

Abstract
We find that the peripheral ER in Saccharomyces cerevisiae forms a dynamic network of interconnecting membrane tubules throughout the cell cycle, similar to the ER in higher eukaryotes. Maintenance of this network does not require microtubule or actin filaments, but its dynamic behavior is largely dependent on the actin cytoskeleton. We isolated three conditional mutants that disrupt peripheral ER structure. One has a mutation in a component of the COPI coat complex, which is required for vesicle budding. This mutant has a partial defect in ER segregation into daughter cells and disorganized ER in mother cells. A similar phenotype was found in other mutants with defects in vesicular trafficking between ER and Golgi complex, but not in mutants blocked at later steps in the secretory pathway. The other two mutants found in the screen have defects in the signal recognition particle (SRP) receptor. This receptor, along with SRP, targets ribosome–nascent chain complexes to the ER membrane for protein translocation. A conditional mutation in SRP also disrupts ER structure, but other mutants with translocation defects do not. We also demonstrate that, both in wild-type and mutant cells, the ER and mitochondria partially coalign, and that mutations that disrupt ER structure also affect mitochondrial structure. Our data suggest that both trafficking between the ER and Golgi complex and ribosome targeting are important for maintaining ER structure, and that proper ER structure may be required to maintain mitochondrial structure.
Keywords: SRP receptor, endoplasmic reticulum, cytoskeleton, mitochondria, yeast
Introduction
The ER usually consists of a polygonal meshwork of membrane tubules and sheet-like cisternae (Palade 1956). These structures are connected with one another and with the continuous membrane of the outer nuclear envelope. Much of the ER, called RER, is involved in protein translocation. It is studded with ribosomes carrying nascent chains that are transferred into or across the ER membrane as they are being synthesized. In some cells, smooth regions of the ER are found that lack ribosomes and have a different structure and function. Little is known about how the complex morphology of the ER is formed and maintained, or what role the structure plays in functions of the ER.
In mammalian cells, the ER tubule network is highly dynamic and is continually being extended to the periphery of the cell (Lee and Chen 1988; Waterman-Storer and Salmon 1998). It is rearranged by tubules sliding along one another and by new tubules emerging from preexisting tubules and fusing with others. The formation and maintenance of the ER network is thought to require microtubules (Terasaki et al. 1986; Terasaki 1990). Depolymerization of microtubules with drugs like nocodazole causes a gradual and reversible contraction of the ER to the center of the cell (Lee et al. 1989). Newly forming tubules are thought to be pulled out of existing tubules by motor proteins migrating along microtubules and/or by being dragged by the tip of polymerizing microtubules. In vitro, microtubules have also been proposed to be required for the formation of ER-like networks of membrane tubules (Dabora and Sheetz 1988). However, the in vitro formation of ER-like tubule networks in the absence of microtubules has recently been reported (Dreier and Rapoport 2000). In addition, maintenance of ER structure does not solely rely on a microtubule network, since ER tubules frequently do not coalign with it (Terasaki et al. 1986; Lee et al. 1989). In plant cells, actin filaments, rather than microtubules, are thought to be important for ER formation and maintenance (Terasaki 1990; Liebe and Menzel 1995).
Little is known about the structure of ER in Saccharomyces cerevisiae. Indirect immunofluorescence and EM have shown that the yeast ER consists primarily of perinuclear and cortical domains, with a few tubules connecting them (Matile et al. 1969; van Rijn et al. 1975; Novick et al. 1980; Rose et al. 1989; Preuss et al. 1991). Structural details of the cortical ER, which is closely apposed to the plasma membrane, remain to be elucidated. In addition, it is unclear which mechanisms or molecules are involved in keeping the ER at the cell periphery, and whether the structure of the peripheral ER remains intact during the cell cycle. Here, we show that the peripheral ER forms a dynamic tubular network throughout the cell cycle and have isolated mutants that dramatically affect ER structure.
Materials and Methods
Media, Plasmids, and Strains
The strains used are listed in Table . Media were prepared according to standard methods (Sherman 1991). The plasmid pJK59 encodes Sec63-GFP, a fusion of the S65T V163A mutant of GFP to the COOH terminus of Sec63p under the SEC63 promoter. It was constructed by amplifying SEC63 and ∼400 bp of its 5′ promoter from pPS599 (from R. Schekman, University of California, Berkeley, CA) with primers 5′-GGTCTAGACTTCAAGTTCCCAGCGAG-3′ and 5′-GGCTCGAGCTTCTGGTGATTCATCATCTTC-3′. The resulting product was cloned between the XbaI and XhoI sites of the CEN/URA3 plasmid, pJK50 (Kahana et al. 1998). The plasmid pWP1055 encodes ss-GFP-HDEL under the MET25 promoter. The DNA encoding green fluorescent protein (GFP) was amplified from pJK59 using oligonucleotides that added sequences encoding the signal sequence of carboxypeptidase Y at the NH2 terminus (5′-AGATACTCTAGAATGAAAGCATTCACCAGTTTACTATGTGG-ACTAGGCCTGTCCACTACACTCGCTAAGCCGCTAGCAAAGGAGAAGAACTC-3′) and HDEL at the COOH terminus of GFP (5′-GATATACGAATCCCAATTCGTCGTGGCAGCCGGATCCTTTGTATAGTTC-3′). The product was then cloned into the CEN/URA3 plasmid p416MET25 (Mumberg et al. 1994) between the XbaI and EcoRI sites.
Table 1.
Yeast Strains Used in This Study
| Strain | Genotype | Source |
|---|---|---|
| W303 | MATa/MATα ade2-1/ade2-1 trp1-1/trp1-1 leu2-3,-112/ leu2-3,-112 his 3-11/his 3-11 ura 3-1/ura 3-1 can1-100/can1-100 | Deshaies and Schekman 1990 |
| FY23 | MATa ura3-52 trp1Δ63 leu2Δ1 | Winston et al. 1995 |
| FY86 | MATα ura3-52 his3Δ200 leu2Δ1 | Winston et al. 1995 |
| RSY455 | MATa his4 trp1-1 leu2-3,112 ura3-52 HOL1-1 sec61-3 | Stirling et al. 1992 |
| YFP338 | MATα leu1-3, 112 ura 3-52 ade2-3 pep4-3 sec61-2 | Rose et al. 1989 |
| KF5 | MATα leu2-3, 112 ura3-521 ade2-3 pep4-3 sec61-2 ssh1::ADE2 | Finke et al. 1996 |
| RSY457 | MATa ura3-52 ade2 trp1-1 leu2-3,-112 sec65-1 | Stirling et al. 1992 |
| RSY1293 | MATa can1-100 leu2-3,-112 his3-11,-15 trp1-1 ura3-1 ade2-1 sec61::HIS3 (pDQ1 [SEC61-his6]) | Pilon et al. 1998 |
| RSY1294 | RSY1293, but sec61-32 | Pilon et al. 1998 |
| RSY1300 | RSY1293, but sec61-11 | Pilon et al. 1998 |
| RSY1303 | RSY1293, but sec61-23 | Pilon et al. 1998 |
| RSY1304 | RSY1293, but sec61-24 | Pilon et al. 1998 |
| SOY162 | W303 MATα srp102::HIS3 | Ogg et al. 1998 |
| BHY116 | MATα trp1 lys2 his3 ura3 ade2srp54::LYS2 | Hann and Walter 1991 |
| SOY36 | W303 MATα srp101::ADE2 | Ogg et al. 1992 |
| MS177 | MATα ura3-52 ade2-101 kar2-159 | Vogel et al. 1990 |
| RSY528 | MATa leu2 ura3 his4 sec62-1 | R. Schekman |
| RSY265 | MATα ura3-52 his4-619 sec13-1 | R. Schekman |
| RSY267 | MATα ura3-52 his4-619 sec16-2 | R. Schekman |
| RSY277 | MATα ura3-52 sec21-1 | R. Schekman |
| RSY281 | MATa ura3-52 his4-619 sec23-1 | R. Schekman |
| RSY1319 | MATa ura3 leu2 his3-Δ200 trp1 lys2 ade2 sec28::HIS3 | R. Schekman |
| RSY769 | MATa ura3-52 leu2-3,-112 sec27-1 | R. Schekman |
| RSY1317 | MATa ura3-52 leu2-3,-112 ret1-3 | R. Schekman |
| JSY2977 | MATa ura3-52 his3Δ200 leu2Δ1 trp1Δ63 fzo1-1 | Hermann et al. 1998 |
| YSB105 | MATa trp1Δ1 leu2 ade2 his3 ura3 lys2 mmm1-1 | R. Jensen |
| WPY152 | FY23 srp102-510 | This study |
| WPY154 | FY86 srp101-47 | This study |
| WPY153 | FY86 sec27-95 | This study |
| NY13 | MATa ura3-52 | P. Novick |
| NY3 | MATa ura3-52 sec1-1 | P. Novick |
| NY130 | MATa ura3-52 sec2-41 | P. Novick |
| NY412 | MATa ura3-52 sec3-2 | P. Novick |
| NY405 | MATa ura3-52 sec4-8 | P. Novick |
| NY17 | MATa ura3-52 sec6-4 | P. Novick |
| NY410 | MATa ura3-52 sec8-9 | P. Novick |
| NY57 | MATa ura3-52 sec9-4 | P. Novick |
| NY64 | MATa ura3-52 sec15-2 | P. Novick |
| DBY4060 | MATα his4-619 mal gal cdc48-3 | D. Botstein |
| WPY107 | MATa ura3-52 his4-619 cdc48-3 | This study |
| MLY2141 | MATα ura3-52 trp1-1 his3-11,-15 ufe::TRP1 (pUT1 [ufe1-1]) | M. Latterich |
Isolation and Characterization of Mutants with Disrupted ER Structure
A collection of 1,016 mutants with growth defects at 37°C was obtained from C. Cole (Dartmouth Medical School, Hanover, NH; Amberg et al. 1992) and transformed with pJK59. This plasmid could not be introduced into 24 of the mutants. The mutants containing pJK59 were grown at 25°C, were shifted to 37°C for 3 h, and were then examined live in a ZEISS Axioplan II fluorescence microscope using a ZEISS 100× Plan Apochromat oil immersion objective. No fluorescent signal could be detected in 27 of the mutants after the shift to nonpermissive temperature.
Microscopy
Cells expressing GFP were mounted in growth medium and examined live. For time course experiments, coverslips were sealed with Valap. Images were acquired using a DeltaVision microscope (Applied Precision Instruments) built around a ZEISS Axiovert microscope with a PXL CCD camera (Photometrics) and a ZEISS 100× Plan Apochromat oil immersion objective. All images shown were deconvolved and scaled using Delta Vision software.
To stain the actin cytoskeleton with Alexa 594 phalloidin (Molecular Probes), cells were fixed in 4% formaldehyde, 50 mM KPO4, pH 6.5, and 1 mM MgCl2 for 2 h at room temperature. They were then washed once with PBS pH 7.4 and resuspended in PBS at 10 OD600 /mL. 92.4 μL of the cell suspension was mixed with 7.6 μL of Alexa 594 phalloidin (6.6 μM stock in methanol) and incubated for 1 h at room temperature in the dark. The cells were washed three times with cold (4°C) PBS and viewed immediately.
Immunofluorescence with antitubulin antibody and rhodamine-conjugated secondary antibody (Molecular Probes) was performed as described in Pringle et al. 1991.
For Fig. 6 B, the cells were grown for 1 h in medium supplemented with 0.1 μg/ml DAPI to visualize nuclear DNA.
Figure 6.
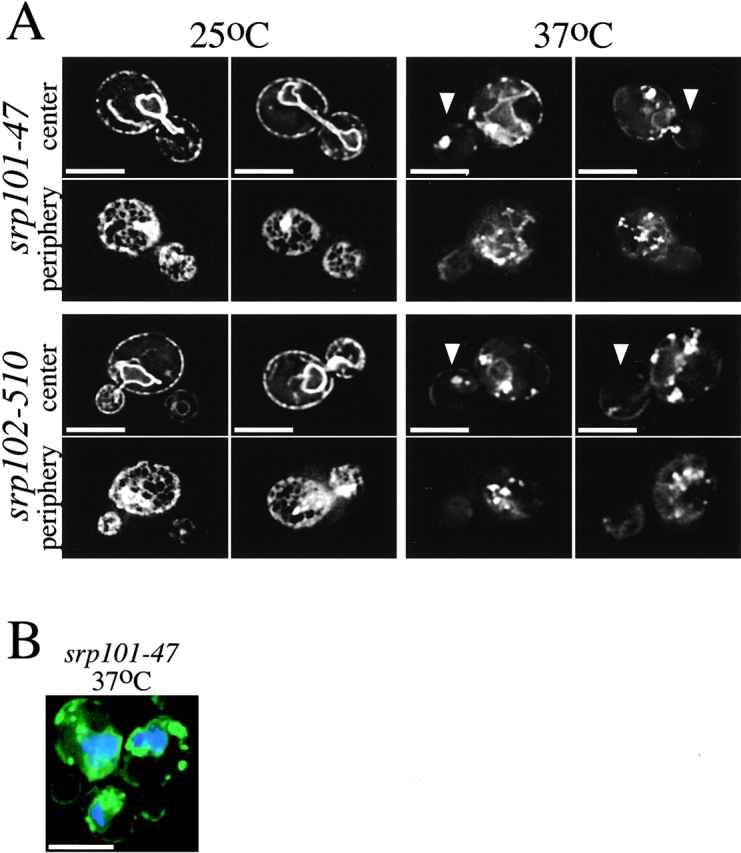
Mutants with altered ER morphology at the nonpermissive temperature. A, Images of srp101-47 and srp102-510 cells expressing Sec63-GFP grown at either 25°C or 2 h after shift to nonpermissive temperature (37°C). Arrows point to the daughter cells in some images. Images were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm. B, srp101-47 cells grown for 2 h at 37°C and stained with DAPI to visualize nuclear DNA (blue). Bar, 5 μm.
Mitochondria were stained with TMR-CH2-Cl (Mitotracker, Molecular Probes) as described in Nunnari et al. 1997.
Electron Microscopy
Cells were fixed in 1.5% KMnO4 for 20 min, poststained with uranylacetate for 4 h, and were dehydrated by incubation with 50, 70, 96, and 100% ethanol for 15 min each. The cells were then incubated in fresh 100% ethanol for 30 min and embedded in Epon 812. Ultrathin sections were cut with a diamond knife and examined in a Philips EM 400. The relative surface area of the ER was calculated with the point counting technique (Weibel and Bolender 1976). The final magnification of the micrographs was 6,000 and the point spacing of the test system was 1.5 cm.
Pulse-labeling and Immunoprecipitation of Kar2p
Pulse-labeling and immunoprecipitation were done essentially as described in Stirling et al. 1992, except that 5 OD600 units of cells were labeled for 7 min with 250 μCi of 35S protein-labeling mix (NEN Life Science Products).
Treatment of Cells with Cycloheximide, Nocodazole, and Latrunculin-A
For the experiments with cycloheximide, cultures were grown logarithmically at 25°C, 1 μM cycloheximide (Sigma-Aldrich) was added to half of each culture, and the cultures were then grown at 37°C for 6 h. This amount of cycloheximide decreased the growth rate of wild-type cells about fivefold and did not significantly affect the growth rate of the srp101-47 and srp102-510 mutants. A portion of the cultures was pulse-labeled and Kar2p was immunoprecipitated as described above. To depolymerize microtubules, cells were grown with 20 μg/mL nocodazole (1 mg/mL stock in DMSO; Sigma-Aldrich) for 2.5 h (Jacobs et al. 1988). Latrunculin-A (Molecular Probes) was added to cultures at 200 μM from a 10 mM stock in DMSO (Ayscough et al. 1997), and cultures were grown for the indicated amount of time.
Ribosome Binding to Sec61p
The fraction of Sec61p bound to ribosomes was determined basically as described in Panzner et al. 1995. Cultures were grown in YPD to an OD600 of 0.5 at 25°C, and then half of each culture was shifted to 37°C for 3 h while the rest continued to grow at 25°C. NaN3 was then added to 10 mM and the cultures cooled in an ice-water bath. The cells were pelleted and resuspended with an equal volume of buffer containing 50 mM potassium acetate, 10 mM magnesium acetate, 20% glycerol, and 100 mM Hepes, pH 7.8, and lysed in a Biospec bead beater. The lysates were spun at 4,000 g to remove unbroken cells and debris, and then at 12,000 g to remove mitochondria and nuclei. Microsomes were pelleted by spinning at 500,000 g for 30 min. The membranes were washed in buffer containing 800 mM potassium acetate, 10 mM magnesium acetate, 10% glycerol, and 50 mM Hepes, pH 7.8, and resuspended in this buffer. Microsomes were solubilized by adding digitonin to 2.5% and incubating at 4°C for 30 min. Ribosomes were pelleted by spinning at 500,000 g for 30 min. To release nascent chains from the microsomes, 2 mM puromycin (Fluka) and 1 mM GTP were added before solubilization with digitonin and the microsomes were incubated for 1 h on ice and 30 min at 30°C (Gorlich and Rapoport 1993). The amount of Sec61p in the fractions was determined by immunoblotting using an 125I-labeled secondary antibody (NEN Life Science Products). All samples were run in triplicate and quantitated using a Fujix PhosphorImager. Samples were run at various dilutions to ensure that the quantification was performed within the linear range of the PhosphorImager.
Results
Saccharomyces cerevisiae ER Forms a Tubular Network
Previous immunofluorescence microscopy with fixed S. cerevisiae cells only allowed the visualization of the peripheral ER as a bright ring close to the plasma membrane (Rose et al. 1989; Preuss et al. 1991). To obtain a more detailed picture of the ER, we used a fusion of the ER protein Sec63p to GFP (Sec63-GFP) and employed high-resolution fluorescence microscopy with live cells. A stack of images with focal planes 0.2-μm apart were obtained with a wide-field optical sectioning microscope and deconvolved. Two of the images from each stack are shown in Fig. 1, with focus planes close to the center and periphery of the cell. With the improved resolution, we were able to demonstrate that the peripheral ER forms a network of interconnected tubules (Fig. 1, bottom left). This network could be seen on either side of the cell in only one or two image planes close to the plasma membrane. The nuclear ER, on the other hand, forms a continuous membrane (Fig. 1, top left). To confirm that the peripheral network is ER, we used a GFP-fusion protein targeted to the ER lumen. We expressed GFP with an NH2-terminal signal sequence and a COOH-terminal ER retention signal (HDEL; Pelham et al. 1988). This fusion protein, called ss-GFP-HDEL, shows a localization pattern similar to Sec63-GFP (Fig. 1, right). Thus, the peripheral ER in yeast looks strikingly similar to the ER network in mammalian cells. The network was seen at all stages of the cell cycle and appeared in the daughter cell as soon as the bud started to form. The ER network is also thought to be maintained during mitosis in higher eukaryotes (Ellenberg et al. 1997).
Figure 1.

The peripheral ER forms a network of interconnected tubules. Fluorescence microscopy of wild-type cells expressing either Sec63-GFP or ss-GFP-HDEL. Images were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm.
The ER Is Dynamic
We wondered if the yeast cortical ER is dynamic as it is in higher eukaryotes. Cells expressing Sec63-GFP were visualized in the fluorescence microscope at three-minute intervals (Fig. 2 A). The peripheral ER in the daughter cell (above the mother cell) is highly dynamic and is completely rearranged in three minutes. To facilitate comparison between the frames, stars have been placed at identical locations in the daughter cell in each image. The structures near the stars are qualitatively quite different at each time point. The ER in the mother cell was somewhat more stable than in the daughter cell, with some regions showing little change over the course of the experiment. The star in the mother cell indicates a region that remains relatively unchanged during the course of the experiment. This qualitative difference in the extent of ER structural alteration in daughter and mother cells was seen consistently in many experiments (not shown).
Figure 2.
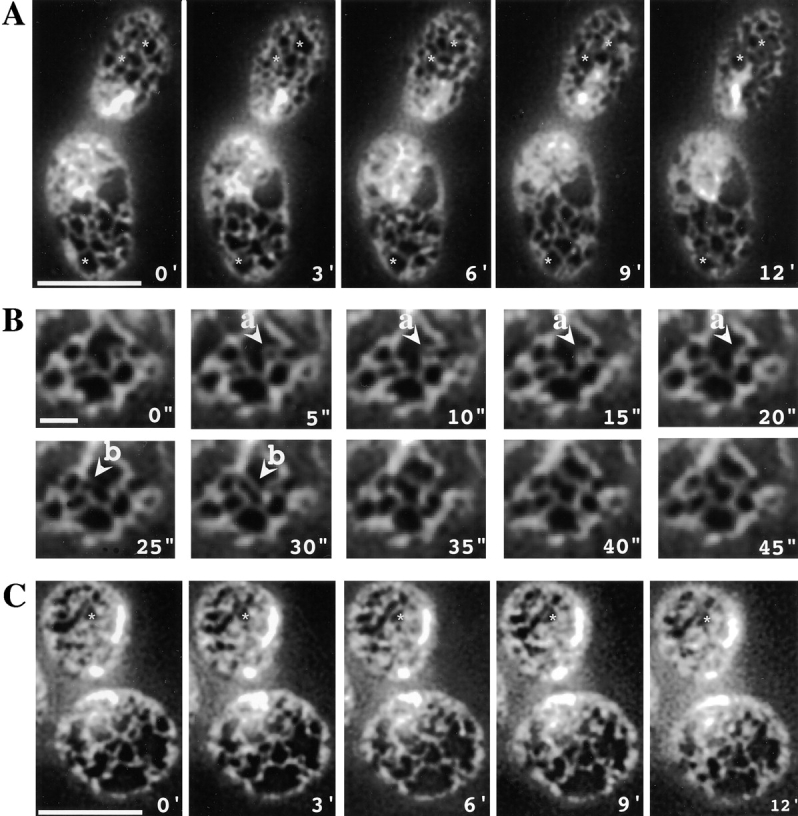
Actin is required for the dynamics of the peripheral ER. Time-lapse images of cortical ER in wild-type cells expressing Sec63-GFP. A, Images of a cell taken at 3-min intervals (mother cell on bottom). Bar, 5 μm. To facilitate comparisons between frames, stars have been placed in identical places in each frame. B, Images of a portion of a cell taken at 5-s intervals. Indicated are an instance of ring closure (a) and formation of a new ER tubule (b). Bar, 1 μm. C, Images of a cell taken at 3-min intervals 10 min after 200 μM latrunculin-A was added to the medium (mother cell on bottom). Bar, 5 μm.
Video microscopy of mammalian cells revealed three types of movement of the ER: tubule branching, ring closure (the contraction of a loop of ER to a single tubule), and tubule sliding (the movement of a junction of ER tubules along a tubule; Lee and Chen 1988). Similar movements occur in S. cerevisiae. Fig. 2 B shows time-lapse microscopy of a portion of a mother cell with pictures taken every five seconds. An example of ring closure occurs between 5 and 20 s (Fig. 2, arrow a points to ring). An instance of tubule branching occurs between 25 and 30 s (Fig. 2, arrow b points to the new tube). We find that tubule branching and ring closure occur at approximately the same rate as in mammals (Lee and Chen 1988). Therefore, ER tubule movements appear to occur similarly in yeast and mammalian cells.
Role of Microtubules and Actin Cytoskeleton in ER Structure
To examine the role of microtubules in ER formation, we incubated yeast in medium containing nocodazole. After two hours, microtubules could not be detected by immunofluorescence with antitubulin antibodies (not shown; Jacobs et al. 1988). Neither the structure of the ER (Fig. 3 A) nor ER dynamics (not shown) were affected by the disruption of microtubules. These results indicate that, unlike the situation in mammals, microtubules play no role in the maintenance of ER structure.
Figure 3.

Neither actin filaments nor microtubules are required to maintain cortical ER structure. A, Images of the ER in wild-type cells expressing Sec63-GFP were taken after 2.5 h of growth in medium with either 20 μg/ml nocodazole or 200 μM latrunculin-A. Images were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm. B, Wild-type cells expressing Sec63-GFP were fixed and stained with Alexa 594 phalloidin to visualize actin. Images were acquired while focusing on the cell cortex. Arrows point to regions in which actin filaments and ER tubules align. Bars, 5 μm.
We next tested the role of actin filaments and patches in the formation and maintenance of the ER. We first assessed the alignment of tubules of the peripheral ER with cortical actin filaments in fixed cells (Fig. 3 B). Although fixation slightly distorts the ER, there are clearly a few regions where ER tubules and actin filaments align (indicated by arrows). However, most ER tubules do not align with any actin filament. Next, we treated yeast cells with latrunculin-A, which results in the disruption of actin structures within minutes, as seen by staining with rhodamine-phalloidin (not shown; Ayscough et al. 1997). However, even after growth for two hours in medium with latrunculin-A, the ER structure was not disrupted, although there appeared to be some minor proliferation of ER tubules (Fig. 3 A). Strikingly, minutes after addition of latrunculin-A, the dynamics of the ER was greatly reduced (Fig. 2, compare C with A, daughter cell on top in both). To facilitate comparison between the frames, stars were placed at identical locations in the daughter cell in each frame. To quantitatively compare the changes of ER structure seen in Fig. 2A and Fig. C, we determined the pixel values at different time points in the same portion of each image of the daughter cell, and calculated an average correlation coefficient. The coefficient for the drug-treated cell (0.779) was clearly higher than for the control cell (0.293), and may have been underestimated because of a slight drift of the sample or Brownian movement. Taken together, our results suggest that the cytoskeleton is not required for maintenance of the ER structure, but actin filaments play a role in ER dynamics.
Isolation of Mutants with Disrupted ER Structure
We used a genetic screen to identify proteins that are required for maintenance of the structure of the cortical ER. Since maintaining ER structure may be essential for cell viability, we screened a collection of temperature-sensitive mutants (Amberg et al. 1992) for those that had altered cortical ER structure or defects in segregating the peripheral ER into the daughter cell. Approximately 1,000 mutants were transformed with the plasmid encoding Sec63-GFP. They were grown at permissive temperature (25°C), shifted to nonpermissive temperature (37°C) for three hours, and examined in a fluorescence microscope. Mutants which displayed ER throughout the cell were numerous and were discarded since it is well known that ER proliferation can be caused by overexpression of membrane proteins or disrupting ER to Golgi complex transport. Three mutants that had disrupted ER structure were identified (WPY152, WPY153, and WPY154).
In all three mutants, the mutations that cause the ER structure defects were recessive, segregated 2:2 as determined by back-crosses with a wild-type strain, and were linked to the temperature-sensitive growth phenotype. We isolated plasmids from genomic libraries that complemented the growth defect of each of the mutants at 37°C. Using a combination of DNA sequencing, subcloning, and integrative mapping, we determined that the mutations responsible for the altered ER structure in the mutants are in SEC27 (WPY153), SRP101 (WPY154), and SRP102 (WPY152). We named the altered alleles of these genes sec27-95, srp101-47, and srp102-510. SEC27 encodes a component of COPI, a coat-forming complex involved in vesicle budding (Duden et al. 1994). SRP101 and SRP102 encode the α and β subunits of the signal recognition particle (SRP) receptor, a heterodimeric complex of GTPases in the ER membrane (Gilmore et al. 1982a,Gilmore et al. 1982b; Meyer et al. 1982; Ogg et al. 1992, Ogg et al. 1998). SRP and SRP receptor target ribosome–nascent polypeptide complexes to the ER membrane for cotranslational protein translocation. We sequenced srp101-47 and srp102-510. The alteration in Srp101p (L422P) affects a residue that is not conserved between species, but is close to two regions likely to be involved in GTP hydrolysis (Ogg et al. 1992; Montoya et al. 1997). The change in Srp102p (L58P) is in a region of unknown function.
ER–Golgi Trafficking Mutants Affect ER Structure
In the sec27-95 mutant found in the screen, the peripheral ER in mother cells was disorganized and appeared to be converted from tubules to sheets. In ∼50% of the cells, the ER in daughter cells was dramatically reduced and disorganized (Fig. 4 A). The phenotype was not simply the result of mislocalization of Sec63-GFP, since ss-GFP-HDEL showed a similar distribution (not shown). In thin-section EM, there was a reduction in the amount of peripheral ER at nonpermissive temperature in both mother and daughter cells. Only small areas of the peripheral ER were seen, in contrast to the situation in wild-type cells in which most of the plasma membrane has ER tubules near it (Fig. 5). To quantitate these differences, we determined the surface area of the ER (both perinuclear and cortical) relative to total cell volume. There was a significant decrease in the relative surface area in sec27-95 cells compared with wild-type cells which became more pronounced at nonpermissive temperature (Table ).
Figure 4.

Mutants with conditional defects in vesicular transport between the ER and Golgi complex have altered ER structure at nonpermissive temperature. Images of cells expressing Sec63-GFP acquired while focusing on either the center or periphery of the cells. Bars, 5 μm. Arrows point to daughter cells in some cells. A, Images of sec27-95 cells grown either at 25°C or 3 h after shift to nonpermissive temperature (37°C). B, Images of sec21-1, sec23-1, and ret1-3 cells 3 h after shift to 37°C. C, Images of ufe1-1 cells 2 h after shift to 37°C and cdc48-3 cells 4 h after shift to 37°C.
Figure 5.
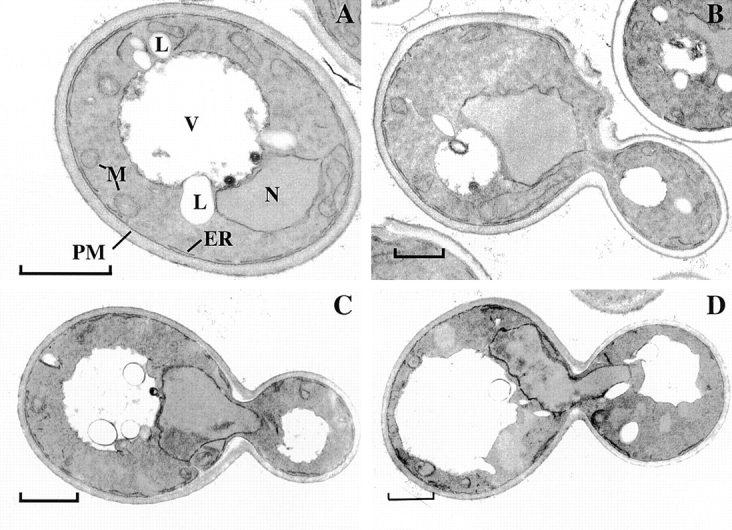
sec27-95 cells have less peripheral ER at nonpermissive temperature. Thin-section EM of wild-type (A and B) and sec27-95 (C and D) cells grown at 25°C (A and C) or 3 h after shift to 37°C (B and D). L, Lipid droplet; M, mitochondrium; N, nucleus; PM, plasma membrane; V, vacuole; ER, endoplasmic reticulum. Bars, 1 μm.
Table 2.
Quantification of the Decrease in Relative ER Surface Area in the Mutants with Disrupted ER Structure
| Strain | Relevant genotype | 25°C | 37°C |
|---|---|---|---|
| FY23 | Wild-type | 1.36 ± 0.12 | 1.32 ± 0.16 |
| WPY154 | srp101-47 | 1.03 ± 0.18 | 0.66 ± 0.08 |
| WPY152 | srp102-510 | 1.07 ± 0.09 | 0.26 ± 0.04 |
| WPY153 | sec27-95 | 1.01 ± 0.10 | 0.80 ± 0.09 |
The relative ER surface area, μm2 ER (both perinuclear and cortical) per μm3 cell volume, was determined by EM on ultrathin sections of KMnO4-fixed cells grown at 25°C, or after a 3-h shift to 37°C. Values are the average of 100 cells ± SD.
A similar phenotype was shared by mutants with conditional defects in other components of COPI (sec21-1 and ret1-3), a mutant with a defect in the functionally related coat complex COPII (sec23-1), and Ufe1p, an ER-localized vesicle soluble N-ethylmaleimide–protein attachment protein receptor (v-SNARE; ufe1-1; Fig. 4B and Fig. C). Mutants with defects in other components of COPI or COPII (sec28Δ::HIS3, sec27-1, sec13-1, and sec16-2) had disorganized ER structure at nonpermissive temperature, but did not have the dramatic reduction in the amount of ER in daughter cells seen in the other mutants (not shown). It is unclear why this phenotype is seen in some mutants with defects in ER–Golgi complex traffic and not in others. Perhaps the rate and extent of inactivation of the mutant proteins differs in these two groups. This might also explain why this phenotype is seen in the sec27-95 mutant we isolated, but not the previously identified sec27-1 mutant. Previous EM studies on sec mutants with ER–Golgi apparatus trafficking defects have also shown that ER structure becomes disorganized at nonpermissive temperature (Novick et al. 1980; Duden et al. 1994). In contrast to these mutants that affect traffic between the ER and Golgi complex, mutants with conditional defects in later steps of the secretory pathway (sec1-1, sec2-41, sec 3-2, sec4-8, sec6-4, sec 8-9, sec9-4, and sec15-1) did not show changes in ER morphology after three hours at nonpermissive temperature (not shown).
Besides its role in vesicular traffic from the Golgi apparatus to the ER (Lewis et al. 1997), Ufe1p has also been implicated in homotypic ER fusion (Patel et al. 1998). Since homotypic ER fusion may well be required to maintain ER structure, we looked at a temperature-sensitive mutant in Cdc48p, another protein implicated in the process (Latterich et al. 1995). Surprisingly, even after four hours at nonpermissive temperature, ER structure remained normal in a cdc48-3 mutant (Fig. 4 C).
SRP Pathway Disruption Affects ER Structure
In the SRP receptor mutants the ER structure was also most disturbed in daughter cells, which often contained one bright fluorescent spot, and in some cases did not contain any detectable ER (Fig. 6 A). However, most mother cells also lacked detectable tubular regions of the ER, and instead contained numerous bright fluorescent spots. Abnormal ER structure was first observed one hour after shift to elevated temperature, and became pronounced after two hours. The structure of the perinuclear ER was not disrupted in the mutants; a ring of Sec63p-GFP was seen around the nucleus stained with 4′,6-diamidino-2-phenylindole (DAPI; Fig. 6 B). Both SRP receptor mutants have normal ER structure at the permissive temperature (Fig. 6 A). We ruled out that the mutants only cause the mislocalization of Sec63-GFP, rather than actually disrupting ER structure, because ss-GFP-HDEL also has an altered distribution in the mutants at the nonpermissive temperature (not shown). Analysis of the mutants by thin-section EM revealed a reduction in the amount of ER at the periphery of the cells, particularly at nonpermissive temperature (Fig. 7 and Table ). In addition, large sheets of membrane near the nucleus can be seen that are not present in wild-type cells (Fig. 5). These structures are likely to be responsible for the brightly fluorescing spots seen in the fluorescence microscope.
Figure 7.
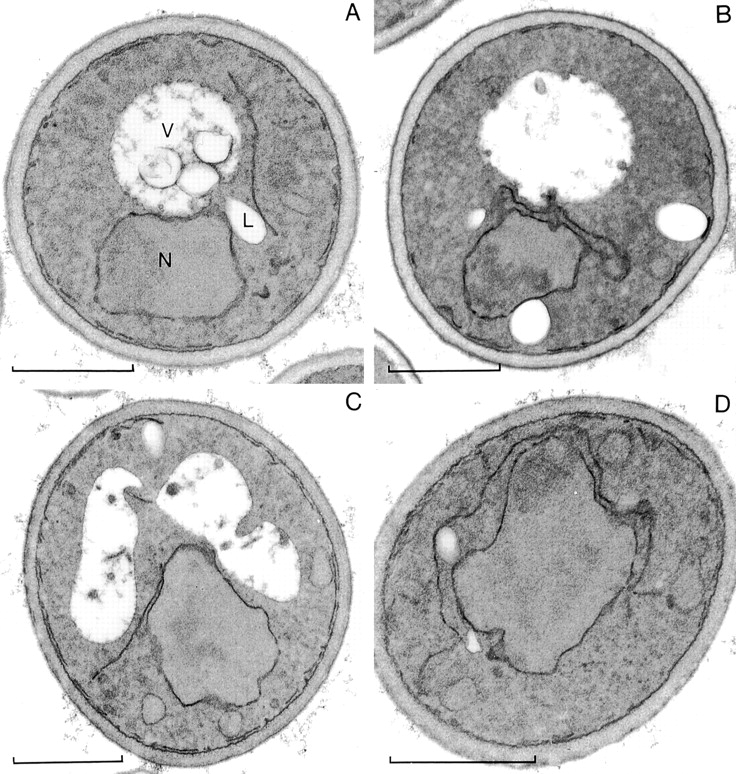
Mutants with conditional defects in SRP receptor have altered peripheral ER morphology at nonpermissive temperature. Thin-section EM of srp102-510 (A and B) and srp101-47 (C and D) cells grown at 25°C (A and C) or 3 h after shift to 37°C (B and D). L, Lipid droplet; M, mitochondrium; N, nucleus; PM, plasma membrane; V, vacuole. Bars, 1 μm.
A conditional mutation in SRP itself also affects ER structure. Yeast cells bearing a mutation in Sec65p, the homologue of the mammalian SRP19 subunit (Stirling and Hewitt 1992), have abnormal ER morphology at the nonpermissive temperature, similar to that seen in the srp101-47 and srp102-510 mutants isolated in the screen (Fig. 8 A).
Figure 8.

Phenotypes of various translocation mutants. A, Images of sec65-1, sec61-3, sec61-2, and sec61-2 Δssh1 cells expressing Sec63-GFP were taken 3 h after shift to nonpermissive temperature (37°C). Images were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm. B, Translocation of Kar2p was tested 1 h after shift to the nonpermissive temperature (37°C). The cells were pulse-labeled for 7 min and Kar2p was immunoprecipitated. The positions of mature Kar2p (m) and of Kar2p that has not undergone signal sequence cleavage (p) are indicated.
Like other mutants with defects in SRP and SRP receptor, srp101-47 and srp102-510 mutants have a protein translocation defect; both mutants accumulate precursor of the ER lumenal protein, Kar2p (Fig. 8 B). However, the translocation defect per se may not be responsible for the altered ER morphology. We tested a number of mutants with temperature-sensitive or cold-sensitive defects in other components of the protein translocation machinery and none of them had abnormal ER structure. Specifically, temperature-sensitive mutations in the major component of the translocation channel, Sec61p (mutants sec61-3 and sec61-2), have only a few minor changes in ER structure three hours after shift to the nonpermissive temperature (Fig. 8 A). These mutants develop a translocation defect less than one hour after shift to elevated temperature (Fig. 8 B; and Rose et al. 1989; Stirling et al. 1992), but continue to grow for a few hours (not shown). We ruled out that differences in strain background account for the difference between the sec61 mutants and the SRP pathway mutants, by moving the sec61 alleles into the same strain background as the SRP receptor mutants we isolated (data not shown). ER structure is also not significantly disrupted in cold-sensitive sec61-11, sec61-23, sec61-24, or sec61-32 mutants after they were grown at nonpermissive temperature for up to six hours (not shown). A sec61-2 mutant that also lacks the Sec61p homologue Ssh1p (Δssh1 sec61-2) has a strong protein translocation defect (Finke et al. 1996), but no ER morphology defect (Fig. 8 A). We also looked at two mutants with temperature-sensitive defects in proteins required for posttranslational translocation of proteins into the ER, a process that does not require the SRP pathway (Panzner et al. 1995). Both mutants (kar2-159 and sec62-1) had only slight ER morphology alterations similar to those seen in the sec61 mutants (not shown). Thus, it seems that the altered ER morphology is not a consequence of a general translocation defect.
Neither SRP101 nor SRP102 are essential genes, but cells lacking them grow approximately five times slower than wild-type cells (Ogg et al. 1992, Ogg et al. 1998). The srp101-47 and srp102-510 mutants we isolated were also able to grow slowly at the nonpermissive temperature. After prolonged growth at 37°C (>24 h) the ER morphology defect was no longer observed (not shown). Similarly, cells with deletions of SRP101, SRP102, or SRP54 (which encodes a component of SRP), showed no ER morphology defect (not shown).
One way the SRP pathway might affect ER structure is via ribosome targeting. We reasoned that slowing translation might restore ribosome binding in mutants with a defect in the SRP pathway by giving ribosome–nascent chain complexes more time to bind to the ER membrane. Sublethal doses of the translation elongation inhibitor cycloheximide were added to cultures of wild-type, srp101-47, and srp102-510 strains immediately before shifting them to the elevated temperature. After six hours, the mutants grown without cycloheximide had ER morphology defects (not shown), whereas those grown in the presence of the drug had almost normal ER (Fig. 9 A). In addition, as described for other translocation mutants (Lee and Beckwith 1986; Ogg and Walter 1995), cycloheximide suppressed or reduced the translocation defect of the mutants (Fig. 9 B). Thus, the role of the SRP pathway in maintaining ER morphology is likely related to its known function in targeting ribosomes to the ER membrane.
Figure 9.

Cycloheximide suppresses both the ER morphology and translocation defects of SRP receptor mutants. Strains expressing Sec63-GFP were grown at 25°C and then for 6 h at 37°C with or without 1 μM cycloheximide. A, Images of the cells grown with cycloheximide. They were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm. B, A portion of the cultures were pulse-labeled for 7 min and lysates were immunoprecipitated for Kar2p. m, Mature Kar2p; p, precursor of Kar2p.
To determine ribosome binding to the ER, we measured the amount of Sec61p that is associated with ribosomes after solubilization of ER membranes in detergent. ER membranes from wild-type, sec61-3, and srp101-47 cells were washed with high salt to remove loosely bound ribosomes not engaged in translocation (Gorlich and Rapoport 1993), and solubilized in digitonin. The ribosomes were pelleted, and the amount of cosedimenting Sec61p was determined by quantitative immunoblotting (Fig. 10). At permissive temperature, ∼30% of the Sec61p was bound to ribosomes in all three strains (Fig. 10, bars 1, 5, and 9). Release of the nascent chains from the ribosomes with puromycin greatly decreased the amount of Sec61p pelleting with the ribosomes (Fig. 10, bar 2; and data not shown), indicating that the association requires the presence of a translocating polypeptide. When the strains were grown for three hours at nonpermissive temperature, the fraction of Sec61p bound to ribosomes reproducibly decreased to ∼20% in the srp101-47 strain (Fig. 10, bar 6), whereas it remained about the same in the sec61-3 (Fig. 10, bar 10) and wild-type strains (Fig. 10, bar 3). The amount of Sec61p per cell in these strains was about the same when they were grown at permissive temperature or after three hours at nonpermissive temperature (data not shown; Biederer et al. 1996). When the srp101-47 mutant was grown at 37°C for prolonged periods, conditions that restore ER morphology, the amounts of Sec61p bound to ribosomes increased to normal levels (Fig. 10, bar 8 vs. 6). A similar correlation between restoration of ER morphology and ribosome binding was seen after treatment of the mutant with cycloheximide (Fig. 10, bar 7 vs. 6). A small increase of ribosome-associated Sec61p was observed when wild-type cells were grown with cycloheximide (Fig. 10, bar 4 vs. 3), which might be expected when the rate of translation is slowed. Taken together, these results suggest that the SRP pathway influences ER morphology through its effect on ribosome binding.
Figure 10.

Ribosome binding to Sec61p. Wild-type, sec61-3, and srp101-47 cells were grown at 25°C and then shifted to 37°C for the indicated number of hours. In some cases, cycloheximide was added to the medium when the cultures were shifted to elevated temperature. ER membranes were isolated, washed with high salt, and solubilized with digitonin. The ribosomes were pelleted and the amount of Sec61p in the pellet determined by quantitative immunoblotting. Each bar is the average of at least three independent determinations. Some samples were treated with puromycin before solubilization to release nascent polypeptides (column 2). The differences between columns 5 and 6 and columns 3 and 4 are statistically significant (P < 0.01).
Connection between ER and Mitochondrial Structures
We found that in the SRP receptor mutants, not only peripheral ER, but also mitochondrial structure, is disrupted. In wild-type cells, mitochondria form branched reticular structures near the cell cortex (Hermann and Shaw 1998; Fig. 11 A). In the SRP receptor mutants we isolated, mitochondria appear to fragment; at nonpermissive temperature, a number of bright spots were seen throughout the cytoplasm (Fig. 11 B). Mitochondrial structure is normal when the mutants are grown at permissive temperature (not shown). Interestingly, when the ER and mitochondria were visualized in the same cell, fragments of the disrupted ER were frequently localized next to fragments of mitochondria (Fig. 11 B). Mutants with defects in ER–Golgi complex trafficking that affect ER structure (e.g., ret1-3), also have altered mitochondrial structure. As with the SRP mutants, the disrupted ER and mitochondrial structures are often near one another (Fig. 11 C). Even in wild-type cells, the tubules of the peripheral ER sometimes coaligned with mitochondrial tubules (Fig. 11 A). These data suggest that the membranes of the ER and mitochondria are linked at certain sites and that the ER may have an effect on the structure of mitochondria. The reverse may not be true. Conditional mutations in two mitochondrial outer membrane proteins, Mmm1p and Fzo1p, cause mitochondria to form large spheres (mmm1-1) or fragment (fzo1-1) at nonpermissive temperature (Burgess et al. 1994; Hermann et al. 1998; Rapaport et al. 1998). ER structure is unaffected in these mutants (Fig. 11D and Fig. E).
Figure 11.

The SRP receptor mutants also disrupt mitochondrial structure. Cells expressing Sec63-GFP were labeled with TMR-CH2Cl to visualize mitochondria and viewed live. Wild-type cells were grown at 30°C. The other strains were grown at 25°C and then shifted to 37°C for 2 h (srp101-47), 2.5 h (ret1-3), 4 h (mmm1-1), or 30 min (fzo1-1). Arrows point to regions of alignment of mitochondria and ER tubules in wild-type cells. Images were acquired while focusing on either the center or periphery of the cells. Bars, 5 μm.
Discussion
A major result of this work is the demonstration that the peripheral ER in yeast, previously viewed simply as a bright ring by immunofluorescence (Rose et al. 1989; Preuss et al. 1991), is a tubular network, similar in structure to the ER in higher eukaryotes. The tubules have a relatively constant diameter and undergo the same kinds of dynamic changes that have been described in mammalian cells. As in mammals, the ER network is maintained during mitosis. Yeast cells can therefore serve as an excellent model system in which to study the mechanisms underlying the formation and maintenance of ER networks.
Our data indicate that mechanisms proposed for maintaining the reticular structure of the ER in mammalian cells are probably not correct for yeast. We found that microtubules are not required to maintain the structure of the peripheral ER, in contrast to their function in mammalian cells. This is perhaps not too surprising, since there are few cytoplasmic microtubules in yeast. While actin does form an extensive cortical network, it is also not required for the maintenance of ER structure. Since there is also relatively little coalignment between actin structures and the ER network, ER tubules are probably stabilized by mechanisms other than association with an underlying cytoskeleton scaffold. The actin cytoskeleton does appear to be required for ER dynamics, which are drastically reduced minutes after addition of latrunculin-A to the medium. However, it is difficult to rule out that the effect may be indirect. Actin filaments are also required for the dynamics of mitochondria in yeast (Simon et al. 1995), but again the mechanism remains to be elucidated.
Given that the nuclear and peripheral ER are structurally distinct and that the cytoskeleton does not play a role in maintaining ER structure in yeast, we decided to use genetics to search for other components that do. In our visual screen, we looked for mutants that affect either ER inheritance or the maintenance of the ER. Only three out of the thousand mutants we screened had such defects, indicating that a general perturbance of cell physiology does not readily lead to these phenotypes. It is clear that the screen has not been saturated, and indeed we have found additional mutants affecting ER structure that were not identified in the screen.
One class of mutants we isolated affects ER structure and the propagation of the ER from the mother to the daughter cell by disrupting vesicular transport between the ER and Golgi complex. Defects later in the secretory pathway did not affect ER structure or inheritance. How exactly vesicular transport between the ER and Golgi complex is required for ER structure is unclear. Resident ER proteins are known to be transported to the Golgi apparatus at some low rate (Teasdale and Jackson 1996), and it therefore seems possible that retrograde transport from the Golgi apparatus to the ER may be required to recycle ER proteins needed to maintain proper structure.
Surprisingly, we find that ER structure is not affected in a temperature-sensitive mutant with a defect in a protein (Cdc48p) required for homotypic ER fusion in vitro (Latterich et al. 1995). Since yeast ER is dynamic, it seems unlikely that ER structure could be maintained in the absence of homotypic ER fusion. It may be that, in vivo, other proteins are able to substitute for the role of Cdc48. Alternatively, it may be that Cdc48p plays a role in the ER–ER fusion required for nuclear division, but is not essential for fusion events in peripheral ER.
Our data show that the SRP pathway plays an important role in maintaining the reticular structure of the ER. The linkage could be direct. Only mutants with defects in the α and β subunits of the SRP receptor and in SRP itself had the ER disruption phenotype; other mutants with translocation defects did not show this effect. In fact, some of the mutations in Sec61p, a component required for transport of all substrates, have stronger translocation defects than the srp101-47 and srp102-510 mutants we isolated (Stirling et al. 1992; Finke et al. 1996; Pilon et al. 1998). In addition, the Sec61p mutants can continue to grow at the nonpermissive temperature for a few hours after onset of the translocation defect, and their ER remains normal. Despite this evidence, it is hard to exclude that the SRP pathway affects ER structure by blocking the translocation of a particular protein. However, we consider it more likely that the SRP pathway affects ER structure via ribosome targeting to the ER membrane. We find a small, but reproducible, reduction in ribosome binding to the ER in a SRP receptor mutant, but not a Sec61p mutant or wild-type strain, when they are grown at elevated temperature. In addition, under conditions where ER morphology is restored, ribosome binding is increased back to wild-type levels. The experiments with cycloheximide are consistent with this interpretation. Slowing translation with sublethal amounts of cycloheximide restored ribosome binding and suppressed both the ER morphology and translocation defects of the SRP receptor mutants. These data show that both phenotypes are related to ribosome targeting and argue against an additional function of the SRP pathway. It is unclear how ribosome binding to the ER might affect its structure. In mammals, membrane-bound ribosomes have been proposed to be connected with each other by a protein network (Kriebich et al. 1983; Amar-Costesec et al. 1984; Marcantonio et al. 1984). Perhaps, ribosome binding to the Sec61p or Ssh1p complexes affects this network. A role for ribosome binding in determining ER structure might also explain the morphological differences between the rough and smooth portions of the ER that have been noted since the organelle was first described (Palade 1956).
The SRP pathway is clearly not essential for ER formation and maintenance since cells with deletions of SRP pathway components have normal ER structure. In addition, both the translocation and ER morphology defects in the SRP receptor mutants we isolated are partially ameliorated during prolonged growth without this pathway. Adaptation of SRP pathway mutants has been noted before and was not found to be the result of suppressing mutations (Hann and Walter 1991; Ogg et al. 1992). While it is possible that suppressing mutations may occur in our mutants when they are grown at high temperature, it is more likely that they also undergo adaptation. Since we found that ribosome binding is restored under these conditions, our data suggest that it is SRP-independent ribosome targeting that restores ER structure. SRP-independent ribosome targeting may explain both the small changes in ribosome binding to the ER in SRP pathway mutants when they are shifted to nonpermissive temperature and the fact that the SRP pathway is not essential for viability (Hann and Walter 1991; Ogg et al. 1992, Ogg et al. 1998). Indeed, SRP-independent, direct binding of ribosome–nascent chain complexes to ER membranes has been demonstrated in vitro (Jungnickel and Rapoport 1995).
Finally, our data point to an interesting connection between the structures of the ER and mitochondria. Both organelles form reticular networks close to the plasma membrane and are often closely apposed to one another. A close association between these two organelles has also been observed in mammalian cells (Rizzuto et al. 1998). In SRP receptor mutants, both structures are disrupted, but maintain their association, indicating that they are physically linked at a number of sites. It is possible that the ER is required for maintaining the mitochondrial reticulum. Interestingly, the reverse may not be true since mutations that disrupt mitochondrial structure leave the ER intact. A structural connection between the ER and mitochondria is consistent with known functional interactions between the two organelles. For example, it has been proposed that direct contact between the ER and mitochondria is required for phospholipid transport between them (Shiao et al. 1998; Achleitner et al. 1999), and Ca2+ released from one organelle can act locally on the other (Rizzuto et al. 1998).
Acknowledgments
We thank R. Schekman, F. Winston, M. Latterich, J. Nunnari, R. Jensen, P. Walter, P. Novick, C. Shamu, D. Botstein, and T. Sommer for strains; C. Shamu, K. Matlack, and L. Dreier for critically reading the manuscript; A. Kram and J. Zagers for help with EM; M. Rolls and J. White for help with quantification of images; and B. Glick for helpful advice.
T.A. Rapoport is a Howard Hughes investigator and W.A. Prinz is supported by a postdoctoral fellowship from the American Cancer Society.
Footnotes
Jason A. Kahana's current address is Ludwig Institute for Cancer Research, CMM-East, Rm 3080, 9500 Gilman Dr., La Jolla, CA 92093-0660.
Abbreviations used in this paper: GFP, green fluorescent protein; SRP, signal recognition particle.
References
- Achleitner G., Gaigg B., Krasser A., Kainersdorfer E., Kohlwein S.D., Perktold A., Zellnig G., Daum G. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 1999;264:545–553. doi: 10.1046/j.1432-1327.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- Amar-Costesec A., Todd J.A., Kriebich G. Segregation of the polypeptide translocation apparatus to regions of the endoplasmic reticulum containing ribophorins and ribosomes. I. functional tests on rat liver microsomal subfractions. J. Cell Biol. 1984;99:2247–2253. doi: 10.1083/jcb.99.6.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg D.C., Goldstein A.L., Cole C.N. Isolation and characterization of RAT1an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- Ayscough K.R., Stryker J., Pokala N., Sander M., Crews P., Drubin D.G. High rates of actin filament turnover in budding yeast and roles for actin in estabishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T., Volkwein C., Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin–proteasome pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Burgess S.M., Delannoy M., Jensen R.E. MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J. Cell Biol. 1994;26:1375–1391. doi: 10.1083/jcb.126.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabora S.L., Sheetz M.P. The microtubule-dependent formation of a tubularvesicular network with characterisitics of the ER from cultured cell extracts. Cell. 1988;54:27–35. doi: 10.1016/0092-8674(88)90176-6. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J., Schekman R. Structural and functional dissection of Sec62p, a membrane-bound component of the yeast endoplasmic reticulum protein import machinery. Mol. Cell. Biol. 1990;10:6024–6035. doi: 10.1128/mcb.10.11.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier L., Rapoport T.A. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J. Biol. Chem. 2000;148:883–898. doi: 10.1083/jcb.148.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden R., Hosobuchi M., Hamamoto S., Winey M., Byers B., Schekman R. Yeast β- and β′-coat proteins (COP) J. Biol. Chem. 1994;269:24486–24496. [PubMed] [Google Scholar]
- Ellenberg J., Siggia E., Moreira J., Smith C., Presely J., Worman H., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cellstargeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke K., Plath K., Panzner S., Prehn S., Rapoport T.A., Hartmann E., Sommer T. A second trimeric complex containing homologs of the Sec61p complex functions in protein transport across the ER membrane of S. cerevisiae . EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:1482–1494. [PMC free article] [PubMed] [Google Scholar]
- Gilmore R., Blobel G., Walter P. Protein translocation across the endoplasmic reticulum. I. Detection in the microsomal membrane of a receptor for the signal recognition particle J. Cell Biol. 95 1982. 463 469a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore R., Walter P., Blobel G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor J. Cell Biol. 95 1982. 470 477b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D., Rapoport T.A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 1993;75:615–630. doi: 10.1016/0092-8674(93)90483-7. [DOI] [PubMed] [Google Scholar]
- Hann B.C., Walter P. The signal recognition particle in S. cerevisiae . Cell. 1991;67:131–144. doi: 10.1016/0092-8674(91)90577-l. [DOI] [PubMed] [Google Scholar]
- Hermann G.J., Shaw J.M. Mitochondrial dynamics in yeast. Annu. Rev. Cell Dev. Biol. 1998;14:265–303. doi: 10.1146/annurev.cellbio.14.1.265. [DOI] [PubMed] [Google Scholar]
- Hermann G.J., Thatcher J.W., Mills J.P., Hales K.G., Fuller M.T., Nunnari J., Shaw J.M. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C.W., Adams A.E.M., Szaniszlo P.J., Pringle J.R. Functions of microtubules in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 1988;107:1409–1426. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungnickel B., Rapoport T.A. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 1995;82:261–270. doi: 10.1016/0092-8674(95)90313-5. [DOI] [PubMed] [Google Scholar]
- Kahana J.A., Schlenstedt G., Evanchuk D.M., Geiser J.A., Hoyt M.A., Silver P.A. The yeast dynactin complex is involved in partitioning the mitotic spindle between mother and daughter cells during anaphase B. Mol. Biol. Cell. 1998;9:1741–1756. doi: 10.1091/mbc.9.7.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriebich G., Marcantonio E.E., Sabiniti D.D. Ribophorins I and IImembrane proteins characteristic of the rough endoplasmic reticulum. Methods Enzymol. 1983;96:520–530. doi: 10.1016/s0076-6879(83)96045-7. [DOI] [PubMed] [Google Scholar]
- Latterich M., Frohlich K.U., Schekman R. Membrane fusion and the cell cycleCdc48p participates in the fusion of ER membranes. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Lee C., Chen L.B. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Lee C., Ferguson M., Chen L.B. Construction of the endoplasmic reticulum. J. Cell Biol. 1989;109:2045–2055. doi: 10.1083/jcb.109.5.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.A., Beckwith J. Suppression of growth and protein secretion defects in Escherichia coli secA mutants by decreasing protein synthesis. J. Bacteriol. 1986;166:878–883. doi: 10.1128/jb.166.3.878-883.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M.J., Rayner J.C., Pelham H.R. A novel SNARE complex implicated in vesicle fusion with the endoplasmic reticulum. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:3017–3024. doi: 10.1093/emboj/16.11.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebe S., Menzel D. Actomyosin-based motility of endoplasmic reticulum and chloroplasts in Vallisneria mesophyll cells. Biol. Cell. 1995;85:207–222. doi: 10.1016/0248-4900(96)85282-8. [DOI] [PubMed] [Google Scholar]
- Marcantonio E.E., Amar-Costesec A., Kriebich G. Segregation of the polypeptide translocation apparatus to regions of the endoplasmic reticulum containing ribophorins and ribosomes. II. Rat liver microsomal subfractions contain equimolar amounts of ribosomes. J. Cell Biol. 1984;99:2254–2259. doi: 10.1083/jcb.99.6.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matile P., Moor H., Rainbow C.F. Yeast cytology. In: Rose A.H., Harrison J.S., editors. The Yeasts. Vol. 1. Academic Press; New York: 1969. pp. 219–302. [Google Scholar]
- Meyer D.I., Louvard D., Dobberstein B. Characterization of molecules involved in protein translocation using a specific antibody. J. Cell Biol. 1982;92:579–583. doi: 10.1083/jcb.92.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya G., Svensson C., Luirink J., Sinning I. Crystal structure of the NG domain from the signal-recoginition particle receptor FtsY. Nature. 1997;385:365–368. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- Mumberg D., Muller R., Funk M. Regulatable promoters of Saccharomyces cerevisiaecomparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Nunnari J., Marshall W.F., Straight A., Murray A., Sedat J.W., Walter P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell. 1997;8:1233–1242. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S.C., Walter P. SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell. 1995;81:1075–1084. doi: 10.1016/s0092-8674(05)80012-1. [DOI] [PubMed] [Google Scholar]
- Ogg S.C., Poritz M.A., Walter P. Signal recognition particle receptor is important for cell growth and protein secretion in Saccharomyces cerevisiae . Mol. Biol. Cell. 1992;3:895–911. doi: 10.1091/mbc.3.8.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S.C., Barz W.P., Walter P. A functional GTPase domain, but not its transmembrane domain, is required for function of SRP receptor β-subunit. J. Cell Biol. 1998;142:341–354. doi: 10.1083/jcb.142.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade G. The endoplasmic reticulum. J. Biophys. Biochem. Cytol. 1956;2:85–97. doi: 10.1083/jcb.2.4.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzner S., Dreier L., Hartmann E., Kostka S., Rapoport T.A. Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell. 1995;81:561–570. doi: 10.1016/0092-8674(95)90077-2. [DOI] [PubMed] [Google Scholar]
- Patel S.K., Indig F.E., Olivieri N., Levine N.D., Latterich M. Organelle membrane fusiona novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell. 1998;95:611–620. doi: 10.1016/s0092-8674(00)81129-0. [DOI] [PubMed] [Google Scholar]
- Pelham H.R., Hardwick K.G., Lewis M.J. Sorting of soluble ER proteins in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:1757–1762. doi: 10.1002/j.1460-2075.1988.tb03005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon M., Romisch K., Quach D., Schekman R. Sec61p serves multiple roles in secretory precursor binding and translocation into the endoplasmic reticulum membrane. Mol. Biol. Cell. 1998;9:3455–3473. doi: 10.1091/mbc.9.12.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss D., Mulholland J., Kaiser C.A., Orlean P., Albright C., Rose M.D., Robbins P.W., Botstein D. Structure of the yeast endoplasmic reticulum using immunofluorescence and immunoelectron microscopy. Yeast. 1991;7:891–911. doi: 10.1002/yea.320070902. [DOI] [PubMed] [Google Scholar]
- Pringle J.R., Adams A.E.M., Drubin D.G., Haarer B.K. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Rapaport D., Brunner M., Neupert W., Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae . J. Biol. Chem. 1998;273:20150–20155. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Misra L.M., Vogel J.P. KAR2, a karyogamy gene, is the yeast homolog of the mammalian Bip/GRP78 gene. Cell. 1989;57:1211–1221. doi: 10.1016/0092-8674(89)90058-5. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Shiao Y.-J., Balcerzak B., Vance J.E. A mitochondrial membrane protein is required for translocation of phosphatidylserine from mitochondria-associated membranes to mitochondria. Biochem. J. 1998;331:217–223. doi: 10.1042/bj3310217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon V.R., Swayne T.C., Pon L.A. Actin-dependent mitochondrial motility in mitotic yeast and cell-free systemsidentification of a motor activity on the mitochondrial surface. J. Cell Biol. 1995;130:345–354. doi: 10.1083/jcb.130.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling C.J., Hewitt E.W. The S. cerevisiae SEC65 gene encodes a component of the yeast signal recognition particle with homology to human SRP19. Nature. 1992;356:534–537. doi: 10.1038/356534a0. [DOI] [PubMed] [Google Scholar]
- Stirling C.J., Rothblatt J., Hosobuchi M., Deshaies R., Schekman R. Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell. 1992;3:129–142. doi: 10.1091/mbc.3.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale R.D., Jackson M.R. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 1996;12:27–54. doi: 10.1146/annurev.cellbio.12.1.27. [DOI] [PubMed] [Google Scholar]
- Terasaki M. Recent progress on structural interactions of the endoplasmic reticulum. Cell. Motil. Cytoskel. 1990;15:71–75. doi: 10.1002/cm.970150203. [DOI] [PubMed] [Google Scholar]
- Terasaki M., Chen L.B., Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn H.J.M., Linnemans W.A.M., Boer P. Localization of acid phosphatase in protoplasts from Saccaromyces cerevisiae . J. Bacteriol. 1975;123:1144–1149. doi: 10.1128/jb.123.3.1144-1149.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J.P., Misra L.M., Rose M.D. Loss of BiP/GRP78 function blocks translocation of secretory proteins in yeast. J. Cell Biol. 1990;110:1885–1895. doi: 10.1083/jcb.110.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer C.M., Salmon E.D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- Weibel E.R., Bolender P. Stereological techniques for electron microscopic morphometry. In: Hayat M.A., editor. Principles and Techniques of Electron Microscopy. van Nostrand Reinhold; New York: 1976. [Google Scholar]
- Winston F., Dollard C., Ricupero-Hovasse S.L. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast. 1995;11:53–55. doi: 10.1002/yea.320110107. [DOI] [PubMed] [Google Scholar]