

Abstract
The Tight skin (Tsk) mutation is a duplication of the mouse fibrillin 1 (Fbn1) gene that results in a larger (418 kD) than normal (350 kD) protein; Tsk/+ mice display increased connective tissue, bone overgrowth, and lung emphysema. Lung emphysema, bone overgrowth, and vascular complications are the distinctive traits of mice with reduced Fbn1 gene expression and of Marfan syndrome (MFS) patients with heterozygous fibrillin 1 mutations. Although Tsk/+ mice produce equal amounts of the 418- and 350-kD proteins, they exhibit a relatively mild phenotype without the vascular complications that are associated with MFS patients and fibrillin 1–deficient mice. We have used genetic crosses, cell culture assays and Tsk-specific antibodies to reconcile this discrepancy and gain new insights into microfibril assembly. Mice compound heterozygous for the Tsk mutation and hypomorphic Fbn1 alleles displayed both Tsk and MFS traits. Analyses of immunoreactive fibrillin 1 microfibrils using Tsk- and species-specific antibodies revealed that the mutant cell cultures elaborate a less abundant and morphologically different meshwork than control cells. Cocultures of Tsk/Tsk fibroblasts and human WISH cells that do not assemble fibrillin 1 microfibrils, demonstrated that Tsk fibrillin 1 copolymerizes with wild-type fibrillin 1. Additionally, copolymerization of Tsk fibrillin 1 with wild-type fibrillin 1 rescues the abnormal morphology of the Tsk/Tsk aggregates. Therefore, the studies suggest that bone and lung abnormalities of Tsk/+ mice are due to copolymerization of mutant and wild-type molecules into functionally deficient microfibrils. However, vascular complications are not present in these animals because the level of functional microfibrils does not drop below the critical threshold. Indirect in vitro evidence suggests that a potential mechanism for the dominant negative effects of incorporating Tsk fibrillin 1 into microfibrils is increased proteolytic susceptibility conferred by the duplicated Tsk region.
Keywords: elastic fibers, microfibrils, Tsk, Marfan syndrome, extracellular matrix
Introduction
Morphologically distinct networks of elastic fibers are present in the extracellular matrix of virtually every organ and are particularly abundant in tissues subjected to periodic stress, such as the respiratory and cardiovascular systems (Mecham and Davies 1994; Ramirez 1996). All elastic fibers are similarly composed of an amorphous core of cross-linked elastin embedded within a peripheral mantle of microfibrils. Microfibrillar aggregates devoid of elastin are also widely distributed in the connective tissue, where they are believed to connect elastic fibers, anchor cells to the matrix, hold organ structures in place, confer tensile strength, and provide structural support (Mecham and Davies 1994; Ramirez 1996). Microfibrils are thought to regulate the assembly and organization of the elastic fibers by providing the scaffold that guides tropoelastin deposition during organogenesis (Mecham and Davies 1994). Electron microscopy has resolved the microfibrils into aggregates of threadlike filaments composed of periodically spaced globular domains (beads) connected by multiple linear arms (Keene et al. 1991; Ren et al. 1991). Additional analyses have demonstrated that the filaments of the beaded structure correspond to parallel fibrillin monomers aligned in a head-to-tail configuration, but have left unresolved the biochemical nature of the intervening beads (Sakai et al. 1991; Reinhardt et al. 1996). In spite of much effort, the molecular mechanisms driving fibrillin polymerization and microfibril assembly are largely unknown. The process can be broadly summarized as consisting of the following two steps: (1) polymerization of fibrillin 1 monomers into microfibrils; and (2) aggregation of fibrillin 1 microfibrils into microfibrillar bundles (see Fig. 1 a). Naturally occurring and artificially generated mutations in humans and mice have shed some light on the contributions of the fibrillins to the formation and function of microfibrillar aggregates (Ramirez 1996; Pereira et al. 1997, Pereira et al. 1999).
Figure 1.


Hypothetical models of fibrillin 1 microfibril assembly (a) and pathogenesis (b–d). The pathogenic models are based on the threshold hypothesis (b) of Pereira et al. 1999, and on the studies of the Tsk mutation reported by Kielty et al. 1998 (c) and described in the present paper (d).
Fibrillins 1 and 2 display superimposable multidomain structures that consist mainly of calcium-binding epidermal growth factor-like (cbEGF) repeats interspersed by a novel cysteine-rich (8-cys) motif, present only in the fibrillins and latent transforming growth factor-β–binding proteins (Ramirez 1996). Heterozygous mutations of fibrillin 1 give rise to the pleiotropic manifestations of Marfan syndrome (MFS), which principally involve the musculoskeletal, ocular, and cardiovascular systems (Dietz et al. 1994). A dissecting aneurysm with subsequent rupture of the aortic wall is the main cardiovascular symptom of MFS and the leading cause of premature death of affected individuals. The initial model of MFS pathogenesis postulated that mutant fibrillin 1 molecules exert a dominant negative activity on elastic fiber formation by interfering with microfibrillar assembly and/or function (Dietz et al. 1994; Ramirez 1996; see Fig. 1 b). Herein, the term “antimorph” will be used to describe the dominant negative activity of structurally defective fibrillin 1 molecules on the wild-type counterparts.
Two lines of mutant fibrillin 1 mice that were created by homologous gene targeting have recently refined and extended this pathogenic model (Pereira et al. 1997, Pereira et al. 1999). The first line of mice (mgΔ) contains a mutation that combines a structural defect of fibrillin 1 with reduced gene expression. As a result, the mgΔ allele produces 5–10% of the normal amount of fibrillin 1 with an internal deletion of 272 amino acids (Pereira et al. 1997). Heterozygous mutant mice are asymptomatic and morphologically normal because the 10-fold excess of wild-type protein overrides the negative effect of the antimorphic mgΔ product. On the other hand, homozygous mgΔ animals die of MFS-like vascular complications within the first month of postnatal life as a result of substantial fibrillin 1 deficiency. The second line of mice (mgR) contains a mutation that produces 15–20% the amount of wild-type fibrillin 1 (Pereira et al. 1999). Whereas mgR/+ mice are normal, mgR/mgR animals produce less than optimal amount of fibrillin 1 microfibrils, gradually develop skeletal abnormalities, and eventually die during early adulthood (4–8 mo) of respiratory distress and MFS-like vascular complications. Herein, the term “hypomorph” will be used to describe fibrillin 1 mutations with weaker expression than the wild-type gene. Taken together, the human and mouse studies indicate that antimorphic mutations (i.e., structural defects) and hypomorphic mutations (i.e., reduced expression) of fibrillin 1 equally lead to a pleiotropic phenotype that includes the biomechanical failure of the aortic wall (see Fig. 1 a). These studies also suggest a threshold effect, whereby the relative abundance of functionally competent microfibrils determines the incremental appearance and degree of severity of distinct MFS traits (Pereira et al. 1999). However, this model is inconsistent with the recent characterization of fibrillin 1 mutation in the Tight skin (Tsk) mouse (Green et al. 1975; Siracusa et al. 1996).
The Tsk mutation is a genomic duplication within the mouse fibrillin 1 (Fbn1) gene that results in the production of normal levels of a mutant protein with 984 additional amino acids and a predicted molecular mass of 418 kD (Fig. 2; Siracusa et al. 1996). The duplication includes the region deleted in the mgΔ allele. According to the dominant negative model of MFS mutations, the Tsk gene product should negatively affect the function of the wild-type molecules and its antimorphic effect should cause vascular complications and the premature death of Tsk/+ mice. Paradoxically, Tsk/+ mice have a normal life span and no evidence of vascular complications (Green et al. 1975). Tsk/+ manifestations include all of the following: thickened skin, bone and cartilage overgrowth, myocardial hypertrophy, lung emphysema, small tendons with tendon sheath hyperplasia, and autoimmunity (Green et al. 1975; Bocchieri and Jimenez 1990). Homozygous Tsk embryos degenerate in utero at ∼8 d of gestation of unknown causes (Green et al. 1975). A recent study has correlated the Tsk/+ phenotype with the segregation of normal and abnormal fibrillin 1 monomers into two morphologically distinct groups of beaded polymers through an undefined assembly process (see Fig. 1 c; Kielty et al. 1998). The authors have argued that the pool of wild-type fibrillin 1 microfibrils is sufficient to sustain normal vascular development of Tsk/+ animals, whereas the Tsk duplication may cause excessive matrix deposition by altering the binding of mutant fibrillin 1 microfibrils to growth factors (Kielty et al. 1998).
Figure 2.

Schematic illustration of the wild-type (WT) and mutant (Tsk) fibrillin 1 proteins with the wild-type and mutant sequences at the NH2-terminal junction of the Tsk duplication (gray segment). The sequence of the peptide used to raise the Tsk-specific antibodies is underlined, and the amino acid change discussed in the text is highlighted by the asterisk. White rectangles and ovals represent cbEGF and 8-cys motifs, respectively.
Here, we report studies of the Tsk mutation using genetic crosses between the various Fbn1 alleles, cell culture models, and antibodies specific for the Tsk protein. Our results exclude the assembly of distinct homotypic fibrillin 1 polymers in Tsk/+ mice and, instead, suggest the following model (see Fig. 1 d). Tsk fibrillin 1 molecules participate in the initial stage(s) of microfibril assembly but cannot properly polymerize into long microfibrils unless they are in the presence of wild-type molecules. Copolymerization of Tsk and wild-type fibrillin 1 in Tsk/+ mice decreases the amount of functional microfibrils below the threshold level of bone overgrowth and lung emphysema, but not of vascular abnormalities. We also present indirect evidence suggesting that the Tsk duplication destabilizes the mutant product, thus, rendering the protein more sensitive to proteolysis than the wild-type molecule.
Materials and Methods
Mice
The Tsk/+ mice used in this study were previously backcrossed onto the C57BL/6J background (Siracusa et al. 1996). The mgΔ and mgR mutations were maintained in the heterozygous state in the mixed C57BL/6J × 129Sv-ter/+ background (Pereira et al. 1997, Pereira et al. 1999). Mice were genotyped by Southern blot hybridization to previously described probes (Siracusa et al. 1996; Kasturi et al. 1997; Pereira et al. 1997, Pereira et al. 1999). Nearly 100 offspring were genotyped in each of the compound heterozygote crosses.
Cell Cultures
Primary dermal fibroblasts were prepared from newborn skin explants of Tsk/+, mgR/+, mgΔ/+, Tsk/mgΔ, Tsk/mgR, and +/+ mice, whereas Tsk/Tsk cells were prepared from 9-d postcoitum embryos. Mouse fibroblasts and human amnion, epithelial-like WISH cells (ATCC CCL-25; American Type Culture Collection) were maintained in DME supplemented with 10% FBS and antibiotics (Gibco Laboratories). Aside from DNA genotyping, cell lines were characterized by protein analysis of metabolically labeled, conditioned medium that was immunoprecipitated and fractionated on SDS-PAGE (see below).
Antibodies and Immunoblotting
pAb8368 was raised against the peptide M-A-E-Y-Q-A-L-C-S-S-G-P-G-M-T-S-A-G-T-K synthesized on a Milligen 9050 peptide synthesizer using standard Fmoc chemistry. The peptide was deprotected, purified by reverse-phase chromatography and the amino acid sequence was confirmed by Edman degradation; the peptide was coupled to keyhole limpet hemocyanin using the COOH-terminal–added lysine residue, and injected into a rabbit in the presence of Freund's adjuvant. Serum was prepared and tested by ELISA against the immunizing peptide. Antibody specificity was established by Western blot analysis. Conditioned medium of fibroblasts cultured 48 h in DME without serum was harvested, briefly centrifuged to clear cell debris, and precipitated with 10% TCA (Sakai and Keene 1994). Proteins were separated by 4.5% SDS-PAGE, transferred onto a nitrocellulose filter and, after treatment with 3% BSA in PBS, the filter was incubated with the pAb9543 or pAb8368 antibodies and with phosphatase-conjugated second antibodies. Antibodies were visualized using nitro-blue tetrazolium (NBT)/5-bromo-4-chloro-3′ indoylphosphate (BCIP) (Pierce Chemical Co.). Molecular mass markers included myosin (203 kD), β-galactosidase (116 kD), and phosphorylase B (87 kD; Bio-Rad Laboratories). pAb9543 was prepared by immunizing a rabbit with the recombinant fibrillin 1 polypeptide rF11, which corresponds to the NH2-terminal half of human fibrillin 1 (Reinhardt et al. 1996). pAb9543 cross-reacts with fibrillin 1 from other species, including mouse, but does not cross-react with fibrillin 2 (Pereira et al. 1997). Preparation and characterization of mAb201, the antibody that recognizes only human fibrillin 1, have been described previously (Sakai et al. 1986; Reinhardt et al. 1996).
Histological and Ultrastructural Analyses
Mouse tissues were processed as previously described (Andrikopoulos et al. 1995; Pereira et al. 1997, Pereira et al. 1999). 6-micron sections were stained with hematoxylin and eosin, and adjacent sections were assayed for elastic tissue or collagen using a commercial kit (Sigma Chemical Co.). Tsk/+ and mgR/mgR mice were 6 mo old, Tsk/mgΔ and Tsk/mgR mice were ∼2 wk old and were analyzed together with age-matched wild-type littermates. Samples of adult Tsk/+ and +/+ mouse skin were subjected to collagenase extraction and molecular sieve chromatography to isolate intact microfibrils according to the protocol of Kielty et al. 1998. Likewise, confluent cells cultured for 3–4 wk were subjected to the same collagenase treatment to purify microfibrils from assembled matrix (Kielty et al. 1998). Samples were prepared and processed for electron microscopy and immunoelectron microscopy according to the protocols of Sakai and Keene 1994.
Immunoprecipitation and Fibrillin 1 Proteolysis
For immunoprecipitation, subconfluent cells were grown for 24 h in Met/Cys-free DME supplemented with 50 μCi/ml of [35S]Cys/Met mixture (ICN Biomedicals; Gayraud et al. 1997; Reinhardt et al. 1997). After collection, the medium was supplemented with protease inhibitors to a final concentration of 2 mM Pefabloc (Roche Molecular Biochemicals) and clarified by centrifugation at 2,000 rpm for 5 min. Individual aliquots of 50 μl of protein G–agarose (Roche Molecular Biochemicals) were incubated with 3 μl of pAb9543 for 1 h at 4°C. Protein G–antibody complexes were washed three times with PBS and incubated with 500 μl of 35S-labeled medium for 1 h. The protein G–immune complexes were washed four times with PBS-Tween 0.05% and twice with PBS and dissociated by boiling for 5 min in SDS-PAGE sample buffer. Immunoprecipitated proteins were separated in 4.5% SDS-PAGE containing 5% β-mercaptoethanol. The same protocol was used for the analysis of fibrillin 1 digestion by plasmin. To reduce the background, fibronectin was depleted from the radiolabeled cell culture by gelatin–Sepharose 4B (Amersham Pharmacia Biotech). To isolate fibrillin 1 proteins, 700 μl of protein G was coupled with 150 μl of pAb9543 and the protein G–antibody complex was incubated overnight at 4°C with 35S-labeled medium (Reinhardt et al. 1997). Immune complexes were extensively washed with 50 mM Tris and 0.15 M NaCl. Fibrillin 1 was eluted with glycine/HCl, pH 2.5, buffered with Tris, and dialyzed for 12 h against 50 mM Tris, 0.15 M NaCl, and 10 mM CaCl2. Fractions obtained after elution were counted in a Beckman, LS 5000TD scintillation counter. Samples containing the same amount of radioactivity were incubated for 14 h at 37°C with increasing amounts (0–2 μg) of bovine plasmin (Sigma Chemical Co.). Samples were loaded in SDS-PAGE sample buffer with 5% β-mercaptoethanol and the degradation products were separated in 4.5% SDS-PAGE. For autoradiography, gels were fixed and treated with Amplify (Amersham Pharmacia Biotech) for 20 min, and then dried before exposure to X-ray film.
Indirect Immunofluorescence
Early passages of primary culture fibroblasts were plated on glass coverslips at a density of 175,000 cell/ml and cultured for 120 h. Cells were washed with PBS, fixed at –20°C with cold methanol for 20 min and rinsed with PBS. The primary (pAb9543, pAb8368, or mAb201) and secondary antibodies (fluorescein isothiocyanate anti-rabbit [Sigma Chemical Co.], or Cy3 anti-rabbit [Jackson ImmunoResearch Laboratories]) were applied for 1 h at room temperature (Sakai et al. 1986, Sakai et al. 1991). In some experiments, nuclei were counterstained with 0.2 μg/ml propidium iodide. In the coculture experiments, 75,000 cells/ml from each cell line were plated onto glass coverslips and cultured for 150 h before being subjected to indirect immunofluorescence. For confocal analysis, cells were cocultured for 2 wk, and the mAb201 antibody was visualized using a secondary anti-mouse antibody coupled to Texas red (Jackson ImmunoResearch Laboratories). Indirect immunofluorescence staining was carried out on 5-μm cryosections of unfixed skin samples from newborn legs following the same protocol and by using 3% BSA in PBS for 1 h before staining (Sakai et al. 1986, Sakai et al. 1991). Fluorescence was monitored with a Zeiss Axiophot microscope using timed exposures at standard magnification and field of view for a reliable assessment of staining intensity and morphology.
Results
Analysis of Compound Heterozygous Mutant Mice
Tsk/+ mice are readily recognizable from wild-type littermates for the tightness of the skin, as early as 5–7 d of age, and, by 3 mo, they display a characteristic hump in the shoulder region. Skin tightness correlates with excessive matrix accumulation and marked hyperplasia in the loose connective tissue (Fig. 3 a), whereas the hunched posture is a consequence of bone overgrowth (Green et al. 1975; Jimenez et al. 1984). Additional distinguishing features of the Tsk/+ mouse are distended air spaces in the lungs and enlarged auricles of the heart, both noticeable by the first month of postnatal life (Green et al. 1975; Martorana et al. 1989). Bone overgrowth and lung emphysema are also shared by the mgR/mgR mice, which, however, show no signs of skin (Fig. 3 b) or myocardial abnormalities (Pereira et al. 1999). Aneurysmal dilation with focal fragmentation of elastic fibers in the aortic wall is the distinguishing vascular feature of the mgR/mgR mice, a trait that becomes more evident between the third and fourth month of life (Pereira et al. 1999). The phenotypic consequences of the Tsk, mgR, and mgΔ alleles can be summarized as follows: connective tissue hyperplasia is only associated with the Tsk allele; dissecting aneurysm is only associated with the mgR and mgΔ alleles; and bone overgrowth and lung emphysema are common traits of the Tsk, mgR, and mgΔ alleles (Table ).
Figure 3.
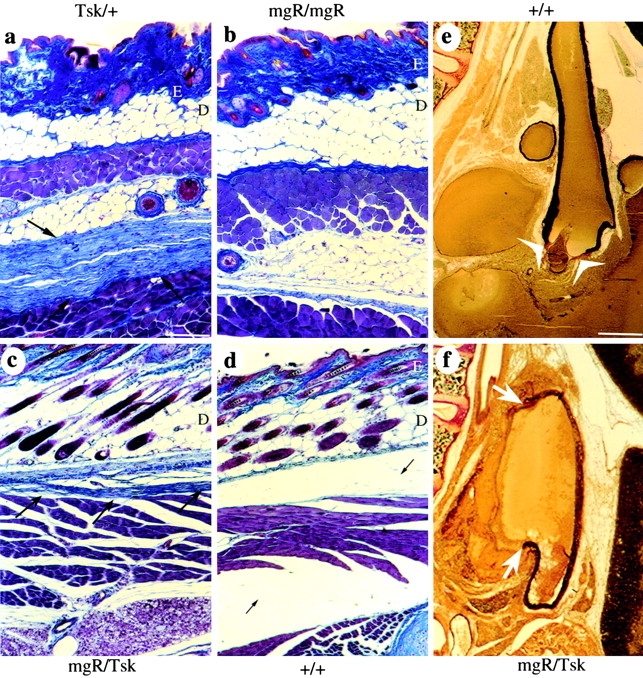
Skin and vascular phenotypes of Fbn1 mutant mice. Trichrome-stained skin sections from adult (a and b) and newborn (c and d) mice heterozygous for the Tsk allele (a), homozygous for the mgR allele (b), compound heterozygous for the Tsk and mgR alleles (c), and from wild-type (+/+) animals (d). Dermis and epidermis are indicated by the letters D and E, respectively. The arrows point to the loose connective tissue that contains excessive collagen fibers and is tightly bound to the muscle layer in the adult Tsk/+, and in newborn Tsk/mgR animals. Cross-section of the ascending aorta stained with van-Gieson from 3-wk-old Tsk/+ (e) and Tsk/mgR (f) mice. Arrows point to the fragmentation of elastic lamellae in the Tsk/mgR sample, whereas the arrowheads indicate the valve leaflets in the cross-section of the Tsk/+ vessel. Bars: (a–d) 140 μm; 520 μm (e and f).
Table 1.
Genotype-phenotype Correlations in Fbn1 Mutant Mice
| Boneovergrowth | Lungemphysema | Vasculardissection | Skinfibrosis | Myocardialhypertrophy | Lifespan | |
|---|---|---|---|---|---|---|
| Tsk/+ | + | + | − | + | + | Normal (n = 100) |
| mgR/mgR | + | + | + | − | − | 3.8 ± 3.4 mo (n = 50) |
| mgΔ/mgΔ | n.a. | + | + | − | − | 3 ± 1 wk (n = 30) |
| mgΔ/+ | − | − | − | − | − | Normal (n = 100) |
| Tsk/mgR | n.a. | + | + | + | + | 16 ± 15 d (n = 40) |
| Tsk/mgΔ | n.a. | + | + | + | + | 11 ± 9 d (n = 40) |
Heterozygous fibrillin 1 mutant mice were crossed to examine the segregation of the distinguishing and common traits in the compound heterozygous Tsk/mgR and Tsk/mgΔ mice. Compound heterozygous Tsk/mgR mice die within the first 3 wk of life of the same vascular complications as mgR/mgR animals, which are absent in the parental Tsk/+ line (Fig. 3e and Fig. f). Moreover, Tsk/mgR animals exhibit the skin abnormalities of the Tsk/+ mice, which are absent in mgR/mgR mice (Fig. 3 c). These results indicate codominant expression of the mutant Tsk and mgR allele; the same conclusion was reached with compound Tsk/mgΔ heterozygotes (Table ). Additionally, Tsk/+ animals are more severely affected than mgΔ/+ mice, in spite of the fact that the two mutant mice make comparable amounts of wild-type fibrillin 1 (Table ). This last observation raised the possibility that the Tsk protein may participate in heterotypic assembly, thus, exerting some antimorphic effect on the normal gene product.
Microfibrillar Assembly Differs in Tsk/Tsk, Tsk/+, and +/+ Cell Cultures
Metabolically labeled polypeptides from the media of cultured fibroblasts with different combinations of Fbn1 alleles were immunoprecipitated using polyclonal antibodies (pAb9543) that recognize the NH2-terminal half of fibrillin 1 (Fig. 2). Since pAb9543 epitopes are present in both wild-type and mutant fibrillin 1 and in both human and mouse proteins, the pAb9543 antibodies will be referred to as α-Fbn. As expected, SDS-PAGE analysis of the α-Fbn immunoprecipitates correlated normal size (350 kD), shorter (331 kD), and longer (418 kD) fibrillin 1 proteins with their respective Fbn1 alleles (Fig. 4 a). It also confirmed the presence of comparable amounts of mutant and wild-type fibrillin 1 (418 and 350 kD, respectively) in the Tsk/+ sample, and the relatively low levels of mgΔ and mgR products (331 and 350 kD, respectively) in the compound heterozygous mutant cell cultures (Fig. 4 a).
Figure 4.
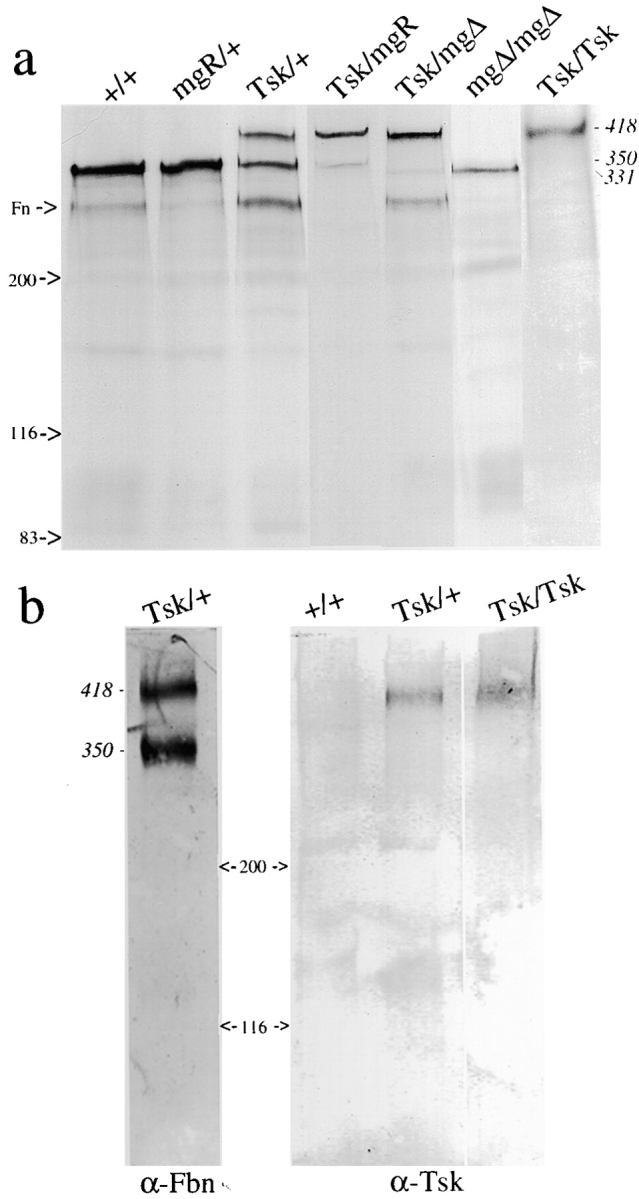
(a) SDS-PAGE analysis under reducing conditions of metabolically labeled fibrillin 1 immunoprecipitated with the pAb9543 antibody from the medium of cultured fibroblasts isolated from wild-type (lane 1), mgR/+ (lane 2), Tsk/+ (lane 3), Tsk/mgR (lane 4), Tsk/mgΔ (lane 5) and mgΔ/mgΔ (lane 6) adult mice, and from Tsk/Tsk (lane 7) embryos. (b) Western blot analysis under native conditions of medium of cell cultures from wild-type (+/+) and heterozygous (Tsk/+) and homozygous (Tsk/Tsk) mutant animals with antibodies against the NH2 terminus of fibrillin 1 (pAb9543 or α-Fbn) and the mutant Tsk fibrillin 1 (pAb8368 or α-Tsk). In both panels, the molecular weights of the fibrillin 1 proteins are indicated along with size markers. Contaminating traces of fibronectin (Fn) are highlighted in a.
Next, the α-Fbn antibodies were used to examine the microfibrillar meshwork assembled in vitro by hyperconfluent fibroblasts. Consistent with quantitation of fibrillin 1 expression in Fig. 4 a, the amount of α-Fbn immunofluorescent material deposited after 120 h of culture was noticeably less in mgR/+ than +/+ cultures (Fig. 5, compare a and b). A less abundant meshwork was also observed in the Tsk/+ sample, and even less in the Tsk/mgR cell cultures (Fig. 5, compare c and d). The same relative decrease in the amount of ordered meshwork was observed with the mgΔ/+ and Tsk/mgΔ cells (data not shown). Taken together, the results suggested that Tsk fibrillin 1 contributes poorly to the assembly of the microfibrillar meshwork that is normally observed in hyperconfluent +/+ fibroblast cultures. To test this postulate, the analysis was repeated with an antibody that recognizes only Tsk fibrillin 1.
Figure 5.
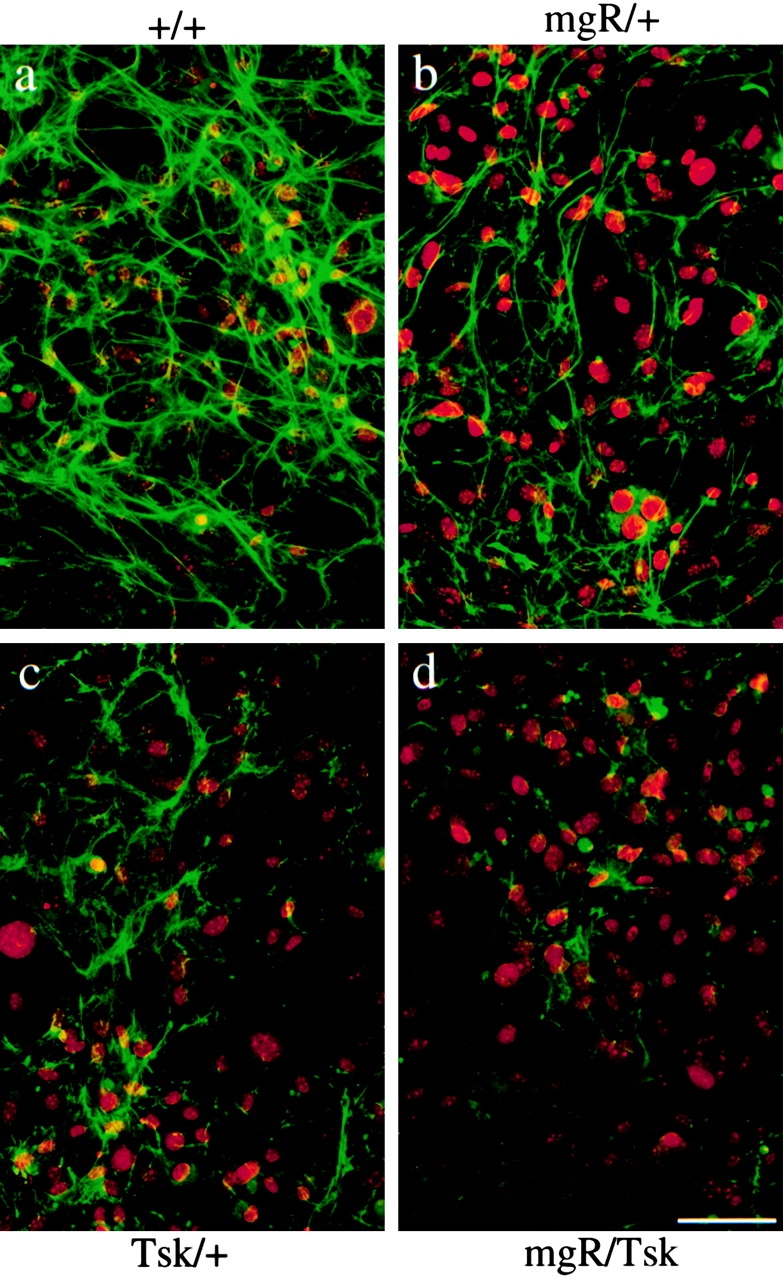
Immunofluorescence of cultured fibroblasts from wild-type (a), mgR/+ (b), Tsk/+ (c) and Tsk/mgR (d) mice using the anti-fibrillin 1 antibody pAb9543 (α-Fbn). Nuclei were stained with propidium iodine to appreciate cell density. Bar, 60 μm.
The Tsk mutation is an in-frame duplication of exons 17–40 and, as a result, the mutant sequence differs from the normal counterpart by interrupting the continuity of the second 8-cys motif of fibrillin 1, which is normally encoded by exons 16 and 17. In turn, this creates a novel peptide with a single cysteine residue (the segment encoded by exon 17) immediately adjacent to the 24th cbEGF motif of fibrillin 1, which is the repeat encoded by exon 40 (Fig. 2; Siracusa et al. 1996). We reasoned that this partial domain sequence may not be properly folded and would be a good target for antipeptide antibodies. Accordingly, a synthetic peptide corresponding to 19 amino acids across the junction sequence encoded by exons 40/17 was used to raise anti-Tsk fibrillin 1 antibodies. The specificity of the resulting antiserum (pAb8368 or α-Tsk) was tested by a Western blot analysis of proteins from conditioned media of +/+, Tsk/+, and Tsk/Tsk cell cultures. Unlike α-Fbn, which recognized both the normal 350-kD and mutant 418-kD products, the α-Tsk antiserum showed specificity only for the longer species (Fig. 4 B). The specificity of α-Tsk was further supported by the lack of immunostaining of +/+ fibroblast cultures (Fig. 6 b). These results indicated that the Tsk peptide sequence is available for antibody binding in native Tsk fibrillin 1 and unavailable in native wild-type fibrillin 1; this, in turn, suggested that the sequence present in the native wild-type fibrillin 1 is probably folded in a manner that prevents antibody binding. Parallel immunostaining of Tsk/+ cell cultures with α-Fbn and α-Tsk antisera visualized both quantitative and morphological differences. Whereas the α-Fbn antibodies revealed an elaborate network of immunoreactive microfibrils, the α-Tsk antiserum yielded a reduced amount of immunoreactive material (Fig. 6c and Fig. d). The same results were obtained with fibroblasts from the compound heterozygous Tsk/mgR and Tsk/mgΔ mutant mice (data not shown). Surprisingly, both antibodies also revealed an immunoreactive meshwork in Tsk/Tsk cell cultures (Fig. 6e and Fig. f). However, in this case, the pattern of the immunoreactive material was more diffuse, less linear, and more punctate than the one generated by the α-Fbn antiserum with +/+ fibroblasts (Fig. 6 a). The morphological differences between the wild-type and mutant fibrils were more apparent when the immunostaining patterns of the +/+ and Tsk/Tsk cultures were compared at a higher magnification (Fig. 6g and Fig. h).
Figure 6.
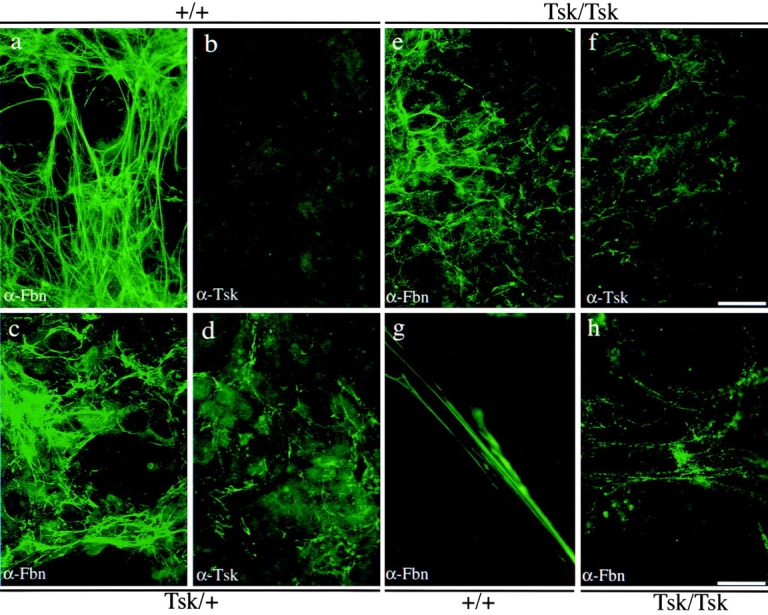
Immunofluorescence of cultured fibroblasts from wild-type (a and b), Tsk/+ (c and d) and Tsk/Tsk (e and f) animals using the pAb9543 (α-Fbn) antibody (a, c, and e) or pAb8368 (α-Tsk) antibody (b, d, and f). Bar, 65 μm. Representative fields at a higher magnification of wild-type (g) and Tsk/Tsk (h) samples immunostained with pAb9543 (α-Fbn) are shown to better appreciate morphological differences, Bar, 15 μm.
To validate the immunostaining data at the ultrastructural level, microfibrils were purified from the matrix elaborated by hyperconfluent +/+ and Tsk/Tsk cells and examined using two distinct electron microscopic techniques, rotary shadowing, and negative staining (Fig. 7). In Tsk/Tsk preparations, the former technique visualized microfibrils that were fewer and significantly shorter than +/+ microfibrils, and which also display irregular arms and globular domains (Fig. 7, compare a to c–f). The gentler technique of negative staining revealed that the shorter Tsk/Tsk microfibrils have beads of different sizes and irregular periodicity (Fig. 7, compare b to g–k). The immunofluorescence and ultrastructural data suggested the following two points. First, Tsk fibrillin 1 molecules by themselves can initiate polymerization, but are incapable of elaborating in vitro the same complex meshwork of microfibrils as wild-type proteins; second, Tsk fibrillin 1 molecules are partially incorporated into the meshwork assembled by the wild-type proteins (Fig. 1 d).
Figure 7.
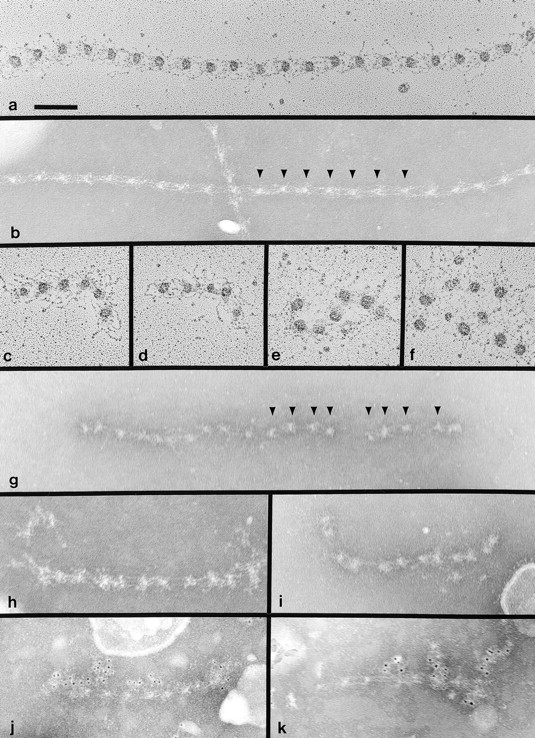
Electron microscopy of fibrillin 1 microfibrils extracted from the matrix of +/+ (a and b) and Tsk/Tsk fibroblasts (c–k). Microfibrils were examined after rotary shadowing (a and c–f) or after negative staining (b and g–k). Tsk/Tsk microfibrils were specifically labeled with pAb8368 (α-Tsk) antibody (j and k). Arrowheads in b indicate typical regular periodicities found between globular beads in normal microfibrils. Arrowheads in g indicate typical irregular periodicities found in Tsk/Tsk microfibrils. Bar, 100 nm.
Wild-type Fibrillin 1 Drives Partial Polymerization Of Mutant Tsk Fibrillin 1
We next tested whether Tsk fibrillin 1 could copolymerize with wild-type fibrillin 1 by assaying cocultures of normal and mutant cells. Recently, we found that human amnion, epithelial-like WISH cells secrete fibrillin 1 proteins into the medium but fail to assemble a microfibrillar network, either because the molecules are present at less than optimal concentration or because the cell line lacks other factors required for fibrillin 1 microfibril assembly. However, coculturing WISH cells together with mouse dermal fibroblasts leads to copolymerization of the human and mouse fibrillin 1 proteins (Sakai, L.Y., unpublished data). This same approach was employed here to test whether the microfibrillar pattern of Tsk/Tsk matrix could be rescued by wild-type human fibrillin 1 secreted by WISH cells.
WISH and Tsk/Tsk cells were cultured separately or together, and each sample was immunostained with antibodies specific for human fibrillin 1 (mAb201 or α-hFbn), human and mouse fibrillin 1 (pAb9543 or α-Fbn), or Tsk fibrillin 1 (pAb8368 or α-Tsk). As expected, no extracellular immunostainable microfibrillar meshwork was observed in the WISH sample with α-hFbn antibodies, and only diffuse and irregular staining was seen in the Tsk/Tsk sample using the α-Fbn antiserum (Fig. 8, a and b). Coculturing the two cell types led to the formation of an elaborate meshwork of microfibrillar bundles in which both α-hFbn and α-Tsk antibodies were colocalized with confocal microscopy (Fig. 8, d–i). Finally, immunoelectron microscopy of the extracellular microfibrils elaborated by the cocultured cells confirmed that the immunostainable meshwork is made of microfibrils containing both human WISH fibrillin 1 and mouse Tsk fibrillin 1 (Fig. 9e and Fig. f). The morphology of the rescued Tsk microfibrils displayed features of the Tsk/Tsk microfibrils, such as abnormal globular domains and irregular periodicities between globules; unlike Tsk/Tsk microfibrils, however, these abnormal structures were embedded within seemingly normal microfibrils (Fig. 9). Therefore, the results of the coculture experiments demonstrated copolymerization between human wild-type and mouse mutant fibrillin 1, and suggested that Tsk monomers participate in the initial steps of fibrillin 1 polymerization. Implicitly, the experiments confirmed that protein concentration and/or cell type–specific factors contribute to the in vitro assembly of fibrillin 1 microfibrils.
Figure 8.
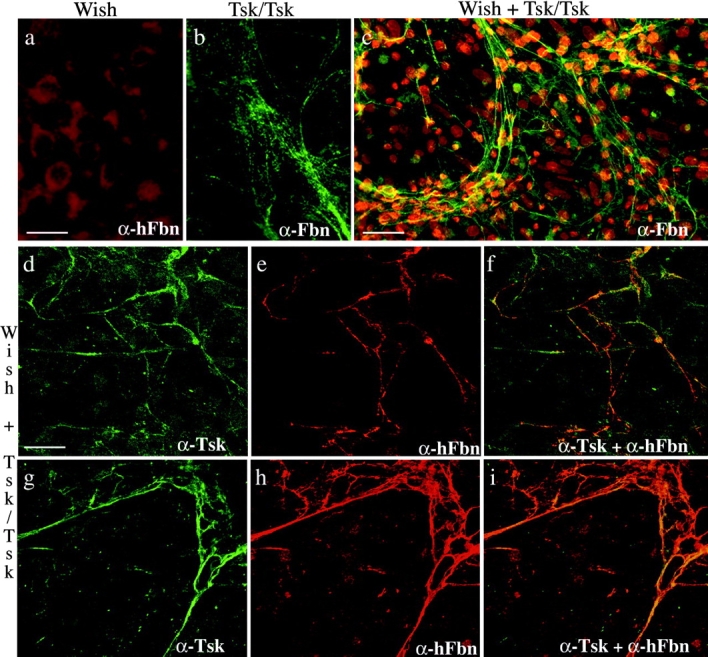
Immunofluorescence of WISH cultures using the mAb201 (α-hFbn) antibody (a), Tsk/Tsk cultures using the pAb9543 (α-Fbn) antibody (b), and WISH and Tsk/Tsk cocultures using the pAb9543 (α-Fbn) (c), pAb8368 (α-Tsk) (d and g), or mAb201 (α-hFbn) antibody (e and h). f and i show the superimposition of the images in d and e and g and h, respectively. The antibody pAb9543 (α-Fbn) was employed to better appreciate the morphology (b) and extent (c) of the microfibrillar aggregates; additionally, nuclei of cocultured cells were stained with propidium iodine to document the level of cell confluency. Bars: (a and b) 20 μm; (c) 60 μm; (d–i) 40 μm.
Figure 9.
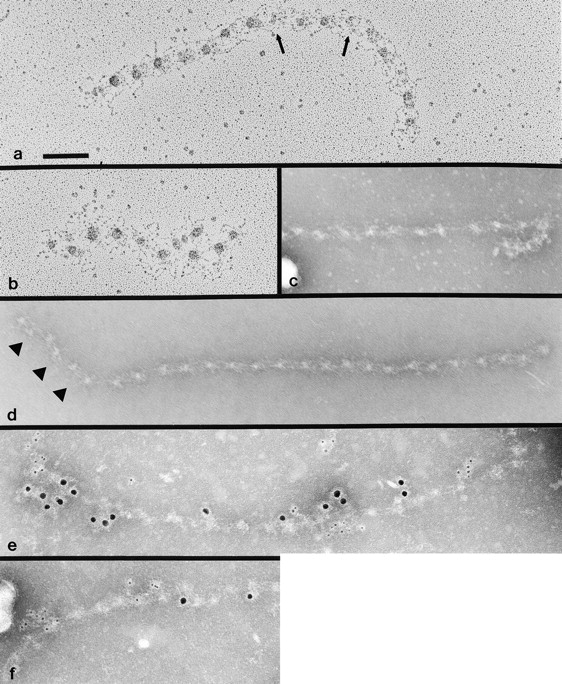
Electron microscopy of fibrillin 1 microfibrils extracted from the matrix of cocultures of WISH and Tsk/Tsk fibroblasts and viewed after rotary shadowing (a and b) or after negative staining (c–f). Arrows in a indicate abnormal globular morphologies. Arrowheads in d indicate irregularities in periodicity. Microfibrils in e and f are labeled with mAb201 (recognized by the large, 10-nm, gold conjugate), specific for human fibrillin 1, and pAb8368 (recognized by the small, 5-nm, gold conjugate), specific for mouse Tsk fibrillin 1. Bar, 100 nm.
Tsk Fibrillin 1 Is More Susceptible to Proteolysis
Rotary shadowing of microfibrils prepared from Tsk/+ skin has allegedly visualized two morphologically distinct beaded structures in equal amounts (Kielty et al. 1998). One fraction was reported to exhibit normal morphology and periodicity, and the other diffuse interbeads and longer periodicity. We repeated the ultrastructural analysis on Tsk/+ skin samples using the same experimental protocol as Kielty et al. 1998 and found no evidence for two different populations of beaded structures (data not shown). Although we occasionally encountered images of abnormal-beaded structures of mixed periodicity, their number was too limited to draw any meaningful conclusion.
Indirect immunofluorescence on Tsk/+ skin with α-Fbn and α-Tsk antibodies demonstrated that 418-kD fibrillin 1 molecules are integrated into the microfibrillar network. However, the images also suggested that the junctional epitopes of Tsk protein may not be as abundant as the epitopes recognized by α-Fbn, particularly in the mature, deeper portions of the dermis (Fig. 10). Immunoelectron microscopy confirmed the presence of mutant fibrillin 1 in the microfibrils of Tsk/+ skin samples (Fig. 11).
Figure 10.

Immunofluorescence of Tsk/+ skin using the pAb9543 (α-Fbn) (a) or pAb8368 (α-Tsk) antibody (b). The immunofluorescence of wild-type skin using the α-Tsk antibody is also shown (c) to appreciate the unspecific nature of the strong epidermis staining of b. Bar, 60 mm.
Figure 11.
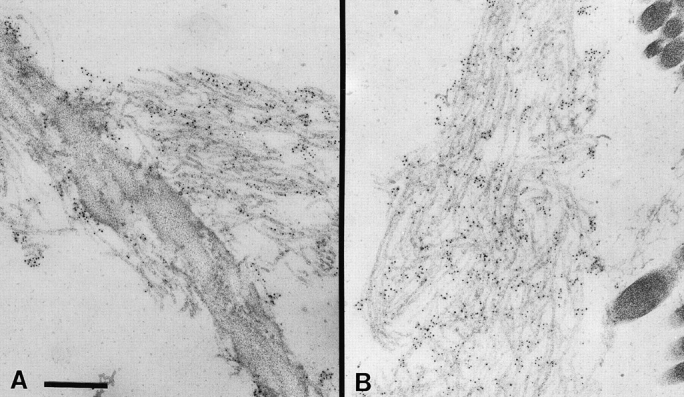
Immunoelectron microscopy of Tsk/+ skin using the anti-Tsk fibrillin 1 antibody (pAb8368) which localizes mutant molecules to elastin associated (A) or isolated (B) microfibrils. Bar, 250 nm.
The last experiment was designed to assess the stability of the Tsk protein using an in vitro assay that compared the resistance of wild-type and mutant molecules to a broad substrate protease. To this end, immunoprecipitates of metabolically labeled fibrillin 1 from cultured Tsk/+ cells were incubated in vitro with increasing amounts of plasmin. SDS-PAGE analysis of the digested products documented the more rapid disappearance of the mutant 418-kD product with respect to the wild-type 350-kD species (Fig. 12).
Figure 12.

SDS-PAGE analysis under reducing condition of radioimmunoprecipitated fibrillin 1 from Tsk/+ fibroblast cultures incubated for 16 h with increasing amounts of plasmin.
Discussion
Our current understanding of the role of fibrillin 1 microfibrils in connective tissue physiology is based primarily on the study of human mutations in MFS and homologous gene targeting in the mouse (Dietz and Pyeritz 1995; Ramirez et al. 1999). The dominant inheritance of MFS has underscored the dominant negative (antimorphic) nature of mutations in structural macromolecules, whereas the MFS-like phenotype of fibrillin 1–deficient (hypomorphic) mice has pointed to the importance of optimal levels of gene expression (Fig. 1 a). Together, these results have indicated that alternative pathogenic pathways can similarly result in microfibrillar incompetence and pleiotropic manifestations that include the biomechanical collapse of the aortic wall (Pereira et al. 1999). These observations have led to the formulation of the threshold hypothesis, which correlates decreasing levels of functionally competent microfibrils with increasing phenotypic diversity and clinical severity of fibrillin 1 mutations (Pereira et al. 1999).
According to the pathogenic model of fibrillin 1 mutations in MFS, one would have predicted a full antimorphic effect of the Tsk mutation leading to the collapse of the elastic lamellae in the aortic wall and the early demise of Tsk/+ animals (Dietz and Pyeritz 1995; Ramirez 1996). As this is not the case, an alternative explanation is that the Tsk gene product may fail to participate in microfibril assembly and, consequently, that the skeletal and lung abnormalities of heterozygous Tsk/+ mice may be indicative of fibrillin 1 deficiency. In line with this idea, Kielty et al. 1998 proposed that the longer Tsk fibrillin 1 molecules polymerize separately from their wild-type counterparts into beaded microfibrils with extended bead-to-bead periodicities. This conclusion contrasts with the results of the present study, which offer an alternative explanation for the metabolic and phenotypic consequences of the Tsk mutation that is based on three distinct lines of experimental evidence.
The first line of evidence relies on the phenotypic features of mutant mice with different combinations of Fbn1 alleles. Tsk/+ mice produce nearly the same amount of normal fibrillin 1 as mgΔ/+ mice and, yet, they are more severely affected. The most likely explanation for this apparent discrepancy is that the greater number of Tsk molecules, compared with mgΔ products, may exert a partial antimorphic effect on the wild-type fibrillin 1 proteins. In this respect, the Tsk allele should be viewed as an antimorphic mutation with partial penetrance.
The second line of evidence in support of our pathogenic model of the Tsk mutation was derived from cell culture experiments. Immunofluorescence analysis of fibrillin 1 microfibrils elaborated by various cell cultures demonstrated decreased immunoreactivity in the following order: +/+, mgR/+ or Tsk/+, and Tsk/mgR. These results indicate that Tsk fibrillin 1 cannot substitute for wild-type fibrillin 1 in this assembly assay, and imply that the mutant protein may be assembled into microfibrils more slowly than the wild-type counterpart. In contrast to the extended linear microfibrils found in wild-type cultures, immunofluorescence of Tsk/Tsk cultures demonstrated a punctate pattern of fibrillin 1 in somewhat focal aggregates. Consistent with these images, electron microscopic analyses revealed short strings of beaded fibrils composed of Tsk fibrillin 1. Compared with wild-type fibrillin 1 microfibrils, the Tsk/Tsk aggregates exhibited clear irregularities in the size and shape of the globular beads, as well as in the distances between beads. These data strongly suggest that Tsk fibrillin 1 can polymerize into microfibril-like structures with abnormal morphologies.
The third and final supporting evidence was based on a novel coculture assay, which documented the ability of Tsk fibrillin 1 to interact with the wild-type counterpart. The resulting fibrillin 1 microfibrils were long and composed of both wild-type human fibrillin 1 and mutant mouse fibrillin 1, thus, demonstrating that Tsk and wild-type molecules can copolymerize into a single microfibril (Fig. 1 d). The microfibrils exhibited focal areas of abnormalities similar to those found in Tsk/Tsk fibrils. Interestingly, abnormal microfibrils were not seen in Tsk/+ cell cultures or tissues. We believe that the reason for this apparent discrepancy reflects intrinsic differences of the two experimental systems. Fibroblasts synthesize and secrete the mutant and wild-type products together before they are assembled extracellularly and a number of factors may influence this coupled process. In the coculture system, the two populations of wild-type and mutant molecules are secreted separately and, subsequently, are assembled by the fibroblasts. Therefore, the cocultured system should be viewed more as a biochemical binding assay to test homopolymerization versus heteropolymerization rather than a physiological model of microfibril assembly. With this reservation in mind, the coculture data indicate that Tsk fibrillin 1, when copolymerized with wild-type fibrillin 1, can participate in microfibril elongation.
The comparison of the mutant Fbn1 mice has also raised an interesting structural consideration. The phenotypic severity of Tsk and mgΔ homozygotes is dramatically different. Tsk/Tsk embryos produce a homogenous population of longer molecules in normal abundance; mgΔ/mgΔ mice produce a homogeneous population of shorter molecules in a much lower abundance. However, Tsk/Tsk embryos die in utero at the peri-implantation stage, and mgΔ/mgΔ embryos complete development (Green et al. 1975; Pereira et al. 1997). Conceivably, these very different outcomes reflect fundamental differences in the assembly potential and/or stability of the two mutant chains. Deletion of exons 19–24 results in no apparent morphological differences between wild-type fibrillin 1 microfibrils and mgΔ/mgΔ fibrillin 1 microfibrils (Pereira et al. 1997). However, duplication of this region and surrounding domains in Tsk fibrillin 1 results in Tsk/Tsk aggregates that display distinct abnormalities. The morphological abnormalities of Tsk/Tsk microfibrils must, therefore, be due to the duplication of the region encoded by exons 17–40. We propose that the region containing the long stretch of cbEGF domains, which is duplicated in Tsk fibrillin 1, is required for proper linear and unbranched alignment of fibrillin 1 molecules within the microfibril and for elongation of microfibrils.
The Tsk duplication begins with exon 17, which encodes the COOH-terminal 18 amino acid residues of the second 8-cys domain (Siracusa et al. 1996). The full 8-cys domain contains 69 residues, including eight cysteines, which are predicted to form four intrachain disulfide bonds. Within the duplicated 18 residues of the 8-cys domain is the final cysteine residue. Our immunochemical data, using antisera raised to this synthetic peptide, demonstrate that this sequence is normally folded in the native molecule in a manner that precludes binding to the antipeptide antibodies. In Tsk fibrillin 1, this region is available for reactivity to antibodies. We speculate that this improperly folded region of Tsk fibrillin 1 may result in two effects. It may form an inappropriate flexible region that could distort the proper linear alignment of fibrillin 1 necessary for elongation of microfibrils; and it may cause inappropriate folding of adjacent domains, leading to a region of increased proteolytic susceptibility. Consistent with the latter prediction, the longer Tsk fibrillin 1 was degraded at a faster rate than wild-type fibrillin 1 in an in vitro assay of Tsk/+ proteins. It is likely that the duplication of a partial domain sequence destabilized adjacent domains, which contain sites (R or K) for plasmin cleavage. In vivo evidence also suggests that Tsk fibrillin 1 may be selectively degraded by tissue proteases. Immunolocalization of Tsk fibrillin 1 in Tsk/+ skin, which is the result of development over some length of time, was noticeably reduced compared with antibodies directed toward wild-type fibrillin 1. Further supporting this contention is the notion that the overall amount of matrix components, including microfibrils, is substantially elevated in Tsk/+ skin as result of fibrosis (Jimenez et al. 1984). The most intense labeling by the α-Tsk antibodies was observed at the dermal-epidermal junction, an area of the skin which is thought to represent the more immature stroma compared with the connective tissue of the deeper dermis.
In conclusion, we propose that incorporation of Tsk fibrillin 1 along with wild-type fibrillin 1 renders all microfibrils more susceptible to proteolysis. According to this model, Tsk/+ animals are more severely affected than mgΔ/+ mice because of the relatively higher ratio of mutant to wild-type molecules in the former compared with the latter. This model is in line with the conclusion of a recent study on the consequences of mutations in cbEGF domains in fibrillin 1 (Reinhardt et al. 2000). This study proposed that proteolysis of conformationally abnormal fibrillin 1 molecules by tissue proteases, which are normally unable to degrade the microfibrils, may lead to destruction of the supramolecular assembly (Reinhardt et al. 2000). A similar pathogenic paradigm has recently emerged to explain the effect of collagen mutations on the long-term stability of the resulting fibers (Fernandes et al. 1998; Weis et al. 1998). Although we have provided a reasonable account for the metabolic and phenotypic consequences of the Tsk mutation, the role of Tsk fibrillin 1 in the genesis of tissue hyperplasia and autoimmunity remains unresolved.
Acknowledgments
The authors are much indebted to Drs. S. Jimenez and L. Siracusa (Jefferson Medical College, Philadelphia, PA) for continued support, Dr. K. Kasturi (Mount Sinai School of Medicine, New York, NY) for useful suggestions, and Dr. D. Rifkin (New York University, Medical Center, New York, NY) for critically reviewing the manuscript. They also wish to thank Ms. N. Charbonneau, S.F. Tufa, and S. Lee for excellent technical assistance and Ms. K. Johnson for typing the manuscript.
This work was supported by grants from the National Institutes of Health (AR42044 and AR32564), Shriners Hospital for Children, the National Marfan Foundation, and the Dr. Amy and James Elster Research Fund (all except Shriners [which is to L. Sakai] are to F. Ramirez).
Footnotes
Abbreviations used in this paper: cbEGF, calcium-binding epidermal growth factor-like; cys, cysteine; Fbn1, mouse fibrillin 1 gene; MFS, Marfan syndrome; Tsk, Tight skin mutation.
References
- Andrikopoulos K., Liu X., Keene D.R., Jaenisch R., Ramirez F. Targeted mutation in the Col5a2 gene reveals a regulatory role for type V collagen during matrix assembly. Nat. Genet. 1995;9:31–36. doi: 10.1038/ng0195-31. [DOI] [PubMed] [Google Scholar]
- Bocchieri M.H., Jimenez S.A. Animal models of fibrosis. Rheum. Dis. Clin. North Am. 1990;16:153–167. [PubMed] [Google Scholar]
- Dietz H.C., Pyeritz R.E. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum. Mol. Genet. 1995;4:1799–1809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- Dietz H.C., Ramirez F., Sakai L.Y. Marfan syndrome and other microfibrillar diseases. Adv. Hum. Genet. 1994;22:153–186. doi: 10.1007/978-1-4757-9062-7_4. [DOI] [PubMed] [Google Scholar]
- Fernandes R.J., Wilkin D.J., Weis M.A., Cohn D.H., Rimoin D.L., Eyre D.R. Incorporation of structurally defective type II collagen in cartilage matrix in Kniest chondrodysplasia. Arch. Biochem. Biophys. 1998;355:282–290. doi: 10.1006/abbi.1998.0745. [DOI] [PubMed] [Google Scholar]
- Gayraud B., Hopfner B., Jassim A., Aumailley M., Bruckner-Tuderman L. Characterization of a 50-kDa component of epithelial basement membranes using GDA-J/F3 monoclonal antibody. J. Biol. Chem. 1997;272:9531–9538. doi: 10.1074/jbc.272.14.9531. [DOI] [PubMed] [Google Scholar]
- Green M.C., Sweet H.O., Bunker L.E. Tight-skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am. J. Pathol. 1975;82:493–512. [PMC free article] [PubMed] [Google Scholar]
- Jimenez S.A., Millan A., Bashey R.I. Scleroderma-like alterations in collagen metabolism occurring in the TSK (tight skin) mouse. Arthritis Rheum. 1984;27:180–185. doi: 10.1002/art.1780270209. [DOI] [PubMed] [Google Scholar]
- Kasturi K.N., Hatakeyama A., Murai C., Gordon R., Phelps R.G., Bona C.A. B-cell deficiency does not abrogate development of cutaneous hyperplasia in mice inheriting the defective fibrillin-1 gene. J. Autoimmunity. 1997;10:505–517. doi: 10.1006/jaut.1997.0158. [DOI] [PubMed] [Google Scholar]
- Keene D.R., Sakai L.Y., Burgeson R.E. Human bone contains type III collagen, type VI collagen, and fibrillintype III collagen is present on specific fibers that may mediate attachment of tendons, ligaments, and periosteum to calcified bone cortex. J. Histochem. Cytochem. 1991;39:59–69. doi: 10.1177/39.1.1983874. [DOI] [PubMed] [Google Scholar]
- Kielty C.M., Raghunath M., Siracusa L.D., Sherratt M.J., Peters R., Shuttleworth C.A., Jimenez S.A. The Tight skin mousedemonstration of mutant fibrillin-1 production and assembly into abnormal microfibrils. J. Cell Biol. 1998;140:1159–1166. doi: 10.1083/jcb.140.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana P.A., van Even P., Gardi C., Lungarella G. A 16-month study of the development of genetic emphysema in tight-skin mice. Am. Rev. Resp. Dis. 1989;139:226–232. doi: 10.1164/ajrccm/139.1.226. [DOI] [PubMed] [Google Scholar]
- Mecham R.P., Davies E.C. Elastic fiber structure and assembly. In: Yurchenco P.D., Birk D.E., Mecham R.P., editors. Extracellular Matrix Assembly and Structure. Academic Press; New York: 1994. pp. 281–314. [Google Scholar]
- Pereira L., Andrikopoulos K., Tian J., Lee S.Y., Keene D.R., Ono R., Reinhardt D.P., Sakai L.Y., Biery N.J., Bunton T., Dietz H.C., Ramirez F. Targeting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- Pereira L., Lee S.Y., Gayraud B., Andrikopoulos K., Shapiro S.D., Bunton T., Biery N.J., Dietz H.C., Sakai L.Y., Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F. Fibrillin mutations in Marfan syndrome and related phenotypes. Curr. Opin. Genet. Dev. 1996;6:309–315. doi: 10.1016/s0959-437x(96)80007-4. [DOI] [PubMed] [Google Scholar]
- Ramirez F., Gayraud B., Pereira L. Marfan Syndromenew clues to genotype-phenotype correlations. Annu. Med. 1999;31:202–207. doi: 10.3109/07853899909115979. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Keene D.R., Corson G.M., Poschl E., Bachinger H.P., Gambee J.E., Sakai L.Y. Fibrillin-1organization in microfibrils and structural properties. J. Mol. Biol. 1996;258:104–116. doi: 10.1006/jmbi.1996.0237. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Ono R.N., Sakai L.Y. Calcium stabilizes fibrillin-1 against proteolytic degradation. J. Biol. Chem. 1997;272:1231–1236. doi: 10.1074/jbc.272.2.1231. [DOI] [PubMed] [Google Scholar]
- Reinhardt D.P., Ono R.P., Notbohm H., Muller P.K., Bachinger H.P., Sakai L.Y. Mutations in calcium-binding EGF modules render fibrillin 1 susceptible to proteolysisa potential disease-causing mechanism in Marfan Syndrome. J. Biol. Chem. 2000;275:12339–12345. doi: 10.1074/jbc.275.16.12339. [DOI] [PubMed] [Google Scholar]
- Ren Z.X., Brewton R.G., Mayne R. An analysis by rotary shadowing of the structure of the mammalian vitreous humor and zonular apparatus. J. Struct. Biol. 1991;106:57–63. doi: 10.1016/1047-8477(91)90062-2. [DOI] [PubMed] [Google Scholar]
- Sakai L.Y., Keene D.R. Fibrillinmonomers and microfibrils. Methods Enzymol. 1994;245:29–52. doi: 10.1016/0076-6879(94)45004-8. [DOI] [PubMed] [Google Scholar]
- Sakai L.Y., Keene D.R., Morris N.P., Burgeson R.E. Type VII collagen is a major structural component of anchoring fibrils. J. Cell Biol. 1986;103:1577–1586. doi: 10.1083/jcb.103.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai L.Y., Keene D.R., Glanville R.W., Bachinger H.P. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J. Biol. Chem. 1991;266:14763–14770. [PubMed] [Google Scholar]
- Siracusa L.D., McGrath R., Ma Q., Moskow J.J., Manne J., Christner P.J., Buchberg A.M., Jimenez S.A. A tandem duplication within the fibrillin 1 gene is associated with the mouse tight skin mutation. Genome Res. 1996;6:300–313. doi: 10.1101/gr.6.4.300. [DOI] [PubMed] [Google Scholar]
- Weis M.A., Wilkin D.J., Kim H.J., Wilcox W.R., Lachman R.S., Rimoin D.L., Cohn D.H., Eyre D.R. Structurally abnormal type II collagen in a severe form of Kniest dysplasia caused by an exon 24 skipping mutation. J. Biol. Chem. 1998;273:4761–4768. doi: 10.1074/jbc.273.8.4761. [DOI] [PubMed] [Google Scholar]