

Abstract
Fibronectin's RGD-mediated binding to the α5β1 integrin is dramatically enhanced by a synergy site within fibronectin III domain 9 (FN9). Guided by the crystal structure of the cell-binding domain, we selected amino acids in FN9 that project in the same direction as the RGD, presumably toward the integrin, and mutated them to alanine. R1379 in the peptide PHSRN, and the nearby R1374 have been shown previously to be important for α5β1-mediated adhesion (Aota, S., M. Nomizu, and K.M. Yamada. 1994. J. Biol. Chem. 269:24756–24761). Our more extensive set of mutants showed that R1379 is the key residue in the synergistic effect, but other residues contribute substantially. R1374A decreased adhesion slightly by itself, but the double mutant R1374A-R1379A was significantly less adhesive than R1379A alone. Single mutations of R1369A, R1371A, T1385A, and N1386A had negligible effects on cell adhesion, but combining these substitutions either with R1379A or each other gave a more dramatic reduction of cell adhesion. The triple mutant R1374A/P1376A/R1379A had no detectable adhesion activity. We conclude that, in addition to the R of the PHRSN peptide, other residues on the same face of FN9 are required for the full synergistic effect. The integrin-binding synergy site is a much more extensive surface than the small linear peptide sequence.
Keywords: RGD, integrin, fibronectin type III, α5β1
Introduction
One of the best characterized integrin–ligand interactions is α5β1 binding to fibronectin (FN). The RGD sequence in the FN III domain 10 (FN10) is the key binding site for α5β1 (Pierschbacher and Ruoslahti 1984), but it also has been found that the FN III domain 9 (FN9) contributes importantly to adhesion. Although FN9 has no adhesive activity by itself, it causes an ∼100-fold increase in cell adhesion to FN via α5β1. Thus, the adhesive region in FN9 has been called the synergy site (Obara et al. 1988; Kimizuka et al. 1991; Aota et al. 1994).
The synergy site in FN9 has been mapped using a series of chimeric FN9-10 fragments in which regions of FN9 were replaced with the equivalent residues from FN8 (Aota et al. 1994). These homology scanning mutants revealed the importance of P1376-N1380 (PHSRN) to the synergy effect, and site-directed mutagenesis showed that R1379 was a key residue in the synergy site. An R1374/ R1379 double mutant also had a dramatic effect on synergy, and the authors concluded that R1374 and PHSRN were the major components of the synergy site (Aota et al. 1994). The integrin αIIbβ3 apparently uses a similar synergy site, since the peptide DRVPHSRNSIT inhibited binding of FN to this integrin (Bowditch et al. 1994).
The requirement of the synergy site for maximum adhesion to the FN via α5β1 is not absolute, however, since fully activated α5β1 binds equally well to fibronectin fragments in which the classic PHSRN synergy site was highly mutated (Danen et al. 1995). This abrogation of the need for the synergy site by integrin activation also extends to matrix assembly. FN with a synergy mutation was shown to be poorly incorporated into matrix, yet exogenous activation of integrins stimulated incorporation of the mutant FN into the extracellular matrix (Sechler et al. 1997). The synergy site has been shown to have a physiological role in Xenopus development, as an antibody against it blocked gastrulation (Ramos and DeSimone 1996).
The three-dimensional structure of the FN III domains 7–10 (FN7–10) revealed that the PHSRN site and the RGD loop are ∼35 Å apart but are on the same side of the FN molecule (Leahy et al. 1996), suggesting that both sites can contact the same integrin. The orientation and positioning of the FN III domains with respect to each other would be critical if the integrin binding includes both sites. Insertion of short (2–6 amino acid) linkers between domains 9 and 10 significantly decreased cell adhesion (Grant et al. 1997). The introduction of the linkers could result in reduced adhesion by altering the distance between the RGD and the synergy site, by allowing more random rotation of the two domains, or by a combination of these effects.
Since the PHSRN and RGD sites are separated by 35 Å, we considered the possibility that amino acids in this intervening surface, other than PHSRN, might contribute to the synergy site. Using the three-dimensional structure of FN7–10 as a guide, we identified candidate amino acids based on the following two criteria: (1) that they face in the same direction as RGD and PHSRN (toward the integrin); and (2) that they project from the protein surface. We made a series of single site-directed mutants, further combined them in pairs and triplets, and tested each of the recombinant proteins for cell adhesion.
We have found that R1379 of the PHSRN site is a major component of the synergy site, but mutating several other residues on the surface of FN9 also significantly reduced the synergy effect. Some residues had no observable effect when mutated singly, yet they reduced cell adhesiveness when combined with other mutations. In all cases, combining mutations augmented the effects of deleterious single mutations, particularly when the combination included R1379A.
Materials and Methods
Cells
K-562 cells (ATCC number CCL-243) and TS2/16 hybridomas were obtained from A. Garcia (Georgia Institute of Technology, Atlanta, GA). K-562 cells were cultured in DME supplemented with 10% calf serum and penicillin/streptomycin. TS2/16 hybridomas were maintained in DME supplemented with 10% FBS and penicillin/streptomycin. K-562 cells were activated for adhesion assays by incubating with the TS2/16 culture supernatant for 30 min at 4°C (Danen et al. 1995; Garcia et al. 1999).
Site-directed Mutagenesis, Expression, and Purification
A plasmid containing FN III domains 7–10 (FN7–10) in the pET11b vector was constructed previously (Aukhil et al. 1993; Leahy et al. 1996). This plasmid was used as a template for site-directed mutagenesis using the Quick-Change mutagenesis kit (Stratagene), as directed by the manufacturer. In brief, complementary primers containing the desired mutation were annealed to the template plasmid and the Pfu polymerase was used to replicate the template, incorporating the mutant primers. Digestion with DpnI was used to fragment the template DNA before bacterial transformation with the PCR mix. After verification of the mutant plasmid sequence, proteins were expressed and purified essentially as described previously (Leahy et al. 1996), except that chromatography on Mono S was replaced by crystallization in 0.02 M sodium formate, pH 4.35. The protein from the Mono Q was dialyzed for 1–8 h against the sodium formate, and the crystalline precipitate was collected by centrifugation. This precipitation gives the same high purity as chromatography on Mono S without the substantial losses that we realize during Mono S chromatography.
Cell Adhesion Assay
Cell adhesion to the fibronectin fragments was quantitated using the centrifugal force assay of McClay and Hertzler 1999, with a few significant modifications. In brief, the wells of a 96-well plate were coated with 20 μg/ml (high FN) or 0.8 μg/ml (low FN) solutions (in PBS) of the proteins to be tested and blocked with 10 mg/ml BSA in PBS. Activated K-562 cells were labeled with rhodamine B isothiocyanate (Sigma Chemical Co.), and 104 cells were plated into each well of the 96-well plate. The wells were filled with medium and sealed with transparent tape (3M book tape). A swinging bucket centrifuge fitted with microplate carriers was used to subject the plate to a force of 50× G for 5 min at 4°C, gently forcing the cells into contact with the substrate. The plate was immediately inverted and the same force was applied for 5 min at 4°C to dislodge the cells that did not adhere to the substrate. The inverted plate was examined by fluorescence microscopy using either a Nikon Axiophot or Axioplan microscope equipped with a Star 1 digital camera (Photometrics Inc.). A digital image of the central 44% of each well was captured and examined using the public domain NIH Image program (developed at the National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/). Thresholding and particle analysis were used to count the number of bound cells in the imaged area of each well. The number of cells bound to a set of four wells (see below) was normalized to the number of cells bound to a set of four FN7–10-coated wells on the same plate.
We used tissue culture–treated polystyrene 96-well plates (Costar #3596; Costar Corp.) for our assays since we could not get reproducibly uniform protein coating of the PVC plates that were used in the original assay. On the PVC plates, we frequently observed a bull's-eye effect: cells bound preferentially around the edge of the well and to a spot in the center, leaving a cell-free annulus between the central spot and the edge. Occasionally, we also noticed a reverse bull's-eye in our BSA-coated control wells: a cell-covered annulus between a cell-free central spot and the cell-free well edge. Both effects were exacerbated by a low protein concentration, and this unpredictably uneven cell distribution prevented any sort of quantitative analysis. On the polystyrene plates, we rarely saw this sort of annular cell distribution. Thanks to the rimmed well design of the polystyrene plates, we were able to plate cells into all 96 wells.
We noticed that the wells on the outside of the plate tended to have cells accumulate toward the outside edge, with a consequent depletion toward the inside edge. This effect can be explained by the radial distribution of G forces. In the center of the plate, the G force is precisely perpendicular to the surface of the well but, at the edges, it acquires a small component parallel to the surface and toward the outside of the plate. This is a small effect, but to eliminate the associated error, we arranged the wells so that each substrate was coated on a set of four wells, on one side of the 8-well row, and we averaged these four wells to give a set average. All data presented here represent a further averaging of triplicate or quadruplicate set averages with the exception of the data from the R1371A/R1379A double mutant that is the average of two set averages. The SEM was calculated for this average of the set averages. For each experiment, the number of cells bound to a set of FN7–10-coated wells was compared with the number of cells bound to a set of wells that were not blocked with BSA (cells bound strongly to this uncoated plastic). The number of cells bound to the high and low FN concentrations was 100 ± 2% and 71 ± 4% of the number of cells bound to the uncoated plastic.
Competitive Inhibition Assay
Our competitive inhibition assay was a slight modification of the cell adhesion assay described above. For this assay, each of the wells of a polystyrene 96-well plate was coated with 50 μl of 20 μg/ml FN7–10 in PBS, and then blocked with 10 mg/ml BSA in PBS. The soluble protein to be tested as a competitor was added to the well immediately before the activated K-562 cells. After the centrifugation steps, the wells were imaged and analyzed as above.
Gradient Sedimentation Analysis
Wild-type FN7–10, FN7–10 mutants, and standard proteins were sedimented through 5-ml linear gradients of 15–40% glycerol in 0.2 M ammonium bicarbonate (Aukhil et al. 1993). Fractions of these gradients were analyzed by SDS-PAGE.
Results
Cell Adhesion Assays
We used the centrifugation assay of McClay and Hertzler 1999 to measure adhesion because it gives a reproducible quantitative measure of receptor–ligand interactions. The forces experienced by the cells during the centrifugation assay are limited to those applied by the centrifuge, unlike the uncharacterized fluid shear forces generated by pipetting and washing during the more traditional assay. In addition, most cell adhesion assays include an incubation at 37°C, usually for 30–60 min, during which the cells spread on the adhesive substrate. The cytoskeletal coupling that occurs during spreading makes the cells more rigid, therefore, more resistant to detachment, even though the number and strength of ligand–receptor interactions may be unchanged. To avoid the effects of cell spreading, we did the adhesion assay at 4°C and completed the assay in 15 min.
Aota et al. 1994 have demonstrated by site-directed mutagenesis that R1379, in the PHSRN sequence, was a critical component of the synergy site, and demonstrated that the nearby R1374 was also important. Based on the three-dimensional atomic structure (Leahy et al. 1996), we selected these two and five additional amino acids in FN9 as likely candidates to contribute to integrin binding, based on a geometry projecting them toward the integrin (Fig. 1). Each of these residues is highly conserved in all seven species for which FN has been sequenced (Fig. 1), and we found all of them to be fully exposed to solvent beyond the beta carbon. All of our mutations replaced the side chain with the single methyl group of alanine. We began by replacing R1374 and R1379 with A, both singly and in tandem. These mutations confirmed that R1379 makes a significant contribution to the synergy effect, as R1379A reduced cell adhesion to ∼75% of that to wild-type FN7–10 (Fig. 2). R1374 makes a smaller contribution to this effect, as R1374A reduced adhesion to ∼85% of the wild type (Fig. 2). We also confirmed the observation of Aota et al. 1994 that the effects of R1374A and R1379A were additive. Adhesion to the double mutant was ∼55% of adhesion to wild-type FN7–10 (Fig. 2).
Figure 1.
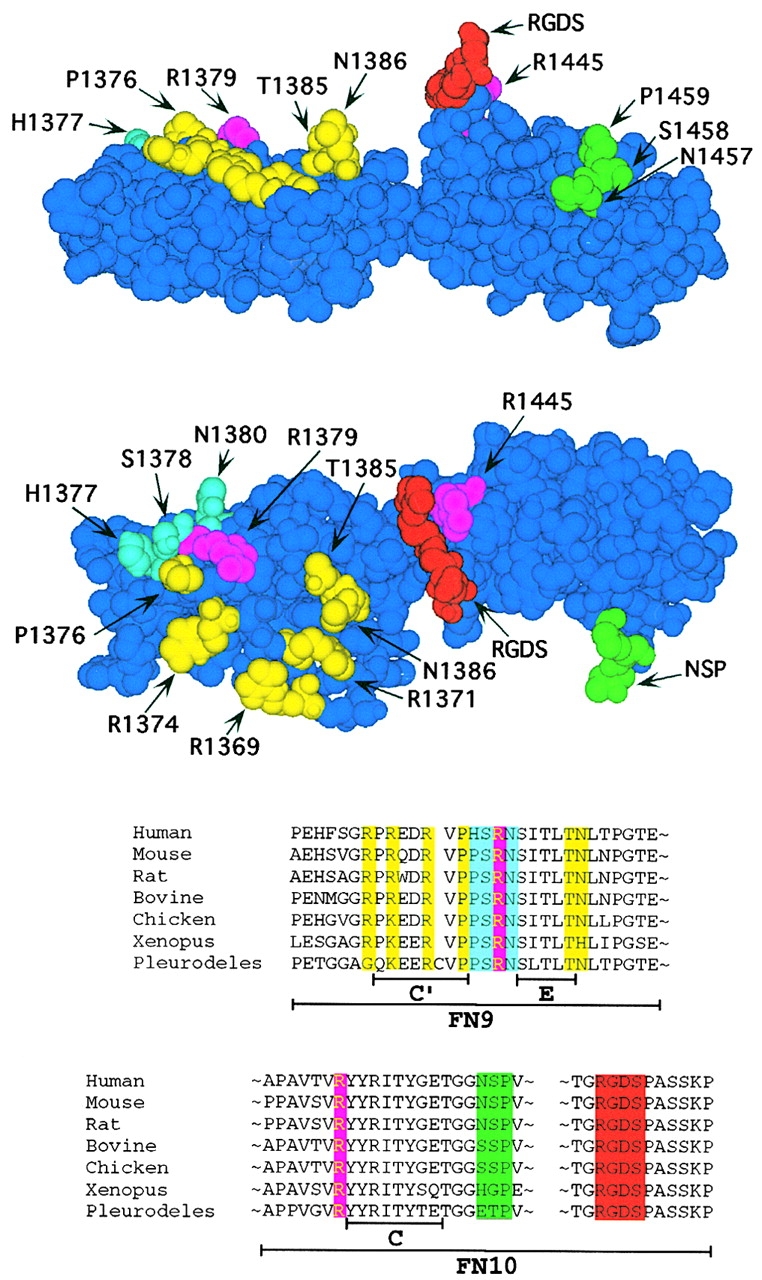
Two views of the three-dimensional structure of FN9-10 (PDB ID: 1fnf; Leahy et al. 1996) and a linear representation of the relevant regions. The RGDS segment is in red, and the two residues with the next strongest effect on adhesion (R1445 and R1379) are in magenta. The remainder of the PHSRN site is shown in light blue (mutation of these residues had little effect on adhesion activity (Aota et al. 1994)). The residues shown in yellow contribute to adhesion, but the effects of these mutations are most obvious when combined with the mutation R1379. The C-C′ loop (NSP), which does not contribute to adhesion, is colored green. A linear representation of the relevant sequences is shown below with residue coloring corresponding to that shown above. The amino acid sequence of FN is shown for the seven species from which it has been cloned.
Figure 2.

Cell adhesion to proteins with mutations in or adjacent to the PHRSN sequence. ΔRGDS and FN10 were included as controls to indicate adhesion in the absence of RGD or FN9. The average number of cells bound to each protein is given as a percentage of the number of cells attached to wild-type FN7–10. Note that in this experiment, the wells were coated with 20 μg/ml protein, which is the high concentration in later experiments.
Although the data of Aota et al. 1994 suggested that only R1379 of the PHRSN sequence was critical for the synergistic effect, all of their mutants contained a proline at either position 1376 or 1377. To test the importance of proline in the PHSRN site, we made the triple mutant R1374A/P1376A/R1379A. Cell adhesion to this mutant protein was essentially destroyed (Fig. 2). For a control, we constructed a protein in which the RGD sequence was deleted (we deleted the four amino acids RGDS, as the three-dimensional structure showed that this deletion would cause minimal distortion of the domain). We also tested FN10, a single domain protein that lacks the synergy domain. Both ΔRGDS and FN10 gave negligible adhesion (Fig. 2).
The atomic structure shows T1385 and N1386 to be the most prominent amino acids protruding from the surface of FN9 between the PHSRN loop and the RGD loop of FN10 (Fig. 1). Surprisingly, neither T1385A nor N1386A had a significant effect on cell adhesion under the assay conditions we were using, specifically to wells coated at 20 μg/ml (Fig. 3, high FN). Therefore, we constructed the double mutant, T1385A/N1386A, and found that this mutant reduced adhesion slightly, to ∼80% of the wild type (Fig. 3, high FN).
Figure 3.

Cell adhesion defects of T1385A and N1386A mutants. We tested cell adhesion to plates coated with two concentrations of wild-type and mutant FN7–10: 20 μg/ml (high FN) or 0.8 μg/ml (low FN). Adhesion to each mutant protein was normalized to the wild type at each coating concentration; adhesion to 0.8 μg/ml wild-type FN7–10 was ∼70% of adhesion to 20 μg/ml. The low concentration of FN gave a more sensitive indication of adhesion defects.
In these initial assays, the wells were coated at 20 μg/ml protein (high concentration). We found that a lower concentration of the coating protein gave a much more sensitive indication of adhesion defects. Fig. 3 also shows the assay with wells coated with 0.8 μg/ml of recombinant protein (low concentration). Although cell adhesion to T1385A or N1386A was indistinguishable from wild type at the high coating concentration, adhesion to T1385A or N1386A was ∼85 or 80% that of the wild-type FN7–10, respectively, at the low concentration (Fig. 3, low FN). Adhesion to the low concentration of the T1385A/N1386A double mutant was reduced to ∼30% of binding to the wild type (Fig. 3, low FN), and adhesion to the low concentration of R1379A was ∼20% that of wild type (data not shown). We also tested adhesion to an R1379A/T1385A/N1386A triple mutant; this showed 30 and 5% of adhesion to wild type at the high and low concentrations (Fig. 3).
Two other surface-exposed residues on the PHSRN face of FN9 were tested, R1369 and R1371. R1369A had no effect on cell adhesion at either high or low protein concentration (Fig. 4). R1371A showed no effect at a high concentration, but at a low concentration adhesion was reduced to 65% that of the wild type (Fig. 4). When either R1369A or R1371A was combined with R1379A, adhesion was dramatically reduced. Adhesion to either of these double mutants was 40% that of the wild type at a high coating concentration, and was completely eliminated (5% of wild type) at a low concentration (Fig. 4). Adhesion to these double mutants was comparable to adhesion to R1374A/R1379A, yet, unlike R1374A, R1369A and R1371A had little or no effect by themselves (compare Fig. 2 with Fig. 4, high).
Figure 4.

Arginine residues 1369 and 1371 were examined for their effect on adhesion. Plates were coated with 20 μg/ml (high FN) or 0.8 μg/ml (low FN) of the indicated mutant FN7–10.
In addition to the synergy residues in FN9, we tested two mutants in FN10. The bulky and charged side chain of R1445, which is distant from the RGD in the linear sequence, but immediately behind the RGD loop in the three-dimensional structure, was replaced by A. This mutation had a dramatic effect on cell adhesion, giving just 60% of binding to the wild type at a high concentration and ∼15% of wild-type binding at a low concentration (Fig. 3). We also tested the role of the loop connecting the C-C′ strands by replacing all three amino acids, NSP, with A. This mutation had no effect on adhesion, suggesting that the C-C′ loop does not make an important contact with the integrin (data not shown).
Competitive Inhibition Assays
Since competitive inhibition has been used as a sensitive indicator of the cell adhesive activity of mutant fibronectin fragments (Aota et al. 1994), we tested several of our recombinant proteins for their ability to block cell adhesion to wild-type FN7–10. For each protein, we tested four different concentrations of soluble protein, from 0.037 to 37 μM in 10-fold dilutions (Fig. 5). We found that T1385A and N1386A inhibited cell adhesion nearly as well as wild-type FN7–10, although the small decrease in competition for the mutants was consistent and probably significant. The competitive activity of R1379A, on the other hand, was ∼100-fold less than wild-type FN7–10. FN10 was equally ineffective in competition. These results confirm that replacing R1379 with A has a devastating effect on cell adhesion, while replacing T1385 or N1386 with A has only a small effect. Since R1379A was essentially inactive in the competition assay, we did not expect to see any enhancement of added mutations, and did not test the double mutants in this assay.
Figure 5.

Competitive inhibition of cell adhesion by soluble proteins. Just before the addition of cells, soluble FN10, FN7–10, or FN7–10 mutant proteins were added to wells coated with 20 μg/ml FN7–10, and the plates were subjected to the centrifugation assay.
Integrity of the Mutant Proteins
To interpret site-directed mutagenesis, it is important to know that the mutation does not simply disrupt the folding of the domain. All of the amino acids that we selected here are seen in the crystal structure to be projecting from the domain surface, and they have minimal buried surface beyond the beta carbon, so we did not expect any of the mutants to affect folding. All mutant proteins were recovered as soluble from the bacterial expression, and were monomeric as determined by sedimentation (see below). We have an important additional criterion for proper folding, in that our final purification step is the formation of a crystalline precipitate in sodium formate, pH 3.35. All of our mutant proteins crystallized identically to the wild-type protein, which is strong evidence that they have an identical three-dimensional structure.
To confirm that the mutant proteins were indeed monomeric, several were further tested by gradient sedimentation. Double and triple mutant proteins were chosen that included each of the seven mutations. The wild-type and all mutant proteins sedimented identically in fraction 8.5, about 3.5 S as determined by comparison to standards (Fig. 6). Even R1374A/P1376A/R1379A, where the proline mutation might have altered the peptide conformation, showed no alteration in sedimentation or in the crystallization step of purification.
Figure 6.

Gradient sedimentation analysis of FN7–10 mutant proteins. The indicated proteins were sedimented through 15–40% glycerol gradients, and the fractions were analyzed by SDS-PAGE. Ovalbumin (OVA, 3.5 S) and bovine serum albumin (BSA, 4.6 S) were used as standards. Lanes are labeled as follows: ST, starting material; 5–10, fractions 5–10; and M, markers (with size indicated on the lower left gel). The 30–40% recovery of protein is typical for this type of gradient because of the loss on the walls of the tubes.
In contrast to these surface amino acids, we have also made mutations of the central tryptophan residues in FN9 and FN10. Here, we made the conservative change W→F, but were still concerned that this might disrupt the structure, since the tryptophan is completely buried in the protein core, and almost totally conserved in FN-III domains. The W→F mutation in FN10 gave soluble protein that sedimented as wild type, and this protein also had wild-type adhesion activity (data not shown). However, the W→F mutation in FN9 gave a protein that was rapidly degraded. FN9 has a melting temperature around 50°C, whereas FN10 melts at 90°C (Litvinovich and Ingham 1995). We conclude that the less stable FN9 is not able to fold at room temperature after the W→F mutation, whereas the more stable FN10 folds normally even with the mutation. The instability seen for this mutant FN9 is a typical indication of misfolding, and provides a contrast to our other mutants, which are properly folded.
Discussion
One of the more general conclusions of our study is that the synergy site in FN9 is not limited to the short PHSRN sequence. The previously identified R1379 in this sequence is indeed the most important residue, but the synergy site receives contributions from several other amino acids. These are distributed over the surface of FN9, projecting from the fibronectin surface in the same direction as the RGD, and presumably making contact with the integrin. The integrin-binding interface spans at least the 41 Å from R1374 to R1445, and involves several residues between and beside these residues, forming an extensive integrin-binding surface. The integrin-binding interface may extend even farther, as we have not tested more distant residues.
In contrast to the atomic detail of the FN side of the interface, there is not much information on the structure of the integrins. Nevertheless, there are several studies that indicate that the synergy site binds primarily or exclusively to the α5 integrin subunit, and that RGD binds to both the α5 and β1 integrins. We discuss the details of these assignments below. A model showing an integrin in scale with the FN9-10 atomic model, and indicating the contacts, is shown in Fig. 7.
Figure 7.

A model for FN9-10 binding to α5β1 integrin. FN residue coloring is as in Fig. 1. The head of the integrin dimer is ∼10–12-nm-wide, based on electron microscopy studies (Carrell et al. 1985; Nermut et al. 1988), and is shown approximately to scale with FN9-10. The contact between the aspartate carboxylate of the RGD and α5 is shown in bold. The contact between the arginine guanidinium and the DDL in β1 is shown as a gray line to indicate that it lies in a plane behind the Asp/α5 contact. Although we have not determined which integrin subunit R1445 contacts, a line illustrates a possible contact with the β subunit. The contacts of the most important synergy residues are shown as solid lines, and the contributory residues are shown as dotted lines.
The integrin site that binds the synergy region has been identified primarily from antibody inhibition experiments, which showed that the synergy- and RGD-binding regions of α5β1 are relatively distant (Mould et al. 1998). This is consistent with the crystal structure of FN7–10 that shows the R1379 and the RGD are separated by ∼32 Å (Leahy et al. 1996). Antibody data also indicated that the synergy site binds to α5, primarily via the third of its seven NH2-terminal repeats (Mould et al. 1997; Burrows et al. 1999).
Several studies show that RGD binds to both the α5 and β1 integrins. Recombinant fragments of α5 containing repeats IV–VII or III–VII have been shown to bind to RGD (Banères et al. 1998, Banères et al. 2000). More specifically, these α5 integrin fragments have been shown to bind to the RGD of fibronectin via the carboxylate group of the aspartate, probably through an intermolecular metal coordination (Banères et al. 2000).
The case is even stronger for RGD binding the β integrin. Cross-linking studies, mutational analysis, and mapping of ligand-blocking antibodies show that the β1 I-like domain, ∼250 amino acids near the NH2 terminus, binds to RGD (for review see Loftus and Liddington 1997). Recent studies have refined this observation to show that β1 binds to the guanidinium of the arginine in the RGD. This binding is possibly to one of the aspartates in the highly conserved (among β integrin subunits) DDL (Banères et al. 2000).
Two points of flexibility in FN may be important for its interaction with the integrin. The solution structures of FN fragments show that the RGD loop is quite flexible (Main et al. 1992; Copie et al. 1998), allowing variation in both its distance from and orientation toward the synergy site. In addition, the crystal structure of FN7–10 showed that the interface between FN9 and FN10 is exceptionally small, suggesting that this junction may be somewhat flexible (Leahy et al. 1996). NMR solution structures provided additional evidence that this junction is flexible in solution (Copie et al. 1998). If flexibility at the FN9-10 hinge is important for adapting FN to different integrins, each integrin may bind FN with a different bend.
The bacterial protein, invasin, binds α5β1 with 100 times higher affinity than does FN, and it binds a range of other β1 integrins. Hamburger et al. 1999 determined the X-ray structure of invasin and suggested a possible mimic of FN, including an aspartate residue that might correspond to the D in RGD, and an arginine and aspartate 32 Å away that might correspond to R1379 and D1373 in the FN synergy region. However, the shapes of FN and invasin between these residues are quite different, FN being concave and invasin convex. Moreover, invasin is completely rigid. A pronounced hump of invasin comprising Y863 and Y885 is positioned similarly to the T1385/N1386 of FN, but it projects much farther. If T1385/N1386 contacts the integrin, as indicated by our data, this same interface could not accommodate the hump of invasin. Even bending FN at the FN9-10 hinge cannot bring a match to the convex surface of invasin. An alternative explanation is that invasin binds to an interface on the integrin that is different from, but partly overlapping the FN binding site.
Our guiding principle in thinking about the FN–integrin interface is that protein–protein interfaces are preformed, rigid, complimentary surfaces that fit together like a lock and key. This principle was originally proposed by Chothia and Janin 1975, and has been confirmed by the X-ray structures of numerous protein–protein complexes. For a simple protein–protein association the interface is usually a contiguous patch of amino acids, ∼20 Å in diameter, with some hot spots contributing most to the interaction (Cunningham and Wells 1993; Clackson and Wells 1995). The FN–integrin interaction may well comprise two interfaces: one for the synergy site on the α integrin subunit, and one for RGD in a cleft between the α and β integrins. The atomic structure of FN has provided important insights into that side of the interface. A complete understanding of the interaction must await a comparable atomic structure of the integrin.
Acknowledgments
We thank David McClay (Duke University) for advice on the cell adhesion assay. We are also grateful to Andres Garcia for his gift of the K-562 and TS2/16 cell lines.
This work was supported by the National Institutes of Health grant CA47056.
Footnotes
Abbreviations used in this paper: FN, fibronectin; FN7–10, fibronectin III domains 7–10; FN9-10, fibronectin III domains 9 and 10, respectively.
References
- Aota S., Nomizu M., Yamada K.M. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 1994;269:24756–24761. [PubMed] [Google Scholar]
- Aukhil I., Joshi P., Yan Y., Erickson H.P. Cell- and heparin-binding domains of the hexabrachion arm identified by tenascin expression proteins. J. Biol. Chem. 1993;268:2542–2553. [PubMed] [Google Scholar]
- Banères J.-L., Roquet F., Green M., Lecalvez H., Parello J. The cation-binding domain from the alpha subunit of integrin α5β1 is a minimal domain for fibronectin recognition. J. Biol. Chem. 1998;273:24744–24753. doi: 10.1074/jbc.273.38.24744. [DOI] [PubMed] [Google Scholar]
- Banères J.-L., Roquet F., Martin A., Parello J. A minimized human integrin α5β1 that retains ligand recognition. J. Biol. Chem. 2000;275:5888–5903. doi: 10.1074/jbc.275.8.5888. [DOI] [PubMed] [Google Scholar]
- Bowditch R.D., Hariharan M., Tominna E.F., Smith J.W., Yamada K.M., Getzoff E.D., Ginsberg M.H. Identification of a novel integrin binding site in fibronectin. Differential utilization by β3 integrins. J. Biol. Chem. 1994;269:10856–10863. [PubMed] [Google Scholar]
- Burrows L., Clark K., Mould A.P., Humphries M.J. Fine mapping of inhibitory anti-α5 monoclonal antibody epitopes that differentially affect integrin-ligand binding. Biochem. J. 1999;344:527–533. [PMC free article] [PubMed] [Google Scholar]
- Carrell N.A., Fitzgerald L.A., Steiner B., Erickson H.P., Phillips D.R. Structure of human platelet membrane glycoproteins IIb and IIIa as determined by electron microscopy. J. Biol. Chem. 1985;260:1743–1749. [PubMed] [Google Scholar]
- Chothia C., Janin J. Principles of protein-protein recognition. Nature. 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- Clackson T., Wells J.A. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Copie V., Tomita Y., Akiyama S.K., Aota S., Yamada K.M., Venable R.M., Pastor R.W., Krueger S., Torchia D.A. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regionscomparison with the human fibronectin crystal structure. J. Mol. Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- Cunningham B.C., Wells J.A. Comparison of a structural and a functional epitope. J. Mol. Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- Danen E.H., Aota S., van Kraats A.A., Yamada K.M., Ruiter D.J., van Muijen G.N. Requirement for the synergy site for cell adhesion to fibronectin depends on the activation state of integrin α5β1. J. Biol. Chem. 1995;270:21612–21618. doi: 10.1074/jbc.270.37.21612. [DOI] [PubMed] [Google Scholar]
- Garcia A.J., Takagi J., Boettiger D. Two-stage activation for α5β1 integrin binding to surface-adsorbed fibronectin. J. Biol. Chem. 1999;273:34710–34715. doi: 10.1074/jbc.273.52.34710. [DOI] [PubMed] [Google Scholar]
- Grant R.P., Spitzfaden C., Altroff H., Campbell I.D., Mardon H.J. Structural requirements for biological activity of the ninth and tenth FIII domains of human fibronectin. J. Biol. Chem. 1997;272:6159–6166. doi: 10.1074/jbc.272.10.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger Z.A., Brown M.S., Isberg R.R., Bjorkman P.J. Crystal structure of invasina bacterial integrin-binding protein. Science. 1999;286:291–295. doi: 10.1126/science.286.5438.291. [DOI] [PubMed] [Google Scholar]
- Kimizuka F., Ohdate Y., Kawase Y., Shimojo T., Taguchi Y., Hashino K., Goto S., Hashi H., Kato I., Sekiguchi K., Titani K. Role of type III homology repeats in cell adhesive function within the cell-binding domain of fibronectin. J. Biol. Chem. 1991;266:3045–3051. [PubMed] [Google Scholar]
- Leahy D.J., Aukhil I., Erickson H.P. 2.0 Å crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- Litvinovich S.V., Ingham K.C. Interactions between type III domains in the 110 kDa cell-binding fragment of fibronectin. J. Mol. Biol. 1995;248:611–626. doi: 10.1006/jmbi.1995.0246. [DOI] [PubMed] [Google Scholar]
- Loftus J.C., Liddington R.C. New insights into integrin-ligand interactions. J. Clin. Invest. 1997;99:2302–2306. doi: 10.1172/JCI119408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main A.L., Harvey T.S., Baron M., Boyd J., Campbell I.D. The three-dimensional structure of the tenth type III module of fibronectinan insight into RGD-mediated interactions. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- McClay D.R., Hertzler P.H. Quantitative measurement of cell adhesiveness Bonifacino J.S., Dasso M., Lippincott-Schwartz J., Harford J.B., Yamada K.M. Current Protocols in Cell Biology 1999. 9 John Wiley and Sons; New York: 2.1–9.2.10. [Google Scholar]
- Mould A.P., Askari J.A., Aota S., Yamada K.M., Irie A., Takada Y., Mardon H.J., Humphries M.J. Defining the topology of integrin α5β1-fibronectin interactions using inhibitory anti-α5 and anti-β1 monoclonal antibodies—evidence that the synergy sequence of fibronectin is recognized by the amino-terminal repeats of the α5 subunit. J. Biol. Chem. 1997;272:17283–17292. doi: 10.1074/jbc.272.28.17283. [DOI] [PubMed] [Google Scholar]
- Mould A.P., Garratt A.N., Puzon-McLaughlin W., Takada Y., Humphries M.J. Regulation of integrin functionevidence that bivalent-cation-induced conformational changes lead to the unmasking of ligand-binding sites within integrin α5β1. Biochem. J. 1998;331:821–828. doi: 10.1042/bj3310821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nermut M.V., Green N.M., Eason P., Yamada S.S., Yamada K.M. Electron microscopy and structural model of human fibronectin receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:4093–4099. doi: 10.1002/j.1460-2075.1988.tb03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara M., Kang M.S., Yamada K. Site-directed mutagenesis of the cell-binding domain of human fibronectinseparable, synergistic sites mediate adhesive function. Cell. 1988;53:649–657. doi: 10.1016/0092-8674(88)90580-6. [DOI] [PubMed] [Google Scholar]
- Pierschbacher M.D., Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Ramos J.W., DeSimone D.W. Xenopus embryonic cell adhesion to fibronectinposition-specific activation of RGD/synergy site-dependent migratory behavior at gastrulation. J. Cell Biol. 1996;134:227–240. doi: 10.1083/jcb.134.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler J.L., Corbett S.A., Schwarzbauer J.E. Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol. Biol. Cell. 1997;8:2563–2573. doi: 10.1091/mbc.8.12.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]