

Abstract
Cortical vesicles (CV) possess components critical to the mechanism of exocytosis. The homotypic fusion of CV centrifuged or settled into contact has a sigmoidal Ca2+ activity curve comparable to exocytosis (CV–PM fusion). Here we show that Sr2+ and Ba2+ also trigger CV–CV fusion, and agents affecting different steps of exocytotic fusion block Ca2+, Sr2+, and Ba2+-triggered CV–CV fusion. The maximal number of active fusion complexes per vesicle, <n\>Max, was quantified by NEM inhibition of fusion, showing that CV–CV fusion satisfies many criteria of a mathematical analysis developed for exocytosis. Both <n\>Max and the Ca2+ sensitivity of fusion complex activation were comparable to that determined for CV–PM fusion. Using Ca2+-induced SNARE complex disruption, we have analyzed the relationship between membrane fusion (CV–CV and CV–PM) and the SNARE complex. Fusion and complex disruption have different sensitivities to Ca2+, Sr2+, and Ba2+, the complex remains Ca2+- sensitive on fusion-incompetent CV, and disruption does not correlate with the quantified activation of fusion complexes. Under conditions which disrupt the SNARE complex, CV on the PM remain docked and fusion competent, and isolated CV still dock and fuse, but with a markedly reduced Ca2+ sensitivity. Thus, in this system, neither the formation, presence, nor disruption of the SNARE complex is essential to the Ca2+-triggered fusion of exocytotic membranes. Therefore the SNARE complex alone cannot be the universal minimal fusion machine for intracellular fusion. We suggest that this complex modulates the Ca2+ sensitivity of fusion.
Keywords: calcium, cytoplasmic vesicles, exocytosis, membrane fusion, secretion
Decades of biochemical and genetic research has culminated in the identification of a set of proteins critical for exocytosis (Pryer et al., 1992; Söllner et al., 1993; Ferro-Novick and Jahn, 1994; Pevsner et al., 1994; Rothman, 1994; Südhof, 1995; Augustine et al., 1996; Calakos and Scheller, 1996). However, the roles of these proteins in the molecular pathway of membrane fusion are poorly understood because most assays fail to differentiate between different steps in the exocytotic pathway. To study the final fusion steps of exocytosis in isolation from concomitant processes, such as vesicle mobilization from reserve pools, priming, and recycling, requires an isolated physiological membrane system that retains the capacity for Ca2+-triggered fusion and is amenable to a variety of biochemical manipulations. Isolated cortical vesicles (CV)1 from the eggs of the sea urchin Strongylocentrotus purpuratus are a high purity, high yield preparation that have proven useful for the study of docking and fusion events (Vogel and Zimmerberg, 1992; Vogel et al., 1992; Tahara et al., 1998). By definition, these CV are fully primed and docked to the plasma membrane (PM) before isolation (Baker and Whitaker, 1978; Moy et al., 1983; Zimmerberg et al., 1985; Whalley and Whitaker, 1988; Zimmerberg and Liu, 1988). Isolated CV retain their Ca2+ sensitivity for fusion, carrying with them all the molecular machinery necessary for docking, Ca2+ sensing, and membrane–membrane fusion (Vogel and Zimmerberg, 1992; Vogel et al., 1992). Use of centrifugation to initiate CV–CV contact before application of Ca2+ supplants the usual cellular mechanisms of transport, targeting, and contact initiation to focus more directly on the membrane constituents essential to docking and fusion.
Interactions between several of the identified components of the exocytotic pathway have been suggested as a general model to explain the specificity of vesicle-to-PM targeting, docking, and fusion (Rothman, 1994; Söllner et al., 1993, Söllner, 1995; Rothman and Söllner, 1997). This general model, the SNARE hypothesis, holds that a heterotrimeric intermembrane “core complex” of the proteins VAMP (on the vesicle membrane), SNAP-25, and syntaxin (on the PM) mediates vesicle targeting and docking to the PM. In detergent extracts, the cytosolic proteins α-/β-/γ-SNAP and the N-ethylmaleimide–sensitive factor (NSF), an ATPase, associate with the SNARE core complex. Hydrolysis of ATP by NSF results in the disruption of the SNARE complex and this has led to the hypothesis that this disruption is an intrinsic, essential event that initiates membrane fusion in the exocytotic pathway (Söllner et al., 1993; Rothman, 1994, 1996; Rothman and Warren 1994; Söllner and Rothman, 1994, 1996; Söllner, 1995; Südhof, 1995; Rothman and Söllner, 1997). It is clear, however, that ATP-driven SNARE complex disruption does not have a role at the membrane fusion step of exocytosis (Vacquier, 1975; Baker and Whitaker, 1978; Whitaker and Baker, 1983; Vilmart-Seuwen et al., 1986; Holz et al., 1989; Parsons et al., 1995; Ungermann et al., 1998). Recent work on the yeast vacuolar fusion system has shown NSF-mediated disruption of SNARE complexes on individual vacuoles to be a priming step for intermembrane SNARE complex formation during vacuole aggregation/docking (Ungermann et al., 1998). Thus, a more recent version of the SNARE hypothesis (SNAREpin; Weber et al., 1998) suggests that intermembrane complex formation provides a minimal fusion machine, ostensibly by ensuring membrane–membrane contact (Jahn and Hanson, 1998). A reduced, physiological model system is required to test whether SNARE complex formation and/or disruption has a role in exocytotic membrane fusion.
We have previously identified the SNARE proteins VAMP, syntaxin, and SNAP-25 by cross-reactivity with antibodies to mammalian VAMP2, syntaxin1A, and SNAP- 25 (Tahara et al., 1998). These proteins were present as both monomers and as constituents of a heterotrimeric core complex on isolated sea urchin egg CV in suspension and between contacting CV. Like the isolated egg cortex consisting of PM with endogenously docked CV, isolated CV require only an increase in free Ca2+ concentration to trigger fusion (Vogel and Zimmerberg, 1992; Vogel et al., 1992; Tahara et al., 1998). Notably, we have shown that free Ca2+ concentrations ([Ca2+]free) triggering maximal CV–CV fusion result in complete disruption of the SNARE complex, and this disruption is inhibited by N-ethylmaleimide (NEM) at concentrations that also inhibit fusion (Tahara et al., 1998). In contrast, lysophosphatidylcholine (LPC), which blocks a late lipid-dependent step in membrane fusion, does not inhibit Ca2+-induced complex disruption; thus, disruption is not a result of membrane fusion but must be a prior or concomitant process, consistent with predictions of the SNARE hypothesis. As Ca2+ disrupts rather than stabilizes the SNARE complex, “zippering-up” of the SNARE proteins (the SNAREpin hypothesis) can not be the mechanism of membrane fusion in the sea urchin CV system (Tahara et al., 1998). What then is the relationship between the SNARE complex and membrane fusion?
The present study involves the testing and characterization of CV–CV fusion as a reduced system for the fusion steps of exocytosis. In addition to Ca2+, both Sr2+ and Ba2+ were tested as alternative triggers for fusion; these divalent cations have long been known to substitute for Ca2+ as triggers for exocytotic fusion in a variety of systems (Miledi, 1966; Dodge et al., 1969; Foreman and Mongar, 1972; Mellow, 1979; Augustine and Eckert, 1984; Blache et al., 1987; TerBush and Holz, 1992; Boonen et al., 1993; Rüden et al., 1993; Barnett and Misler, 1995), including cortical preparations of the sea urchin egg (Whitaker and Aitchison, 1985). As Sr2+ and Ba2+ are known to mimic some but not all Ca2+–protein and Ca2+–lipid interactions, they too satisfy the minimal requirements for triggering the final step(s) in exocytosis. This reduced system is amenable to a number of experimental manipulations that have allowed us to directly and extensively analyze the relationship between membrane fusion and the formation, presence and disruption of SNARE complexes. We find no correlation between the fusion step and these different states of the SNARE complex. Rather, SNARE complexes may modulate the Ca2+ sensitivity of fusion.
Materials and Methods
Materials
ATP, dithiothreitol (DTT), protease inhibitors, and NP-40 purified for membrane research were purchased from Boehringer Mannheim (Indianapolis, IN). Bovine serum albumin was from ICN Biochemicals (Costa Mesa, CA). Peroxidase-conjugated goat anti–rabbit IgG and enhanced chemiluminescence reagents were from Amersham (Little Chalfont, UK). Trypsin (7,120 U/mg) and high purity calcium, strontium, and barium (chloride salts) were purchased from Fluka (Ronkonkoma, NY). All other reagents were of analytical grade and were purchased from Sigma (St. Louis, MO). Anti-VAMP2 antibody (Pevsner et al., 1994) was generously supplied by R. Scheller (Stanford University, Stanford, CA).
Preparation of Sea Urchin Egg Cortical Vesicles
Sea urchins (S. purpuratus) were purchased from Marinus (Long Beach, CA) or Westwind (Victoria, BC) and maintained at 10°C in tanks containing artificial sea water. Intracoelomic injection of 0.5M KCl was used to recover eggs which were then dejellied by passage through 90-μm nylon mesh. CV were prepared according to Crabb and Jackson (1985), with modifications. In brief, to prepare cell surface complexes (CSC), eggs were kept on ice and gently homogenized in IM buffer (220 mM K-glutamate, 500 mM glycine, 10 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 1 mM benzamidine HCl, 2.5 mM ATP, 2 mM DTT, 2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, pH 6.7), as previously described (Whalley and Sokoloff, 1994). After thorough washing, CSC were either used directly in fusion experiments (Vogel et al., 1996) or for protein isolation, or they were resuspended in high pH vesicle isolation buffer (450 mM KCl, 2 mM EGTA, 50 mM NH4Cl, 1 mM benzamidine HCl, pH 9.1) and kept on ice for 1 h during which time vesicles detached from the PM. CV were isolated from PM fragments by centrifugation at 700 g for 2 min at 4°C. This was repeated, and the final supernatant containing the CV was then centrifuged at 2,000 g for 5 min at 4°C. This final CV pellet was resuspended in IM buffer and maintained on ice until used in fusion assays or for protein isolation (within 1–2 h). All stages of the preparation were monitored under a light microscope and the final CV suspension corresponded to single, isolated vesicles ∼1 μm in diameter; any evidence of CV clumping resulted in the preparation being discarded. In some experiments, PKME buffer (425 mM KCl, 10 mM MgCl2, 5 mM EGTA, 50 mM Pipes, pH 6.7) was used throughout, rather than IM buffer (Whalley and Sokoloff, 1994).
Isolation and Analysis of Membrane Proteins
Membrane proteins were extracted and isolated from CV or CSC as described previously (Tahara et al., 1998). Samples for protein isolation were always treated in parallel with samples used for fusion assays. After concentration and resuspension in SDS sample buffer (50 mM Tris-HCl, pH 6.8, 1.5% SDS, 10 mM DTT, 2 mM EDTA, 11% sucrose, and 0.01% bromophenol blue), proteins were separated by electrophoresis, transferred to PVDF membrane, and then analyzed by Western blotting and densitometric scanning (Tahara et al., 1998).
Measuring CV–CV Fusion
Exocytosis from CSC was measured as described (Vogel et al., 1996). CV– CV fusion was measured as described by Vogel and Zimmerberg (1992). All experiments were carried out at room temperature. In brief, CV were suspended in IM to give an A405 of 0.2–0.3 U, measured using a microtiter plate reader (ThermoMax; Molecular Devices, Menlo Park, CA). Aliquots of CV suspension (100 μl) were dispensed into 96-well, flat bottom microtiter plates (Costar, Cambridge, MA) and CV–CV contact initiated by centrifugation (1,000 g, 10 min). Turbidity (A405) of the resulting sheets of CV was measured. Fusion was triggered by addition of an equal volume of Ca2+-, Sr2+-, or Ba2+-IM buffer, designed to give the desired free divalent concentration, and the plates were centrifuged again. A final turbidity measurement was made and the extent of fusion calculated as ΔOD/ODinitial, corrected for a background, which was determined by lysing CV with distilled water. In some experiments a third centrifugation step was added to check for an effect on fusion. Two to ten determinations per condition were always made within an experiment, and determinations were often repeated two or three times over the course of an experiment.
The standard assay is therefore summarized as follows: CV → centrifuge → read A405 → add Ca2+ → centrifuge → read A405. The final free Ca2+ concentrations were verified in mock samples using a Ca2+-sensitive electrode (World Precision Instruments, Sarasota, FL) and were also calculated using the computer program MaxChelator (WINMAXC v.1.70, BERS constants; Bers et al., 1994). The measured (m) and calculated (c) pCa values were linearly correlated, with intercept and slope not significantly different from 0 and 1, respectively; pCam = a + b(pCac), a = 0.01 ± 0.50, b = 0.98 ± 0.09, r 2 = 0.93. This program was used to calculate the free Sr2+ and Ba2+ concentrations in experimental samples using the EGTA and ATP binding constants from Martell and Smith (1974a ,b). To facilitate comparisons between experiments all exocytosis and CV–CV fusion data were normalized so that the lowest free divalent concentration used in an experiment (∼100 nM) gave 0% fusion, and the mean fusion triggered by 150-1,000 μM [Ca2+]free corresponded to 100% (Tahara et al., 1998). The data presented are mean ± SEM of the normalized data from a total of 58 separate CV preparations made over the course of two seasons; parallel experiments with Sr2+ and Ba2+ were done in 24 and 21 of these preparations, respectively. In all fusion assays, cumulative log-normal fits were made to the total data (scatter plots) for any given treatment; these fits are shown superimposed on the mean data. Constancy in the shape but not the midpoints of these sigmoidal activity curves (translational invariance) is best demonstrated by comparing the fitting parameters obtained when data sets have the same numbers of cases and data points. Since these varied with our experimental conditions, an alternative method was adopted. First, the ED50 was determined using linear interpolation between data points above and below 50% fusion. Second, ΔpMe2+ (offset) was calculated by subtracting the ordinate values from the ED50. Translational invariance is indicated by the overlap of the ΔpMe2+ data sets. All determinations of significance were at P < 0.05.
Two variations of the CV–CV fusion assay were also examined. One variation was used to test whether CV membrane constituents themselves could initiate membrane contact that would support fusion without centrifugation. This assay is similar to that described above except that CV were permitted to settle into contact for 60 min before Ca2+ addition, and then further processed using a single centrifugation step to facilitate the dispersal of vesicle contents from the collapsed, fused CV. To test the effects of exposing CV to Ca2+ during the initiation of CV–CV contact rather than after contact had already been established, 100-μl aliquots of CV suspension were added directly to 100-μl aliquots of the cation-IM buffer stocks and initial A405 was immediately measured. The microtiter plates containing the suspended CV were then centrifuged twice for comparison to results obtained in the standard assay format and the turbidity was measured.
Fusion and Shear Assays on Egg Cortices
Sea urchin egg cortices were prepared as described (Zimmerberg et al., 1985; Blank et al., 1998) and experiments were carried out in a rapid perfusion chamber (Kaplan et al., 1996; Blank et al., 1998) permitting constant monitoring of the preparations both by light scattering and by direct visual inspection. Low and high shear conditions correspond to volume flow rates of ∼1 ml/s and ∼8 ml/s, respectively; low shear presents laminar solution flow whereas high shear is turbulent. All shear experiments used IM buffer containing [Ca2+]free of ∼100 nM, 20 μM or 100 μM, in the absence or presence of 100 μM LPC. Light scattering signals were normalized such that [Ca2+]free of ∼100 nM and 1.0 mM gave 0% and 100% fusion, respectively.
Analysis of Exocytosis
At CV–PM docking sites, the number of active fusion complexes per CV is a Poisson distributed random variable in which the average number of active fusion complexes, <n>, varies between 0 and ∼9 depending upon the [Ca2+]free (Vogel et al., 1996). The relationship between extent of fusion,%F, and <n> is: %F = 100 * (1 − exp (−<n>)).
Values of <n> greater than three, corresponding to >95% fusion, can not be accurately determined from the extent of fusion as small changes in %F correspond to large variations in <n>. Higher values of <n>, including <n> Max, the maximum number of active fusion complexes, can only be determined using inhibitors of fusion having exponential temporal and concentration dependence. Back extrapolation to either zero time or concentration is then used to determine <n>.
Results
Homotypic CV Fusion and Exocytosis In Vitro
The abilities of Ca2+, Sr2+, and Ba2+ to trigger membrane fusion between contacting CV was compared (Fig. 1 a). CV–CV fusion was exquisitely sensitive to increasing [Ca2+]free. The Ca2+ activity curve was parameterized using a cumulative log-normal distribution,%F = 50 * erfc ((pCa + log (10−6 M)) / (√ 2 * W)), where erfc is the complimentary error function and W is the distribution width (Blank et al., 1998). This characteristically sigmoidal activity curve, fit to a scatter plot of the total data, had midpoint M = 4.21 ± 0.06 μM Ca2+ and distribution width W = 0.16 ± 0.01. An additional centrifugation step did not alter this activity curve; as such, the energy of centrifugation per se does not drive fusion (Fig. 1 a). A comparable activity curve for Ca2+-triggered exocytosis in vitro (CV–PM fusion) overlapped this CV–CV fusion curve. Sr2+ was only slightly more efficacious than Ba2+ in triggering CV–CV fusion or exocytosis, and both were much less effective than Ca2+ (Fig. 1 a). When normalized to the highest [Ca2+]free tested (0.15–1.0 mM), the midpoints and distribution widths for Sr2+- and Ba2+-triggered CV–CV fusion were M = 2.3 ± 0.1 and 8.0 ± 0.3 mM, and W = 0.24 ± 0.02 and 0.33 ± 0.02, respectively. 15–20 mM [Sr2+]free caused almost the same extent of fusion as high [Ca2+]free; 20–45 mM [Ba2+]free triggered ∼80% of the fusion seen at high [Ca2+]free. At higher free concentrations of Sr2+ and Ba2+ (>95 mM), there was an increase in the measured turbidity. This results from a failure of vesicle contents to properly disperse at high concentrations of these cations and not to a decrease in CV–CV fusion; after a CV–CV fusion experiment, replacing buffers containing >95 mM free Sr2+ or Ba2+ with calcium free artificial sea water resulted in a decrease in turbidity which we interpret as the complete dispersal of contents from collapsed, fused CV (data not shown). Although shifted, the similarity in the shapes of the Ca2+, Sr2+, and Ba2+ activity curves for CV– CV fusion and exocytosis were suggestive of a common underlying process (Blank et al., 1998). The differences in W for Ca2+, Sr2+, and Ba2+ reflect different amounts of data in the transition region of the activity curves, and the different total number of experiments carried out using these three cations. Since W can not be well characterized when only two data points span the transition region (Sr2+ and Ba2+), an alternative analysis was used to compare the shapes of the three activity curves (offset; Materials and Methods). Consistent with a common underlying mechanism, the results of this analysis (Fig. 1 b) show that the activity curves for Ca2+-, Sr2+-, and Ba2+-triggered exocytosis and CV–CV fusion superimpose on the well characterized Ca2+ activity curve for exocytosis in the isolated planar cortex (Blank et al., 1998), demonstrating translational invariance. The slight deviation between CV–CV fusion and CV–PM fusion seen at the highest free cation concentrations tested is the result of differences in normalization of the data in these two preparation types, as the reduced CV–CV system relies on centrifugation to facilitate content dispersal after fusion (see below), a step not necessary with CV endogenously docked to the PM.
Figure 1.
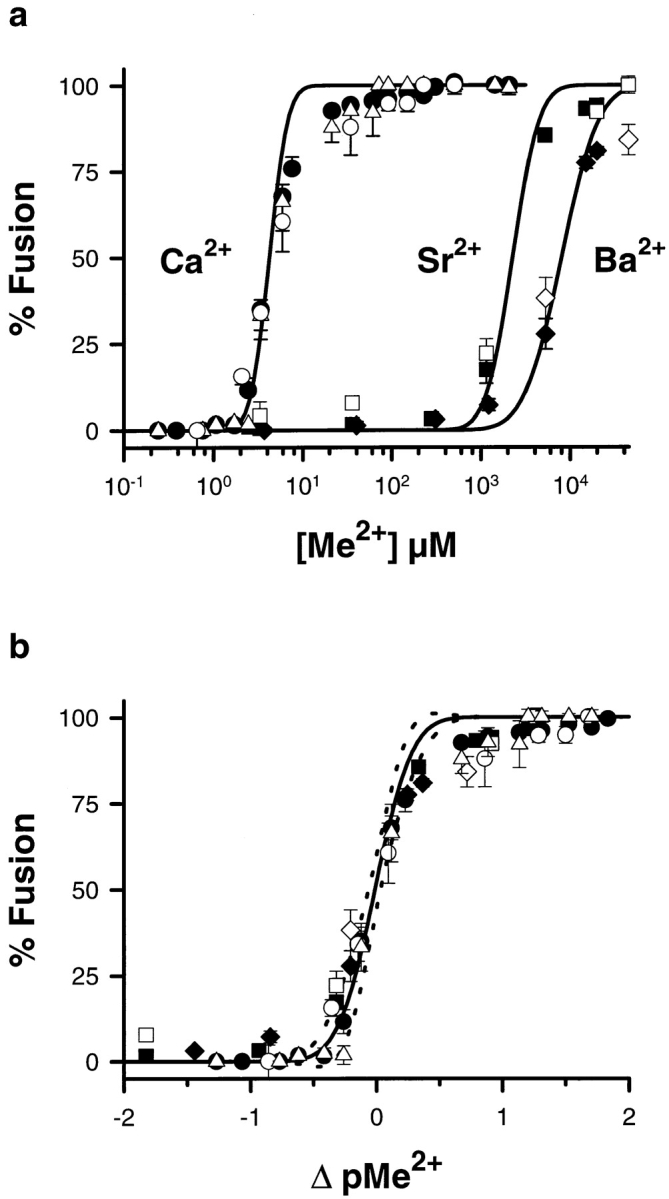
Comparing CV–CV fusion and exocytosis. (a) CV and CSC suspensions were separately tested for fusion in response to Ca2+, Sr2+, and Ba2+ using standard assay formats (Materials and Methods). For CV–CV fusion, each point is the mean ± SEM of 15–104 separate determinations for Ca2+, 28–35 for Sr2+, and 22– 26 for Ba2+; exocytosis data from CSC represent four or five separate determinations for Ca2+, and two determinations each for Sr2+ and Ba2+. For both CV–CV fusion and exocytosis, the results for Sr2+ and Ba2+ have been normalized to the peak Ca2+-induced fusion. Solid symbols, CV–CV fusion data for the indicated cations; open symbols, comparable CSC data; open triangles, CV– CV fusion data obtained after an additional (third) centrifugation step following the standard assay (Materials and Methods). Solid lines, cumulative log-normal fits to the scatter plots of all the CV– CV fusion data (Materials and Methods). (b) Ca2+, Sr2+, and Ba2+ activity data from a for CV–CV fusion and exocytosis exhibit translational invariance (Materials and Methods) and superimpose on a comparable curve for Ca2+-triggered exocytosis from the planar cortex (Blank et al., 1998). Dashed lines delimit the 99% confidence interval for the cortex data.
To further test that the mechanism of fusion triggered by Sr2+ and Ba2+ is the same as that by Ca2+, the effects of known inhibitors of Ca2+-triggered CV–PM fusion (Chernomordik et al., 1993; Sasaki, 1984; Vogel and Zimmerberg, 1992; Vogel, et al., 1993) were compared with respect to their effects on CV–CV fusion initiated by Ca2+, Sr2+, or Ba2+ (Table I). For NEM, trypsin, and chaotropic anions, which are protein-directed inhibitors, and for lauroyl-LPC (LPC), which blocks a late lipid merger step in fusion (Chernomordik, 1996; Chernomordik et al., 1993; Vogel et al., 1993), the extent of inhibition at peak Ca2+-, Sr2+-, or Ba2+-triggered fusion was identical. Thus, as suggested by the activity curves (Fig. 1), fusion triggered by ∼20 mM [Sr2+]free and by ∼45 mM [Ba2+]free is also comparable to that triggered by >0.1 mM [Ca2+]free in terms of susceptibility to various inhibitors. In addition, these concentrations of all three divalent cations were found to trigger CV–liposome fusion (data not shown). Together, the results indicate that Sr2+ and Ba2+ trigger fusion via the same common mechanism as does Ca2+.
Table I.
Effects of Inhibitors on Ca2+-, Sr 2+-, and Ba2+-triggered CV–CV Fusion
| Inhibitor | Inhibition of fusion | |||||
|---|---|---|---|---|---|---|
| Ca2+ | Sr2+ | Ba2+ | ||||
| % | % | % | ||||
| NEM | 90 ± 2 (7) | 92 ± 2 (5) | 96 ± 4 (3) | |||
| Trypsin | 97 ± 1 (6) | 96 ± 4 (4) | 93 ± 2 (3) | |||
| 1 h, 37°C (PKME buffer) | 96 ± 3 (7) | 100 ± 1 (2) | 93 (1) | |||
| LPC | 95 ± 0 (4) | 93 ± 1 (4) | 98 ± 4 (3) | |||
Samples of CV suspension were incubated in the absence or presence of the indicated inhibitors and subsequently challenged with 100 μM Ca2+, 20 mM Sr2+, or 45 mM Ba2+ in standard fusion assays (Materials and Methods; Fig. 1). The percent inhibition of fusion in treated samples was calculated relative to the maximum extent of fusion seen in the parallel untreated controls. The inhibitor treatments were as follows: NEM (5 mM) and trypsin (5 mg/ml) were added for 1 h at room temperature before fusion assays (Vogel and Zimmerberg, 1992); LPC (100 μM) was equilibrated for 2 min and was present throughout the fusion assay; and the combined chaotropic anion-temperature treatment involved isolation and subsequent incubation (1 h, 37°C) of CV in PKME buffer before assaying fusion at room temperature. Parentheses, number of determinations. For each inhibitor, analysis of variance indicates that there is no significant difference in the extent of inhibition of Ca2+-, Sr2+-, or Ba2+-triggered CV–CV fusion, P ≥ 0.254.
Since the experimental evidence indicated a good correlation between CV–CV fusion and the known features of exocytosis, we sought to apply a more quantitative test to the characterization of homotypic CV fusion. A mathematical analysis has been developed which satisfactorily describes key features of sea urchin exocytosis in vitro (Vogel et al., 1996). This analysis treats docked CV on the PM as having a pool of randomly distributed active fusion complexes; the average number of active fusion complexes per vesicle, <n>, is thus a critical quantity in this analysis. If CV–CV fusion proceeds through the same fusion complexes as exocytosis, it should also be fully described by the same analysis. This is of critical importance as the analysis defines the fusogenic entity (the fusion complex) based only on fusion criteria. To determine the maximum average number of fusion complexes per CV, <n>Max, NEM was used to irreversibly inactivate fusion complexes on CV over time. CV in suspension were treated with 5 mM NEM, and Ca2+-triggered CV–CV fusion was subsequently tested in the standard assay format. As with NEM-treated cortices, there is a decrease in CV fusion over time (Fig. 2 a). This loss of function corresponded to a decrease in <n> as an exponential function of time (Fig. 2 b). By back extrapolation, the value of <n>Max (at t = 0) for CV– CV fusion is 6.8 ± 0.3 (n = 5; weighted average), comparable to the value determined for CV docked to the PM (Fig. 3, inset) (Vogel et al., 1996). Furthermore, as with CV–PM fusion, the relationship between Ca2+-triggered CV–CV fusion and <n> is sigmoidal (Fig. 3). The sigmoidal relationship between <n> and [Ca2+]free, while shifted, is comparable to that observed for CV–PM fusion (Vogel et al., 1996; Blank et al., 1998). Thus, CV–CV fusion can be described by the mathematical analysis developed for CV– PM fusion, yielding comparable fitting parameters. Together with the experimental results, this suggests that CV–CV fusion is an effective system with which to analyze the molecular mechanism of exocytotic fusion.
Figure 2.
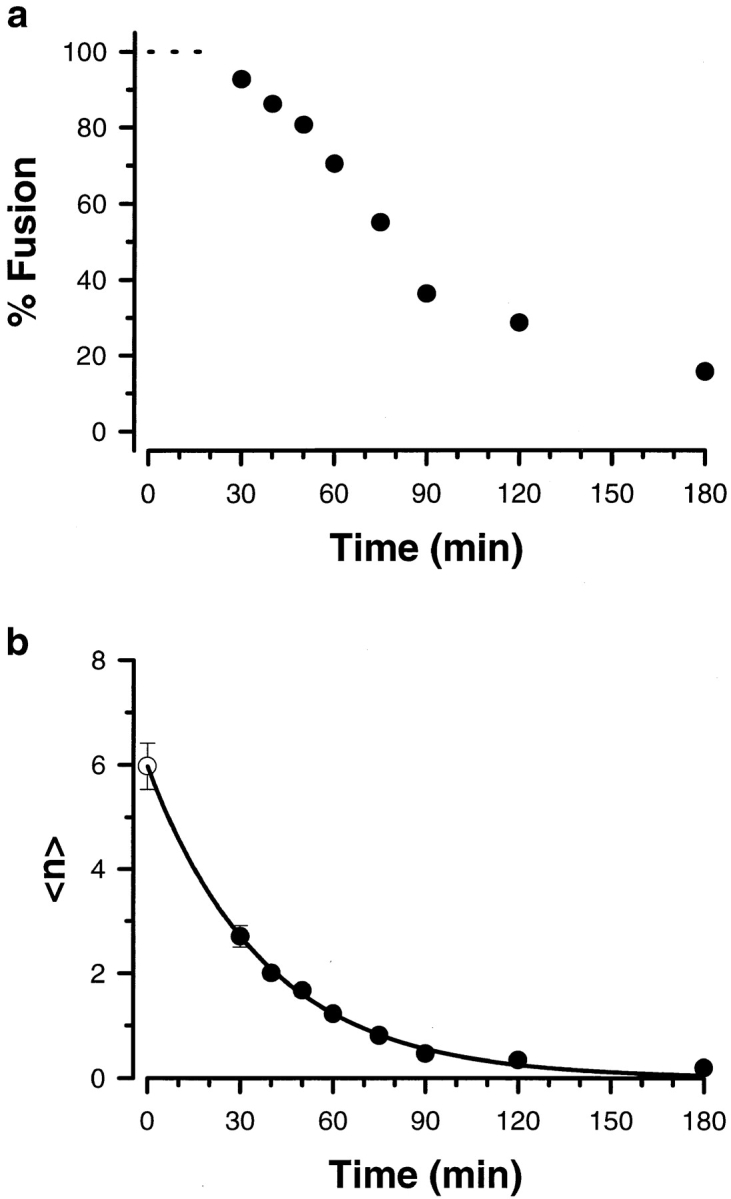
NEM inactivation of CV–CV fusion. The time course for the inhibition of CV–CV fusion by NEM was characterized. Isolated CV in suspension were incubated with 5 mM NEM and, at the indicated times, aliquots were quenched by dilution in IM buffer containing excess DTT. (a) The extent of fusion in response to a maximal dose of Ca2+ (1 mM) was determined using the standard assay format (Materials and Methods). There was no detectable effect of NEM on fusion during the first 20 min (dashed line). (b) The average number of active fusion complexes per vesicle was calculated from the results in (a) as described in Materials and Methods. Open circle, back-extrapolated (t = 0) value for <n>Max determined using the best fit single exponential (solid line) to the values for <n> calculated from the NEM inactivation of fusion (solid circles).
Figure 3.

Analyzing the Ca2+ dependence of active fusion complexes, <n>, in CV–CV fusion. <n>Max in CV–CV fusion is comparable to that observed in the cortex (dashed line). Open circles, <n> extrapolations from NEM inactivation experiments; solid circles, derived from Ca2+ dose-response curves for CV–CV fusion (see Fig. 1). The log–log inset of the same data highlights differences in <n\> at low Ca2+ concentrations.
SNARE Complex Is Not Fusogenic
We are using the CV–CV system to identify membrane constituents essential to the fusion steps of exocytosis. Since we could readily observe and trigger the disruption of the ∼70-kD sea urchin egg SNARE complex with Ca2+ (Tahara et al., 1998), we tested the SNARE hypothesis by analyzing the relationship between the complex and membrane fusion. We first tested whether disruption of this intermembrane complex is involved in the initiation of membrane fusion. Using two methods of detecting SNARE complexes—SDS-PAGE of unboiled TX-114–extracted CV proteins (Figs. 4–6) and sucrose gradients of NP-40– solubilized CV (data not shown)—we have reproduced the finding that [Ca2+]free > 100 μM, which cause maximal fusion, result in complete disruption of CV SNARE complexes (Tahara et al., 1998). Treating contacting CV with 150 μM [Ca2+]free caused a complete loss of SNARE complexes, but Sr2+ and Ba2+ concentrations eliciting 94 ± 1% (n = 34) and 81 ± 1% (n = 26) fusion, respectively, had little effect on the complexes (Fig. 4 a). This lack of comparable SNARE complex disruption during fusion triggered by Sr2+ or Ba2+, while directly demonstrating that divalent cation-triggered fusion does not require SNARE complex disruption, might be interpreted to indicate that these cations effect fusion by an alternate pathway from that triggered by Ca2+. None of our experimental results support the existence of a separate, parallel Sr2+- and Ba2+-triggered fusion pathway (Fig. 1 and Table I).
Figure 4.

SNARE complex disruption has different sensitivities to Ca2+, Sr2+, and Ba2+. (a) The sensitivity of SNARE complex disruption to concentrations of Ca2+, Sr2+, or Ba2+ that trigger maximal fusion was compared. Contacting CV were exposed to either IM buffer alone (Control), 150 μM [Ca2+]free (Ca2 +), 20 mM [Sr2+]free (Sr2 +), or 20 mM [Ba2+]free (Ba2 +). (b) The sensitivity of SNARE complex disruption to increasing concentrations of Ca2+ was examined. Contacting CV were exposed to either IM buffer alone (zero calcium), or to the indicated [Ca2+]free. In all cases, proteins were extracted with Triton X-114, dissolved in SDS sample buffer without boiling and then analyzed by SDS-PAGE on 12% gels followed by immunoblotting. Immunoblots were probed with anti-VAMP antibody to visualize both VAMP monomers (∼19 kD; data not shown) and the SNARE complex (∼70 kD; Tahara et al., 1998).
Figure 6.

There is no direct correlation between exocytosis and SNARE complex disruption. (a) The Ca2+ sensitivity of SNARE complex disruption on CSC was examined. CSC in suspension were exposed to either IM buffer alone (zero calcium) or to the indicated [Ca2+]free in assays analogous to those used to measure exocytosis from CSC (Fig. 1 a). Proteins were extracted and analyzed as described in Fig. 4. (b) Ca2+-triggered exocytosis was measured in the well-characterized egg cortex preparation using a double Ca2+ challenge paradigm. Egg cortices were isolated in a KCl-based buffer and exocytosis assayed in a rapid perfusion microscope chamber, as described (Blank et al., 1998). Cortices were first challenged with 14 μM [Ca2+]free (short arrow) and subsequently with 24 μM [Ca2+]free (tall arrow). The resulting light-scattering data were normalized such that ∼100 nM [Ca2+]free gave 0% fusion and 300 μM [Ca2+]free gave 100% fusion (Blank et al., 1998).
To verify this separation of fusion from SNARE complex disruption, and to further test the relationship between Ca2+-triggered fusion and the SNARE complex, we assayed the amount of complex present at different points along the Ca2+ activity curve (Fig. 4 b). In these experiments, 3.5 μM [Ca2+]free, that caused 35 ± 3% fusion (n = 104), did not affect the amount of SNARE complex relative to controls. A slightly higher [Ca2+]free (6 μM) caused a complete loss of SNARE complexes, as did higher doses of Ca2+. However, 6 μM [Ca2+]free caused only 68 ± 4% fusion (n = 79). With 150 μM [Ca2+]free there was 98 ± 1% fusion (n = 59) and again, complete loss of SNARE complexes. In summary, membrane fusion was triggered at a lower [Ca2+]free than was detectable SNARE complex disruption, and SNARE complex disruption was complete at lower [Ca2+]free than that required for 100% fusion. Thus, there was no stoichiometric correlation between the extents of Ca2+-, Sr2+-, or Ba2+-triggered fusion and the presence or the loss of heterotrimeric complexes.
As another analysis of the relationship between fusion and SNARE complex formation or disruption, we tested for an effect of Ca2+-induced SNARE complex disruption using fusion-incompetent CV. Despite centrifuging CV into contact, incubation in a chaotropic buffer at 37°C for 1 h resulted in the complete block of Ca2+-triggered CV–CV fusion (Table I and Fig. 5 A). Untreated controls maintained on ice for 1 h before the CV–CV fusion assay remained fully responsive to Ca2+ (Fig. 5 A) with a characteristic sigmoidal activity curve comparable to that obtained for CV–CV fusion in IM buffer (Fig. 1). Although the CV treated at 37°C were no longer fusogenic, these contacting CV had the same amount of SNARE complex as the controls (Fig. 5 B), and these SNARE complexes were still competent to undergo complete disruption in response to Ca2+. Thus, there is no direct correlation between fusion and SNARE complex formation and disruption.
Figure 5.

The SNARE complex remains Ca2+ sensitive on fusion-incompetent CV. The Ca2+ sensitivity of SNARE complex disruption was tested with CV rendered nonfusogenic by a 1-h exposure to chaotropic buffer at 37°C. (A) After incubation in PKME buffer at 37°C or on ice (Control), CV suspensions were tested for fusion in response to Ca2+ using the standard assay format (Materials and Methods); each point is the mean ± SEM from three to seven separate determinations from five CV preparations. (B) Parallel samples were exposed to either buffer alone (−), or to 100 μM [Ca2+]free (+). CV in PKME buffer had previously been incubated either on ice for 1 h (Control), or at 37°C (37°C). Proteins were extracted with Triton X-114, dissolved in SDS sample buffer without boiling, and then analyzed by SDS-PAGE on 12% gels followed by immunoblotting. Immunoblots were probed with anti-VAMP antibody.
It might be argued that we changed the Ca2+ sensitivity of the SNARE complex by using centrifugation in our standard assay. Perhaps SNARE complexes at endogenous CV–PM docking sites have a range of Ca2+ sensitivities more comparable to those of fusion, and different from that of the isolated, centrifuged CV (Fig. 4 b). Although not done routinely because of the large amounts of cortex that must be extracted to satisfactorily visualize the SNARE complex, a limited analysis of Ca2+-triggered SNARE complex disruption in the CV–PM system was undertaken as another approach to addressing the question of possible centrifugation effects in our assays. These data for the CV–PM system show complete SNARE complex disruption by 6 μM [Ca2+]free (Fig. 6 a), comparable to the Ca2+ sensitivity determined in isolated, contacting CV. However, 6 μM [Ca2+]free triggered only 60 ± 9% (n = 5) exocytosis (Fig. 1 a). Thus, for endogenously docked CV at the PM, complete disruption of SNARE complexes did not correlate with the extent of Ca2+-triggered fusion. This was also demonstrated in the isolated planar cortex using a double Ca2+ challenge paradigm (Blank et al., 1998). In the example shown (Fig. 6 b), 14 μM [Ca2+]free, sufficient to fully disrupt SNARE complexes, elicited 50% fusion; subsequent challenge with 24 μM [Ca2+]free yielded an additional 26% fusion. These data, obtained using a well-characterized preparation exhibiting in vitro Ca2+-triggered exocytosis of CV in vitro, which were docked and primed in vivo, confirm our prediction from the CV–CV system that neither the presence nor the disruption of SNARE complexes is essential to exocytotic membrane fusion.
As a final test, the relationship between SNARE complex disruption and membrane fusion was also analyzed mathematically. Using the values for Ca2+-triggered fusion and SNARE complex disruption determined experimentally (Fig. 1 and Fig. 4 b), we predicted the consumption of fusion machines at critical points on the CV–CV Ca2+ activity curve (Fig. 7). With a level of reserve complexes, <n>Max = 6.8 ± 0.3 (Fig. 3), the analysis predicts a loss of only 3 and 10% of active fusion complexes at 3.5 and 6.0 μM [Ca2+]free, respectively; these Ca2+ concentrations induced 35 and 68% fusion. Although it might be argued that a 3% loss could escape detection, the 100% loss of SNARE complexes detected at 68% fusion (Fig. 4 b) is incompatible with the 10% loss predicted by the analysis, if SNARE complex disruption is equivalent to the utilization of fusion complexes. Therefore, Ca2+-triggered SNARE complex disruption does not cause fusion.
Figure 7.

The Ca2+ sensitivity of SNARE complex disruption does not correlate with either the observed extent of fusion or the derived number of active fusion complexes (<n>). Line Fusion, the same curve fit to the Ca2+ data in Fig. 1 a; line Fusion Complex, predicted loss of functionally defined active fusion complexes (100 * (1 − <n>/<n>Max)), normalized by <n>Max for CV–CV fusion (see Fig. 3). Complete disruption of SNARE core complexes occurs between 3.5 and 6 μM [Ca2+]free (dashed line) corresponding to 35–68% fusion, on average (open circles). Therefore, ∼35% fusion occurs in the absence of complex disruption (solid horizontal line at 100%) and an additional ∼30% fusion proceeds despite the complete loss of complexes (solid horizontal line at 0%). At 6 μM [Ca2+]free, <n> analysis predicts only a 10% consumption of fusion complexes, but the data indicate 100% disruption of SNARE complexes (Fig. 4 b); complex disruption does not correlate with the predicted consumption of fusion complexes.
What then is the role of the SNARE complex? The SNARE hypothesis also suggests a docking function. The data indicated that an essential role in docking was unlikely, as both CV–CV fusion and exocytosis (Figs. 1 and 6) were still functional at [Ca2+]free higher than required for SNARE complex disruption. We sought to verify these findings using another assay on the CV–PM system.
SNARE Complex Alone Does Not Mediate CV–PM Docking
100 μM [Ca2+]free caused 97 ± 2% fusion (n = 5) in isolated, perfused egg cortex preparations. In the presence of 100 μM LPC, exocytotic fusion was blocked (Fig. 8, 0–100 s). However, even under the highest shear stress conditions possible with the flow chamber used, CV remained attached to the PM as evidenced both by the light scattering signal (Fig. 8, 66–71 s) and by direct microscopic observation. The <3% change in light scattering detected during high shear results from the loss of small pieces of membrane from the coverslips, not from a loss of CV. Thus, CV remained more firmly attached to the PM than the latter did to the poly-l-lysine–coated coverslips. That CV remained docked and fusion-competent was shown by delivery of a solution lacking LPC and containing 1 mM [Ca2+ ]free (Fig. 8, arrow), resulting in 100% exocytosis; we saw the appearance of “fusion domes” (Zimmerberg and Liu, 1988), verifying vectorial discharge of granule contents across the PM. Comparable results were obtained using 20 μM [Ca2+]free, which is sufficient to disrupt all SNARE complexes (Fig. 4 b and Fig. 6), but triggered only 42 ± 8% fusion (n = 6) in controls, demonstrating that docking is not modified by LPC. As suggested by the findings for CV–CV (Fig. 4 b) and CV–PM fusion (Fig. 6), the SNARE complex cannot be the sole determinant of docking. Additional docking components, most probably localized to the CV, must be present.
Figure 8.

CV remain firmly attached to the plasma membrane despite SNARE complex disruption. Isolated planar cortices from sea urchin eggs were monitored by quantitative light scattering in a rapid-flow perfusion chamber. After treatment of the isolated cortices for 10 min with 100 μM LPC in the presence of low [Ca2+]free, cortices were perfused successively with (i) LPC plus 100 μM [Ca2+]free, sufficient to fully disrupt SNARE complexes (low flow, 0–60 s); (ii) LPC plus 100 μM [Ca2+]free under high shear conditions (8 ml/s solution delivery, 65–70 s); and (iii) 1 mM [Ca2+]free at low flow (beginning at arrow). Representative of nine experiments.
SNARE Complex Affects the Ca2+ Sensitivity of Fusion
To confirm that CV membranes contained additional docking machinery to mediate the tight membrane–membrane coupling necessary for fusion, and to test for a modulatory role of the SNARE complex, we used two variations on the standard fusion assay. First, in the absence of cytosolic factors and without centrifugation, CV were allowed to settle into contact for 60 min before the application of Ca2+. These assays still yielded extensive Ca2+-triggered fusion, but with an activity curve significantly right-shifted (M = 27.3 ± 2.6 μM, W = 0.88 ± 0.06; Fig. 9 a) relative to that obtained using the standard, centrifuged assay format. Centrifugation of these “settled” CV samples resulted in a significant leftward shift in the Ca2+ activity curve (M = 13.4 ± 0.9 μM, W = 0.30 ± 0.03). This activity curve for CV centrifuged after “settling” and the application of Ca2+ was therefore only slightly right-shifted relative to results obtained with the standard assay format. This latter activity curve was remarkably similar to that obtained for CV–PM fusion (M = 18.2 ± 0.6 μM, W = 0.23 ± 0.02; Blank et al., 1998), as shown by its translational invariance with respect to this Ca2+-triggered exocytosis in the cortex (Fig. 9 c).
Figure 9.
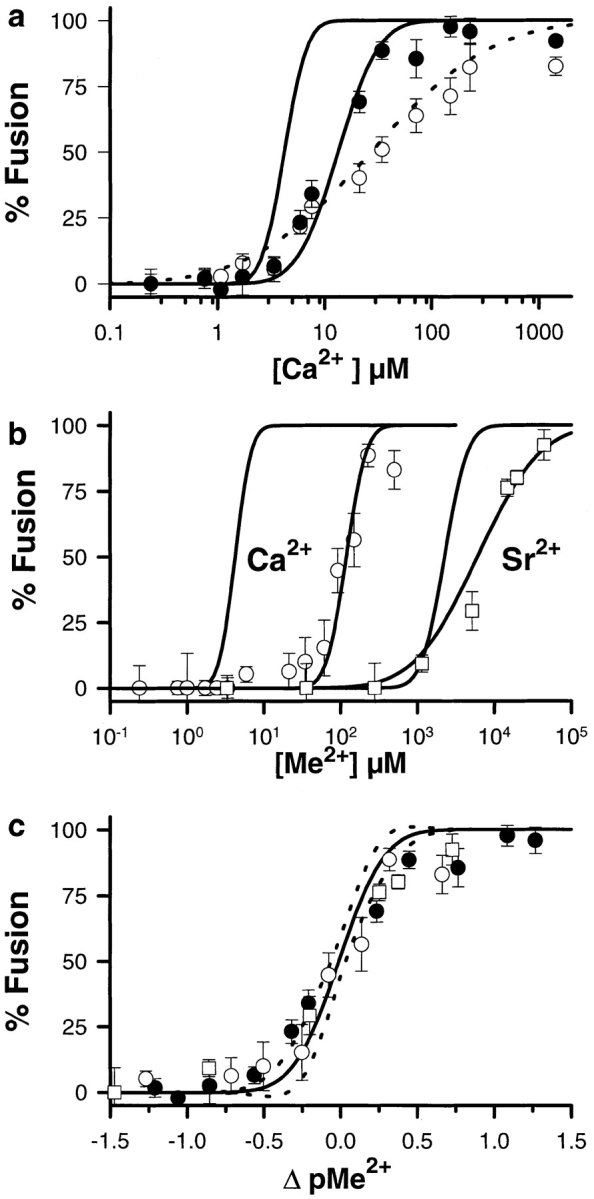
CV membrane components mediate docking and the SNARE complex may enhance the Ca2+ sensitivity of fusion. (a) CV allowed to settle into contact for 60 min before Ca2+ application (open circles; dotted line fit) undergo extensive fusion; centrifugation of these samples (solid circles; solid line fit) results in a curve slightly right-shifted relative to samples assayed using the standard format (curve to left, from Fig. 1 a). Each point is the mean ± SEM of 3–14 separate determinations, done in parallel with standard assays. (b) Exposure to Ca2+ while bringing CV into contact results in a significant loss of Ca2+ sensitivity for fusion. CV suspensions were added directly to cation buffer stocks before centrifugation (Materials and Methods). There is a significant rightward shift in the Ca2+ activity curve (open circles) relative to the standard assay (curve to left, from Fig. 1 a). Comparable assays involving Sr2+ (open squares) yield a less pronounced shift relative to the standard assay (Sr2+ curve from Fig. 1 a). Each point is the mean ± SEM of 3 –15 separate determinations for Ca2+ and eight or nine for Sr2+, done in parallel with standard assays. In all experiments, results have been normalized to the peak Ca2+-induced fusion in parallel standard assays. (c) The Ca2+ activity curves for the CV–CV fusion assays described in a (solid circles) and b (open symbols) show translational invariance and superimpose on a comparable curve for Ca2+-triggered exocytosis from the planar cortex (Blank et al., 1998). Dashed lines delimit the 99% confidence interval for the cortex data.
As a test for possible Ca2+-dependent inactivation of fusion complexes or other modulatory influences of the SNARE complex, CV were exposed to Ca2+ before the establishment of CV–CV contact. Such exposure to Ca2+ during the initiation of contact produced an ∼30-fold rightward shift in the Ca2+ activity curve for CV–CV fusion (M = 119 ± 8 μM, W = 0.2 ± 0.04; Fig. 9 b). Thus, although the ED50 for Ca2+-triggered fusion was now ∼120 μM [Ca2+]free, compared with the ∼4 μM determined for CV brought into contact before application of the Ca2+ trigger (standard assay format), the CV did still fuse. The results of this fusion assay are also translationally invariant with respect to Ca2+-triggered exocytosis (Fig. 9 c). CV exposed to >100 μM [Ca2+]free but then subjected to chelation of [Ca2+]free back to ∼100 nM before the establishment of CV–CV contact (see Fig. 6 in Tahara et al., 1998) yielded fusion curves comparable to controls not pretreated with Ca2+ (data not shown). Notably, exposure to Sr2+ (Fig. 9 b) or Ba2+ (data not shown) before and during the initiation of CV–CV contact had substantially less pronounced effects than seen with Ca2+. The ED50 for Sr2+ increased less than threefold relative to that determined using the standard assay, indicating that the effect seen with Ca2+ correlated with a loss of SNARE complexes.
Discussion
By many criteria, CV–CV fusion proceeds via the same molecular mechanism as the fusion stage of exocytosis. The data indicate that the heterotrimeric SNARE protein core complex is neither necessary nor sufficient for fusion in this system, and therefore is not the universal minimal fusion machine. This complex does appear to enhance the Ca2+-sensitivity for fusion.
CV–CV Fusion Retains Many Essential Features of the Membrane Fusion Stage of Exocytosis
A key question in this work is whether or not CV–CV fusion and exocytosis use the same mechanistic pathway. We cannot directly analyze the final, fusion steps of exocytosis without an isolated, native membrane system capable of Ca2+-triggered membrane fusion and amenable to a variety of biochemical manipulations. This system must recapitulate the essential features of the fusion stage of exocytosis and possess the components critical to this mechanism without modulatory cytosolic factors, or active concurrent processes (reserve vesicle mobilization, recycling/endocytosis). Homotypic fusion of sea urchin egg CV, isolated from their fully primed, docked, release-ready state on the PM, is such a homogeneous, stage-specific system. Isolated CV retain sufficient molecular machinery for efficient docking, Ca2+-sensing, and fusion (Vogel et al., 1992). Furthermore, unlike other preparations in which priming and docking are readily reversible (Mayer et al., 1996; Steyer et al., 1997), CV of isolated cortices remain stable and docked to the PM for hours, and isolated CV remain fully primed and fusion-competent for over 16 h (Tahara et al., 1998).
We have recently shown that fusion competent CV have SNARE proteins and that an intermembrane complex forms between these proteins on opposed CV (Tahara et al., 1998). By several criteria, this is the same SNARE complex as that described in other secretory systems. In addition, working with a fully primed, isolated native membrane system has allowed us to identify a novel feature of this SNARE complex: its stability is Ca2+-sensitive in the native membrane, but not in detergent extracts (Tahara et al., 1998).
Using a buffer system more like the intracellular milieu of the egg, and thus minimizing the concentration of chaotropic ions, we first show that CV–CV fusion has a Ca2+ sensitivity comparable to that of exocytotic fusion in the isolated cortex. In addition to Ca2+, Sr2+, and Ba2+ also trigger CV–CV and CV–PM fusion, as demonstrated for exocytosis in other secretory systems including the cortex (Whitaker and Aitchison, 1985). Furthermore, by showing that CV–CV fusion is described by a mathematical model that uses fusion criteria alone to describe the fusion mechanism, we confirmed that CV–CV fusion is mediated by the same number of operationally defined “fusion complexes” as in exocytosis (Vogel et al., 1996). That is, despite obvious differences in the two systems such as the sensitivity to the different ionic triggers, global differences in the composition of the fusing membranes, differences in the milieu at the time of docking (cytosol versus buffered ionic solutions), and the presence or absence of ATP and GTP, CV–CV fusion appears to use the same fusion pathway as Ca2+-triggered CV–PM fusion (Fig. 1 b). Like exocytosis, the reduced homotypic fusion system retains sigmoidal Ca2+ sensitivity (submaximal responses), triggering by Sr2+ and Ba2+, the Ca2+ dependency of <n>, <n>Max, Ca2+-sensitive SNARE complex disruption, and sensitivity to identified inhibitory agents. These findings are all inconsistent with the existence of an alternate fusion pathway, and consistent with CV–CV fusion proceeding by the same molecular mechanism as exocytotic fusion, as suggested by similarities in the Ca2+ activity curves for a wide range of secretory systems (Knight and Scrutton, 1986). Similarly, comparable subpopulations of CV enter the fusion pathway for given incremental increases in the concentrations of the different divalent cations (ΔpMe2+; Fig. 1 b). That these curves overlap a similar curve for Ca2+-triggered CV–PM fusion (exocytosis) suggests that a common molecular mechanism is being used to overcome the energy barrier to fusion (Blank et al., 1998). Differences in the sensitivity of divalent cation binding sites therefore allow for selectivity as to which vesicles enter the fusion pathway.
The reduced CV–CV system also shares another feature with exocytosis in vitro: there is no Ca2+-induced inactivation (Blank et al., 1998). Treatment of isolated CV in suspension with high [Ca2+]free has no effect on the resulting fusion activity curve provided that the Ca2+ is chelated back to basal levels before the establishment of CV–CV contact (data not shown) (also see Whalley and Whitaker, 1988). Exposure to Ca2+ immediately before and during the initiation of CV–CV contact does not result in an inability to fuse, but does substantially shift the Ca2+ sensitivity of fusion (Fig. 9 b). Together, these results argue against Ca2+-induced inactivation (Blank et al., 1998). Variations on the standard fusion assay have been used to determine the extent to which the Ca2+ sensitivity of the fusion mechanism can be modulated. CV that are allowed to settle into contact before application of the Ca2+ trigger have an activity curve that is right shifted relative to CV that are centrifuged into contact (approximately sevenfold shift in the ED50). Thus, CV membrane components are sufficient to establish the tight intermembrane contact necessary to support Ca2+-triggered fusion. Subsequent centrifugation of the “settled”, Ca2+-challenged samples yields a sigmoidal activity curve that is only slightly right shifted (approximately threefold shift in the ED50) relative to CV that are initially centrifuged into contact (Fig. 9 a). As the energy of centrifugation does not drive fusion, determined using the standard assay (Fig. 1 a), and because CV not already in contact do not readily fuse upon subsequent exposure to Ca2+ and centrifugation (Fig. 9 b), we interpret the change in the settled samples to reflect the ability of centrifugation to enhance the dispersal of CV contents after fusion. This activity curve of settled, Ca2+-challenged and subsequently centrifuged CV samples (Fig. 9 a) is remarkably similar to that of Ca2+-challenged cortices (Blank et al., 1998). That initially centrifuging CV into contact (the standard assay) results in a slightly enhanced Ca2+-sensitivity suggests the hypothesis that contact area modulates Ca2+ sensitivity, perhaps via SNARE coupling between opposed CV (see below). This interpretation is consistent with the fact that CV settled into contact have only a slightly reduced Ca2+ sensitivity for fusion (Fig. 9 a), whereas CV exposed to conditions promoting SNARE complex disruption have a markedly reduced Ca2+ sensitivity, despite subsequent centrifugation into contact (Fig. 9 b). Notably, although contact seems to modulate Ca2+ sensitivity, fusion appears to proceed via the same mechanism in all cases, including the exocytosis of vesicles endogenously docked to the PM (Fig. 1 b and Fig. 9 c). Together, the results suggest that the exocytotic mechanism uses a balanced combination of contact area, fusion pore expansion (Scepek et al., 1998), and postfusion regulation of vesicular content extrusion (Rahamimoff and Fernandez, 1997), most probably involving membrane tension (Chizmadzhev et al., 1995; Dai and Sheetz, 1995), to effect Ca2+-triggered secretion. Thus, in the CV–CV system, centrifugation serves to enhance membrane-membrane contact and “normalize” content dispersal after fusion. This latter function is necessary as vesicle contents do not disperse as efficiently after CV–CV fusion as is characteristic of dispersal following CV–PM fusion in the planar cortex. This may also indicate the loss during isolation of modulatory components associated with the PM in vivo.
The SNARE Complex Is Not Essential to Membrane Fusion
By comparing the inhibitory effects of NEM and LPC on Ca2+-triggered fusion and SNARE complex disruption (Tahara et al., 1998), we previously concluded that disruption must occur immediately before or during fusion, consistent with one SNARE hypothesis (Rothman, 1994) but not another (Weber et al., 1998). Using the CV–CV fusion system, we have now tested the SNARE hypotheses by directly analyzing the relationship between the SNARE complex and membrane fusion. Using new tools, including dose-response analyses, alternate divalent cations, inactivation of fusion, shear treatments, and mathematical analyses, we significantly extend the previous findings to show that there is no correlation whatsoever between the fusion step and the formation, presence, or disruption of the SNARE complex. Ca2+ could induce fusion in the absence of complex disruption, and Ca2+ could still induce fusion even under conditions causing the complete loss of complexes, in both isolated CV and on cortices. Ca2+ could also induce complex disruption without causing fusion, even though vesicles were centrifuged into contact (Fig. 5); this insensitivity to chaotropic anions, which block fusion (Table I), is further indication that the SNARE complex is not the fusogenic entity. Our mathematical analysis, which we have used to predict the consumption of fusion complexes, shows that the SNARE complex does not function in a manner predicted by any of the SNARE hypotheses (Fig. 7). Furthermore, neither Sr2+ nor Ba2+ cause SNARE complex disruption although they support near-maximal fusion, comparable with data on divalent-triggered exocytosis in neurons and other secretory cells (Miledi, 1966; Dodge et al., 1969; Mellow, 1979; TerBush and Holz, 1992; Boonen et al., 1993; Rüden et al., 1993; Barnett and Misler, 1995). Although these cations may differ in their point of entry into the fusion pathway, the Ca2+, Sr2+, and Ba2+ activity curves (Fig. 1 a) are all translationally invariant with respect to CV–CV fusion and CV–PM fusion (Fig. 1 b); the simplest hypothesis is that these cations all trigger a final common mechanism. Further corroboration for this hypothesis comes from inhibitor studies (Table I). In all cases tested, including lipid as well as protein-directed agents, the same inhibition was seen regardless of whether CV–CV fusion was triggered by Ca2+, Sr2+, or Ba2+. These results do not support the existence of a separate Sr2+- or Ba2+-triggered fusion pathway. However, Sr2+ and Ba2+ may simply bypass early steps in the fusion pathway (SNARE complex disruption), triggering only late, critical Ca2+ sites.
In addition, we also used chaotropic anions to test the SNARE hypothesis (Fig. 5). Loss over time of factors “essential” to exocytosis occurs with all secretory systems in vitro, including the egg cortex, and the effect is particularly pronounced in buffers that are more chaotropic in nature (Sasaki, 1984). This was the reason for our use of glutamate-glycine–based IM buffer, which better preserved native function (Fig. 1). The loss of function in chaotropic buffers is also accelerated by temperature. CV incubated for 1 h at 37°C in PKME buffer were no longer fusogenic but SNARE complexes were still present and responsive to Ca2+. Resuspension of these CV in IM buffer for up to 1 h at room temperature, or on ice, did not result in a recovery of Ca2+- or Sr2+-triggered fusion (data not shown), arguing against a simple, reversible reorganization of the membrane lipids. These results support our findings that there is no direct correlation between fusion and the formation or disruption of SNARE complexes.
Thus, in these systems SNARE complex formation is not sufficient for fusion, and complex disruption does not initiate fusion, is not inherently fusogenic, nor is it intrinsic to the process of membrane–membrane fusion. Fusion must be the result of steps subsequent to complex disruption. Even if intermembrane complex disruption and fusion are at all related, fusion must be the result of steps well removed from disruption (Calakos and Scheller, 1996). Therefore, neither version of the SNARE hypothesis (Rothman, 1994; Weber et al., 1998) can effectively explain membrane fusion in this system, and several proposed models for SNARE complex function are effectively ruled out (Weis and Scheller, 1998). The SNARE complex may have a secondary role in docking, or a modulatory role that enhances Ca2+-sensitivity (Fig. 9), but these roles are before the fusion step. The minimal docking and fusion components are physically separate from the heterotrimeric SNARE complex. However, as SNARE complex disruption is likely to occur concurrently with fusion in response to local transients in [Ca2+]i occurring during exocytosis in vivo, this disruption and effective removal of the intermembrane complex from the vicinity of the fusion site may be at least one of the reasons for the high efficacy of Ca2+ in triggering exocytotic fusion, particularly in comparison to other divalent cations (Fig. 1 a).
SNARE Complex Function
The idea of a common, conserved exocytotic mechanism is well supported by experimental evidence (Dan and Poo, 1992; Steinhardt et al., 1994; Ferro-Novick and Jahn, 1994; Miyake and McNeil, 1995; Coorssen et al., 1996). The SNARE hypothesis has promoted the concepts of docking and fusion as two separate but intimately related mechanistic processes: there cannot be fusion without efficient and “proper” molecular level interactions of the membranes involved. However, the exact function of the SNARE complex and its constituent proteins remains unresolved. The formation of an intermembrane SNARE complex may be necessary for efficient, sensitive Ca2+-triggered fusion, but it is not, in and of itself, sufficient for fusion. Perhaps the complex helps to more fully stabilize intermembrane contact and thus to promote formation of a focal site preferential for membrane fusion or to optimize such a site by recruiting or orienting other critical components. Therefore, the complex could define a local site of close membrane apposition conducive to high affinity Ca2+ binding between phospholipid headgroups. The elegant work of Feigenson (1989) shows that Ca2+ binds to closely spaced anionic (phosphatidylserine) lipid headgroups over the range of physiologically relevant [Ca2+]free. Such interbilayer Ca2+ binding results in dehydration of lipid headgroups and the subsequent fusion and collapse of the membranes (Feigenson, 1989; Coorssen and Rand, 1995). This may occur in defined, localized fusion domains to which exocytotic vesicles are targeted and docked, perhaps delimited by a ring of SNARE complexes, ensuring a focal site of close inter-membrane interaction as in viral fusion (Chernomordik et al., 1998). We note that if these putative domains were composed of purely anionic lipids, the dehydration energy would exceed that of membrane integrity; including the zwitterionic lipids of cellular membranes in such a putative domain could help to regulate the dehydration, and to promote lipid coalescence during the ensuing fusion (Coorssen and Rand, 1995). Alternately, the release of SNARE monomers could be a preparatory step for the endocytosis that occurs subsequent to regulated exocytosis (Whalley et al., 1995; Bock and Scheller, 1997; Littleton et al., 1998).
Docked, Primed, and Release-Ready Prefusion States Exist Downstream of Complex Disruption
There is no evidence that the SNARE complex is essential to CV–PM docking or fusion, since under conditions of high shear stress and complete SNARE complex disruption, CV remain firmly attached to the PM and retain full competence for Ca2+-triggered exocytotic fusion (Fig. 8). Our results are consistent with recent findings in both synaptic vesicles (Butz et al., 1998) and yeast ER vesicles (Cao et al., 1998) showing the existence of other tethering or docking components in addition to the SNARE proteins. If such additional tethering/docking machinery is localized on CV, then CV brought into contact under conditions favoring SNARE complex disruption should still fuse in response to Ca2+. This is indeed the case (Fig. 9 b).
We found that intermembrane SNARE complexes may promote the Ca2+ sensitivity of fusion. When intermembrane SNARE complexes are not allowed to form, CV still dock to one another sufficiently well to undergo Ca2+-triggered fusion, albeit at [Ca2+]free higher than required in the presence of SNARE complexes (Fig. 9 b) (Tahara et al., 1998). This would suggest that clostridial toxins inhibit exocytosis, in part, by reducing the Ca2+ sensitivity of the fusion mechanism. If so, vesicles are not actually rendered nonfusogenic by toxins, but have a vastly reduced probability to fuse due to their diminished Ca2+ sensitivity. Three recent studies are also consistent with this idea. The elegant work of Rettig et al. (1997) indicates that synaptic vesicles incapable of localization to Ca2+ channels still dock to the PM, and can still fuse in response to [Ca2+]free comparable to that near open Ca2+ channels. Thus, SNARE proteins may help to localize vesicles at Ca2+ channels but may not be essential to Ca2+-triggered exocytosis. Similarly, Lawrence et al. (1996) and Capogna et al. (1997) demonstrated that increasing [Ca2+] (or Sr2+ or Ba2+) can partially overcome the effects of botulinum intoxication, but not the effects of tetanus toxin, where “rescue” of triggered fusion required Mg-ATP. These latter results might in part be explained by incomplete cleavage of the SNAREs, with NSF/ATP then “activating” residual syntaxin to form new intermembrane SNARE complexes (Ungermann et al., 1998). All of these results are also consistent with a model in which Ca2+ triggers an early, high-affinity SNARE complex-dependent step in the fusion pathway, but that at higher concentrations (mimicked by Sr2+ and Ba2+), Ca2+ can also directly trigger a later, critical, SNARE complex-independent fusion step. This might also explain why delivery of a sufficiently high [Ca2+]free can trigger significant exocytosis in epithelial cells and fibroblasts (Coorssen et al., 1996; Ninomiya et al., 1996).
As the heterotrimeric SNARE complex acts upstream of fusion, other identified SNARE protein interactions (Chapman et al., 1996; Mehta et al., 1996; Schiavo et al., 1997; Shao et al., 1997) could serve in docking or fusion. The core complex might then function as a source of reserve monomers. Complex disruption would release SNAREs to interact with counterparts on an opposing membrane, resulting in enhanced membrane–membrane contact areas and an increased Ca2+ sensitivity for fusion. This would imply the involvement of specific SNARE proteins in a putative second stage of docking that optimizes membrane–membrane contact and the Ca2+-sensitivity of fusion. Whether these protein interactions might also somehow be more directly involved in fusion must be considered. Such a model is consistent with the finding that syntaxin appears to be at least partly responsible for defining a “reserve” or “regulated” pool of vesicles (Bittner et al., 1996; Nagamatsu et al., 1996), that Drosophila syntaxin knockouts lack constitutive and regulated release (Schulze et al., 1995), and that syntaxin also appears to be the important target for activation by NSF in yeast vacuolar membranes (Ungermann et al., 1998). These results are consistent with the fact that a substantial fraction (up to 95%) of the total VAMP can be completely removed from our system without affecting fusion (Tahara et al., 1998). VAMP may have a role in recycling, or in initiating, enhancing, or stabilizing membrane–membrane contact that we bypass by centrifugation in our CV–CV fusion system. As such, our findings are also consistent with genetic studies involving SNARE mutants (Nonet et al., 1993, 1998; Broadie, 1996; Deitcher et al., 1998; Reist et al., 1998). Another SNARE protein, perhaps synaptotagmin, syntaxin, SNAP-25, some complex of these proteins, or an as yet unidentified membrane component associated with the SNAREs, may confer Ca2+ sensitivity on the fusion machinery. The Sr2+ and Ba2+ sensitivity of several synaptotagmin isoforms has been demonstrated (Li et al., 1995). However, if these proteins only function to localize vesicles near Ca2+ microdomains (a function bypassed in our reduced system), then the removal of SNARE protein monomers should not seriously affect Ca2+-triggered CV– CV fusion, provided sufficient monomers of the types possibly necessary to dock opposing membranes are still present and that sufficient [Ca2+]free is supplied. Possible roles for identified SNARE proteins therefore require critical evaluation in a reduced physiological system.
In summary, the heterotrimeric SNARE complex is not universally necessary nor sufficient for docking or fusion. Another factor or factors, perhaps other SNAREs, synaptotagmin, or as yet unidentified components, must be involved in membrane fusion. At least one must be proteinaceous, since NEM and trypsin inhibit CV–CV fusion (Table I) (Vogel and Zimmerberg, 1992), but ultimately the lipids themselves must fuse. Overall, results of studies to date emphasize the need for a molecular level definition of docking and a dissection of the docking stages required for optimal membrane–membrane contact and efficient fusion. Minimal biological systems such as CV–CV fusion will be useful for investigating the roles of SNAREs and other protein and lipid components in the final steps of Ca2+-triggered exocytosis.
Acknowledgments
The authors wish to thank S. Vogel (Medical College of Georgia, Augusta, GA) for invaluable advice and discussions during the initiation of this work. We thank K. Timmers, P. Backlund, L. Chernomordik, E. Leikina (LCMB, NICHD, NIH), and T. Whalley (University of Stirling, Stirling, UK) for critical discussions during the course of this work.
Abbreviations used in this paper
- [Ca2+]free
free Ca2+ concentration
- CSC
cell surface complex
- CV
cortical vesicles
- LPC
lysophosphatidylcholine
- <n>
average number of active fusion complexes
- NEM
N-ethylmaleimide
- NSF
N-ethylmaleimide-sensitive factor
- PM
plasma membrane
- pMe2+
−log[divalent cation]free
- SNAP
soluble NSF attachment protein
- SNARE
SNAP receptor
- VAMP
vesicle-associated membrane protein
Note Added in Proof
A lack of direct SNARE complex involvement in yeast vacuole homotypic fusion has also been demonstrated. (Ungermann, C., K. Sato, and W. Wickner. 1998. Defining the functions of trans-SNARE pairs. Nature. 396:543–548.)
Footnotes
M. Tahara's present address is Department of Obstetrics and Gynecology, Kaizuka Municipal Hospital, Osaka 597-0015, Japan.
References
- Augustine GJ, Eckert R. Divalent cations differentially support transmitter release at the squid giant synapse. J Physiol. 1984;346:257–271. doi: 10.1113/jphysiol.1984.sp015020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PF, Whitaker MJ. Influence of ATP and calcium on the cortical reaction in sea urchin eggs. Nature. 1978;276:513–515. doi: 10.1038/276513a0. [DOI] [PubMed] [Google Scholar]
- Barnett DW, Misler S. Coupling of exocytosis to depolarization in rat pancreatic islet β-cells: effects of Ca2+, Sr2+ and Ba2+-containing solutions. Pflügers Arch Eur J Physiol. 1995;430:593–595. doi: 10.1007/BF00373898. [DOI] [PubMed] [Google Scholar]
- Bers D, Patton C, Nuccitelli R. A practical guide to the preparation of Ca2+buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- Bittner MA, Bennett MK, Holz RW. Evidence that syntaxin1A is involved in storage in the secretory pathway. J Biol Chem. 1996;271:11214–11221. doi: 10.1074/jbc.271.19.11214. [DOI] [PubMed] [Google Scholar]
- Blache D, Ciavatti M, Ojeda C. Platelet aggregation and endogenous 5-HT secretion in presence of Ca2+, Sr2+ and Ba2+. Effects of calcium antagonists. Thromb Res. 1987;46:779–791. doi: 10.1016/0049-3848(87)90070-3. [DOI] [PubMed] [Google Scholar]
- Blank PS, Cho M-S, Vogel SS, Kaplan D, Kang A, Malley J, Zimmerberg J. Sub-maximal responses in calcium-triggered exocytosis are explained by differences in the calcium sensitivity of individual secretory vesicles. J Gen Physiol. 1998;112:559–567. doi: 10.1085/jgp.112.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JB, Scheller RH. A fusion of new ideas. Nature. 1997;387:133–135. doi: 10.1038/387133a0. [DOI] [PubMed] [Google Scholar]
- Boonen GJJC, VanSteveninck J, Elferink JGR. Strontium and barium induce exocytosis in electropermeabilized neutrophils. Biochim Biophys Acta. 1993;1175:155–160. doi: 10.1016/0167-4889(93)90018-k. [DOI] [PubMed] [Google Scholar]
- Broadie KS. Molecular mechanisms of neurotransmitter release. Biochem Soc Trans. 1996;24:639–645. doi: 10.1042/bst0240639. [DOI] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Südhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking, and fusion: a molecular description. Physiol Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- Cao X, Ballew N, Barlowe C. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO (Eur Mol Biol Organ) J. 1998;17:2156–2165. doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, Gähwiler BH, Thompson SM. Ca2+ or Sr2+partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, An S, Edwardson JM, Jahn R. A novel function for the second C2 domain of synaptotagmin. Ca2+-triggered dimerization. J Biol Chem. 1996;271:5844–5849. doi: 10.1074/jbc.271.10.5844. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV. Non-bilayer lipids and biological fusion intermediates. Chem Phys Lipids. 1996;8:203–213. doi: 10.1016/0009-3084(96)02583-2. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Vogel SS, Sokoloff A, Onaran HO, Leikina EA, Zimmerberg J. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS (Fed Eur Biochem Soc) Lett. 1993;318:71–76. doi: 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 1998;140:1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizmadzhev YA, Cohen FS, Shcherbakov A, Zimmerberg J. Membrane mechanics account for fusion pore dilation in stages. Biophys J. 1995;69:2489–2500. doi: 10.1016/S0006-3495(95)80119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorssen JR, Rand RP. Structural effects of neutral lipids on divalent cation-induced interactions of phosphatidylserine-containing bilayers. Biophys J. 1995;68:1009–1018. doi: 10.1016/S0006-3495(95)80276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorssen JR, Schmitt H, Almers W. Ca2+triggers massive exocytosis in Chinese hamster ovary cells. EMBO (Eur Mol Biol Organ) J. 1996;15:3787–3791. [PMC free article] [PubMed] [Google Scholar]
- Crabb JH, Jackson RC. In vitro reconstitution of exocytosis from plasma membrane and isolated secretory vesicles. J Cell Biol. 1985;101:2263–2273. doi: 10.1083/jcb.101.6.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Sheetz MP. Regulation of endocytosis, exocytosis, and shape by membrane tension. Cold Spring Harb Symp Quant Biol. 1995;60:567–571. doi: 10.1101/sqb.1995.060.01.060. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Quantal transmitter secretion from myocytes loaded with acetylcholine. Nature. 1992;359:733–736. doi: 10.1038/359733a0. [DOI] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophilagene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Miledi R, Rahamimoff R. Strontium and quantal release of transmitter at the neuromuscular junction. J Physiol. 1969;200:267–283. doi: 10.1113/jphysiol.1969.sp008692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson GW. Calcium ion binding between lipid bilayers: the four-component system of phosphatidylserine, phosphatidylcholine, calcium chloride, and water. Biochemistry. 1989;28:1270–1278. doi: 10.1021/bi00429a048. [DOI] [PubMed] [Google Scholar]
- Ferro-Novick S, Jahn R. Vesicle fusion from yeast to man. Nature. 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- Foreman JC, Mongar JL. The role of the alkaline earth ions in anaphylactic histamine secretion. J Physiol. 1972;224:753–769. doi: 10.1113/jphysiol.1972.sp009921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz RW, Bittner MA, Peppers SC, Senter RA, Eberhard DA. MgATP-independent and MgATP-dependent exocytosis. J Biol Chem. 1989;264:5412–5419. [PubMed] [Google Scholar]
- Jahn R, Hanson PI. Membrane fusion. SNAREs line up in new environment. Nature. 1998;393:14–15. doi: 10.1038/29871. [DOI] [PubMed] [Google Scholar]
- Kaplan D, Bungay P, Sullivan J, Zimmerberg J. A rapid-flow perfusion chamber for high-resolution microscopy. J Microsc. 1996;181:286–297. doi: 10.1046/j.1365-2818.1996.103383.x. [DOI] [PubMed] [Google Scholar]
- Knight DE, Scrutton MC. Gaining access to the cytosol: the technique and some applications of electropermeabilization. Biochem J. 1986;234:497–506. doi: 10.1042/bj2340497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence GW, Foran P, Dolly JO. Distinct exocytotic responses of intact and permeabilized chromaffin cells after cleavage of the 25-kDa synaptosomal-associated protein (SNAP-25) or synaptobrevin by botulinum toxin A or B. Eur J Biochem. 1996;236:877–886. doi: 10.1111/j.1432-1033.1996.00877.x. [DOI] [PubMed] [Google Scholar]
- Li CL, Davletov A, Südhof TC. Distinct Ca2+ and Sr2+binding properties of synaptotagamins. J Biol Chem. 1995;270:24898–24902. doi: 10.1074/jbc.270.42.24898. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- Martell, A.E., and R.M. Smith. 1974a. Critical Stability Constants. Vol. 1. Amino Acids. Plenum Press, New York. 469pp.
- Martell, A.E., and R.M. Smith. 1974b. Critical Stability Constants. Vol. 6. Second Supplement. Plenum Press, New York. 643 pp.
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Mehta PP, Battenberg E, Wilson MC. SNAP-25 and synaptotagmin involvement in the final Ca2+-dependent triggering of neurotransmitter exocytosis. Proc Natl Acad Sci USA. 1996;93:10471–10476. doi: 10.1073/pnas.93.19.10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellow AM. Equivalence of Ca2+ and Sr2+ in transmitter release from K+-depolarised nerve terminals. Nature. 1979;282:84–85. doi: 10.1038/282084a0. [DOI] [PubMed] [Google Scholar]
- Miledi R. Strontium as a substitute for calcium in the process of transmitter release at the neuromuscular junction. Nature. 1966;212:1233–1234. doi: 10.1038/2121233a0. [DOI] [PubMed] [Google Scholar]
- Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy GW, Kopf GS, Gache C, Vacquier VD. Calcium-mediated release of glucanase activity from cortical granules of sea urchin eggs. Dev Biol. 1983;100:267–274. doi: 10.1016/0012-1606(83)90221-x. [DOI] [PubMed] [Google Scholar]
- Nagamatsu S, Fujiwara T, Nakamichi Y, Watanabe T, Katahira H, Sawa H, Akagawa K. Expression and functional role of syntaxin 1/HPC-1 in pancreatic beta cells. Syntaxin 1A, but not 1B, plays a negative role in regulatory insulin release pathway. J Biol Chem. 1996;271:1160–1165. doi: 10.1074/jbc.271.2.1160. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Miyashita Y, Kasai H. Ca2+-dependent exocytotic pathways in Chinese hamster ovary fibroblasts revealed by a caged-Ca2+compound. J Biol Chem. 1996;271:17751–17754. doi: 10.1074/jbc.271.30.17751. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegansmutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis eleganssynaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons T, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Pevsner J, Hsu S-C, Braun JEA, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Pryer NK, Wuestehube LJ, Schekman R. Vesicle-mediated protein sorting. Annu Rev Biochem. 1992;61:471–516. doi: 10.1146/annurev.bi.61.070192.002351. [DOI] [PubMed] [Google Scholar]
- Rahamimoff R, Fernandez JM. Pre- and postfusion regulation of transmitter release. Neuron. 1997;18:17–27. doi: 10.1016/s0896-6273(01)80043-x. [DOI] [PubMed] [Google Scholar]
- Reist NE, Buchanan J, Li J, DiAntonio A, Buxton EM, Schwarz TL. Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. . J Neuroscience. 1998;18:7662–7673. doi: 10.1523/JNEUROSCI.18-19-07662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann C, Ashery U, Sheng Z-H, Yokoyama CT, Catterall WA, Neher EJ. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+channel/syntaxin interaction. Neuroscience. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE. The protein machinery of vesicle budding and fusion. Protein Sci. 1996;5:185–194. doi: 10.1002/pro.5560050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Söllner TH. Throttles and dampers: controlling the engine of membrane fusion. Science. 1997;276:1212–1213. doi: 10.1126/science.276.5316.1212. [DOI] [PubMed] [Google Scholar]
- Rüden LV, Garcia AG, López MG. The mechanism of Ba2+-induced exocytosis from single chromaffin cells. FEBS (Fed Eur Biochem Soc) Lett. 1993;336:48–52. doi: 10.1016/0014-5793(93)81606-z. [DOI] [PubMed] [Google Scholar]
- Sasaki H. Modulation of calcium sensitivity by a specific cortical protein during sea urchin egg cortical vesicle exocytosis. Dev Biol. 1984;101:125–135. doi: 10.1016/0012-1606(84)90123-4. [DOI] [PubMed] [Google Scholar]
- Scepek S, Coorssen JR, Lindau M. Fusion pore expansion in horse eosinophils is modulated by Ca2+and protein kinase C via distinct mechanisms. EMBO (Eur Mol Biol Organ) J. 1998;17:4340–4345. doi: 10.1093/emboj/17.15.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Stenbeck G, Rothman JE, Söllner TH. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc Natl Acad Sci USA. 1997;94:997–1001. doi: 10.1073/pnas.94.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophilasyntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- Shao X, Li C, Fernandez I, Zhang X, Südhof TC, Rizo J. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron. 1997;18:133–142. doi: 10.1016/s0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- Söllner T. SNAREs and targeted membrane fusion. FEBS (Fed Eur Biochem Soc) Lett. 1995;369:80–83. doi: 10.1016/0014-5793(95)00594-y. [DOI] [PubMed] [Google Scholar]
- Söllner T, Rothman JE. Neurotransmission: harnessing fusion machinery at the synapse. Trends Neurosci. 1994;17:344–348. doi: 10.1016/0166-2236(94)90178-3. [DOI] [PubMed] [Google Scholar]
- Söllner T, Rothman JE. Molecular machinery mediating vesicle budding, docking and fusion. Cell Struct Function. 1996;21:407–412. doi: 10.1247/csf.21.407. [DOI] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Tahara M, Coorssen JR, Timmers K, Blank PS, Whalley T, Scheller R, Zimmerberg J. Calcium can disrupt the SNARE protein complex on sea urchin egg secretory vesicles without irreversibly blocking fusion. J Biol Chem. 1998;273:33667–33673. doi: 10.1074/jbc.273.50.33667. [DOI] [PubMed] [Google Scholar]
- TerBush DR, Holz RW. Barium and calcium stimulate secretion from digitonin-permeabilized bovine adrenal chromaffin cells by similar pathways. J Neurochem. 1992;58:680–687. doi: 10.1111/j.1471-4159.1992.tb09771.x. [DOI] [PubMed] [Google Scholar]
- Ungermann C, Nichols BJ, Pelham HRB, Wickner W. A vacuolar v_t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacquier VD. The isolation of intact cortical granules from sea urchin eggs: calcium ions trigger granule discharge. Dev Biol. 1975;43:62–74. doi: 10.1016/0012-1606(75)90131-1. [DOI] [PubMed] [Google Scholar]
- Vilmart-Seuwen J, Kersken H, Stürzl R, Plattner H. ATP keeps exocytosis sites in a primed state but is not required for membrane fusion: an analysis with Paramecium cells in vivo and in vitro. J Cell Biol. 1986;103:1279–1288. doi: 10.1083/jcb.103.4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SS, Zimmerberg J. Proteins on exocytotic vesicles mediate calcium-triggered fusion. Proc Natl Acad Sci USA. 1992;89:4749–4753. doi: 10.1073/pnas.89.10.4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SS, Blank PS, Zimmerberg J. Poisson-distributed active fusion complexes underlie the control of the rate and extent of exocytosis by calcium. J Cell Biol. 1996;134:329–338. doi: 10.1083/jcb.134.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SS, Chernomordik LV, Zimmerberg J. Calcium-triggered fusion of exocytotic granules requires proteins in only one membrane. J Biol Chem. 1992;267:25640–25643. [PubMed] [Google Scholar]
- Vogel SS, Leikina EA, Chernomordik LV. Lysophosphatidylcholine reversibly arrests exocytosis and viral fusion at a stage between triggering and membrane merger. J Biol Chem. 1993;268:25764–25768. [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westernmann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weis WI, Scheller RH. SNARE the rod, coil the complex. Nature. 1998;395:328–329. doi: 10.1038/26354. [DOI] [PubMed] [Google Scholar]
- Whalley T, Whitaker M. Exocytosis reconstituted from the sea urchin egg is unaffected by calcium pretreatment of granules and plasma membrane. Biosci Rep. 1988;8:335–343. doi: 10.1007/BF01115224. [DOI] [PubMed] [Google Scholar]
- Whalley T, Sokoloff A. The N-ethylmaleimide–sensitive protein thiol groups necessary for sea urchin egg cortical-granule exocytosis are highly exposed to the medium and are required for triggering by Ca2+ . Biochem J. 1994;302:391–396. doi: 10.1042/bj3020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalley T, Terasaki M, Cho M-S, Vogel SS. Direct membrane retrieval into large vesicles after exocytosis in sea urchin eggs. J Cell Biol. 1995;131:1183–1192. doi: 10.1083/jcb.131.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker MJ, Baker PF. Calcium-dependent exocytosis in an in vitro secretory granule plasma membrane preparation from sea urchin eggs and the effects of some inhibitors of cytoskeletal function. Proc Royal Soc Lond B. 1983;218:397–413. doi: 10.1098/rspb.1983.0047. [DOI] [PubMed] [Google Scholar]
- Whitaker MJ, Aitchison M. Calcium-dependent polyphosphoinositide hydrolysis is associated with exocytosis in vitro. FEBS (Fed Eur Biochem Soc) Lett. 1985;182:119–124. doi: 10.1016/0014-5793(85)81167-4. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Liu J. Ionic and permeability requirements for exocytosis in vitro in sea urchin eggs. J Membrane Biol. 1988;101:199–207. doi: 10.1007/BF01872835. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Sardet C, Epel D. Exocytosis of sea urchin egg cortical vesicles in vitro is retarded by hyperosmotic sucrose: kinetics of fusion monitored by quantitative light-scattering microscopy. J Cell Biol. 1985;101:2398–2410. doi: 10.1083/jcb.101.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]