a postinternalization step in rcy1Δ cells.
Figure 3.

Degradation of α-factor pheromone and the plasma membrane protein uracil permease is defective in the rcy1Δ mutant cells. Internalization (A and B) and degradation (C–F) of α-factor (A, C, and E) and uracil permease (B, D, and F) was studied. (A) Internalization of α-factor was followed as the ratio of internalized radio-labeled α-factor (resistant to pH 1.1 wash) divided by the total cell-associated cpm (internalized plus surface receptor-bound α-factor retained after pH 6 wash). rcy1Δ cells (solid line) internalized [S35]α-factor at rates similar to wild-type cells (dashed line). (B) Transport of [3H]uracil into yeast cells was used as a measure for the relative amounts of uracil permease exposed at the cell surface. Upon addition of cycloheximide (100 μg/ml), uracil permease levels at the cell surface decreased. Uracil uptake is plotted as the percentage activity at the initial timepoint. The decrease of uracil transport over time is identical in the rcy1Δ (solid line) and wild-type cells (dashed line). (C) Degradation of α-factor. Cells were allowed to take up radiolabeled α-factor as described in A. Membrane traffic was stopped and cells were washed in cold buffer, pH 1.1, at the indicated timepoints in minutes. Cells were extracted and cell-associated radioactivity was analyzed by TLC and autoradiography. The disappearance of intact (i) α-factor was quantified (E). In wild-type cells, degraded α-factor (d) was already observed 20 min after internalization at 24°C had been initiated. (D) Degradation of uracil permease. At the indicated times after addition of cycloheximide (100 μg/ml), total proteins were extracted. The intensity of the uracil permease band as detected on the Western blot was quantified and the values presented as the relative amounts of the signal before adding cycloheximide (F). (E and F) Wild-type (dashed line) and rcy1Δ (solid line).
In addition to these quantitative endocytic assays, we also followed the α-factor receptor Ste2p during endocytosis at the single cell level by immunofluorescence, to determine whether Ste2p travels to the enlarged compartment of rcy1Δ mutant cells. Consistent with previous studies (Hicke et al. 1997), in the absence of α-factor, Ste2p is mainly present at the plasma membrane in wild-type cells (Fig. 4 A, right column). After ligand binding, Ste2p endocytosis is strongly stimulated (Hicke et al. 1998). The receptor first localizes to peripheral punctuate structures (Fig. 4 A, 4 min) and then accumulates in larger structures close to the vacuole at later timepoints (16 min). Strikingly, in the rcy1Δ mutant cells, Ste2p was localized during the entire timecourse to a large compartment that did not resemble the compartments observed in the wild-type cells (Fig. 4 A, left column). This compartment was present in ∼40% of the rcy1Δ cells (Fig. 4 B). Smaller Ste2p-positive compartments were also observed but could not be distinguished from the Ste2p-positive compartments that can be seen in wild-type cells after the addition of α-factor. In agreement with the localization of LY-positive membranes (Fig. 1), the most intensely labeled Ste2p-positive compartments were found in the bud, close to the forming bud, and bud neck (Fig. 4A and Fig. C). We conclude that Ste2p is transported to an endosomal compartment and that traffic out of this compartment to the vacuole is reduced, leading to Ste2p accumulation there in the rcy1Δ strain. The fact that Ste2p was already observed in this internal compartment in the absence of α-factor indicates that constitutive (not induced by ligand) endocytosis of Ste2p is sufficient to lead to accumulation of the receptor in this compartment. The rate-limiting endocytic step in the rcy1Δ mutant cells appears to be transport out of this endosomal compartment. Despite the accumulation of internal Ste2p, the receptor was still in sufficient quantity at the cell surface to mediate normal internalization of α-factor (Fig. 3 A), and can be seen at the cell surface at early timepoints (Fig. 4 A, 0 min and Fig. 4 C). An alternative interpretation for the presence of Ste2p on the enlarged compartment of rcy1Δ cells would have been missorting of plasma membrane proteins during secretion. To address this possibility, double immunofluorescence of Ste2p with the plasma membrane ATPase (Pma1p) was performed. Pma1p colocalized with Ste2p on the plasma membrane but not on intracellular compartments of the mutant cells (Fig. 4 C). The absence of Pma1p on these membranes favors the view that plasma membrane proteins traffic normally to the cell surface and that Ste2p was present on the cell surface before its presence on intracellular membranes. The absence of Pma1p from this compartment is probably due to its very long half-life (>10 h) at the plasma membrane (Benito et al. 1991).
Figure 4.
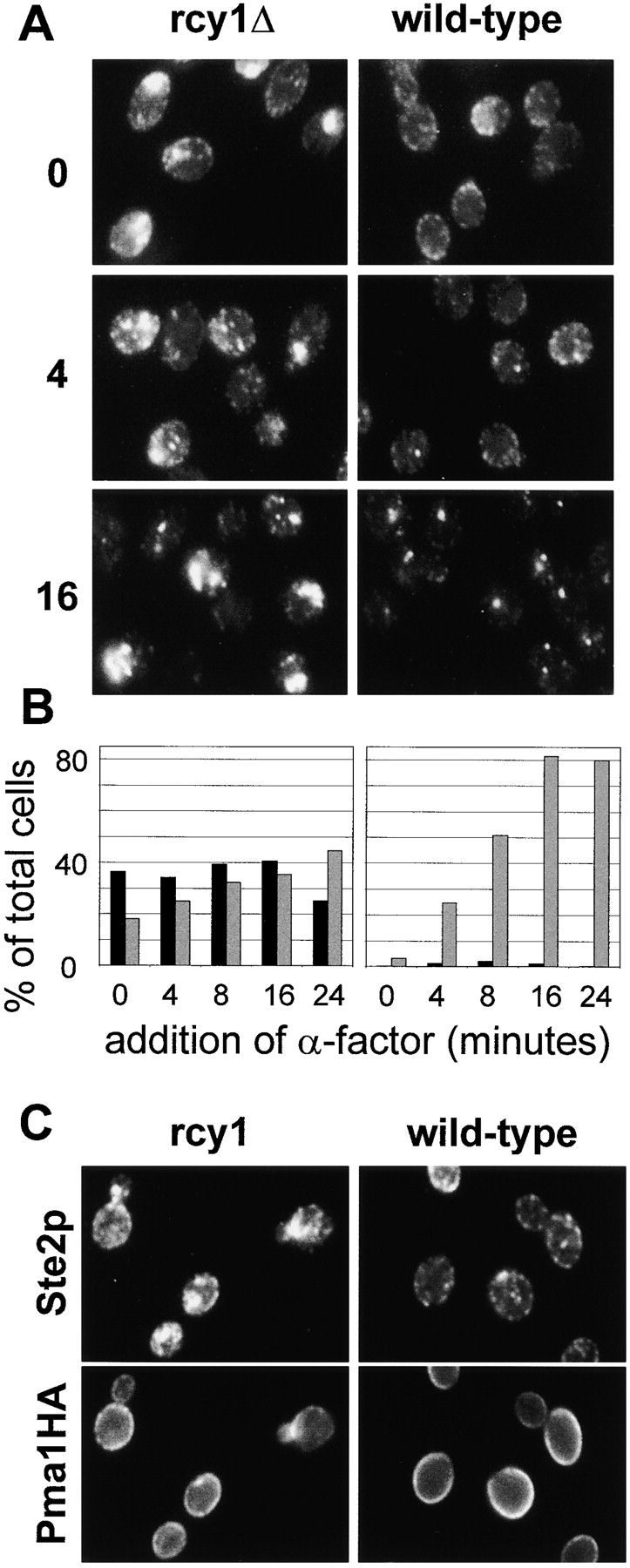
Ste2p traffic is blocked in an endocytic compartment. (A) Ligand-induced endocytosis of Ste2p was followed over time. rcy1Δ (left column) and wild-type cells (right column) were exposed to α-factor (10−7 M) for the indicated times in minutes. Cells were fixed and stained using a polyclonal antibody against Ste2p. Note the large Ste2p-positive membranes in the rcy1Δ strain. (B) For comparison of rcy1Δ (left) and wild-type strains (right) the cells were quantified for the presence of Ste2p-positive compartments. The sizes of Ste2p-positive compartments were judged by immunofluorescence (not real size of compartments). The percentage of total cells with Ste2p-positive structures with an apparent size >1 μm at its largest diameter is represented by the black bars. The grey bars show the percentage of cells with smaller round Ste2p-positive compartments of ∼0.5–1 μm in diameter. (C) Pma1p does not colocalize to the internal Ste2p-positive membranes. A wild-type strain (right panels) and the rcy1Δ mutant strain (left panels) expressing a genomic hemagglutinin-tagged version of Pma1p were labeled for Ste2p as in A (top panels) and Pma1HA using a monoclonal anti-hemagglutinin (bottom panels) by double immunofluorescence. No cross-bleeding was observed when either of the two primary antibodies was omitted (not shown).
In summary, the rcy1Δ mutant cells can internalize endocytic markers normally but are defective in a subsequent step of the pathway leading to the delivery of the uracil permease and α-factor to the vacuole. The defect correlates with the enlargement of an endocytic compartment in which the α-factor receptor accumulates.
vps and Secretion Is Normal in the rcy1Δ Mutant Cells
As the endocytic and vacuole biogenesis pathways have been shown to overlap (Piper et al. 1995), we investigated a possible vps defect in the rcy1Δ mutant strain. To this end, the maturation of the vacuolar protein CPY was monitored. In the Golgi, oligosaccharides are added onto the core glycosylated ER form of CPY (p1CPY,) to form p2CPY. Upon arrival in the vacuole, p2CPY is proteolytically processed to give rise to the mature form of CPY (mCPY) (Bryant and Stevens 1998). Maturation of CPY was followed in pulse-chase experiments and subsequent immunoprecipitation. Analysis of the mutant cells showed that transport from ER to Golgi and further on to the vacuole occurs at normal rates (Fig. 5 A).
Figure 5.

CPY targeting and invertase secretion are normal in the rcy1Δ mutant. (A) Maturation of CPY occurs at a normal rate in rcy1Δ mutant cells. Wild-type (left lanes) and rcy1Δ (right lanes) cells were pulsed for 4 min with 150 μCi/ml of [35S]methionine/cysteine labeling mixture and chased for the indicated times (see Materials and Methods). CPY was immunoprecipitated from total protein extracts. The ER form of CPY (p1CPY) and the Golgi form of CPY (p2CPY) disappear in both strains at approximately equal rates. (B) Like wild-type cells (WT), rcy1Δ cells do not secrete CPY. Secretion of CPY was assayed on nitrocellulose filters. vps mutants secrete a fraction of CPY, which binds to nitrocellulose filters and can be detected by probing with a CPY-specific antibody. Due to inefficient sorting of CPY in the vps mutant (vps1) and the class E vps mutant (vps4), a fraction of CPY is secreted, which results in a strong signal on the membrane. (C) Invertase secretion occurs at normal rates from rcy1Δ cells. Cells grown in YPUAD (2% glucose) were washed and resuspended in a medium containing 2% sucrose and 0.05% glucose at time 0. Samples were taken at different timepoints after this induction and assayed for secreted invertase activity (see Materials and Methods). Both the onset and the extent of invertase secretion from rcy1Δ cells (solid line) and wild-type cells (dashed line) were similar.
Some class E vps mutants are capable of maturing CPY at nearly normal rates, despite a defect in the vps pathway, because vacuolar proteases are matured in the class E compartment. However, class E mutants secrete a significant fraction of CPY into the medium (Raymond et al. 1992). Secreted CPY can be detected using a colony blot assay (see Materials and Methods). In contrast to the two vps mutants tested as positive controls, including a class E mutant (vps4/end13), rcy1Δ cells did not secrete significant amounts of CPY (Fig. 5 B). We conclude that disruption of RCY1 does not affect vps. As traffic from the PVC to the Golgi is required for proper sorting of CPY (Piper et al. 1995), the rcy1Δ strain probably shows no defect in this transport step. The only difference we noted using CPY as a marker was that it migrated at a slightly lower apparent molecular weight on SDS-PAGE gels, which could be the result of reduced glycosylation of the protein (Fig. 5 A). However, the cause of this phenotype is not clear, because the apparent molecular weight of invertase was normal (data not shown).
To determine whether protein secretion to the plasma membrane is affected in rcy1Δ cells, secretion of the periplasmic enzyme invertase was followed. Invertase cleaves sucrose, and its expression is strongly induced when yeast cells are switched from medium containing high levels of glucose to a medium containing sucrose as the main carbon source. In secretory mutants, invertase secretion is inhibited and the enzyme accumulates in the lumen of secretory compartments. Extracellular invertase activity can be measured from intact cells once the enzyme is secreted into the periplasm. Total invertase activity can be measured after lysis of the cells. The rcy1Δ mutant and congenic wild-type cells secrete invertase at the same rate (Fig. 5 C). In several independent experiments the onset of secretion was at ∼20 min at 24°C. Internal invertase, as determined by subtracting secreted invertase activity from total invertase activity, did not accumulate in the rcy1Δ cells (data not shown). Taken together, we conclude that rcy1Δ cells do not have major defects in the secretory or vps pathways.
Defects of FM4-64 Endocytosis in rcy1Δ Cells
To study endocytosis in the rcy1Δ strain, we also used the fluorescent dye FM4-64. This dye binds to membranes and its fluorescence is greatly enhanced in lipid environments (Betz et al. 1996). Due to these properties, FM4-64 has been used in yeast to label vacuoles and smaller endocytic membranes (Vida and Emr 1995; Holthuis et al. 1998b). Wild-type and rcy1Δ mutant cells were incubated with FM4-64 for 10 min, then washed, and subsequently incubated for up to 30 min. After the 10-min pulse of FM4-64, the dye labeled small punctate endocytic membranes in both wild-type and mutant cells at approximately equal intensities (Fig. 6 A) (Holthuis et al. 1998b). During the chase, the dye moved to the vacuolar membrane in wild-type cells (Fig. 6 A) (Vida and Emr 1995). In rcy1Δ cells, FM4-64 was less efficiently transported to the vacuole (Fig. 6A and Fig. B). Strikingly, when the two strains were compared at later timepoints, the intensity of the fluorescence signal decreased greatly for wild-type cells, but remained very strong in the rcy1Δ mutant cells (Fig. 6). These results suggest that the reduction of fluorescence intensity in wild-type cells over time could be the result of resecretion/recycling of the dye out of the cell. If this hypothesis were correct, rcy1Δ mutant cells might be defective in a recycling pathway and would therefore retain FM4-64 in an endosomal compartment distinct from the vacuole (Fig. 6 B).
Figure 6.
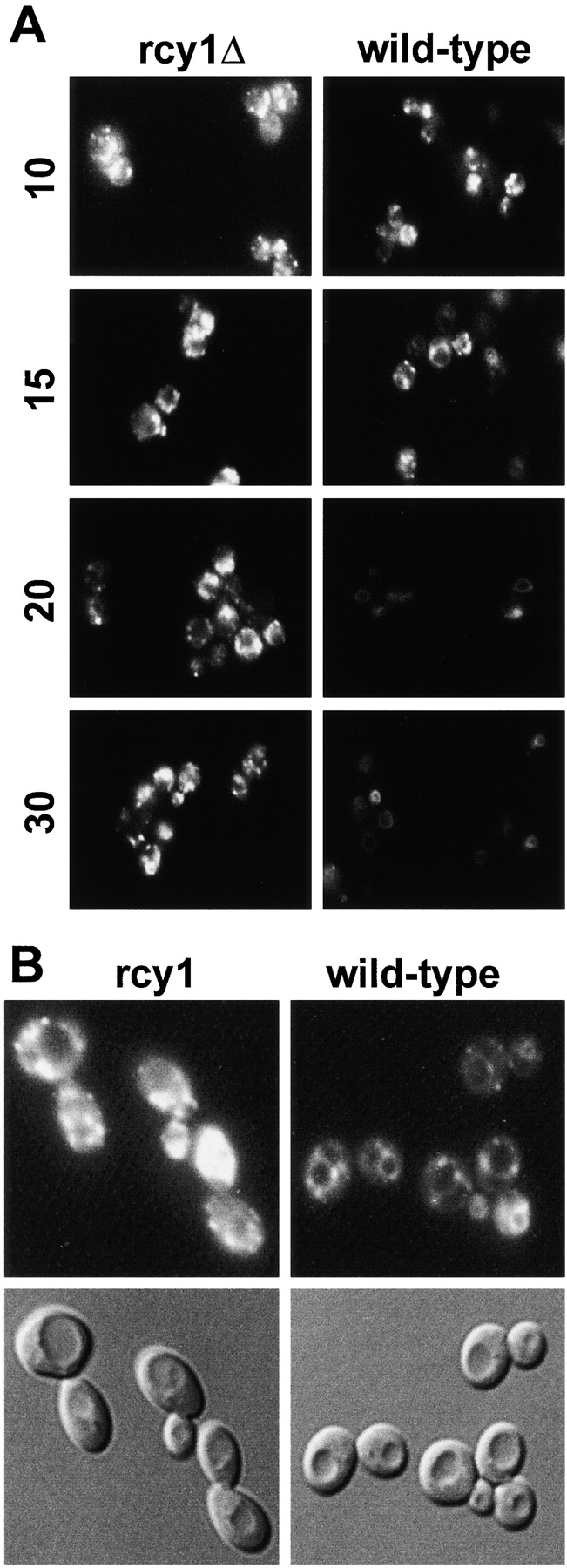
Differences of FM4-64 traffic in rcy1Δ (left) and wild-type (right) strains. (A) Cells were incubated for 10 min in YPUAD in the presence of 20 μM FM4-64. The cells were then washed in fresh medium to remove the surface-bound dye and further incubated. Samples were taken and stopped in cold buffer containing 10 mM NaN3 and 10 mM NaF. The times on the left of the figure indicate the minutes after addition of the dye. The cells were visualized using fluorescence optics. (B) For comparison of vacuolar FM4-64 staining (top), Nomarski images (bottom) are shown for the 20-min timepoint.
Quantitative, Real-time Measurements of Recycling in Yeast
Two additional previous findings suggested that recycling may be affected in rcy1Δ mutant cells. First, the enlarged compartment of rcy1Δ mutant cells was always located close to the site of cell growth (Fig. 1 and Fig. 2). Second, Tlg1p, which appears to be required for the recycling of Chs3p back to specific sites along the plasma membrane (Holthuis et al. 1998b) is localized in this compartment. We therefore attempted to address the question directly of whether recycling of endocytosed material back to the plasma membrane is affected in the mutant.
In the neurosciences, the dye FM1-43 has been used to study synaptic vesicle recycling at the single cell level (Betz and Bewick 1992). FM1-43 belongs to the same family of styryl dyes as FM4-64. These styryl dyes are only weakly fluorescent in water and their quantum yield increases several hundred-fold when bound to lipids (Betz et al. 1996). Fusion of synaptic vesicles and release of the dye into the media therefore leads to a decrease in total fluorescence. In the case of S. cerevisiae, it is more convenient to follow this process from a whole cell population. We developed an assay (see Materials and Methods) in which recycling of FM4-64 can be followed. Cells are allowed to take up FM4-64 for 5–15 min (uptake: 12 min in the results presented in Fig. 7, Fig. 8, and Fig. 9). The cells were then washed at 4°C to remove surface-bound dye and to prevent further membrane traffic. Subsequently, the cells were resuspended in warm medium and the fluorescence associated with the cells was measured continuously in a spectrofluorometer (Fig. 7, Fig. 8, and Fig. 9).
Figure 7.

Measuring recycling in yeast. Cells were allowed to take up FM4-64 (40 μM) for 12 min as described in Fig. 6. The cells were then washed in ice-cold SD minimal medium. Thoroughly washed cells were resuspended in SD medium at 24°C. FM4-64 fluorescence in the cell suspension was measured over time in a spectrofluorometer (see Materials and Methods). (A) Fluorescence associated with wild-type cells (black line) decreases over time. This decrease was blocked when the cells were resuspended in SD medium containing NaN3 and NaF (grey line).
Figure 8.
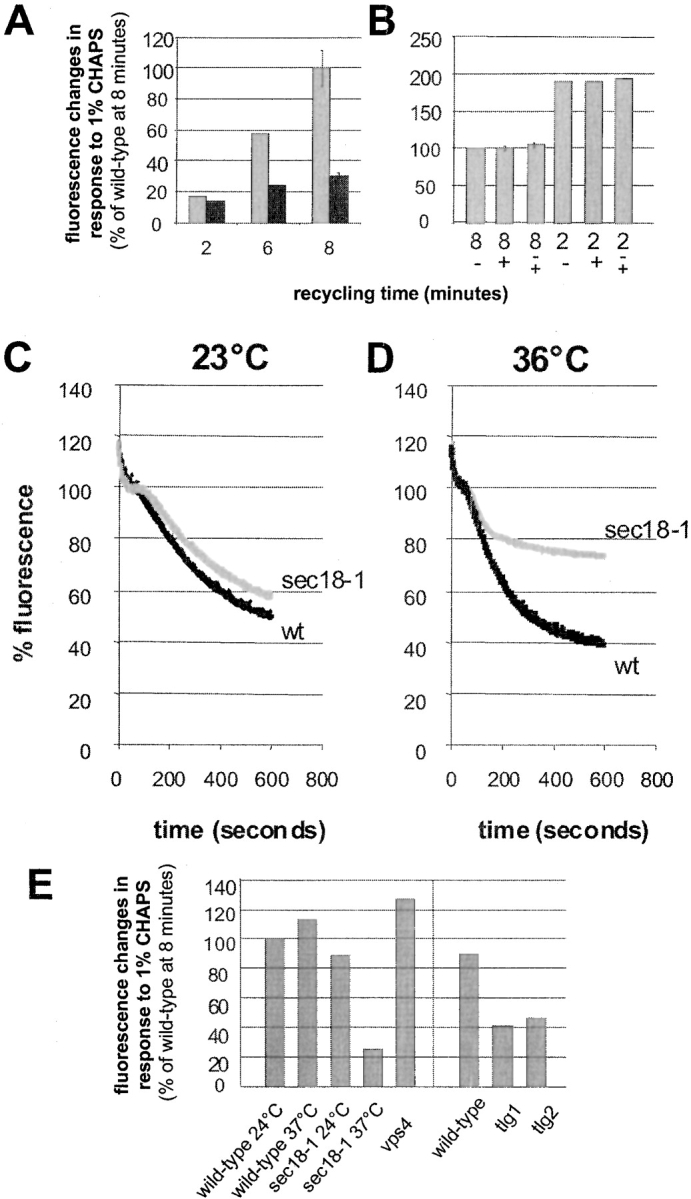
FM4-64 is recycled into the medium in a SEC18-dependent manner. (A) Wild-type cells (grey bars) or rcy1Δ cells (black bars) were loaded with FM4-64 at 24°C, washed, and incubated for the indicated time. Relative fluorescence changes seen after addition of 1% CHAPS were quantified. The fluorescence change seen after 8 min with wild-type cells was set to 100%. (B) Aliquots of recycling cells prepared as in A were treated with (+) or without (−) 1% CHAPS for 1 min. Then, 10 mM sodium azide and 10 mM sodium fluoride were added. After centrifugation, the fluorescence inside the cells was measured and divided by the cell number. After resuspension of cells that did not receive CHAPS (−) in fresh medium, 1% CHAPS was added (−/+). No increase in total fluorescence was observed upon addition of CHAPS under these conditions. In agreement with continuous measurements (Fig. 7), the cell pellet analyzed at 2 min contained about twice as much fluorescence as the cells that had recycled for 8 min. The fluorescence in the pellet of wild-type cells at 8 min was set to 100%. (C and D) Wild-type (black lines) and sec18-1 (grey lines) mutant cells were resuspended in SD medium prewarmed to 23°C (B) or 36°C (C). At 36°C, the recycling of FM4-64 was strongly inhibited in the sec18-1 strain compared with the wild-type. (E) Quantitation of the fluorescence changes in response to 1% CHAPS of the different mutants used in this study. The average of two independent experiments is shown for each mutant and condition. The fluorescence change due to addition of CHAPS seen after 8 min with wild-type cells was set to 100%. Standard error bars are indicated where more than two independent experiments have been performed.
Figure 9.
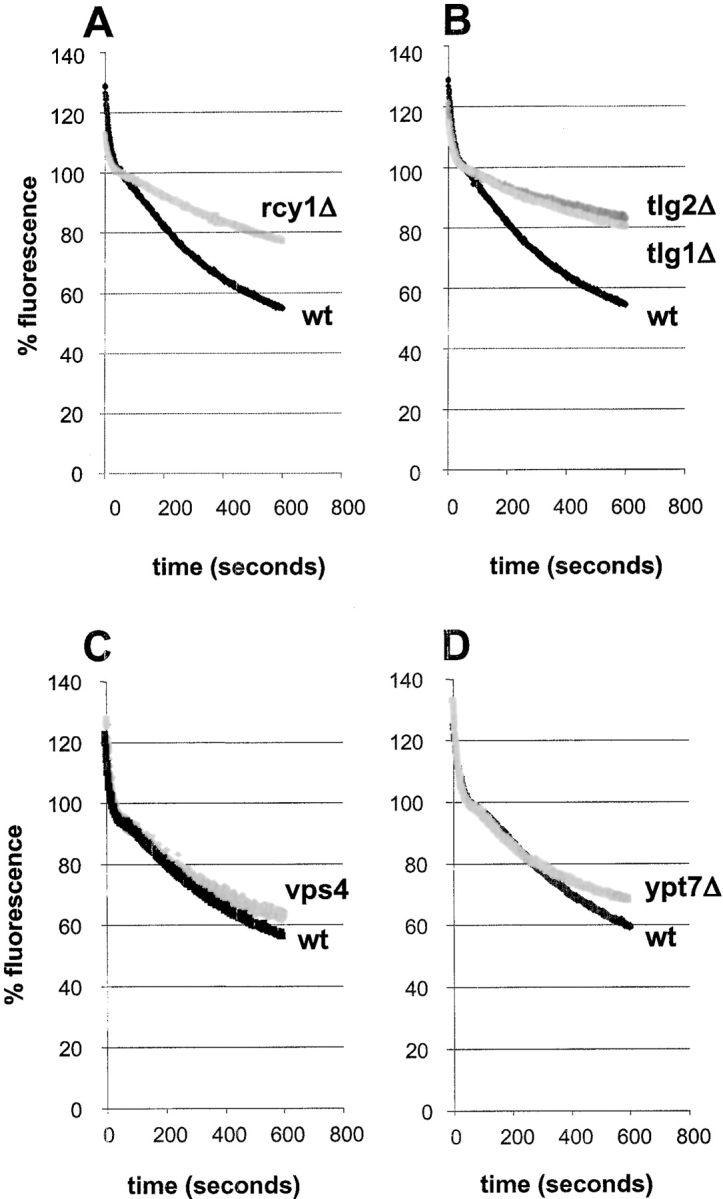
FM4-64 recycling is inhibited in rcy1Δ, tlg1Δ, and tlg2Δ cells. All FM4-64 recycling measurements were done as described in Fig. 7. (A and B) Recycling of FM4-64 at 24°C is strongly inhibited in rcy1Δ (A, grey line), tlg1Δ (B, grey line), and tlg2Δ (B, dark grey line) compared with their corresponding wild-type (black lines) cells. vps4 (C, grey line) and ypt7 (D, grey line) recycle FM4-64 at similar rates as the corresponding wild-type cells (black lines).
In wild-type cells, the fluorescence changes observed can be divided into several steps. Fluorescence decreases very rapidly for the first 30 s. These early changes most likely reflect dissociation of surface-bound FM4-64 that was not removed during the washes at 4°C, because addition of metabolic poisons (see below and Fig. 7, wt + N3 and F−) to deplete intracellular ATP did not inhibit this decrease. In a second phase (30–90 s), total fluorescence remained more or less constant. After this variable lag phase, a decrease in fluorescence occurred for up to 10 min after warming the cells. Fluorescence decreased to ∼55% of the initial value (determined at 50 s) by 10 min (Fig. 7, Fig. 8, and Fig. 9). This process is rapid. The slope of fluorescence decrease is largest between 3 and 5 min after the release has been started at 24°C and corresponds to ∼8% of the internalized FM4-64 per minute under these conditions in the wild-type strain (Fig. 7). The decrease that was observed between 2 and 10 min after resuspending the cells would fit the prediction that cells which have taken up FM4-64 secrete a large proportion of the dye back into the medium, similar to the data obtained for recycling synaptic vesicles at the nerve terminal. Alternatively the fluorescence decrease could also be due to the destruction of the fluorophore or its quenching in the vacuolar system.
To rule out the latter alternatives we used the detergent CHAPS, which has been used previously to detect low concentrations of FM4-64 in solution. As little as 1 nM of FM4-64 could be detected in our medium in the presence of 1% CHAPS, whereas in the absence of the detergent, 1 μM of the dye was essentially nonfluorescent (data not shown). Cells were prepared for the recycling assay as described above, except that the cells were washed at 4°C until no fluorescence could be detected in the wash medium after adding CHAPS. The cells were then resuspended in prewarmed medium and the fluorescence of these cell suspensions was measured after 2 or 8 min. CHAPS was then added to the cell suspension at each timepoint to a final concentration of 1%. CHAPS addition led to an immediate increase of the fluorescence signal (Fig. 8 A). This increase corresponds to ∼70% of the fluorescence changes observed during a continuous recycling measurement. This result indicates that the fluorophore was not destroyed during the recycling experiment because its fluorescence could be recovered by adding CHAPS to the medium. The fluorescence increases in response to CHAPS were quantified and shown to be time-dependent (Fig. 8 A). To control that CHAPS allowed only detection of secreted dye and did not extract dye from the cells nor relieve intracellular quenching of fluorescence, two further control experiments were performed. Cells that had previously internalized FM4-64 and were allowed to recycle the dye for 2 or 8 min were washed and then incubated in the presence or absence of 1% CHAPS. After centrifugation, the cell pellets from CHAPS-treated or untreated cells contained equal amounts of fluorescence showing that CHAPS was unable to extract FM4-64 from the inside of yeast cells (Fig. 8 B). Furthermore, if cells were separated from their recycling medium and resuspended in fresh medium, the addition of CHAPS did not change the total fluorescence (Fig. 8 B). This demonstrates that CHAPS addition had no effect on the fluorescence of FM4-64 within the cells. In summary, these observations demonstrate that the addition of CHAPS to the medium can be used to obtain a relative measure of the amount of FM4-64 dye that was recycled back out of the cells.
The sec18-1 mutant was used to demonstrate that the fluorescence decrease in the continuous recycling assay depends on vesicular traffic. Sec18p is the yeast homologue of NSF (N-ethylmaleimide–sensitive factor), a protein required for many, and perhaps most, membrane fusion events in yeast (Graham and Emr 1991). For our purposes, the sec18-1 mutant was particularly useful because the mutant protein is inactivated very quickly at elevated temperature. At 23°C the changes of fluorescence of the sec18-1 strain paralleled the wild-type strain (Fig. 8 C). However, at nonpermissive temperature (36°C), the sec18-1 strain released FM4-64 for ∼3 min, then release came to an abrupt halt (Fig. 8 D). The fluorescence decrease in wild-type cells lasted longer and was clearly more pronounced (Fig. 8 D). The 3-min time period most likely reflects the time required to inactivate Sec18p function and terminate Sec18-dependent events where Sec18p had already acted. Thus FM4-64 release can be blocked by inactivation of Sec18p function, demonstrating that the decrease in fluorescence intensity is due to membrane trafficking. This assay allowed us to test directly whether different mutants are defective for recycling of FM4-64.
rcy1Δ, tlg1Δ, and tlg2Δ Mutants Are Defective in the Recycling of FM4-64
Experiments with the rcy1Δ mutant showed that recycling of FM4-64 is clearly slower than for wild-type cells (Fig. 9 A). Cell-associated fluorescence of rcy1Δ cells decreased at slow rates with an almost constant slope over the 10-min measurement. Addition of 1% CHAPS to aliquots of rcy1Δ cells led to increases of fluorescence, which changed only slowly with time. After 8 min of recycling, the changes of fluorescence observed for the rcy1Δ mutant were about threefold smaller than those observed with wild-type cells (Fig. 8 A).
As additional controls for and characterization of the assay, other viable deletion mutants that affect vesicular traffic in the endocytic pathway, vps4Δ and ypt7Δ, were examined. The vps4Δ/end13 mutant has been classified as a class E vps mutant. Mutant cells are defective for endocytosis (Munn and Riezman 1994), and accumulate a large compartment composed of stacked cisternae (Babst et al. 1997). Vps4p is probably required for traffic from the PVC back to the Golgi complex, and from the PVC to the vacuole (Piper et al. 1995; Babst et al. 1997). Ypt7p is a small GTPase that regulates traffic from late endosomes to the vacuole. Neither of these mutants had an effect on the rate of recycling by our assay (Fig. 9C and Fig. D). These results provide evidence that recycling of FM4-64 under our conditions does not require several events of the vps pathway, nor late trafficking steps of the endocytic pathway.
Previously, two t-SNARES Tlg1p and Tlg2p were shown to be involved in the recycling of Chs3p. Both, Tlg1p and Tlg2p are necessary for wild-type localization of Chs3p to the bud neck (Holthuis et al. 1998b). As Tlg1p localizes to the enlarged compartment in the rcy1Δ mutant (Fig. 2), we investigated whether recycling of FM4-64 might also be affected in tlg1Δ and tlg2Δ cells. As shown in Fig. 9, tlg1Δ and tlg2Δ cells released FM4-64 with much slower kinetics than wild-type cells (Fig. 9 B). The fluorescence decrease is very similar to that observed in the rcy1Δ cells. By measuring fluorescence before and after the addition of 1% CHAPS, it was found that recycling of the dye was about twofold reduced in both the tlg1Δ and tlg2Δ mutant. A summary of the changes induced by the addition of CHAPS to the sec18-1, vps4, tlg1Δ, and tlg2Δ is shown in Fig. 8 E.
Based on this novel assay we have identified a new recycling mutant rcy1Δ and have been able to quantify the recycling defects in this mutant as well as in tlg1Δ and tlg2Δ mutant strains. Endocytic recycling is rapid and a quantitatively important pathway.
Discussion
The rcy1Δ mutant was identified in a newly developed screen of disruption mutants for defects in accumulation of LY in the vacuole. The rcy1Δ mutant has the surprising phenotype of accumulating LY in punctuate nonvacuolar structures near sites of new membrane insertion. This LY phenotype has not been observed previously. At the ultrastructural level, an enlarged compartment was detected that is likely to correspond to the structures accumulating LY. Tlg1p was present in this compartment (Fig. 2), which allowed us, tentatively, to study the morphology of the corresponding compartment in wild-type cells. The main difference in Tlg1p-bearing structures between mutant and wild-type cells is that the elongated and sometimes horseshoe-shaped membranes in mutant cells had a larger lumenal space. The clear differences in the lumenal volume in the wild-type and the rcy1Δ mutant could explain why the fluid-phase marker LY is only detected in this compartment in mutant cells. The malformed compartment found in rcy1Δ cells does not resemble the morphology of compartments that have been shown to accumulate in sec, vps, or end mutants (Novick et al. 1980; Rieder et al. 1996; Babst et al. 1997).
Membrane Trafficking Defects in the rcy1Δ Cells
The rcy1Δ mutant has defects that involve the endocytosis and recycling pathway. Mutant cells showed normal internalization of endocytic markers, but reduced vacuolar accumulation of LY and FM4-64. Assays following the α-factor receptor and uracil permease show that endocytosis is inhibited at a postinternalization step (Fig. 3). rcy1Δ cells have no vps phenotype, indicating that the postinternalization defect in endocytosis in the rcy1Δ strain lies at a membrane trafficking step before the intersection of the endocytic and vps pathways (Fig. 10, [1]). According to this hypothesis, the enlargement of the Tlg1p-positive compartment may be the consequence of the inhibition of the endocytic pathway. An alternative interpretation would be that the enlarged compartments are derived from Golgi membranes and that the endocytic phenotype is only a secondary effect of a Golgi defect. The presence of Tlg1p on the enlarged compartment of rcy1Δ cells did not help to rule out either possibility, because Tlg1p has been shown previously to localize to late Golgi and/or endosomal membranes (Holthuis et al. 1998a; Lewis et al. 2000). However, several results argue that the absence of Rcy1p affects the endocytic pathway directly and that the enlarged compartment are endosomes. First, secretion of invertase was normal in the mutant. If the Golgi complex was enlarged in this mutant, an effect on the onset of invertase secretion would have been expected. Second, Pma1p did not localize to the Ste2p-positive compartment. If retention in the Golgi apparatus was the reason for the localization of Ste2p to the intracellular compartment, the plasma membrane protein Pma1p would have been expected to localize to these membranes as well. It is thus more likely that both proteins reach the cell surface but that only Ste2p, which has a much higher rate of constitutive endocytosis than Pma1p, is internalized efficiently enough to accumulate in this compartment. Third and foremost, this compartment can be labeled efficiently and very rapidly with externally added endocytic markers.
Figure 10.

Model for membrane traffic in the vacuolar system. (1) Traffic from the postulated sorting endosome to the PVC is inhibited in the rcy1Δ mutant. (2) Two possible pathways that contribute to recycling from the sorting endosome to the plasma membrane, one passing through the Golgi and the other directly to the plasma membrane. (3) Traffic steps blocked in class E vps mutants, which do not affect recycling. (4) Traffic from the late endosome to the vacuole, which is also not required for recycling.
In addition to this endocytic phenotype, we also demonstrate that recycling of FM4-64 is strongly inhibited in the rcy1Δ mutant. We do not know whether Rcy1p function affects both recycling and endocytosis directly (Fig. 10, [1] and [2]). It is possible that one of these two defects is the consequence of the other. For example, if recycling from the endosome is reduced, proteins destined to the plasma membrane or the late Golgi compartment would remain in this endosomal intermediate compartment. The presence of such proteins on these membranes could, in turn, prevent further maturation of the organelle or the formation of intermediate transport vesicles from this compartment. Alternatively, as described for class E mutants, two traffic steps could be affected in a single mutant (Piper et al. 1995; Bryant and Stevens 1998). In the class E mutants, transport to the vacuole and traffic back to the Golgi from the PVC are blocked (Fig. 10, [3]). In the rcy1Δ mutant cells, transport from the Tlg1p-positive compartment (early/sorting endosome) to the vacuole and traffic into the recycling pathway could both be affected directly (Fig. 10, [1] and [2]).
Measuring Recycling in Yeast
We have developed a novel recycling assay by following the endocytic marker FM4-64. The recycling process is fast, and a major fraction of endocytosed dye is recycled under the present conditions (∼50% in 10 min) (Fig. 7, Fig. 8, and Fig. 9). At this time we do not know many details about the recycling pathway in yeast. It is highly unlikely that recycling passes through late stages of the endocytic pathway because the vps4Δ (Fig. 8 C and Fig. 10, [3]) and ypt7Δ (Fig. 8 D and Fig. 10, [4]) mutants had no quantitative effect on the overall recycling efficiency. Membrane material could recycle directly from the sorting endosome to the plasma membrane and/or may first travel to the Golgi complex and be secreted by previously identified secretory vesicles (Fig. 10, [2]). Heuristically, it seems that proteins that need to be used in the secretory pathway would recycle from the sorting endosome to the Golgi for reuse. In fact, the t-SNARE Snc1p seems to recycle by this pathway and depends upon TLG1 (Lewis et al. 2000). If this pathway is used it must be distinct from the pathway for recycling from the PVC to the Golgi defined by the vps4Δ mutant.
In mammalian cells, recycling of transferrin is very rapid and occurs exclusively through endosomal compartments (Ghosh and Maxfield 1995; Sheff et al. 1999). However, no rapidly recycling proteins have been identified thus far in yeast, nor is there any evidence for nutrient uptake systems that resemble those used for transferrin. It is possible that pheromone receptors or plasma membrane permeases could follow such a rapid recycling pathway. Independent of the exact route taken, we could show that the recycling pathway(s) are blocked in the presence of azide and fluoride or in a sec18-1 mutant. This shows that all recycling depends upon membrane traffic, i.e., membrane fusion events. In the rcy1Δ, tlg1Δ, and tlg2Δ cells, recycling of FM4-64 was only partially blocked. This could reflect a partial requirement for these proteins in recycling. However, we prefer the idea of two recycling pathways, similar to the two pathways described above, only one of which would be affected in the individual mutants. It is interesting to note that even though tlg1Δ and tlg2Δ are also reduced for accumulation of LY in the vacuole, they do not show detectable LY accumulation in nonvacuolar structures like those observed in rcy1Δ cells (Seron et al. 1998; data not shown). In electron micrographs of tlg1Δ and tlg2Δ mutant cells, no enlarged compartments were observed (Holthuis et al. 1998a). These two mutants are therefore unlikely to be blocked in the same way as the rcy1Δ mutant. Furthermore, the tlg1Δ and rcy1Δ mutants are synthetically lethal (no double mutants could be found after dissection of 15 tetrads from a heterzygous diploid). One interpretation of this synthetic lethality is that both recycling pathways are defective in the double mutant and that recycling is an essential process.
Function of Recycling in Yeast
In higher eukaryotes recycling of plasma membrane proteins is used as a mechanism to protect recycling proteins from degradation and allows them to undergo repeated cycles of endocytosis and resecretion (Ghosh and Maxfield 1995; Mellman 1996). In addition, recycling is also needed to properly localize proteins. It plays an important role in the sorting of proteins to the apical and basolateral domain of polarized cells (Sheff et al. 1999). In yeast, no plasma membrane protein has been described that mediates repeated cycles of transport of ligands to the endosomal system. However, a major function of the recycling pathway in yeast may be to sort plasma membrane proteins to specialized subdomains along the cell surface as proposed for Chs1p and Chs3p localization to the birth scar or the incipient bud site respectively (Chuang and Schekman 1996; Ziman et al. 1996).
Another major function of the recycling pathway in animal cells is to retrieve plasma membrane proteins and other membrane components from the endocytic pathway (Mayor et al. 1993; Mukherjee et al. 1999). The use of FM4-64 in the yeast recycling assay described in this study should allow us to obtain a more detailed understanding of how membranes are sorted away from the endocytic pathway to be recycled. The Tlg1p-positive compartment is likely to be the site in yeast where a decision is made on the further destination of endocytosed material, i.e., whether it is recycled to the plasma membrane or transported to the vacuole. We therefore propose the name early/sorting endosome for the novel compartment described here (Fig. 10).
Motifs and Domains of Rcy1p
Rcy1p is a rather large protein of 840 amino acids in length. The primary sequence reveals that RCY1 encodes a protein that contains several interesting motifs and domains. At the very COOH terminus it has a CAAX box. This motif has been found in prenylated proteins (Omer and Gibbs 1994), where this lipid modification appears to facilitate membrane binding. The presence of a CAAX box in Rcy1p indicates that this protein may be prenylated.
At the NH2 terminus, Rcy1p contains a domain known as the F-box (Bai et al. 1996). The F-box is a motif of ∼40 amino acids in length that was found in several proteins and defined as an interaction site with the protein Skp1p (Patton et al. 1998b). Well studied members of the F-box family of proteins such as Cdc4, Grr1, and Met30 have been shown to mediate ubiquitination of substrate proteins as components of SCF ubiquitin ligase complexes (Skowyra et al. 1997; Patton et al. 1998a). Besides its described function in degradation of proteins by the proteasome, ubiquitination also plays a role in endocytosis. In yeast, ubiquitination is a signal required for the internalization of plasma membrane proteins that does not require proteasome function (Galan et al. 1996; Hicke and Riezman 1996). Our results suggest that this ubiquitination step does not depend on Rcy1p, because the internalization step of endocytosis occurs normally in rcy1Δ cells (Fig. 3A and Fig. B). For several plasma membrane proteins, including Fur4p, ubiquitination is dependent on the ubiquitin ligase, Rsp5p (Galan et al. 1996). In mammalian cells, postinternalization steps of endocytosis have been suggested to depend on ubiquitination (Jeffers et al. 1997; Strous and Govers 1999; Waterman et al. 1999). Our results suggest a similar situation in yeast, but more experiments will be necessary to address the molecular function of Rcy1p in membrane trafficking.
Two proteins from other yeast species show some sequence homology to Rcy1p. The proteins, from Schizosaccharomyces pombe (25% identity) and Yarrowia lipolytica (26% identity) (Boisrame et al. 1999), both contain a CAAX box and an NH2-terminal F-box, indicating that they could be functional homologues of Rcy1p. The strongest homology between the three proteins was found along the COOH-terminal third of the proteins, 41% identity vs. 26% identity along the entire length. Most intriguing is the COOH-terminal region of ∼270 amino acids, which is also homologous to the COOH-terminal domain of Sec10p and its human homologue (Guo et al. 1997). Since most F-box proteins identified have protein–protein interaction domains whose role is to bind substrates for targeting to the SCF–ubiquitin ligase complex (Patton et al. 1998b), the COOH terminus of Rcy1p could be of particular interest. A future study on Rcy1 protein function and possible interaction partners will be necessary to understand the mechanistic contribution of Rcy1p to membrane trafficking.
Acknowledgments
We would like to thank H.R.B. Pelham and R. Schekman for providing antibodies and strains. We also thank A. Heese-Peck and K. D'Hondt for critical reading of the manuscript, and F. Wohnsland for technical assistance during fluorescence measurements.
This work was supported by the European Community (EUROFAN II within the framework of the Biotech program BIO4-CT95-0080 to R. Haguenauer-Tsapis, including the grant for support of S. Avaro), and by grants from the Swiss National Science Foundation and the Swiss Federal Office for Education and Science (to H. Riezman).
Footnotes
Abbreviations used in this paper: CHAPS, (3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate); CPY, carboxypeptidase Y; LY, Lucifer yellow carbohydrazide; ORF, open reading frame; PVC, prevacuolar compartment; SCF, SKP1/cullin/F-box; vps, vacuole protein sorting; SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
References
- Babst M., Sato T.K., Banta L.M., Emr S.D. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J.W., Elledge S.J. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Banfield D.K., Lewis M.J., Rabouille C., Warren G., Pelham H.R. Localization of Sed5, a putative vesicle targeting molecule, to the cis-Golgi network involves both its transmembrane and cytoplasmic domains. J. Cell Biol. 1994;127:357–371. doi: 10.1083/jcb.127.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito B., Moreno E., Lagunas R. Half-life of the plasma membrane ATPase and its activating system in resting yeast cells. Biochim. Biophys. Acta. 1991;1063:265–268. doi: 10.1016/0005-2736(91)90381-h. [DOI] [PubMed] [Google Scholar]
- Betz W.J., Bewick G.S. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Betz W.J., Mao F., Smith C.B. Imaging exocytosis and endocytosis. Curr. Opin. Neurobiol. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Boisrame A., Beckerich J.M., Gaillardin C. A mutation in the secretion pathway of the yeast Yarrowia lipolytica that displays synthetic lethality in combination with a mutation affecting the signal recognition particle. Mol. Gen. Genet. 1999;261:601–609. doi: 10.1007/s004380050002. [DOI] [PubMed] [Google Scholar]
- Bryant N.J., Stevens T.H. Vacuole biogenesis in Saccharomyces cerevisiaeprotein transport pathways to the yeast vacuole. Microbiol. Mol. Biol. Rev. 1998;62:230–247. doi: 10.1128/mmbr.62.1.230-247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang J.S., Schekman R.W. Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p [published erratum appears in J. Cell Biol. 1996. 135:1925] J. Cell Biol. 1996;135:597–610. doi: 10.1083/jcb.135.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvatchko Y., Howald I., Riezman H. Two yeast mutants defective in endocytosis are defective in pheromone response. Cell. 1986;46:355–364. doi: 10.1016/0092-8674(86)90656-2. [DOI] [PubMed] [Google Scholar]
- Davis N.G., Horecka J.L., Sprague G.F., Jr. Cis- and trans-acting functions required for endocytosis of the yeast pheromone receptors. J. Cell Biol. 1993;122:53–65. doi: 10.1083/jcb.122.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V., Egerton M., Elguindi I., Raths S., Singer B., Riezman H. Yeast endocytosis assays. Methods Enzymol. 1991;194:697–710. doi: 10.1016/0076-6879(91)94051-d. [DOI] [PubMed] [Google Scholar]
- Galan J.M., Moreau V., Andre B., Volland C., Haguenauer-Tsapis R. Ubiquitination mediated by the Npi1p/Rsp5p ubiquitin-protein ligase is required for endocytosis of the yeast uracil permease. J. Biol. Chem. 1996;271:10946–10952. doi: 10.1074/jbc.271.18.10946. [DOI] [PubMed] [Google Scholar]
- Geli M.I., Riezman H. Role of type I myosins in receptor-mediated endocytosis in yeast. Science. 1996;272:533–535. doi: 10.1126/science.272.5261.533. [DOI] [PubMed] [Google Scholar]
- Geli M.I., Riezman H. Endocytic internalization in yeast and animal cellssimilar and different. J. Cell Sci. 1998;111:1031–1037. doi: 10.1242/jcs.111.8.1031. [DOI] [PubMed] [Google Scholar]
- Ghosh R.N., Maxfield F.R. Evidence for nonvectorial, retrograde transferrin trafficking in the early endosomes of HEp2 cells. J. Cell Biol. 1995;128:549–561. doi: 10.1083/jcb.128.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R.N., Gelman D.L., Maxfield F.R. Quantification of low density lipoprotein and transferrin endocytic sorting HEp2 cells using confocal microscopy. J. Cell Sci. 1994;107:2177–2189. doi: 10.1242/jcs.107.8.2177. [DOI] [PubMed] [Google Scholar]
- Gietz D., St. Jean A., Woods R.A., Schiestl R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A., Lampen J.O. Beta-D-fructofuranoside fructohydrolase from yeast. Methods Enzymol. 1975;42:504–511. doi: 10.1016/0076-6879(75)42159-0. [DOI] [PubMed] [Google Scholar]
- Graham T.R., Emr S.D. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J. Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Roth D., Gatti E., De Camilli P., Novick P. Identification and characterization of homologues of the Exocyst component Sec10p. FEBS Lett. 1997;404:135–139. doi: 10.1016/s0014-5793(97)00109-9. [DOI] [PubMed] [Google Scholar]
- Hicke L., Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Hicke L., Zanolari B., Pypaert M., Rohrer J., Riezman H. Transport through the yeast endocytic pathway occurs through morphologically distinct compartments and requires an active secretory pathway and Sec18p/N-ethylmaleimide-sensitive fusion protein. Mol. Biol. Cell. 1997;8:13–31. doi: 10.1091/mbc.8.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L., Zanolari B., Riezman H. Cytoplasmic tail phosphorylation of the alpha-factor receptor is required for its ubiquitination and internalization. J. Cell Biol. 1998;141:349–358. doi: 10.1083/jcb.141.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J.C., Nichols B.J., Dhruvakumar S., Pelham H.R. Two syntaxin homologues in the TGN/endosomal system of yeast EMBO (Eur. Mol. Biol. Organ.) J. 17 1998. 113 126a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J.C.M., Nichols B.J., Pelham H.R.B. The syntaxin Tlg1p mediates trafficking of chitin synthase III to polarized growth sites in yeast Mol. Biol. Cell. 9 1998. 3383 3397b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers M., Taylor G.A., Weidner K.M., Omura S., Vande Woude G.F. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 1997;17:799–808. doi: 10.1128/mcb.17.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubler E., Riezman H. Actin and fimbrin are required for the internalization step of endocytosis in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:2855–2862. doi: 10.1002/j.1460-2075.1993.tb05947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M.J., Nichols B.J., Prescianotto-Baschong C., Riezman H., Pelham H.R.B. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell. 2000;11:23–38. doi: 10.1091/mbc.11.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S., Presley J.F., Maxfield F.R. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J. Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Moreau V., Madania A., Martin R.P., Winson B. The Saccharomyces cerevisiae actin-related protein Arp2 is involved in the actin cytoskeleton. J. Cell Biol. 1996;134:117–132. doi: 10.1083/jcb.134.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Soe T.T., Maxfield F.R. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Biol. 1999;144:1271–1284. doi: 10.1083/jcb.144.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn A.L., Riezman H. Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeastidentification of six new END genes. J. Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Oliver S. A network approach to the systematic analysis of yeast gene function. Trends Genet. 1996;12:241–242. doi: 10.1016/0168-9525(96)30053-x. [DOI] [PubMed] [Google Scholar]
- Omer C.A., Gibbs J.B. Protein prenylation in eukaryotic microorganismsgenetics, biology and biochemistry. Mol. Microbiol. 1994;11:219–225. doi: 10.1111/j.1365-2958.1994.tb00302.x. [DOI] [PubMed] [Google Scholar]
- Patton E.E., Willems A.R., Sa D., Kuras L., Thomas D., Craig K.L., Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box protein complexes that regulate cell division and methionine biosynthesis in yeast [published erratum appears in Genes Dev. 1998. 12:3144] Genes Dev. 12 1998. 692 705a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton E.E., Willems A.R., Tyers M. Combinatorial control in ubiquitin-dependent proteolysisdon't Skp the F-box hypothesis Trends Genet. 14 1998. 236 243b [DOI] [PubMed] [Google Scholar]
- Pelham H.R. SNAREs and the secretory pathway-lessons from yeast. Exp. Cell Res. 1999;247:1–8. doi: 10.1006/excr.1998.4356. [DOI] [PubMed] [Google Scholar]
- Piper R.C., Cooper A.A., Yang H., Stevens T.H. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae . J. Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescianotto-Baschong C., Riezman H. Morphology of the yeast endocytic pathway. Mol. Biol. Cell. 1998;9:173–189. doi: 10.1091/mbc.9.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raths S., Rohrer J., Crausaz F., Riezman H. end3 and end4two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae . J. Cell Biol. 1993;120:55–65. doi: 10.1083/jcb.120.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond C.K., Howald-Stevenson I., Vater C.A., Stevens T.H. Morphological classification of the yeast vacuolar protein sorting mutantsevidence for a prevacuolar compartment in class E vps mutants. Mol. Biol. Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder S.E., Banta L.M., Kohrer K., McCaffery J.M., Emr S.D. Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant. Mol. Biol. Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seron K., Tieaho V., Prescianotto-Baschong C., Aust T., Blondel M.O., Guillaud P., Devilliers G., Rossanese O.W., Glick B.S., Riezman H. A yeast t-SNARE involved in endocytosis. Mol. Biol. Cell. 1998;9:2873–2889. doi: 10.1091/mbc.9.10.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J.A., Mol P.C., Bowers B., Silverman S.J., Valdivieso M.H., Duran A., Cabib E. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 1991;114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff D.R., Daro E.A., Hull M., Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J. Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer-Kruger B., Frank R., Crausaz F., Riezman H. Partial purification and characterization of early and late endosomes from yeast. Identification of four novel proteins. J. Biol. Chem. 1993;268:14376–14386. [PubMed] [Google Scholar]
- Skowyra D., Craig K.L., Tyers M., Elledge S.J., Harper J.W. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Springael J.Y., Andre B. Nitrogen-regulated ubiquitination of the Gap1 permease of Saccharomyces cerevisiae . Mol. Biol. Cell. 1998;9:1253–1263. doi: 10.1091/mbc.9.6.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R.M., Brodie S.E., Cohn Z.A. Membrane flow during pinocytosis. A stereologic analysis. J. Cell Biol. 1976;68:665–687. doi: 10.1083/jcb.68.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strous G.J., Govers R. The ubiquitin-proteasome system and endocytosis. J. Cell Sci. 1999;112:1417–1423. doi: 10.1242/jcs.112.10.1417. [DOI] [PubMed] [Google Scholar]
- van Tuinen E., Riezman H. Immunolocalization of glyceraldehyde-3-phosphate dehydrogenase, hexokinase, and carboxypeptidase Y in yeast cells at the ultrastructural level. J. Histochem. Cytochem. 1987;35:327–333. doi: 10.1177/35.3.3546482. [DOI] [PubMed] [Google Scholar]
- Vida T.A., Emr S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volland C., Urban-Grimal D., Geraud G., Haguenauer-Tsapis R. Endocytosis and degradation of the yeast uracil permease under adverse conditions. J. Biol. Chem. 1994;269:9833–9841. [PubMed] [Google Scholar]
- Wach A., Brachat A., Pohlmann R., Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae . Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Waterman H., Levkowitz G., Alroy I., Yarden Y. The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J. Biol. Chem. 1999;274:22151–22154. doi: 10.1074/jbc.274.32.22151. [DOI] [PubMed] [Google Scholar]
- Wendland B., McCaffery J.M., Xiao Q., Emr S.D. A novel fluorescence-activated cell sorter-based screen for yeast endocytosis mutants identifies a yeast homologue of mammalian eps15. J. Cell Biol. 1996;135:1485–1500. doi: 10.1083/jcb.135.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki R., Roganti T., Van Dyck E., de Kerchove D'Exaerde A., Foury F. Disruption and basic phenotypic analysis of 18 novel genes from the yeast Saccharomyces cerevisiae . Yeast. 1999;15:165–171. doi: 10.1002/(SICI)1097-0061(19990130)15:2<165::AID-YEA351>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Ziman M., Chuang J.S., Schekman R.W. Chs1p and Chs3p, two proteins involved in chitin synthesis, populate a compartment of the Saccharomyces cerevisiae endocytic pathway. Mol. Biol. Cell. 1996;7:1909–1919. doi: 10.1091/mbc.7.12.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]