

Abstract
Intracellular deposition of misfolded protein aggregates into ubiquitin-rich cytoplasmic inclusions is linked to the pathogenesis of many diseases. Why these aggregates form despite the existence of cellular machinery to recognize and degrade misfolded protein and how they are delivered to cytoplasmic inclusions are not known. We have investigated the intracellular fate of cystic fibrosis transmembrane conductance regulator (CFTR), an inefficiently folded integral membrane protein which is degraded by the cytoplasmic ubiquitin-proteasome pathway. Overexpression or inhibition of proteasome activity in transfected human embryonic kidney or Chinese hamster ovary cells led to the accumulation of stable, high molecular weight, detergent-insoluble, multiubiquitinated forms of CFTR. Using immunofluorescence and transmission electron microscopy with immunogold labeling, we demonstrate that undegraded CFTR molecules accumulate at a distinct pericentriolar structure which we have termed the aggresome. Aggresome formation is accompanied by redistribution of the intermediate filament protein vimentin to form a cage surrounding a pericentriolar core of aggregated, ubiquitinated protein. Disruption of microtubules blocks the formation of aggresomes. Similarly, inhibition of proteasome function also prevented the degradation of unassembled presenilin-1 molecules leading to their aggregation and deposition in aggresomes. These data lead us to propose that aggresome formation is a general response of cells which occurs when the capacity of the proteasome is exceeded by the production of aggregation-prone misfolded proteins.
Keywords: ubiquitin, proteasome, intermediate filaments, protein aggregation, presenilin
Protein folding is the process by which a linear polymer of amino acids is converted to a unique three-dimensional structure that comprises a functional protein molecule. Folding requires selection of one structure out of a constellation of sterically available but incorrect conformers. In cells, protein folding may fail because of amino acid misincorporation resulting from genetic mutation or errors in transcription, mRNA processing, or translation. Alternatively, environmental factors such as thermal stress (Vidair et al., 1996), osmotic and oxidative stress (Buchner, 1996), or interference from viral gene products (Wiertz et al., 1996a ; Levitskaya et al., 1997) can interfere with the folding of nascent polypeptides. Finally, unequal synthesis of subunits of heterooligomeric protein complexes can lead to the production of unassembled polypeptide chains (Bonifacino et al., 1989). Many misfolded and unassembled proteins inappropriately expose hydrophobic surfaces that are normally buried in the protein's interior or at the interface with other subunits (Wetzel, 1994). Integral membrane proteins possess extensive hydrophobic stretches, which are normally imbedded in the lipid bilayer. Failure to correctly translocate and integrate such proteins could result in exposure of these hydrophobic side chains to the aqueous environment of the cytosol. Such inappropriate exposure of hydrophobic sequences can lead to the adoption of alternate, nonnative conformations that can interact to form aggregates.
Protein aggregates are generally difficult to unfold or to degrade; their formation in cells is tightly linked to the etiologies of several common aging-related degenerative diseases including amyloidoses, slow virus encephalopathies, and Alzheimer's disease (Wetzel, 1994; Carrell and Lomas, 1997). In fact, many neurodegenerative diseases are defined histopathologically by characteristic intracellular inclusions composed of aggregated, ubiquitinated protein and abnormal deposition of intermediate filament (IF)1 protein (Mayer et al., 1989a ). However, the mechanism by which protein aggregates can kill cells, or indeed, whether the presence of aggregates in diseased neurons is a cause or a consequence of underlying cellular pathology, has yet to be convincingly established.
Given the potentially pathogenic consequences of intracellular protein aggregation, it is not surprising that eukaryotic cells have evolved mechanisms to prevent their formation or accumulation. Molecular chaperones bind nonnative protein conformations, sequestering misfolded protein or aggregation-prone folding intermediates from the cytosol, thereby reducing the likelihood of aggregate formation (Hartl, 1996). However, many misfolded proteins are targeted by covalent attachment of multiubiquitin chains for degradation by the proteasome (Haas and Siepmann, 1997). This pathway serves to degrade misfolded cytoplasmic proteins as well as membrane and secretory proteins that are unable to fold in the ER (Kopito, 1997; Sommer and Wolf, 1997). Such proteins must be dislocated across the ER membrane for degradation by cytoplasmic proteasomes. Recent genetic and biochemical evidence suggests that dislocation proceeds by “retrotranslocation” of the misfolded polypeptide through a membrane channel composed of the Sec61p translocon (Wiertz et al., 1996b ; Plemper et al., 1997), and that this process is coupled to the activity of the 26S proteasome (Mayer et al., 1998). Coupling of membrane dislocation and proteolysis in this manner is important because even transient exposure of hydrophobic integral membrane proteins to the cytosol could pose a serious aggregation risk to the cell. In this study we investigated the effects of proteasome inhibition on the intracellular fate of two integral membrane proteins, cystic fibrosis transmembrane conductance regulator (CFTR) and presenilin-1 (PS1).
CFTR is an ∼165-kD polytopic glycoprotein which encodes a Cl− ion channel in the plasma membrane of epithelial cells (Riordan et al., 1989). Loss-of-function mutations in the gene encoding CFTR cause the autosomal recessive disease, cystic fibrosis (reviewed in Welsh and Smith, 1993). The most common CF-causing allele, ΔF508, drastically interferes with the protein's ability to fold, effectively barring it from being functionally expressed. Previously, we (Ward and Kopito, 1994) and others (Lukacs et al., 1994) have demonstrated that wild-type CFTR is an inefficiently folded protein with only 20–40% of nascent chains being able to mature beyond the ER. Newly synthesized CFTR and ΔF508 molecules that fail to fold productively are rapidly eliminated from cells by a process requiring covalent modification with ubiquitin and degradation by the proteasome (Jensen et al., 1995; Ward et al., 1995). Inhibition of CFTR and ΔF508 degradation with proteasome inhibitors such as acetyl-leucyl-leucyl-norleucinal (ALLN), lactacystin (Ward et al., 1995), ZL3H, or MG132 (Ward, C.L., and R.R. Kopito, unpublished data) fails to increase the folding yield, and instead leads to the intracellular accumulation of detergent-insoluble, multiubiquitinated high molecular weight CFTR aggregates.
PS1 is an ∼43-kD integral membrane protein which, like CFTR, spans the lipid bilayer multiple times (Doan et al., 1996). PS1 is ubiquitously expressed at low levels in mammalian cells, where it is predominantly localized to the ER. PS1 has been also reported to be present in the vicinity of the Golgi apparatus (Kovacs et al., 1996; De Strooper et al., 1997; Lah et al., 1997; Zhang et al., 1998) and the centrosome (Li et al., 1997). Mutations in the PS1 gene cosegregate with autosomal dominant early-onset familial Alzheimer's disease (Tanzi et al., 1996; Cruts et al., 1998). Endogenous PS1 is endoproteolytically processed into ∼27-kD NH2-terminal and 17-kD COOH-terminal fragments (Thinakaran et al., 1996). Transfection of PS1 (Thinakaran et al., 1996) or the highly homologous PS2 (Kim, 1997) into mammalian cells leads to increased steady-state accumulation of processed and unprocessed forms, suggesting that the cellular machinery required for presenilin processing may be limiting under conditions of overexpression. Kim et al. (1997) have reported that excess unprocessed (i.e., full-length) PS2 is targeted for proteasomal degradation by multiubiquitination and that inhibition of proteasome activity in PS2 transfected cells leads to the accumulation of high molecular weight, multiubiquitinated PS2 derivatives. These and other (Thinakaran et al., 1997) data suggest that limiting cellular factors restrict the extent to which newly synthesized presenilins are processed and that unprocessed forms may be (like CFTR) substrates for degradation by the ubiquitin-proteasome pathway.
In this study we have characterized the cellular and molecular response to the formation of aggregates of misfolded CFTR and PS1. Our data indicate that aggregation occurs when the capacity of the proteasome degradation pathway is exceeded, either by an increase in substrate expression or by a decrease in proteasome activity. Once formed, these aggregates become refractory to intracellular proteolysis and are delivered to an ubiquitin-rich structure at the microtubule (MT)-organizing center (MTOC) where they are ensheathed by the IF protein vimentin. We have termed this structure the aggresome. Our data show that aggresomes can be formed from aggregates of several different integral membrane proteins and lead us to propose that aggresomes are a general cellular response to the presence of aggregated, undegraded protein.
Materials and Methods
Antibodies and Plasmids
The rabbit polyclonal anti-CFTR antibody C1468 has been described previously (Ward and Kopito, 1994). The following antibodies were used in this study: rabbit polyclonal anti–α-mannosidase II (anti-manII; M. Farquhar, University of California at San Diego, San Diego, CA; Velasco et al., 1993), polyclonal anti–β-COP (S. Pfeffer, Stanford University), monoclonal γ-tubulin (clone: GTU-88; Sigma Chemical Co., St. Louis, MO), monoclonal α-tubulin (DM1a; T. Stearns, Stanford University), rabbit polyclonal antibody to PS1NT (1–65 aa) (G. Thinakaran, Johns Hopkins University, Baltimore, MD; Thinakaran et al., 1996), mouse monoclonal anti–human lysosomal membrane protein-1 (LAMP-1, BB6; M. Fukuda, Burnham Institute, La Jolla, CA; Carlsson et al., 1988), polyclonal anti-ubiquitin (A. Haas, Medical College of Wisconsin, Milwaukee, WI), monoclonal c-myc (monoclonal Ab-1; Oncogene Science, Manhasset, NY), polyclonal and monoclonal anti-GFP (Clontech, Palo Alto, CA), and monoclonal vimentin (clone V9; Sigma Chemical Co.). pRC/CMV CFTR and ΔF508-CFTR, as well as c-myc-ubiquitin plasmids, have been described previously (Ward et al., 1995). GFP-CFTR–expressing plasmid (B. Stanton, Dartmouth Medical School, Hanover, NH; Moyer et al., 1998) was used as a template for construction of the GFP-ΔF508 by site-directed mutagenesis with the megaprimer method (Landt et al., 1990; Sarkar and Sommer, 1990). pcDNA3 PS1– and A246E-expressing plasmids have been described elsewhere (Group, 1995).
Cell Lines and Cell Culture
Human embryonic kidney 293 (HEK) cells were maintained in DME and transfected as described previously (Ward et al., 1995). Stable GFP-CFTR–expressing cell lines were maintained in the same media with the addition of 2 mg/ml G418. Transient transfections were carried out by adding plasmid DNA as a calcium phosphate precipitate (Graham and Eb, 1973). Stable cell lines were prepared by transfection with Lipofectamine (GIBCO BRL, Gaithersburg, MD) followed after 48 h by selection of transformed cells by growth in 1 mg/ml Geneticin (G418). These G418 resistant cells were then analyzed by flow cytometry and the top 20% fluorescent cells were isolated. This population of cells was then expanded and assayed for expression by immunoblotting with antibodies to GFP and CFTR.
Pulse–Chase Experiments and Immunoblotting
Cells were pulse-labeled with [35S]methionine/cysteine for 15 min, harvested, and immunoprecipitated as described previously (Ward and Kopito, 1994). Loading volumes were normalized to the sample with the lowest TCA precipitable cpm. For immunoblotting, cell pellets from washed and transfected HEK cells were lysed in 250 μl of ice-cold IPB buffer (10 mM Tris-HCl, pH 7.5, 5 mM EDTA, 1% NP-40, 0.5% deoxycholate, and 150 mM NaCl) plus protease inhibitors (100 μM TLCK, 100 μM TPCK, and 1 mM PMSF) for 30 min on ice. Insoluble material was recovered by centrifugation at 13,000 g for 15 min and solubilized in 50 μl 10 mM Tris-HCl, 1% SDS for 10 min at room temperature. After addition of 200 μl IPB, samples were sonicated for 20 s with a tip sonicator. Endoglycosidase H (Boehringer Mannheim, Mannheim, Germany) digestion was performed on immunoprecipitates overnight at 37°C in the buffer supplied by the manufacturer. Gel autoradiograms were quantified as described previously (Ward and Kopito, 1994). For immunoblotting, cell fractions normalized for total protein were separated on 7% SDS-PAGE and electroblotted. Chemiluminescent detection was carried out with the Renaissance detection kit (New England Nuclear, Boston, MA).
Fluorescence Microscopy
Cells were seeded onto No. 1 coverslips. For drug treatments, ALLN (Calbiochem, La Jolla, CA) 5–10 μg/ml and nocodozole (Sigma Chemical Co.) 10 μg/ml in DMSO were added to the culture medium 12 h before fixation. Cells were fixed in –20°C methanol (6 min), 50% methanol/50% acetone (6 min at room temperature), 4% paraformaldehyde (followed by a 0.5% Triton X-100 permeabilization for 15 min), with no observable differences in aggresome morphology, or relative position to markers described in the results. After fixation, cells were washed extensively in PBS and blocked with 10% BSA for 10 min. Primary antibody was incubated for 30 min to 12 h. Cells were washed 5 × 5 min in PBS and incubated with fluorophore conjugated secondary antibodies at a 1:100 dilution of 1 mg/ml stock. Cells were washed again (10 × 2 min each) and then incubated for 3 min in PBS + bisbenzamide (Sigma Chemical Co.) at 10 μg/ml to stain the DNA. Cells were washed a final time, mounted onto slides in 50% PBS/ 50% glycerol and viewed at 40×/1.25 NA or 63×/1.4 NA with a Zeiss Axiophot microscope. Images were obtained with Metamorph software (Universal Imaging) on a PC workstation. Montages of images were prepared by using Photoshop 4.01 (Adobe). Deconvolved microscope images were obtained by collecting images on an Olympus microscope with a 63×/1.25 NA objective, and deconvolved using Deltavision software (Applied Precision) on a Silicon Graphics workstation. 8 μm of optical sections 0.2 μm thick was analyzed. The stereo pair image in Fig. 5 was obtained by a 6° tilt of an entire 8-μm stack of images.
Figure 5.
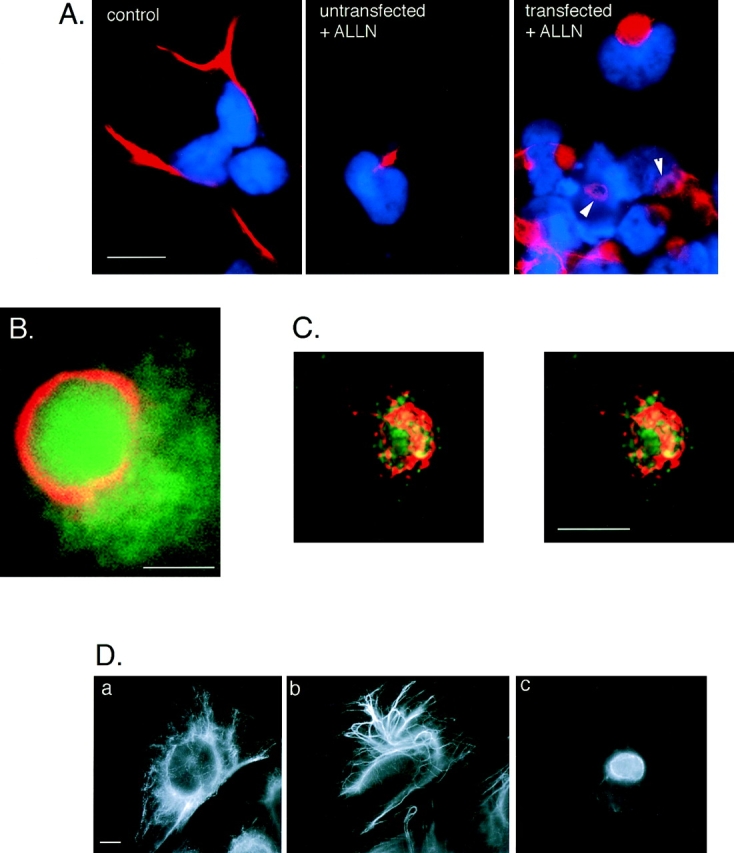
Aggresome formation is accompanied by a reorganization of the vimentin cytoskeleton. (A) Effect of ALLN on the distribution of vimentin (red) with respect to the nucleus (blue) in untransfected (left and middle) and CFTR transfected (right) HEK cells. Cells were either untreated (left) or incubated for 12 h in 10 μg/ml ALLN (middle and right). Note ring-like appearance of vimentin fluorescence in ALLN-treated CFTR-expressing cells (arrows). (B) Colocalization of GFP-CFTR (green) and vimentin (red) in ALLN-treated cells. (C) Stereo pair of optical sections from deconvolved stack of images from ALLN-treated cells showing GFP-CFTR (green autofluorescence) and vimentin (red immunofluorescence). Nuclear fluorescence signal has been omitted. (D) Distribution of vimentin in drug-treated CHO cells. CHO cells stably expressing GFP-CFTR were treated with carrier (a), 10 μg/ml nocodozole (b), or 10 μg/ml ALLN (c) for 12 h and then immunostained with antibodies to vimentin. Note the condensation and bundling of vimentin in b, as compared with the complete collapse of vimentin in c. Bars, 15 μm (A), 2.5 μm (B), and 5 μm (C and D).
EM
Cells were grown to 85% confluence in 100-mm dishes and treated overnight with 10 μg/ml ALLN, washed once in PBS, and then fixed in freshly made AMT buffer (100 mM Hepes, pH 7.4, 5 mM MgCl2, 1% glycerol, and 2% glutaraldehyde [EMS]) for 35 min at room temperature. Cells were washed twice in phosphate buffer (100 mM phosphate buffer, pH 7.5), scraped, and collected by centrifugation for 10 min at 10,000 g. The fixed pellet of cells was then postfixed for 45 min in 2% OsO4, and then washed extensively in water. En bloc staining in 1% UA for 1 h was followed by a series of graded ethanol dehydrations (25, 50, 75, 85, 95, and 100%) followed by overnight incubation in 1:1 100% ethanol/LR White medium grade resin, or Polybed resin (Polysciences, Inc., Warrington, PA). After two changes of fresh 100% resin, the cell pellets were transferred to gelatin capsules and polymerized in fresh resin overnight at 45–60°C. Gold sections were cut and collected onto copper grids and stained with 1% UA for 3 min followed by 5 min in lead citrate to generate final contrast, before viewing in a JOEL electron microscope. For immunogold EM experiments, isolated aggresomes (see below) were incubated for 2 h in primary antibody at 1: 50 (anti-GFP) and 1:10 (antivimentin) by rotating end-over-end at 4°C. The aggresome material was then washed two to three times in PBS by sedimentation at 2,000 g. After the third wash, material was resuspended in 1:10 diluted gold-conjugated antibodies and rotated again for 1 h at 4°C. Labeled aggresomes were washed as before (three times in PBS collected by sedimentation at 2,000 g for 3 min), with the final wash followed by resuspension in 2% glutaraldehyde in PBS for 20 min. Fixed material was then collected at 10,000 g for 10 min. The resulting pellet was washed three times in PBS and postfixed en block in 2% OsO4 for 20 min at room temperature. The pellet was washed extensively in water and then contrasted for 1 h in 1% UA. Ethanol dehydration and embedding was as described above. Gold sections were allowed to dry on nickel grids and were viewed with no further contrasting to maximize visualization of gold particles. Virtually no gold particles were observed when either primary antibody was omitted.
Preparation of Aggresomes
Because the aggresome consists of a juxtanuclear cap of vimentin, the technique of Starger (Starger and Goldman, 1977) was modified to optimize coisolation of GFP-CFTR and IF. In brief, cells were grown to 85% confluency in 100-mm dishes and treated with 10 μg/ml ALLN for 12 h before isolation. Cells were washed twice in PBSa (6 mM sodium-potassium phosphate buffer, 170 mM NaCl, 3 mM KCl), scraped, and collected for 3 min at 2,500 g. Washed cells were resuspended in 1 ml PBSa per 100-mm plate and passaged through a 25-gauge needle three to four times until bright-field microscopy revealed the majority of cells were disrupted. This material was washed by resuspension and sedimentation at 2,000 g three times in PBSa. The resulting material was examined by fluorescence microscopy for the presence of GFP-containing isolated aggresomes. This resulting cellular fraction enriched for aggresomes was collected a final time at 2,000 g and resuspended in 200 μl of PBS/1% BSA. This served as the starting material for the immunoelectron microscopy techniques described above.
Results
Misfolded CFTR Molecules Form Stable Aggregates
Formation of CFTR aggregates was induced in ΔF508-expressing HEK cells either by incubation with proteasome inhibitor (Fig. 1 A) or by overexpression (Fig. 1 B). When expressed at low levels (Fig. 1 A), ΔF508 was barely detectable as a 140-kD core-glycosylated band that was largely detergent-soluble (band B). Exposure to the proteasome inhibitors ALLN (Fig. 1 A) or lactacystin (data not shown) led to a massive increase in the total amount of ΔF508 present in these cells, consistent with the fact that this mutant form of CFTR cannot fold and is quantitatively degraded with t 1/2 = 30–40 min (Lukacs et al., 1994; Ward and Kopito, 1994). Therefore, the low steady-state level of ΔF508 in the absence of proteasome inhibitor must reflect the fact that the rate of ΔF508 degradation in these cells is only slightly exceeded by the rate of its synthesis. Incubation with proteasome inhibitor increased primarily the levels of detergent-insoluble ΔF508 forms which, as we have shown previously (Ward et al., 1995), correspond to aggregated and multiubiquitinated protein. Similarly, a 40-fold increase in ΔF508 expression (Fig. 1 B) even in the absence of proteasome inhibitor led to an enormous increase in the level of insoluble ΔF508. This material could be resolved into a band of mobility of unglycosylated ΔF508 (band A; see below) and, to a lesser extent, a high molecular weight smear corresponding to aggregated, multiubiquitinated forms (the blot in Fig. 1 B was deliberately underexposed to reveal the relative mobilities of bands A and B, and therefore the absolute levels of expression cannot be compared with Fig. 1 A). Incubation of ΔF508-overexpressing cells with proteasome inhibitor led to an even further increase in the levels of insoluble forms of the protein. Similar results, both at high and low expression levels, were obtained for wild-type CFTR (data not shown), which is folded with only 30–40% efficiency (Lukacs et al., 1994; Ward and Kopito, 1994). The glycosylation status of the material labeled bands A and B was confirmed by susceptibility to endoglycosidase H (Fig. 1 C). Pulse–chase analysis of overexpressed ΔF508 revealed that it is initially synthesized as a core-glycosylated, endoglycosidase H–sensitive band (band B) that is nearly completely soluble in nonionic detergent. After a 90-min chase, a significant fraction of this material is converted to a detergent-insoluble form (band A) that is insensitive to endoglycosidase H and migrates with the mobility of the unglycosylated protein. That this material is unglycosylated is also supported by its inability to bind to the lectin concanavalin A (data not shown).
Figure 1.




Misfolded CFTR molecules form stable aggregates. (A and B) Immunoblots showing steady-state accumulation of core-glycosylated (band B), unglycosylated (band A), and high molecular weight forms of ΔF508 in HEK cells expressing low (A) or high (B) levels of the protein. Transfected cells were incubated in the presence of ALLN (10 μg/ml) or carrier for 16 h before harvesting. Cells were lysed and separated into detergent-soluble (s) and -insoluble (i) fractions as described in Materials and Methods. Blot in B was deliberately underexposed to emphasize the mobilities of the accumulated species; therefore, the intensity cannot be directly compared with the blot in A. (C) Deglycosylation of CFTR. HEK cells overexpressing ΔF508 (as in B) were pulse labeled with [35S]Met for 15 min and chased with unlabeled medium for 90 min. At the indicated time points, cell lysates were separated into detergent-soluble and -insoluble fractions before immunoprecipitation with CFTR antibody. Half of each sample was digested with endoglycosidase H (endoH) as indicated, before SDS-PAGE and autoradiography. (D) Extended pulse–chase of detergent-soluble and -insoluble ΔF508. HEK cells overexpressing ΔF508 (as in B) were pulse labeled with [35S]Met for 15 min and chased with unlabeled medium for the times indicated. At each time point, the cell lysate was separated into detergent-soluble and -insoluble fractions before immunoprecipitation with CFTR antibody. (E) Kinetics of degradation of GFP-CFTR. HEK cells expressing CFTR or GFP-CFTR were pulse labeled with [35S]Met for 15 min and chased with unlabeled medium for the times indicated. The detergent-soluble fractions were immunoprecipitated with CFTR antibody, separated by SDS-PAGE; the data were scanned and quantified from autoradiographic images.
The stability of the aggregated protein was examined by extended pulse–chase analysis of cells overexpressing ΔF508 (Fig. 1 D). After a 15-min pulse and a 2-h chase, the protein partitioned into a detergent-soluble fraction, containing mostly core-glycosylated ΔF508 (band B), and a detergent-insoluble fraction. The latter contained a mixture of unglycosylated ΔF508 (band A) and aggregated high molecular weight species. Subsequent chase revealed that, whereas the core-glycosylated form disappeared rapidly (the t 1/2 of this species is only 20–30 min; Ward and Kopito, 1994), the insoluble forms decayed slowly, exhibiting a t 1/2 in the range of 12–15 h. Together, these data suggest that intracellular accumulation of unglycosylated, detergent-insoluble, high molecular weight forms of ΔF508 can be induced either by inhibition or saturation of proteasome activity. Moreover, our data suggest that this process is associated with the conversion of core glycosylated, short-lived ΔF508 protein into unglycosylated stable aggregates.
Misfolded CFTR Molecules Cluster in Juxtanuclear Structures
To determine the intracellular distribution of aggregated and nonaggregated forms of ΔF508, fluorescence microscopy was used to monitor the expression of GFP-tagged ΔF508 after exposure to proteasome inhibitor (Fig. 2 A). The presence of the GFP tag has no detectable effect on the ability of GFP-CFTR to traffic normally through the secretory and the endocytic pathways (Moyer et al., 1998). Moreover, control experiments indicate that the presence of an NH2-terminal GFP tag does not alter the kinetics of ΔF508 or CFTR degradation (Fig. 1 E). In untreated cells, faint GFP fluorescence was detected in a perinuclear and reticular distribution (Fig. 2 A, 0 h), coincident with localization of the ER markers BiP and calnexin (data not shown). These data confirm our immunoblot data (Fig. 1 A) which indicated that, at low expression levels, ΔF508 is mostly in the core-glycosylated, ER form. Exposure to the proteasome inhibitor, ALLN (Fig. 2 A, 4–14 h), increased total cellular GFP fluorescence and increased the fluorescence signal at a distinct site adjacent to the nucleus. After 14 h of exposure to the inhibitor, nearly all of the GFP fluorescence was present in a single large, juxtanuclear structure that appears to impinge upon and to distort the contour of the nuclear envelope. Identical results were obtained in HEK cells expressing GFP-CFTR or transiently expressing wild-type (i.e., nonfusion) CFTR or ΔF508 (data not shown), confirming that the redistribution of the fluorescent signal in response to proteasome inhibitor was not unique to ΔF508 nor was it an artifact of the GFP fusion. Indistinguishable results were obtained using the more specific proteasome inhibitors, MG132 or lactacystin (data not shown). Finally, similar structures were observed in CHO cells expressing CFTR and GFP-CFTR, indicating that this response is not unique to HEK cells (data not shown).
Figure 2.

Ubiquitinated CFTR molecules accumulate in aggresomes. (A) Time course of ΔF508 accumulation. HEK cells stably expressing low levels of GFP-ΔF508 were incubated in the presence of ALLN (10 μg/ml) for the times indicated. Note the absence of plasma membrane staining consistent with the inability of the ΔF508 to traffic to the cell surface. (B) Colocalization of ΔF508 with epitope-tagged ubiquitin. HEK cells were transiently cotransfected with ΔF508 and c-myc-ubiquitin cDNA and incubated in the presence of ALLN (10 μg/ ml) for 12 h and processed for immunofluorescence with antibodies to CFTR and c-myc. The left panel shows immunolocalization of ΔF508 and the right panel shows the same field examined for c-myc immunolocalization. (C). Optical section of an aggresome. HEK cells stably expressing GFP-ΔF508 were treated with ALLN for 12 h. Serial 0.2-μm optical sections were recorded with a cooled CCD camera and deconvolved as described in Materials and Methods. The image shown is a single deconvolved optical section indicating GFP (green) and DNA (blue) localization. Bar, 5 μm.
We found this structure to be extremely stable. For example, washout of the proteasome inhibitor for up to 8 h after overnight proteasome inhibition failed to significantly disrupt the juxtanuclear structure, even when the washout was conducted in the presence of cycloheximide to preclude addition of new GFP-ΔF508. These observations suggest that the juxtanuclear structure described in this paper is remarkably stable and is, by both biochemical and morphological criteria, composed of misfolded CFTR molecules, which are long-lived.
Because aggregated, detergent-insoluble forms of ΔF508 (and CFTR) that accumulate after proteasome inhibition are multiubiquitinated (Ward et al., 1995), we investigated whether or not ubiquitin was present in the juxtanuclear ΔF508-containing structure that is formed in response to proteasome inhibition or overexpression. Staining of proteasome-inhibited cells transiently expressing ΔF508 with antibodies to CFTR and to ubiquitin revealed a colocalization of ubiquitin and CFTR immunoreactivity (data not shown). To confirm this result, cells were transiently transfected with ΔF508 cDNA together with excess plasmid encoding His6-c-myc-tagged ubiquitin and treated for 16 h with proteasome inhibitor before fixation and analysis by immunofluorescence microscopy using antibodies to the COOH terminus of CFTR and c-myc (Fig. 2 B). The majority of c-myc–immunoreactive protein was strictly colocalized with ΔF508. In control non–ALLN-treated cells, His6-c-myc-ubiquitin was present in a diffuse cytoplasmic distribution (data not shown). Since we have shown previously that this His6-c-myc–tagged ubiquitin can be coimmmunoprecipitated with ΔF508 or CFTR (Ward et al., 1995), these data strongly suggest that at least a fraction of the ΔF508 protein that accumulated in the juxtanuclear structure is ubiquitinated.
To more closely examine the juxtanuclear GFP-ΔF508– containing structure, single optical sections were obtained from digitally deconvolved images (Fig. 2 C). These images reveal that the cluster of fluorescent ΔF508 near the nucleus is composed of a congression of many small, discrete, substructures of variable sizes. The image in Fig. 2 C also reveals clearly the relationship between the GFP-ΔF508 aggregate and the edge of the nucleus, indicating the extent to which the nuclear envelope is distorted. In sum, the above data suggest that ΔF508 molecules that are spared from proteasomal degradation form relatively stable, ubiquitinated aggregates that converge on a single juxtanuclear structure in the cell.
We propose that this structure is formed in response to undegradable (or slowly degraded) protein aggregates and thus propose to name this structure the aggresome. The extent of aggresome formation, as assessed by the size of the inclusion and the relative distortion of the nuclear envelope, is dependent on the level of expression of a poorly folded proteasome substrate and the length of time of exposure to proteasome inhibitor. In cells overexpressing CFTR or ΔF508 (as in Figs. 3 A and 4, A and B), well-defined aggresomes form spontaneously, in the absence of proteasome inhibitor. In cells expressing low levels of the protein (as in Fig. 2 A), aggresomes are observed only after chronic exposure to proteasome inhibitor.
Figure 3.

Aggresomes are distinct from Golgi and lysosomal markers. GFP-CFTR (green) visualized together with immunofluorescent staining (red) for Golgi markers manII (A), β-COP (B), and LAMP (C). Cells in B and C were treated overnight with ALLN and digitally imaged using conventional optics. The image in A is a 0.2-μm optical section obtained as described in the legend to Fig. 2 C. Bar, 15 μm.
Figure 4.

Aggresomes form at the MTOC and require MT for formation. HEK cells stably expressing GFP-CFTR were immunostained with antibody to γ-tubulin and imaged for GFP fluorescence (top), γ-tubulin immunofluorescence (middle), and DNA (A, bottom). (A) CFTR and γ-tubulin and DNA in a field of cells. (B) Effect of ALLN. Cells were incubated for 12 h in the presence of untreated (left) or 10 μg/ml ALLN (right). (C) Effect of MT disruption. Cells were incubated for 12 h in the presence of ALLN (10 μg/ml) together with nocodazole (10 μg/ml). Arrows denote the location of the centrosome. The images in A and C were obtained using conventional optics whereas the images in B are single 0.2-μm optical sections. GFP and γ-tubulin fluorescence were digitally overlaid in the bottom panels in B and C. Bars, 15 μm (A), 5 μm (B and C).
Aggresomes Form at the MTOC
The singularity and juxtanuclear position indicates that the aggresome is in the region of the cell that also contains the Golgi apparatus. However, comparison of GFP-CFTR (Fig. 3) or GFP-ΔF508 (data not shown) fluorescence with immunostaining for the Golgi markers manII and β-COP reveals that aggresomes are distinct from the Golgi apparatus. Whereas the manII staining was present in characteristic, discrete structures reflecting the localization of these markers to Golgi vesicles and cisternae (Velasco et al., 1993; Stamnes et al., 1995), the GFP fluorescence was typically found as a single discrete bright focus, often surrounded by fine, diffuse granular particles (this diffuse appearance is especially evident in the images shown in Fig. 3 C). In some cases (e.g., Fig. 3 B), the aggresome appears to be surrounded by the Golgi apparatus. Therefore, these data suggest that the aggregated forms of CFTR and ΔF508 are not in or associated with the cisternae of the Golgi apparatus. This finding is consistent with our biochemical data showing that detergent-insoluble ΔF508 and CFTR is not glycosylated (Fig. 1), and with previous studies which show that ΔF508 molecules are unable to mature to the Golgi apparatus (Lukacs et al., 1994; Ward and Kopito, 1994). Together with the lack of effect of brefeldin A on aggresome structure or formation (data not shown), these data argue strongly that aggresomes are distinct from and are not associated with the Golgi apparatus.
We also compared the distribution of the GFP-CFTR– containing structure with that of LAMP-1, a marker for lysosomes, which also congregates in the Golgi region (Fig. 3 C) (Matteoni and Kreis, 1987). LAMP-1 immunofluorescence was found in discrete vesicular structures throughout the cytoplasm, with the highest density in the juxtanuclear/Golgi region. Although the distribution of LAMP-1 immunofluorescence was close to that of GFP-CFTR aggregates, the patterns were clearly distinct and nonoverlapping, suggesting that aggresomes are distinct from lysosomes. Moreover, treatment of the cells with the lysosomal inhibitor NH4Cl or 3-methyladenine, an inhibitor of autophagy (Vielhaber et al., 1996; Jia et al., 1997), failed to reveal any overlap.
Since the GFP-CFTR– and GFP-ΔF508–containing structures were consistently in the proximity of but not coincident with markers for either Golgi apparatus or lysosomes, we considered the relationship of aggresomes to the centrosome/MTOC. In cells stably overexpressing GFP-CFTR, fluorescence was detectable in the plasma membrane and in small aggresomes that formed spontaneously in the absence of proteasome inhibitor (as in Fig. 1 B). Fig. 4 A shows a strong correlation between aggresomal GFP-CFTR fluorescence and staining for γ-tubulin, a centrosome marker (Dictenberg et al., 1998). Closer examination of the relationship between aggresomes and centrosomes, using digitally deconvolved images (Fig. 4 B) resolved two distinct centrosomes that are in effect surrounded by GFP-CFTR. Treatment of these cells with proteasome inhibitor produced characteristic enlarged aggresome, and also appeared to force γ-tubulin into a more diffuse distribution (Fig. 4 B). These data demonstrate that aggregates of misfolded protein that escape degradation by the proteasome are targeted to and accumulated at the MTOC. It is possible that the accumulation of ubiquitinated protein aggregates at the MTOC could lead to a disruption of the MT cytoskeleton. Therefore, we examined the organization of MT in aggresome-containing cells. However, no significant abnormalities were observed in aggresome-containing HEK cells stained with antibody to α-tubulin (data not shown). Because HEK cells are not flat and hence are not well suited for imaging of MT, we also imaged MT in ALLN-treated CHO cells stably transfected with GFP-CFTR (data not shown). Those data confirmed that formation of even large aggresomes had no discernible effect on MT.
MT Are Required for Aggresome Formation
The consistent presence of aggresomes at the MTOC suggests a directed process in their formation. Retrograde transport on MT could provide one mechanism for the delivery of misfolded proteins to the MTOC. Intact MT are required to establish and maintain the distribution of organelles like lysosomes and Golgi cisternae to this region (Matteoni and Kreis, 1987; Presley et al., 1997; Lippincott-Schwartz, 1998). MT depolymerization causes these structures to become dispersed to the periphery. Therefore, we evaluated the role of MT in aggresome formation, by treating GFP-ΔF508–expressing cells with MT-disrupting drugs together with proteasome inhibitor (Fig. 4 C). Strikingly, treatment with nocodazole completely abrogated the ALLN-induced accumulation of GFP-ΔF508 at a single large juxtanuclear aggresome. Instead, GFP fluorescence was observed at multiple sites throughout the cytoplasm with no obvious concentration near the MTOC, although the juxtanuclear localization of γ-tubulin was not disrupted. Nocodazole also prevented the characteristic distortion of the nuclear envelope that accompanies aggresome formation. Identical results were obtained with MT-disrupting drugs colchicine and vinblastine (data not shown). Disruption of actin with cytochalasin B did not prevent aggresome formation (data not shown), confirming that the effect of nocodazole was specific for MTs. In the absence of proteasome inhibitor, MT-disrupting agents had no effect on either the stability or detergent solubility of GFP-ΔF508 or GFP-CFTR (data not shown). These data demonstrate that an intact MT-based cytoskeleton is required for the formation of aggresomes. However, we observed that preexisting aggresomes cannot be effectively dispersed by MT disruption, even after prolonged (>6 h) incubation with nocodazole. This observation suggests that intact MT are not required for the maintenance of aggresomes and raises the possibility that interactions with other components at the MTOC must contribute the long-term stability of the aggresome.
Reorganization of IF Accompanies Aggresome Formation
The data presented above implicate the MT cytoskeleton in aggresome formation. Therefore, it was important to assess whether formation of aggresomes resulted in changes in the organization of the major cytoskeletal elements, actin, and IF. No significant alterations in the distribution of actin (data not shown) were observed in ΔF508- or CFTR-expressing cells, even after overnight incubation with proteasome inhibitors. In sharp contrast, aggresome formation was highly correlated with profound changes in the distribution of the IF protein, vimentin (Fig. 5). In control cells, vimentin antibodies decorate long, fibrous elements that extend into cellular processes (Fig. 5 A, left), consistent with previous descriptions of the distribution of this IF protein in interphase cells (Franke et al., 1978; Rosevear et al., 1990). By contrast, in essentially 100% of ALLN-treated, GFP-CFTR–expressing cells (Fig. 5 A, right) vimentin was found to be completely redistributed to the aggresomal region. In these cells, vimentin formed a ring-like halo surrounding the core of the aggresome (Fig. 5 A, arrows, and B). This vimentin ring was further resolved by three-dimensional reconstruction of serial optical sections, which reveal that the aggresome consists of a core of GFP-CFTR particles surrounded by a cage-like structure composed of condensed vimentin-positive fibers (Fig. 5 C). The numerous spots of coincidence between GFP and vimentin fluorescence in this image suggest an intimate contact between the vimentin fibers and GFP-CFTR within the aggresome. This aggresomal redistribution of vimentin is distinct from the “collapse” of vimentin to a juxtanuclear cap (Franke et al., 1978) or whorl (Starger and Goldman, 1977) that is observed after treatment of cells with MT-disrupting agents (Fig. 5 D). We also observed an identical redistribution of vimentin to form ring-like structures around pericentriolar aggresomes formed entirely by overexpression of CFTR in the absence of exogenously added proteasome inhibitors (data not shown), indicating that aggresome formation can result from saturation of the ubiquitin-proteasome pathway and does not require direct loss of proteasome function. Interestingly, overnight treatment with proteasome inhibitor induced vimentin to redistribute to a small juxtanuclear spot in a fraction (25–50%) of untransfected cells (Fig. 5 A, middle), suggesting either that aggresomes can form by aggregation of endogenous proteasome substrates or, alternatively, that pericentriolar vimentin redistribution may reflect an indirect effect of proteasome inhibition, possibly on the activity of regulatory factors which control IF dynamics. Future experiments will be needed to discriminate between these two possibilities. Taken together, these data show that aggresome formation is consistently accompanied by a massive redistribution of vimentin to a pericentriolar and periaggresomal location.
Ultrastructure of Aggresomes
We used transmission EM to resolve the structure of aggresomes and to assess their relationship to the centrosome and to IF (Fig. 6). In cells expressing ΔF508 and treated overnight with proteasome inhibitor, aggresomes appear as massive accumulations of closely packed electron-dense particles in the region of the centrosome. Three clusters of particles are evident in the cell shown in Fig. 6 A, the largest of these is nearly round and is immediately adjacent to a cluster in which the particles surround a single centriole (shown at higher magnification in Fig. 6 B). The entire region containing electron-dense particles appears to be “wrapped” by bundles of filamentous material. Both the electron-dense particles and the filamentous structures were consistently observed in proteasome-inhibited cells expressing ΔF508, CFTR, GFP-CFTR, or GFP-ΔF508, but never in untreated, untransfected cells (data not shown). However, in untransfected, proteasome-inhibited cells, we consistently observed a juxtanuclear condensation of IF in the virtual absence of electron-dense particles (data not shown), consistent with the data in Fig. 5 A showing that proteasome inhibition alone is sufficient to induce vimentin redistribution. These observations, together with the distribution of GFP-ΔF508 fluorescence (Figs. 2–5), suggest that the electron-dense particles are composed of aggregates of misfolded CFTR molecules, a conclusion that is strongly supported by immunogold labeling (see below). At higher magnification (Fig. 6 B) this electron-dense, ΔF508-containing material can be resolved into roughly spherical membrane-free particles ranging in diameter from 60 to 80 nm and surrounding a clearly visible centriole. At this magnification, it was possible to determine that the fibrous region consisted of individual filaments 8–10 nm in diameter (Fig. 6 C). These filaments are organized into roughly parallel bundles around the periphery of the aggresome and appear as disordered tangles at the core. Frequently, mitochondria, electron lucent vesicles, and multivesicular bodies appear to be trapped at the periphery of the filamentous meshwork of the aggresome.
Figure 6.
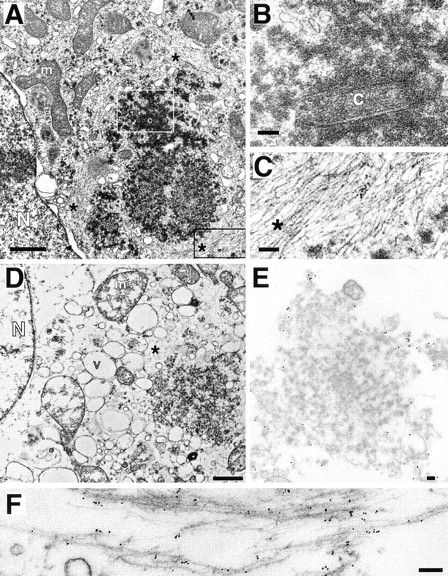
Transmission EM of aggresomes. (A–C) Transmission EM of HEK cell overexpressing ΔF508 and treated overnight with ALLN. B and C are higher magnification views of sections indicated by white and black boxes, respectively. (D–F) Transmission EM of crude aggresome fraction from HEK cells expressing GFP-CFTR. Swollen vesicular structures (v) and mitochondria (m) are the result of hypotonic conditions used during the preparation. Crude aggresomes were incubated with antibodies to GFP (E; 15-nm gold particles) and vimentin (F; 6-nm gold particles) before embedding and sectioning as described in Materials and Methods. m, mitochondrion; N, nucleus; c, centriole; v, vesicle; *, filamentous material ∼8–10 nm per filament. Bars for A and D, 1 μm; B, C, E, and F, 100 nm.
To further characterize aggresomes and to confirm the identity of the electron-dense particles and filaments, a crude fraction of aggresomes was prepared from homogenates of GFP-CFTR–expressing cells (Fig. 6 D) and immunogold labeled with antibodies to GFP and to vimentin (Fig. 6, E and F). Aggresomes isolated in this manner were usually organized around a centriole or centriole pair and frequently cofractionated with an intact nucleus, suggesting a tight association among these structures (Fig. 6 D). The periphery, but not the interior, of the aggresome was strongly immunoreactive with antibodies to GFP (15-nm gold particles; Fig. 6 E) and to vimentin (6-nm gold particles; Fig. 6 F), confirming the localization of these proteins to aggresomes, and suggesting (not surprisingly) that the close packing of filaments and electron-dense particles in the aggresome effectively excludes penetration of 6–15-nm gold. At higher magnification, decoration of the 8–10-nm filaments and filament bundles with 6-nm gold is evident, confirming that they are composed of vimentin. It is also apparent that the 15-nm gold particles (directed against GFP) label primarily the 60–80-nm electron-dense particles, confirming that they are composed of misfolded CFTR protein. Significantly, no membranes are observed in the CFTR-containing particles, consistent with our observation (Fig. 1) that the insoluble forms of CFTR and ΔF508 are not glycosylated. In conclusion, these data demonstrate that aggresomes are clusters of 60– 80-nm misfolded CFTR particles entangled in and surrounded by a meshwork of vimentin IF.
Aggresome Formation Is a General Cellular Response to Misfolded Protein
To assess the generality of the aggresome response, we evaluated the effect of proteasome inhibition on the intracellular distribution of another integral membrane protein, PS1 (Fig. 7). Immunoblot analysis of untransfected HEK cells with antibody to the NH2 terminus of PS1 (PS1NT; Thinakaran et al., 1996) revealed a low level of expression of both the ∼43-kD full-length and the 27-kD NH2-terminal fragment of this ubiquitously expressed protein (Fig. 7 a). Transfection of PS1 and A246E dramatically increased the steady-state levels of both of these forms. Incubation of PS1- or A246E-expressing cells with ALLN for 16 h led to a substantial increase in steady-state level of full-length protein and to the appearance of detergent-insoluble full-length and high molecular weight forms. These data suggest that, like PS2 (Kim, 1997), overexpressed PS1 molecules are degraded in a proteasome-dependent fashion. Immunofluorescence microscopy was used to determine the intracellular site of PS1 accumulation in transfected HEK cells (Fig. 7 b). In control cells not treated with proteasome inhibitor, PS1 (data not shown) and A246E (Fig. 7 b, panels A–C) both appeared in a diffuse, reticular pattern, in agreement with other reports (Kovacs et al., 1996; De Strooper et al., 1997) indicating the predominant localization of PS1 to the ER. By contrast, in proteasome-inhibited cells, PS1 (data not shown) and A246E (Fig. 7 b, panel D) were largely redistributed to aggresomal structures, as judged by their colocalization with GFP-CFTR in cells coexpressing A246E and GFP-CFTR (Fig. 7 b, panel D–G) and with c-myc-ubiquitin in cells cotransfected with A246E and myc-tagged ubiquitin (Fig. 7 b, panels H–K). These data, together with the observation that the mutant and wild-type PS1 aggresomes are also associated with redistributed vimentin (data not shown), strongly support the conclusion that aggresome formation is not limited to CFTR.
Figure 7.

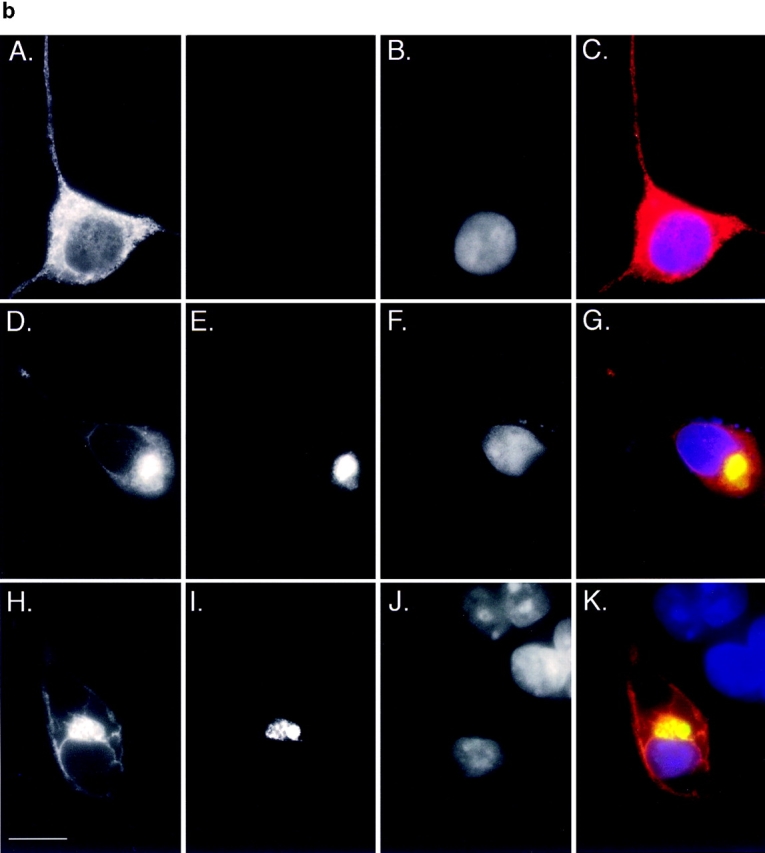
Accumulation of PS1 and A246E after treatment with ALLN. HEK cells transfected with vector alone, PS1, or A246E were incubated for 12 h in the presence or absence of ALLN (10 μg/ml) as indicated and processed for immunoblotting (a) or immunofluorescence (b). (a) Immunoblot. Cells were lysed and separated into detergent-soluble (s) or -insoluble (i) fractions as described in Materials and Methods and probed with antibody to the NH2 terminus of PS1. (b) Immunofluorescence localization of A246E in HEK cells. HEK cells expressing A246E were either untreated (A–C) or treated (D–K) with ALLN. In D–G A246E was transfected into HEK cells that stably express GFP-CFTR. In H–K, A246E was cotransfected with c-myc-ubiquitin into naive HEK cells. Cells were prepared for immunocytochemistry as described in Materials and Methods and stained with antibody to the NH2 terminus of PS1 (A, D, and H), antibody to c-myc (I), or with bisbenzimide to visualize nuclei (B, F, and J). GFP-CFTR fluorescence was imaged in E. C, G, and K are digital overlays. Bar, 15 μm.
Discussion
In this paper we describe a novel structure, the aggresome, which is formed when a cell's capacity to degrade misfolded proteins is exceeded. We define the aggresome as a pericentriolar membrane-free, cytoplasmic inclusion containing misfolded, ubiquitinated protein ensheathed in a cage of IF. Rather than appearing as amorphous globs that might form by diffusion-limited aggregation of denatured protein, aggresomes exhibit a surprising degree of structural organization. Instead of forming at random sites within the cytoplasm, which would be predicted by a “seed” based model of inclusion body formation (Lansbury, 1997), aggresomes are formed specifically at the MTOC by an ordered process requiring intact MT and accompanied by a dramatic reorganization of the IF cytoskeleton. Our data indicate that different proteins, including CFTR and PS1, can form the aggresomal core, leading us to propose that aggresome formation is a general response of cells which is induced when the capacity of the proteasome is exceeded by the production of aggregation-prone misfolded proteins. Below, we propose a model for aggresome formation and discuss implications for understanding the toxicity associated with protein aggregation.
A Model for Aggresome Formation
A model for aggresome formation is illustrated in Fig. 8. Integral membrane proteins like CFTR and PS1 that are cotranslationally translocated and glycosylated at the ER (step 1) can either fold (step 1a) or be multiubiquitinated and targeted for rapid degradation by the proteasome (steps 2 and 3). The data in this paper show that aggresome formation results either from overexpression of inefficiently folded proteins like CFTR or ΔF508 (increasing steps 1 and 2), or from inhibiting proteasomes (preventing steps 3, 7a, and 7b), suggesting that the formation of aggresomes is a cellular stress response pathway for misfolded protein.
Figure 8.

Model for aggresome formation. Numbers indicate various steps in the aggresome biogenesis pathway and are explained in the Discussion. Membrane proteins are cotranslationally translocated to the membrane of the ER (1). Some molecules fold to adopt a maturation-competent conformation (1a). Others misfold and are dislocated from the ER membrane (2). Some proteins may escape the translocation machinery and be delivered directly to the cytoplasm (4). Dislocated, ubiquitinated, misfolded protein can either be rapidly degraded by cytosolic proteasomes (3) or aggregate (5 and 6). Because aggregates are difficult to unfold, they are likely to be slowly degraded by the proteasome (7a and 7b). Misfolded, aggregated protein is transported to the MTOC by MT where it becomes entangled with collapsed IF (8). In the absence of MT, protein aggregates coalesce at dispersed sites throughout the cytoplasm (8a). N, nucleus; Ubn, ubiquitin conjugates; +, orientation of MT in the cell.
Proteasomal degradation has now been demonstrated for a host of misfolded polytopic and monotopic integral membrane proteins in addition to CFTR (Hampton et al., 1996; Hughes et al., 1997; Huppa and Ploegh, 1997; Yu et al., 1997). Delivery of core N-glycosylated integral membrane polypeptides to cytoplasmic proteasomes requires their dislocation across the ER membrane by a Sec61p-dependent process and is usually accompanied by deglycosylation, presumably by cytoplasmic N-glycanase (step 2) (Kopito, 1997). In this paper we show that the detergent-insoluble forms of CFTR and ΔF508 that accumulate in aggresomes are not glycosylated (band A). That these molecules have become dislocated is strongly supported by pulse–chase studies showing that nascent ΔF508 and CFTR are initially detergent-soluble and core-glycosylated (band B) and by the observation that the 60–80-nm CFTR-containing aggresomal particles are not membrane associated. The observation that misfolded forms of CFTR and ΔF508 are chased into a deglycosylated form was confirmed in a recent report that also provided evidence from cell fractionation studies suggesting that some misfolded CFTR molecules are cytosolic (Bebök et al., 1998). However, it is also possible that, in our experiments as well as those of Bebök et al. (1998), some unglycosylated, misfolded CFTR and ΔF508 could enter the cytoplasmic pool directly as a result of failure or inefficiency of the translocation machinery (step 4).
Irrespective of the delivery pathway to the aqueous cytoplasmic compartment, hydrophobic forces would rapidly drive a dislocated or mis-translocated integral membrane protein like CFTR or PS1 into an alternative conformation in which the transmembrane stretches of apolar amino acids are maximally buried. It is likely that the presence of a large hydrophobic surface, as in a polytopic integral membrane protein, might drive the dislocated protein to assume a micelle-like oligomeric structure (steps 5 and 6). We speculate that the 60–80-nm spheroidal cytoplasmic particles that accumulate in proteasome-inhibited cells may represent such micelle-like formations. The presence of CFTR in these particles is strongly supported by immunolocalization with antibodies to CFTR and to GFP and by control experiments showing that these particles are largely absent from proteasome-inhibited cells that do not express CFTR. However, our data do not eliminate the possibility that, in addition to CFTR, other proteins may also be present in aggresomal particles. Future investigations will be required to purify the particles and to assess their biochemical and biophysical properties.
Binding of molecular chaperones like Hsp70 or Hsp90 to surface-exposed hydrophobic domains on nascent or misfolded proteins can serve to minimize protein aggregation (Hartl, 1996). Indeed, persistent association of misfolded CFTR molecules with Hsp70 has been reported (Yang et al., 1993). It has been suggested that the presence of multiubiquitin chains could also help to increase the solubility of misfolded proteins (Beal et al., 1998). However, the most effective way to prevent cytoplasmic aggregation of misfolded integral membrane proteins would be to directly couple their dislocation (step 2) to degradation by the proteasome (step 3). Moreover, because proteasomes can only degrade proteins in an unfolded open-chain conformation (Baumeister et al., 1997) such coupling of dislocation and degradation would increase the efficiency of proteolysis by eliminating the need for unfolding. Although there is some evidence suggesting that dislocation and degradation may indeed be coupled (Mayer et al., 1998), the cytoplasmic accumulation of dislocated deglycosylated, integral membrane proteins like CFTR, TCR-α (Huppa and Ploegh, 1997; Yu et al., 1997), or MHC class I heavy chains (Hughes et al., 1997) in the presence of proteasome inhibitors argues that such coupling is not obligatory.
Our data suggest that retrograde transport on MT is used to deliver misfolded protein aggregates from the peripheral sites where they are generated to a central location at the MTOC (step 8). In support of this hypothesis is the observation that MT disruption prevents delivery of CFTR-containing particles to a single juxtanuclear aggresome (step 8a). Instead, when proteasomal degradation is blocked in the presence of MT-depolymerizing drugs, heterogeneous CFTR aggregates are observed at multiple foci throughout the cytoplasm (Fig. 4 C). Transmission EM of these cells reveals that the peripheral CFTR foci are composed of 60–80-nm particles indistinguishable from aggresomal particles (data not shown), indicating that particle translocation to the MTOC, but not particle formation, requires intact MT. While these data do not demonstrate a direct role for MT or retrograde motors in the delivery of misfolded proteins to the aggresome, preliminary studies showing that taxol-stabilized MT from proteasome-inhibited CFTR-expressing cells can be specifically decorated with immunogold-tagged antibodies to CFTR, suggest that dislocated CFTR molecules do indeed bind to MT (Johnston, J.A., and R.R. Kopito, unpublished data). However, further study will be needed to determine the role of MT, molecular motors, and protein adapters in aggresome formation.
Our data demonstrate that, once formed, aggresomes are quite stable structures. Pulse–chase studies indicate that aggresome formation is accompanied by a nearly 50-fold increase in the half-life of CFTR or ΔF508. Compact, juxtanuclear aggresomes persist for hours after washout of proteasome inhibitors, even in the presence of MT-disrupting drugs, suggesting that aggresomes are not highly dynamic structures held together by a balance between MT-dependent assembly and rapid disassembly. We speculate that the dense network of IF that encircle and enmesh the aggresome serve to stabilize it by restricting the diffusion of aggregated protein particles. Our data demonstrate that vimentin is a component of these filaments, although we cannot exclude the possibility that some filaments may be formed by amyloid-like polymerization of alternative conformers of CFTR or other misfolded proteins.
We observe that an early and invariant step in aggresome formation is the redistribution of vimentin to a single pericentriolar site. In interphase cells, IF form a branching cytoskeletal network that extends to the cell periphery (Houseweart and Cleveland, 1998). Disruption of MT leads in many cell types to collapse of vimentin to form various alternative structures, including perinuclear whorls (Franke et al., 1978) and frequently a large juxtanuclear cap composed of bundles of 10-nm vimentin filaments (Starger and Goldman, 1977). It is important to note that the aggresomal vimentin redistribution observed in the present work differs fundamentally from that which occurs upon MT disruption. Aggresomal vimentin consistently appears as a tight ring- or cage-like structure surrounding the pericentriolar focus of aggregated protein, whereas, in nocodazole-treated cells, vimentin appears as elongated bundles of filaments capping the nucleus (Fig. 5 D). Moreover, the vimentin collapse observed upon aggresome formation cannot be due to MT disruption because aggresome formation is blocked by MT-disrupting drugs (Fig. 4 C). Finally, our data show that, despite deposition of protein at the MTOC, aggresome formation in HEK or CHO cells is not accompanied by a gross disruption of the MT cytoskeleton.
Previous studies have reported that IF undergo dynamic changes in their organization during the cell cycle (Blose and Bushnell, 1982; Rosevear et al., 1990) that appear to be regulated, at least in part, by site-specific vimentin phosphorylation (Inagaki et al., 1987). Vimentin is a substrate for phosphorylation in vivo by cell cycle–regulated kinases including Cdc2 (Chou et al., 1990) and Rho kinase (Goto et al., 1998). During mitosis, vimentin filaments form cage- or basket-like (Zieve et al., 1980) structures which surround the spindle and spindle pole (Blose and Bushnell, 1982). These structures resemble the pericentriolar cages that encircle aggresomes. Since the cell cycle is regulated by kinases and phosphatases which are themselves controlled at the level of ubiquitin-dependent proteolysis (Hershko, 1997; Krek, 1998), and since treatment of cells with proteasome inhibitors can inhibit cell cycle progression (Tsubuki et al., 1996; Fenteany and Schreiber, 1998), it is likely that the conditions which lead to aggresome formation would result in cell cycle disregulation. It is possible that the collapse of vimentin around unduplicated interphase centrosomes during aggresome formation may reflect phosphorylation of this IF by inappropriately activated mitotic kinases. Additional studies will be needed to test this hypothesis and to assess the effect of aggresome formation on proteasome function and cell cycle regulation.
In this paper we demonstrate that unprocessed PS1 molecules accumulate in aggresomes indistinguishable from those containing misfolded CFTR. At the expression levels used in this study, mutant and wild-type PS1 have an ER distribution, consistent with other studies that have localized both PS1 and PS2 to ER membranes (Kovacs et al., 1996; De Strooper et al., 1997; Lah et al., 1997; Zhang et al., 1998). In the functional absence of proteasomes, we find that PS1 and A246E redistribute to a juxtanuclear site which, based on colocalization with CFTR, vimentin, and γ-tubulin, is an aggresome. Studies in which PS1 or PS2 have been overexpressed have suggested that, in addition to the ER, these proteins may also be present in the Golgi (Kovacs et al., 1996; De Strooper et al., 1997; Lah et al., 1997; Zhang et al., 1998). It is possible that overexpression of PS1 in some cell types, in the absence of proteasome inhibitors, results in deposition of some PS1 in aggresomes, which could, in the absence of careful controls and high resolution imaging, be mistaken for the Golgi apparatus. Our findings may also help to explain the observation that endogenous PS1 and PS2 can be identified in the centrosomes of cultured primary fibroblasts (Li et al., 1997), which suggest that, even in the absence of overexpression, a fraction of PS1 may be targeted to the aggresomal pathway. The demonstration that PS2 (Kim et al., 1997) and PS1 (this study) are substrates of the cytoplasmic ubiquitin-proteasome pathway, together with our data demonstrating that PS1 can accumulate in aggresomes, may also have significance for the pathogenesis of spontaneous Alzheimer's disease, particularly in light of the observation that PS1 is present in neurofilament-rich cytoplasmic inclusion bodies and dystrophic neurites in Alzheimer's disease (Busciglio et al., 1997; Chui et al., 1998).
Aggresomes are not restricted to misfolded forms of CFTR. In addition to CFTR and PS1, we have also found that undegraded α chains of the oligomeric T cell antigen receptor (TCR-α) colocalize with CFTR in aggresomes when their degradation by the proteasome is blocked (Yu, H., J. Johnston, and R. Kopito, unpublished data). Like CFTR, inhibition of TCR-α degradation with proteasome inhibitors leads to cytoplasmic accumulation of detergent-insoluble, deglycosylated forms of the protein (Yu et al., 1997). Taken together, these similarities lead us to propose that aggresomes are a general cellular response to cytoplasmic accumulation of misfolded protein. Because aggresomal localization could be mistaken for the Golgi apparatus, our findings suggest that caution should be exercised in interpreting the cellular localization of overexpressed proteins.
Implications for Cell Physiology and Pathology
The experimental conditions used to generate aggresomes in this study, overexpression and proteasome inhibition, impose a significant stress on the cell's degradative capacity. Like the use of other stressors which induce protein misfolding such as heat-shock or incorporation of amino acid analogues (Thomas et al., 1982), or tunicamycin treatment (Marquardt and Helenius, 1992), our experiments reveal the existence of an underlying cellular homeostatic pathway. The data in this paper lead us to speculate that retrograde transport on MT serves to clear the cytoplasm of potentially toxic aggregates of misfolded proteins which have escaped degradation by the ubiquitin-proteasome pathway. In addition to serving a sequestration role, it is possible that the aggresome formation provides a “staging ground” for the incorporation of protein aggregates into autophagic structures (Earl et al., 1987), possibly by facilitating interaction with endosomes and lysosomes which are also delivered by MT to the same region of the cell (Matteoni and Kreis, 1987). Consistent with this notion, we frequently observe double-membrane vesicular structures in the immediate vicinity of the aggresome (data not shown).
Many human neurodegenerative diseases are characterized by the presence of morphologically and biochemically distinct intracellular inclusions which contain deposits of aggregated, ubiquitinated proteins that are often bundled with hyperphosphorylated and disordered IF (Mayer et al., 1989a ,b). The striking morphological and biochemical similarity between experimentally induced aggresomes and many of the neuronal and glial lesions observed in idiopathic, familial, and animal models of degenerative disease (Mayer et al., 1991) suggests that the misfolded protein response pathway described in this paper may participate in pathogenesis. Although aggresome formation may provide a cytoprotective role, the long-term accumulation of aggregates and IF near the MTOC could explain the defects in MT-based axonal transport that are associated with neurological disease (Zhang et al., 1997). Alternatively, because aggregates are inherently difficulty to unfold, they could act as competitive inhibitors of the 26S proteasome, resulting in progressive loss of proteasome activity and, ultimately, in a derangement of critical cell regulatory factors that are normally controlled by proteolysis.
Acknowledgments
We thank N. Ghori and L. Howard for technical help with the preparation of specimens for EM. We also thank C. Canfield and S. Palmieri at Stanford's Cell Sciences Imaging Facility for assistance with deconvolution microscopy. A. Haas, M. Farquhar, M. Fukuda, T. Stearns, G. Thinakaran, S. Sisodia, and S. Pfeffer graciously contributed antibodies. We thank B. Wolozin and J. Hardy for contributing the PS1 and A246E cDNA and N. Bence for the stable GFP-CFTR expressing CHO cell line. We are grateful to R.D. Simoni, J. Frydman, M. Rexach, P. Jackson, and to the members of the Kopito lab for stimulating discussion of the data.
This work was supported by a grant from the National Institutes of Health (DK43994). This research was conducted while R.R. Kopito was an Established Investigator of the American Heart Foundation.
Abbreviations used in this paper
- ALLN
acetyl-leucyl-leucyl-norleucinal
- CFTR
cystic fibrosis transmembrane conductance regulator
- HEK
human embryonic kidney 293
- IF
intermediate filaments
- LAMP
lysosomal membrane protein
- manII
α-mannosidase II
- MT
microtubules
- MTOC
microtubule-organizing center
- PS
presenilin
References
- Baumeister W, Cejka Z, Kania M, Seemuller E. The proteasome: a macromolecular assembly designed to confine proteolysis to a nanocompartment. Biol Chem. 1997;378:121–130. doi: 10.1515/bchm.1997.378.3-4.121. [DOI] [PubMed] [Google Scholar]
- Beal RE, Toscano-Cantaffa D, Young P, Rechsteiner M, Pickart CM. The hydrophobic effect contributes to polyubiquitin chain recognition. Biochemistry. 1998;37:2925–2934. doi: 10.1021/bi972514p. [DOI] [PubMed] [Google Scholar]
- Bebök Z, Mazzochi C, King SA, Hong JS, Sorscher EJ. The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61β and a cytosolic, deglycosylated intermediary. J Biol Chem. 1998;273:29873–29878. doi: 10.1074/jbc.273.45.29873. [DOI] [PubMed] [Google Scholar]
- Blose SH, Bushnell A. Observations on the vimentin-10-nm filaments during mitosis in BHK21 cells. Exp Cell Res. 1982;142:57–62. doi: 10.1016/0014-4827(82)90408-6. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner J. Supervising the fold: functional principles of molecular chaperones. FASEB (Fed Am Soc Exp Biol) J. 1996;10:10–19. [PubMed] [Google Scholar]
- Busciglio J, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA. Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J Neurosci. 1997;17:5101–5107. doi: 10.1523/JNEUROSCI.17-13-05101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson SR, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J Biol Chem. 1988;263:18911–18919. [PubMed] [Google Scholar]
- Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350:134–138. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- Chou YH, Bischoff JR, Beach D, Goldman RD. Intermediate filament reorganization during mitosis is mediated by p34cdc2 phosphorylation of vimentin. Cell. 1990;62:1063–1071. doi: 10.1016/0092-8674(90)90384-q. [DOI] [PubMed] [Google Scholar]
- Chui DH, Shirotani K, Tanahashi H, Akiyama H, Ozawa K, Kunishita T, Takahashi K, Makifuchi T, Tabira T. Both N-terminal and COOH-terminal fragments of presenilin 1 colocalize with neurofibrillary tangles in neurons and dystrophic neurites of senile plaques in Alzheimer's disease. J Neurosci Res. 1998;53:99–106. doi: 10.1002/(SICI)1097-4547(19980701)53:1<99::AID-JNR10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St. George-Hyslop PH, Hofman A, Van Broeckhoven C. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer's disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Zimmerman W, Sparks CA, Young A, Vidair C, Zheng Y, Carrington W, Fay FS, Doxsey SJ. Pericentrin and γ-tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol. 1998;141:163–174. doi: 10.1083/jcb.141.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- Earl RT, Mangiapane EH, Billett EE, Mayer RJ. A putative protein-sequestration site involving intermediate filaments for protein degradation by autophagy. Studies with transplanted Sendai-viral envelope proteins in HTC cells. Biochem J. 1987;241:809–815. doi: 10.1042/bj2410809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Schreiber SL. Lactacystin, proteasome function, and cell fate. J Biol Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- Franke WW, Schmid E, Osborn M, Weber K. Different intermediate-sized filaments distinguished by immunofluorescence microscopy. Proc Natl Acad Sci USA. 1978;75:5034–5038. doi: 10.1073/pnas.75.10.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Kosako H, Tanabe K, Yanagida M, Sakurai M, Amano M, Kaibuchi K, Inagaki M. Phosphorylation of vimentin by Rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J Biol Chem. 1998;273:11728–11736. doi: 10.1074/jbc.273.19.11728. [DOI] [PubMed] [Google Scholar]
- Graham FL, Eb AJvd. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- Group ADC. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Alzheimer's Disease Collaborative Group. Nat Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- Haas AL, Siepmann TJ. Pathways of ubiquitin conjugation. FASEB (Fed Am Soc Exp Biol) J. 1997;11:1257–1268. doi: 10.1096/fasebj.11.14.9409544. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of the 26s proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- Houseweart MK, Cleveland DW. Intermediate filaments and their associated proteins: multiple dynamic personalities. Curr Opin Cell Biol. 1998;10:93–101. doi: 10.1016/s0955-0674(98)80091-4. [DOI] [PubMed] [Google Scholar]
- Hughes EA, Hammond C, Cresswell P. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc Natl Acad Sci USA. 1997;94:1896–1901. doi: 10.1073/pnas.94.5.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Ploegh HL. The alpha chain of the T cell antigen receptor is degraded in the cytosol. Immunity. 1997;7:113–122. doi: 10.1016/s1074-7613(00)80514-2. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Nishi Y, Nishizawa K, Matsuyama M, Sato C. Site-specific phosphorylation induces disassembly of vimentin filaments in vitro. Nature. 1987;328:649–652. doi: 10.1038/328649a0. [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98:673–685. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- Kim T-W, Pettingel WH, Hallmark OG, Moir RD, Wasco W, Tanzi RE. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J Biol Chem. 1997;272:11006–11010. doi: 10.1074/jbc.272.17.11006. [DOI] [PubMed] [Google Scholar]
- Kopito RR. ER quality control: the cytoplasmic connection. Cell. 1997;88:427–430. doi: 10.1016/s0092-8674(00)81881-4. [DOI] [PubMed] [Google Scholar]
- Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, et al. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- Krek W. Proteolysis and the G1-S transition: the SCF connection. Curr Opin Genet Dev. 1998;8:36–42. doi: 10.1016/s0959-437x(98)80059-2. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt O, Grunert HP, Hahn U. A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene. 1990;96:125–128. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- Lansbury PT., Jr Structural neurology: are seeds at the root of neuronal degeneration? . Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci USA. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J. Cytoskeletal proteins and Golgi dynamics. Curr Opin Cell Biol. 1998;10:52–59. doi: 10.1016/s0955-0674(98)80086-0. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO (Eur Mol Biol Organ) J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Helenius A. Misfolding and aggregation of newly synthesized proteins in the endoplasmic reticulum. J Cell Biol. 1992;117:505–513. doi: 10.1083/jcb.117.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer RJ, Lowe J, Lennox G, Doherty F, Landon M. Intermediate filaments and ubiquitin: a new thread in the understanding of chronic neurodegenerative diseases. Prog Clin Biol Res. 1989a;317:809–818. [PubMed] [Google Scholar]
- Mayer RJ, Lowe J, Lennox G, Landon M, MacLennan K, Doherty FJ. Intermediate filament-ubiquitin diseases: implications for cell sanitization. Biochem Soc Symp. 1989b;55:193–201. [PubMed] [Google Scholar]
- Mayer RJ, Arnold J, Laszlo L, Landon M, Lowe J. Ubiquitin in health and disease. Biochim Biophys Acta. 1991;1089:141–157. doi: 10.1016/0167-4781(91)90002-4. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO (Eur Mol Biol Organ) J. 1998;17:3251–3257. doi: 10.1093/emboj/17.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer BD, Loffing J, Schwiebert EM, Loffing-Cueni D, Halpin PA, Karlson KH, Ismailov II, Guggino WB, Langford GM, Stanton BA. Membrane trafficking of the cystic fibrosis gene product, cystic fibrosis transmembrane conductance regulator, tagged with green fluorescent protein in Madin-Darby canine kidney cells. J Biol Chem. 1998;273:21759–21768. doi: 10.1074/jbc.273.34.21759. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA [published erratum appears in Science.1989. 245:1437] Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rosevear ER, McReynolds M, Goldman RD. Dynamic properties of intermediate filaments: disassembly and reassembly during mitosis in baby hamster kidney cells. Cell Motil Cytoskeleton. 1990;17:150–166. doi: 10.1002/cm.970170303. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- Sommer T, Wolf DH. Endoplasmic reticulum degradation: reverse protein flow of no return. FASEB (Fed Am Soc Exp Biol) J. 1997;11:1227–1233. doi: 10.1096/fasebj.11.14.9409541. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Craighead MW, Hoe MH, Lampen N, Geromanos S, Tempst P, Rothman JE. An integral membrane component of coatomer-coated transport vesicles defines a family of proteins involved in budding. Proc Natl Acad Sci USA. 1995;92:8011–8015. doi: 10.1073/pnas.92.17.8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starger JM, Goldman RD. Isolation and preliminary characterization of 10-nm filaments from baby hamster kidney (BHK-21) cells. Proc Natl Acad Sci USA. 1977;74:2422–2426. doi: 10.1073/pnas.74.6.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer's disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Thomas GP, Welch WJ, Mathews MB, Feramisco JR. Molecular and cellular effects of heat-shock and related treatments of mammalian tissue-culture cells. Cold Spring Harbor Symp Quant Biol. 1982;46:985–996. doi: 10.1101/sqb.1982.046.01.092. [DOI] [PubMed] [Google Scholar]
- Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem (Tokyo) 1996;119:572–576. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- Velasco A, Hendricks L, Moremen KW, Tulsiani DR, Touster O, Farquhar MG. Cell type–dependent variations in the subcellular distribution of alpha-mannosidase I and II. J Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidair CA, Huang RN, Doxsey SJ. Heat shock causes protein aggregation and reduced protein solubility at the centrosome and other cytoplasmic locations. Int J Hyperthermia. 1996;12:681–695. doi: 10.3109/02656739609027676. [DOI] [PubMed] [Google Scholar]
- Vielhaber G, Hurwitz R, Sandhoff K. Biosynthesis, processing, and targeting of sphingolipid activator protein (SAP), precursor in cultured human fibroblasts. Mannose 6-phosphate receptor-independent endocytosis of SAP precursor. J Biol Chem. 1996;271:32438–32446. doi: 10.1074/jbc.271.50.32438. [DOI] [PubMed] [Google Scholar]
- Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Wetzel R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 1994;12:193–198. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996a;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996b;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH. Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem. 1998;273:12436–12442. doi: 10.1074/jbc.273.20.12436. [DOI] [PubMed] [Google Scholar]
- Zieve GW, Heidemann SR, McIntosh JR. Isolation and partial characterization of a cage of filaments that surrounds the mammalian mitotic spindle. J Cell Biol. 1980;87:160–169. doi: 10.1083/jcb.87.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]