

Abstract
In this report, we have analyzed the potential role and mechanisms of integrin signaling through FAK in cell cycle regulation by using tetracycline-regulated expression of exogenous FAK and mutants. We have found that overexpression of wild-type FAK accelerated G1 to S phase transition. Conversely, overexpression of a dominant-negative FAK mutant ΔC14 inhibited cell cycle progression at G1 phase and this inhibition required the Y397 in ΔC14. Biochemical analyses indicated that FAK mutant ΔC14 was mislocalized and functioned as a dominant-negative mutant by competing with endogenous FAK in focal contacts for binding signaling molecules such as Src and Fyn, resulting in a decreases of Erk activation in cell adhesion. Consistent with this, we also observed inhibition of BrdU incorporation and Erk activation by FAK Y397F mutant and FRNK, but not FRNKΔC14, in transient transfection assays using primary human foreskin fibroblasts. Finally, we also found that ΔC14 blocked cyclin D1 upregulation and induced p21 expression, while wild-type FAK increased cyclin D1 expression and decreased p21 expression. Taken together, these results have identified FAK and its associated signaling pathways as a mediator of the cell cycle regulation by integrins.
Keywords: FAK, cell cycle, inducible expression, integrin signaling, focal contacts
Focal adhesion kinase (FAK)1 is a cytoplasmic protein tyrosine kinase that has been implicated to play an important role in integrin-mediated signal transduction pathways (Clark and Brugge, 1995; Schwartz et al., 1995; Parsons, 1996). FAK becomes activated and tyrosine phosphorylated in integrin-mediated cell adhesion and is colocalized with integrins and other cytoskeletal proteins in focal contacts in a variety of adherent cells (Schwartz et al., 1995). Although the mechanism of FAK activation by integrins is not fully understood at present, physical interactions of FAK with the integrin cytoplasmic domain as well as cytoskeletal proteins talin, paxillin, and/ or tensin have been proposed to play a key role in FAK activation by facilitating its oligomerization and transphosphorylation (Giancotti, 1997; Guan, 1997).
Upon activation and autophosphorylation at Y397, FAK associates with another tyrosine kinase Src by binding to its SH2 (Src homology 2) domain (Chan et al., 1994; Cobb et al., 1994; Schaller et al., 1994; Xing et al., 1994). Phosphorylation of Y925 of FAK by Src then recruits the Grb2 adaptor protein, which has been proposed to trigger downstream signaling pathways leading to activation of extracellular signal–regulated kinase (Erk) (Schlaepfer et al., 1994; Schlaepfer and Hunter, 1996). The FAK/Src complex has been shown to phosphorylate additional substrates including paxillin and p130cas, which have also been proposed to mediate downstream functions of FAK (Turner and Miller, 1994; Schaller and Parsons, 1995; Vuori et al., 1996; Schlaepfer et al., 1997). In addition, the phosphorylated Y397 has been mapped as the major binding site for the SH2 domains of the regulatory subunit (p85) of phosphatidylinositol 3-kinase (PI 3-kinase) and FAK association with PI 3-kinase in cell adhesion could trigger activation of PI 3-kinase and its signaling pathways (Chen et al., 1996). Together, FAK interactions with these and potentially other proteins are believed to mediate FAK's functions in integrin-dependent signal transduction.
Recent studies have suggested that integrin signaling through FAK plays an important role in regulation of cell spreading and migration. Inhibition of FAK by overexpression of its non-catalytic COOH-terminal domain resulted in decreased rate of cell spreading and cell migration (Richardson and Parsons, 1996; Gilmore and Romer, 1996). Furthermore, cultured fibroblasts isolated from FAK null mice, which demonstrated an embryonic lethal phenotype, showed markedly reduced cell motility in vitro (Ilic et al., 1995). Finally, we have shown that stable overexpression of FAK in CHO cells resulted in increased migration (Cary et al., 1996). Using this system, we are beginning to dissect the role of FAK interactions with various other signaling molecules and downstream pathways in cell migration (Cary et al., 1996, 1998).
Several recent reports also raised the possibility that FAK may regulate cell proliferation and survival. Expression of a membrane-anchored FAK that is constitutively active in suspended cells prevented cell detachment–induced apoptosis of MDCK cells (Frisch et al., 1996). Conversely, inhibition of FAK by microinjection of either the COOH-terminal fragment or an mAb caused cell cycle arrest and apoptosis (Gilmore and Romer, 1996; Hungerford et al., 1996). However, the biochemical mechanisms on the downstream pathways upon FAK inhibition were not investigated in these studies because of the limitation of the microinjection method. Furthermore, other studies have shown that FAK gene knockout did not affect proliferation of FAK−/− fibroblasts (Ilic et al., 1995) and that some integrins control cell cycle progression by coupling to Shc but not FAK (Wary et al., 1996, 1998).
To investigate potential roles of FAK in cell cycle regulation and their molecular mechanisms, we have obtained high level and inducible expressions of exogenous FAK and its mutant in NIH3T3 cells using the tetracycline-regulated expression system. Using this inducible expression system as well as other approaches, we show that FAK serves as a mediator of cell cycle regulation by integrins and provide evidence suggesting that FAK/Src complex formation in the focal contacts, Erk activation, and cyclin D1 and/or p21 are some of the important FAK downstream events in the signaling pathway leading to cell cycle regulation.
Materials and Methods
Antibodies
The mouse mAb 12CA5 (α-HA), the rabbit polyclonal α-PY antibodies and the rabbit polyclonal α-FAK serum have been described previously (Chen and Guan, 1994; Chen et al., 1995). Rabbit α-p21, α-p27, and α-cyclin D1 are gifts from Dr. Y. Xiong (University of North Carolina, Chapel Hill, NC). Rabbit α-integrin α5 is a gift from Dr. R. Hynes (Massachussets Institute of Technology, Cambridge, MA). The following antibodies were purchased as indicated: α-BrdU mouse mAb from Sigma (St. Louis, MO); α-phosphotyrosine mouse mAb PY20 from Transduction Laboratories (Lexington, KY); rabbit α-Src, α-Fyn, α-Erk, α-cyclin A, α-cyclin E, α-Cdk2, α-His (His-Probe), and mouse mAb α-Shc from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); rabbit α-Shc from UBI (Lake Placid, NY); and rabbit α-phospho-MAPK from New England Biolabs, Inc. (Beverly, MA).
Transient Transfection with FAK and Mutants
Expression vectors encoding epitope-tagged WT FAK (pKH3-FAK) and Y397F mutant (pKH3-Y397F) were as described previously (Chen et al., 1996). The plasmid encoding ΔC14 mutant (pKH3-ΔC14) was generated by insertion of an XbaI linker containing stop codons into the ClaI site near the 3′ of FAK coding sequence. The FRNK sequence of FAK was PCR amplified from pGEX2T-FAK.KC (Chen and Guan, 1994) using the following forward and reverse primers: 5′-GAATGAGAATGGGATCCAGGCGACAAG-3′ and 5′-CCGGGAGCTGCATGTGTCAGAGG-3′. The PCR reaction was carried out using Deep Vent DNA polymerase, digested with BamHI and EcoRI, and cloned into pKH3 to generate pKH3-FRNK. The plasmid encoding FRNK with ΔC14 mutant (pKH3-FRNKΔC14) was generated by replacing the NdeI–EcoRI fragment in pKH3-FRNK with the corresponding fragment from FAKΔC14 in the pTet-Splice vector (see below). Human foreskin fibroblasts (HFF) cells were maintained in DMEM supplemented with 10% FCS (Life Technologies, Inc., Gaithersburg, MD). NIH3T3 cells were maintained in DME supplemented with 10% calf serum (CS; Life Technologies, Inc.). The cells were transfected with expression plasmids (pKH3-FAK, pKH3-Y397F, pKH3-ΔC14, pKH3-FRNK, or pKH3-FRNKΔC14) or pKH3 (control) using LipofectAMINE (Life Technologies, Inc.), as described previously (Chen et al., 1996). One day after transfection, cells were processed for analysis of BrdU incorporation as described below.
Generation of Cell Lines with Inducible FAK Expression
The 5′ end segment of FAK cDNA with the triple hemagglutinin (HA) epitope tag and the 3′ end segment of FAK cDNA were excised as a SalI– NdeI fragment from pKH3-FAK (Chen et al., 1996) and a NdeI–EcoRV fragment from pBS-FAK (Cary et al., 1998), respectively. They were then ligated into the cloning site of pTet-Splice (Life Technologies, Inc.). HA epitope–tagged FAK cDNAs lacking the last 14 residues were excised as SalI–ClaI fragments from pKH3-FAK and pKH3-Y397F (Chen et al., 1996), respectively, and inserted into pTet-Splice.
The plasmids encoding FAK, ΔC14, and ΔC14F were transfected into NIH3T3 cells along with the plasmids pTet-tTAk (Life Technologies, Inc.) and pSV2neo as described previously (Chen et al., 1996) using LipofectAMINE (Life Technologies, Inc.). The cells were then selected in 0.5 mg/ml G418 in the medium with 0.4 μg/ml tetracycline. Several clones were isolated after 2–3 weeks, which exhibited inducible expression of the transfected FAK upon removal of tetracycline. They are designated as WT FAK, ΔC14, and ΔC14F cells, respectively. The mock cells were generated similarly with pTet-Splice vector without insert. All cell lines were maintained in DME with 10% CS, 0.5 mg/ml G418 and 0.4 μg/ml tetracycline to suppress exogenous FAK expression until experiments as indicated.
Analysis of 5-Bromodeoxyuridine Incorporation
Subconfluent cells were serum starved for 48 h in DME with 0.5% CS, 0.5 mg/ml G418, and 0.4 μg/ml tetracycline. They were then washed twice with DME and incubated for 16 h or as indicated with 100 μM 5-bromodeoxyuridine (BrdU) (Sigma) in DME plus 10% CS, 0.5 mg/ml G418, and with (uninduced) or without (induced) 0.4 μg/ml tetracycline. Transiently transfected HFF cells and NIH3T3 cells were serum starved and incubated with BrdU similarly but without G418 or tetracycline. Cells were then processed for immunofluorescent staining with α-BrdU mAb, α-FAK, and Hoechst (Sigma), as described below. The percentage of BrdU (+) cells was determined for ∼500 cells in multiple fields in each independent experiment. For transiently transfected cells, ∼40–50 positively transfected cells in multiple fields were scored for BrdU staining in each independent experiment. Statistical analyses were performed by Minitab Release 10.5 Xtra (Minitab Inc., State College, PA).
FACS® Analysis
Serum-starved cells (0 h) were washed twice with DME and incubated for 16 h in DME plus 10% CS, 0.5 mg/ml G418, and with (uninduced) or without (induced) 0.4 μg/ml tetracycline. They were then trypsinized and washed twice with DME plus 10% CS. The cells were lysed and the nuclei were stained by incubation in a hypotonic propidium iodide solution (0.05 mg/ml propidium iodide, 0.1% Triton X-100, and 0.1% sodium citrate) on ice for 30 min. 104 nuclei from each sample were applied to Becton Dickinson FACSCalibur® (Lincoln Park, NJ) for determination of DNA content using CELLQuest™ software (version 1.2), as described previously (Nicoletti et al., 1991). Statistical analyses were performed by Minitab Release 10.5 Xtra (Minitab Inc.).
Immunoprecipitations and Western Blots
For most experiments, cells were washed twice with ice-cold PBS and then lysed with NP-40 lysis buffer (20 mM Tris, pH 8.0, 137 mM NaCl, 1% NP-40, 10% glycerol, 1 mM NaVO4, 1 mM PMSF, 10 μg/ml aprotinin, and 20 μg/ml leupeptin) as described previously (Chen et al., 1996). For the Erk activation and Shc phosphorylation experiments, cell lysates were prepared in ice-cold modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Hepes, pH 7.4, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1 mM sodium vanadate, 10 mM sodium pyrophosphate, 10 mM NaF, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecylsulfate, 10 μg/ml leupepetin, 10 μg/ml aprotinin, and 1 mM PMSF). As a positive control for Shc activation, cells were stimulated with recombinant PDGF-BB (25 ng/ml; UBI) before lysis.
Lysates were cleared by centrifugation and total protein concentrations were determined using Bio-Rad Protein Assay (Hercules, CA). Immunoprecipitations were carried out at 4°C by incubating cell lysates for 2 h with α-HA, α-Src, α-Fyn, α-Shc, α-His, or α-phospho-MAPK as indicated, followed by incubation for 1 h with protein G–Plus (Sigma) or protein A–Sepharose (Sigma). After washing, the beads were then resuspended in SDS-PAGE sample buffer, boiled for 5 min, and resolved by SDS-PAGE. The Western blots were performed with α-HA (1:1,000), α-FAK (1:5,000), α-Erk (1:1,000), α-p21 (1:1,000), α-p27 (1:2,000), α-cyclin D1 (1:5,000), α-Cdk2 (1:5,000), α-cyclin A (1:2,000), α-cyclin E (1:2,000), α-His (1:1,000), α-Shc (1:1,000), α-PY (1:2,000), or PY20 (1:2,000) using the enhanced chemiluminescence (ECL) system (Amersham Life Science, Inc., Arlington Heights, IL), as described previously (Xing et al., 1994). In some experiments, equal amounts of protein lysates were analyzed directly by Western blotting.
Immunofluorescence Staining
Cells were processed for immunofluorescence staining as described (Guan et al., 1991). The primary antibodies used were α-HA (1:50), α-integrin α5 (1:200), α-BrdU (1:300), and α-FAK (1:300). The secondary antibodies used were FITC-conjugated anti–mouse or anti–rabbit antibodies (1:300; Sigma) and rhodamine-conjugated anti–mouse or anti–rabbit antibodies (1:200; Sigma). In α-BrdU staining experiments, cellular DNA was digested with DNase I (0.5 units/ml; New England Biolabs, Inc., Beverly, MA) for 20 min at 37°C before staining with the primary antibodies and nuclei were stained with Hoechst (0.5 μg/ml) for 10 min at room temperature before mounting for analyses by immunofluorescent microscopy.
In Vitro Kinase Assays
For FAK kinase assays, cell lysates were immunoprecipitated by α-HA as described above. The immune complexes were washed and subjected to in vitro kinase assays with or without exogenous substrates poly(Glu,Tyr) (E4Y1), as described previously (Cary et al., 1996). For Src and Fyn kinase assays, cell lysates were immunoprecipitated by α-Src or α-Fyn as described above. The immune complexes were washed and subjected to in vitro kinase assays with acid-denatured enolase as substrates, as described previously (Schlaepfer et al., 1997). For mitogen-activated protein kinase (MAPK) (Erk1/2) assays, cell lysates were immunoprecipitated by α-His (for transiently transfected His-tagged Erk2, a kind gift of Dr. M. Roberson, Cornell University, Ithaca, NY) or α-phospho-MAPK (for endogenous Erks1/2) as described above. The immune complexes were washed three times with ice-cold modified RIPA buffer and twice with kinase buffer (50 mM Tris, pH 7.4, and 10 mM MgCl2). They were then incubated in 48 μl kinase buffer containing 5 μg myelin basic protein (MBP), 25 μM ATP and 10 μCi [γ-32P]ATP for 30 min at 30°C. The kinase reactions were stopped by addition of SDS sample buffer, boiled for 5 min, and then resolved on a 15% SDS-PAGE. The gel was dried and subjected to autoradiography. The phosphorylated MBP bands were excised from the gel and the 32P incorporation was determined by Cerenkov counting.
Results
Analysis of Cell Cycle Regulation by FAK in HFF and NIH3T3 Cells Using Transient Transfection
To investigate the potential role of FAK in the cell cycle regulation, we transiently transfected primary HFF cells or NIH3T3 cells with expression vectors encoding wild-type (WT) FAK or its mutants and then analyzed the effects on cell cycle progression by measuring BrdU incorporation, as described in Materials and Methods. As shown in Fig. 1 A, overexpression of WT FAK resulted in an increase in new DNA synthesis in comparison with the control HFF cells. In contrast, both Y397F mutant (with Y397 converted to F) and ΔC14 mutant (lacking the COOH-terminal 14 amino acids of FAK) inhibited BrdU incorporation under the same conditions. Fig. 1 B shows that overexpression of WT FAK or Y397F did not result in any alterations of new DNA synthesis under the experimental conditions in NIH3T3 cells (P = 0.42 and 0.24 compared with values from control cells, respectively). However, ΔC14 caused a significant reduction of BrdU incorporation in these cells. Taken together these results suggested that FAK may play a role in cell cycle regulation although its effects on primary HFF cells are more pronounced than in NIH3T3 cells whose cell cycle control may have become less dependent on FAK through the countless number of passages. They also suggested that ΔC14 functioned as a stronger inhibitor of cell cycle progression than Y397F and that it could work in both primary HFF cells and NIH3T3 cells under the experimental conditions.
Figure 1.

Inhibition of new DNA synthesis by Y397F and ΔC14. (A) HFF cells were transiently transfected with expression vectors encoding WT FAK, ΔC14, or Y397F, as indicated. The control cells (C) were transfected by pKH3 vector alone. They were then analyzed for BrdU incorporation as described in Materials and Methods. The percentage of BrdU (+)/positively transfected cells was determined by analyzing 40–50 positively transfected cells for each transfection in multiple fields. The results show mean + SE for at least three independent experiments. *P = 0.0041 and 0.036 in comparison to value from control cells for ΔC14 and Y397F, respectively. **P = 0.014 in comparison to value from control cells. (B) Similar experiments were performed with transiently transfected NIH3T3 cells. The results show mean + SE for at least three independent experiments. *P = 0.046 in comparison to value from control cells.
Analysis of Inhibition of Cell Cycle Progression by ΔC14 Using an Inducible Expression System
To investigate the mechanisms by which ΔC14 inhibited cell cycle progression, we generated inducible overexpression of WT FAK, ΔC14 and another mutant ΔC14F in NIH3T3 cells using the tetracycline-regulated expression system (Freundlieb et al., 1997). ΔC14F is a double mutant lacking the COOH-terminal 14 amino acids of FAK and with Y397 converted to F. Fig. 2 shows the regulated expression of ΔC14 by removal of tetracycline in the media. Lysates were prepared from cells at various times after removal of tetracycline and subjected to Western blot analysis by α-FAK or mAb α-HA that recognizes the triple HA epitope tags fused to the NH2 terminus of FAK and mutants. Expression of ΔC14 was detected by α-HA 4–8 h after tetracycline removal and it reached a maximal level 12 h after induction (Fig. 2 b). Western blotting with α-FAK confirmed expression of ΔC14 and densitometric analysis indicated an approximate eightfold level of expression of ΔC14 in comparison with the endogenous FAK in uninduced cells (Fig. 2 d). No expression of ΔC14 was detected in uninduced cells by either α-HA (Fig. 2 a) or α-FAK (Fig. 2 c).
Figure 2.

Induction of ΔC14 expression by removal of tetracycline. ΔC14 cells were incubated in media in the presence (uninduced, U) or absence (induced, I) of 0.4 μg/ml tetracycline. At the indicated times, lysates were prepared and analyzed by Western blot with α-HA or α-FAK.
The effect of induced expression of ΔC14 on cell cycle progression was examined by measuring BrdU incorporation in response to serum stimulation of quiescent cells. As shown in Fig. 3 A, the majority of the uninduced cells entered S phase as detected by α-BrdU staining at 16 hr after serum stimulation (Fig. 2, a and b). In contrast, induction of ΔC14 expression led to a significant inhibition of new DNA synthesis (Fig. 2, c and d). Similar inhibition of BrdU incorporation was also observed when ΔC14 expression was induced in asynchronous cells (data not shown). Quantitative analysis of two different clones in three independent experiments showed that induction of ΔC14 expression decreased new DNA synthesis by ∼40–50% compared with uninduced cells (Fig. 3 B). At 24 and 36 h after serum stimulation, more cells with induced expression of ΔC14 entered S phase. However, ∼20% cells did not show new DNA synthesis even at 36 h after serum stimulation. Under both uninduced and induced conditions, ∼90% of Mock cells entered S phase at 16 h after serum stimulation and nearly 100% had incorporated BrdU at 24 and 36 h after serum stimulation.
Figure 3.
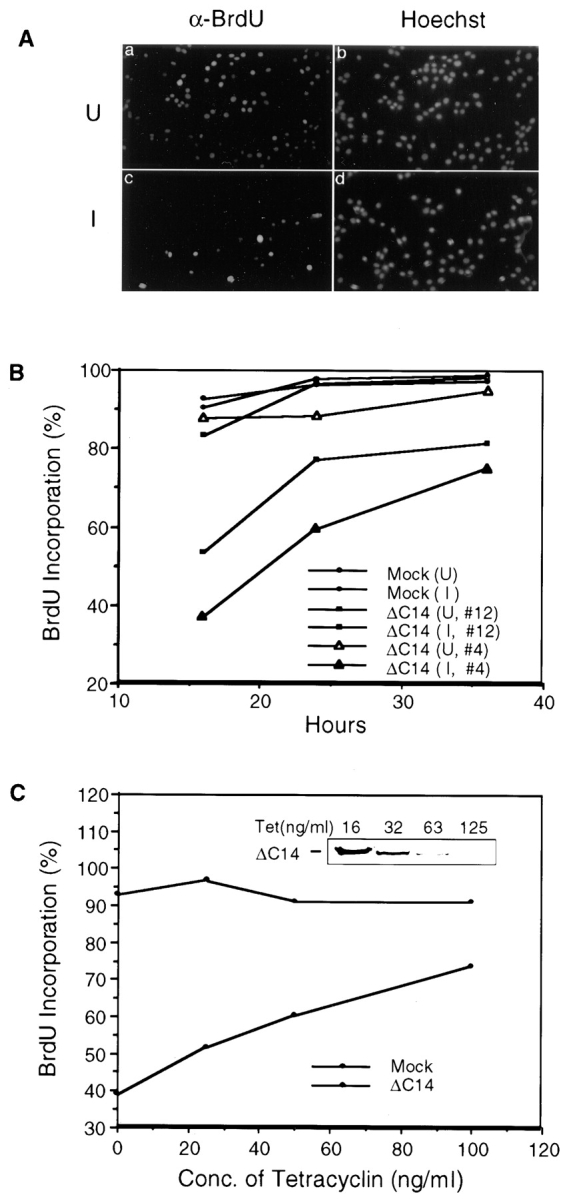
Inhibition of new DNA synthesis by ΔC14. (A) Quiescent ΔC14 cells were stimulated with 10% CS in the presence (uninduced, U) or absence (induced, I) of 0.4 μg/ml tetracycline. 16 hr after stimulation, the cells were processed for immunofluorescent staining with α-BrdU and Hoechst dye staining, as described in Materials and Methods. (B) Similar experiments were performed for Mock cells and two clones of ΔC14 cells, as described in A. The percentage of BrdU (+) cells at 16, 24, and 36 h after stimulation was determined by analyzing ∼500 cells in multiple fields. (C) The percentage of BrdU (+) Mock (▵) and ΔC14 (○) cells at 16 h after serum stimulation in media containing indicated concentrations of tetracycline. The inset shows the induced expression of ΔC14 at various tetracycline concentrations, as detected by α-HA Western blot.
The inhibitory effect of ΔC14 on DNA synthesis was further examined by varying its expression levels using different concentrations of tetracycline. Fig. 3 C shows the decrease in inhibition of BrdU incorporation as the concentration of tetracycline was increased. As expected, decreasing expression levels of ΔC14 were observed under these conditions (Fig. 3 C, inset). ΔC14 inhibited new DNA synthesis by ∼50% at the maximal expression level (16 ng/ml tetracycline), while the inhibition was only ∼20% at the low expression level (125 ng/ml tetracycline). Tetracycline at these concentrations had no effect on new DNA synthesis of Mock cells. Taken together, these data suggested that overexpression of the FAK mutant ΔC14 inhibited cell cycle progression, which are consistent with data obtained from HFF cells and NIH3T3 cells by transient transfection assays.
ΔC14 Inihibits Cell Cycle Progression by Interfering with Endogenous FAK
To determine whether ΔC14 inhibited cell cycle progression by interfering with endogenous FAK function or by other mechanisms, we analyzed the effects on cell cycle by overexpression of WT FAK and ΔC14F mutant. Fig. 4 A shows expression of the exogenous FAK and mutants upon induction by removal of tetracycline in the media, as detected by α-HA (Fig. 4 A, lanes I, top). Western blotting with α-FAK confirmed overexpression of the exogenous FAK and mutants at similar levels (Fig. 4 A, bottom). No expression of the exogenous FAK or the mutants was detected in the presence of tetracycline (Fig. 4 A, lanes U).
Figure 4.
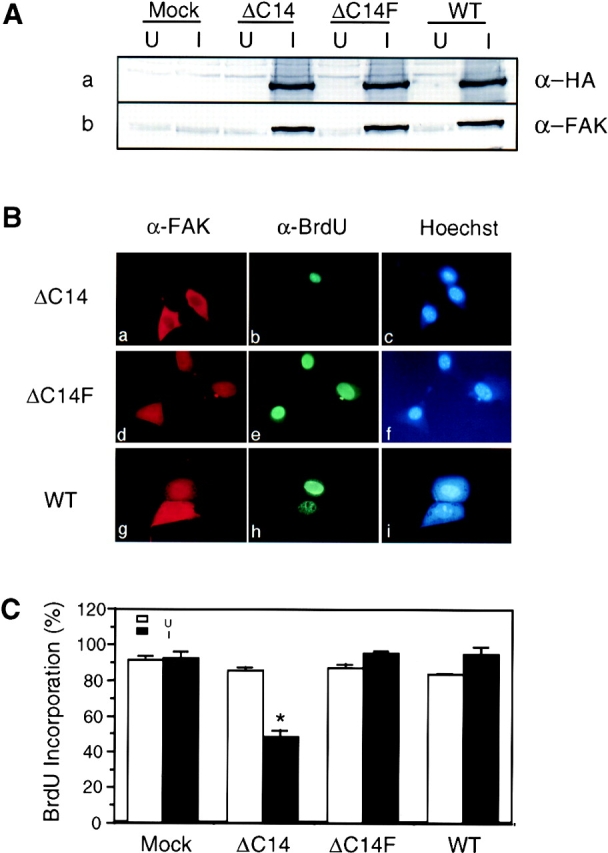
Effects on cell cycle progression by overexpression of FAK and mutants. (A) α-HA or α-FAK Western blot of whole cell lysates prepared from cells expressing the indicated proteins under uninduced (lanes U) and induced (lanes I) conditions. (B) α-FAK immunofluorescence (a, d, g), α-BrdU immunofluorescence (b, e, h) and Hoechst dye staining (c, f, i) of cells expressing ΔC14 (a–c), ΔC14F (d–f), and WT (g–i). (C) The percentage of BrdU (+) cells expressing the indicated proteins at 16 h after serum stimulation under uninduced (open bars) and induced (closed bars) conditions. The results show mean + SE for at least three independent experiments. *P = 0.0022 in comparison to value from uninduced cells.
The effects of induced expression of FAK and the mutants on cell cycle progression were then examined by measuring BrdU incorporation 16 h after serum stimulation of quiescent cells (Fig. 4 B). As expected, expression of ΔC14 led to efficient inhibition of new DNA synthesis (Fig. 4 B, a–c). In contrast, expression of ΔC14F or WT FAK did not block BrdU incorporation under the same conditions (Fig. 4 C, d–i). Quantitative analysis of multiple experiments showed that induction of ΔC14 expression decreased new DNA synthesis by ∼40–50% compared with uninduced cells or Mock cells under either induced or uninduced conditions (Fig. 4 C). Similar analysis indicated that ΔC14F had no effect on BrdU incorporation. Interestingly, such quantitative analysis revealed that overexpression of WT FAK resulted in a small, but statistically significant (P = 0.013) increase in new DNA synthesis under these conditions. Analysis of BrdU incorporation at 12 h after serum stimulation confirmed a positive effect of WT FAK on cell cycle progression (Fig. 5). Approximately 15% of cells with induced WT FAK expression entered S phase in comparison with only 3–4% of the uninduced cells or the Mock cells under either induced or uninduced conditions.
Figure 5.

WT FAK stimulation of cell cycle progression. The percentage of BrdU (+) Mock or WT FAK cells at 12 h after serum stimulation under uninduced (open bars) and induced (closed bars) conditions. The results show mean + SE for three independent experiments. *P = 0.0059 in comparison to value from uninduced cells.
As an independent measure to evaluate the role of FAK in cell cycle progression, we analyzed the DNA content after induction of exogenous FAK expression by FACS®. Fig. 6 A shows that in response to serum stimulation, similar fractions of Mock cells exited the G1 phase as measured by DNA content under the induced and uninduced conditions (Fig. 6 A, a–c). However, induction of ΔC14 expression significantly decreased the fraction of cells exiting G1 phase in comparison to cells under uninduced conditions (Fig. 6 A, d–f). Conversely, expression of WT FAK accelerated G1 exit (Fig. 6 A, j–l) while induction of ΔC14F expression did not affect cells to exit G1 (Fig. 6 A, g–i). Fig. 6 B shows quantitative analysis of results from three such experiments, which are consistent with that obtained from the BrdU incorporation assays. Because increased expression of FAK promoted cell cycle progression, these results strongly suggested that FAK mutant ΔC14 inhibited the cell cycle at G1 phase by interfering with the endogenous FAK function rather than by other mechanisms. They also suggested that Y397 was necessary for the dominant negative effect of ΔC14.
Figure 6.
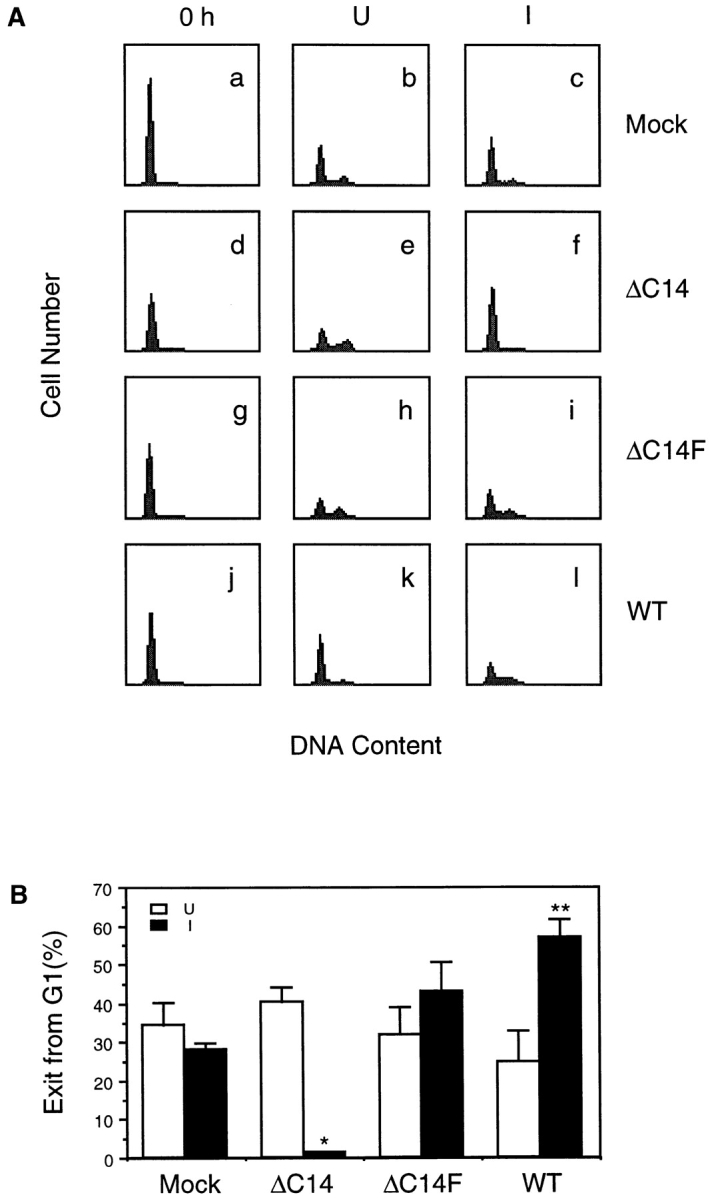
Cell cycle distribution of cells expressing exogenous FAK and mutants. (A) Quiescent (0 h) cells were stimulated with 10% CS in the presence (uninduced, U) or absence (induced, I) of 0.4 μg/ml tetracycline for 16 h. The cells were then analyzed for DNA content by staining with propidium iodide followed by FACS®, as described in Materials and Methods. (B) The decrease in the proportion of cell in G1 at 16 h from that at 0 h was determined for cells expressing the indicated proteins under uninduced (open bars) and induced (closed bars) conditions. The results show mean + SE for at least three independent experiments. *P = 0.0011 and **P = 0.013 in comparison to value from uninduced cells.
Competition of Mislocalized ΔC14 with Endogenous FAK
Focal contacts localization and associations with a variety of intracellular signaling molecules are critical for FAK functions (Hildebrand et al., 1993; Parsons, 1996; Guan, 1997). The COOH-terminal 14 residues of FAK have been shown to be necessary for focal contacts localization of a GST fusion protein containing the COOH-terminal third of FAK (Tachibana et al., 1995). It is therefore possible that ΔC14 is not localized in focal contacts and behaves as a dominant-negative mutant by competing with the endogenous FAK in focal contacts in binding with other cellular signaling molecules. To test this possibility, we first compared subcellular localization of ΔC14 with that of WT FAK in the induced NIH3T3 cells (Fig. 7). Immunofluorescent staining with α-HA showed a diffuse cytoplasmic distribution of ΔC14 (Fig. 7 a) and a typical focal contacts localization of WT FAK (b). Co-staining with α-integrin α5 showed normal focal contacts of both cells with induced FAK or ΔC14 expressions (Fig. 7, c and d).
Figure 7.

Subcellular localization of ΔC14 and WT FAK. α-HA (a, b) and α-integrin α5 (c, d) immunofluorescent staining of cells expressing ΔC14 (a, c) and WT (b, d). Examples of focal contacts are indicated by arrowheads.
We next analyzed tyrosine phosphorylation of the exogenous FAK and its association with intracellular signaling molecules. Lysates were prepared from ΔC14, ΔC14F, and WT FAK or Mock cells after induction of exogenous FAK expression by removing tetracycline. They were immunoprecipitated with α-HA, and the immune complexes were Western blotted with α-FAK to verify similar expression levels (Fig. 8 A) or with an anti-phosphotyrosine antibody, PY20, to determine the in vivo phosphotyrosine levels of the exogenous FAK (Fig. 8 B). As expected, the wild-type FAK was phosphorylated on tyrosine residues in vivo (Fig. 8 B, lane 4). Surprisingly, ΔC14 was also highly phosphorylated in vivo (Fig. 8 B, lane 2) even though it was not targeted to focal contacts (see Fig. 7). The double mutant ΔC14F was not phosphorylated (Fig. 8 B, lane 3), suggesting that ΔC14 was capable of autophosphorylation on residue Y397. In vitro kinase assays of the α-HA immune complexes indicated that ΔC14 (Fig. 8 B, lane 2) and WT FAK (lane 4) had comparable autophosphorylation activity (Fig. 8 C) and similar activity for the exogenous substrate E4Y1 (D). Consistent with the in vivo phosphorylation data, the double mutant ΔC14F was not autophosphorylated in in vitro kinase assays (Fig. 8 C, lane 3). It also showed a reduced activity for E4Y1 (Fig. 8 D, lane 3), possibly resulting from the lack of association with Src and Fyn (see below).
Figure 8.

Biochemical analysis of FAK and mutants. Whole cell lysates were prepared from cells that had been induced to express the indicated proteins. They were immunoprecipitated by α-HA (A–D), α-Src (E, G) or α-Fyn (F, H) followed by Western blot with α-FAK (A), PY20 (B), or α-HA (E, F), or in vitro kinase assay in the presence of E4Y1 (D) or enolase (G, H), or nothing (C). The results show mean + SE of relative kinase activities (normalized to expression levels in each experiment) normalized to WT FAK from at least three independent experiments (D).
To detect FAK association with Src and Fyn, lysates prepared from the induced cells were immunoprecipitated with anti-Src (Fig. 8 E) or anti-Fyn (F) antibodies, and the immune complexes were Western blotted with α-HA. As expected, WT FAK was associated with Src and Fyn (Fig. 8, lane 4) while the ΔC14F mutant was not (lane 3). ΔC14 was associated with Src and Fyn to a similar extent as WT FAK (compare lanes 2 and 4). These results showed that the FAK mutant ΔC14 was not localized in focal contacts, but was phosphorylated and associated with Src and Fyn. Consistent with the requirement of Y397 for the dominant negative effects, these results indicated that ΔC14 functioned as a dominant negative mutant by competing with the endogenous FAK in focal contacts for binding signaling molecules like Src and/or Fyn.
Inhibition of Src-family kinases has previously been shown to cause cell cycle arrest (Twamley-Stein et al., 1993; Roche et al., 1995). To determine if overexpression of FAK or mutants affected the total activity of Src or Fyn in these cells, the anti-Src or anti-Fyn immunocomplexes were subjected to in vitro kinase assays using denatured enolase as a substrate. Fig. 8 G showed that the total Src kinase activities were similar in Mock, WT, ΔC14, and ΔC14F cells. Similarly the total Fyn activities were comparable in these cells (Fig. 8 H). These results further support our hypothesis that ΔC14 functioned as a dominant negative mutant by reducing FAK-associated Src and/or Fyn in focal contacts rather than by altering the total activities of Src or Fyn in these cells.
Role of Erk Activation in Cell Cycle Regulation by FAK
To further explore the mechanisms by which FAK mutant ΔC14 inhibited cell cycle progression, we examined its effect on activation of Erk in integrin-mediated cell adhesion. Mock or ΔC14 cells under either uninduced or induced conditions were removed from the plates and either maintained in suspension (Fig. 9, A–C, lanes Sus) or replated on fibronectin (FN; Fig. 9, A–C, lanes FN). The cells were then lysed and tested for FN adhesion-induced Erk activity, as shown in Fig. 9. Consistent with previous observations (Chen et al., 1994; Schlaepfer et al., 1994; Zhu and Assoian, 1995), cell adhesion to FN caused Erk activation in these cells (Fig. 9 B). However, induction of ΔC14 expression significantly reduced the increase of Erk activation in cell adhesion when compared with uninduced cells or Mock cells under both induced and uninduced conditions (Fig. 9, compare lane 7 with lanes 1, 3, and 5). Western blotting with α-HA confirmed the induced expression of ΔC14 (Fig. 9 A). Blotting with anti-Erk antibody confirmed similar expression levels of Erks under all conditions (Fig. 9 C). Quantitation of Erk activity from three independent experiments indicated an ∼50% reduction in Erk activation by ΔC14 expression (Fig. 9 D).
Figure 9.
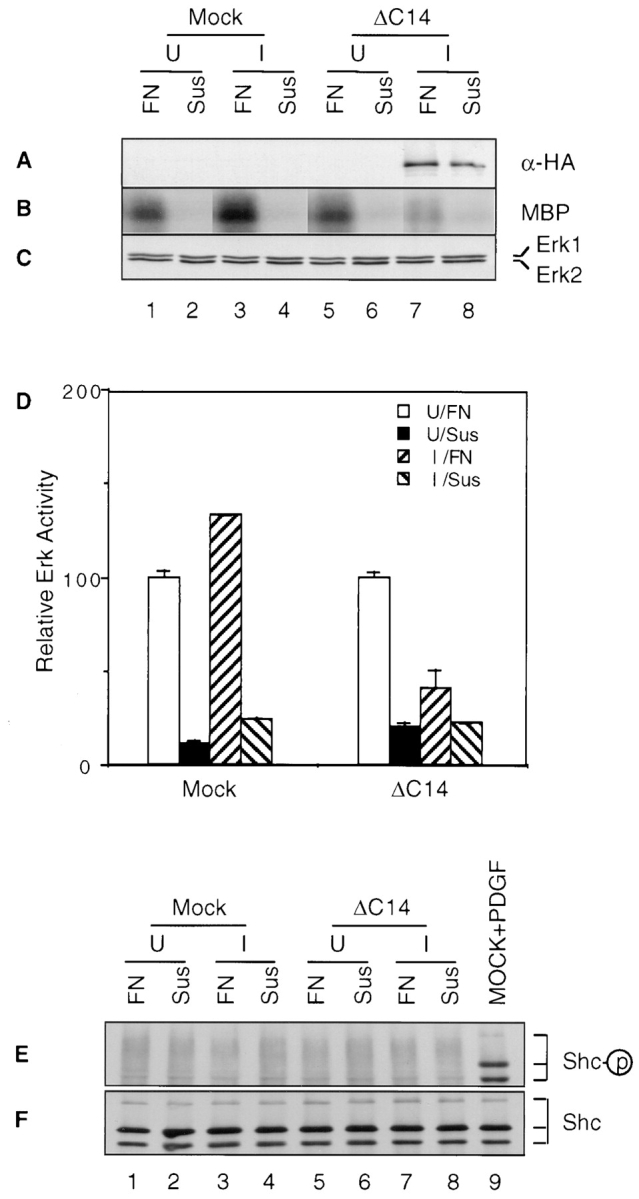
Effects of ΔC14 on Erk activation and Shc phosphorylation. Quiescent Mock or ΔC14 cells under uninduced (U) or induced (I) conditions were detached by trypsin/EDTA (A–D) or EDTA alone (E, F) and either maintained in suspension (Sus) or replated on fibronectin (FN), as described in Materials and Methods. Whole cell lysates were western blotted with α-HA (A) or α-Erk (C), or immunoprecipitated by α-phospho-Erk followed by in vitro kinase assay with MBP as substrate (B). (D) The average and standard deviation of relative Erk activites were obtained from three independent experiments. The relative activities were normalized to uninduced cells plated on FN. Alternatively, the lysates were immunoprecipitated by α-Shc followed by Western blotting with anti-PY (E) or anti-Shc (F). Lysates from PDGF-stimulated cells were also used as a positive control in E and F.
Wary et al. (1996, 1998) have recently observed that some integrins could activate Erk by coupling through Shc, but not FAK. Since FAK has also been suggested to mediate Shc activation by integrins (Schlaepfer et al., 1998), it is possible that ΔC14 inhibited Erk by interfering with Shc activation in cell adhesion. To examine this possibility, lysates were prepared from suspended (Fig. 9, lanes Sus) or FN-attached (lanes FN) Mock or ΔC14 cells under either uninduced or induced conditions. They were immunoprecipitated with anti-Shc followed by Western blotting with anti-PY to detect tyrosine-phosphorylated Shc, as described previously (Wary et al., 1996, 1998). Surprisingly, Fig. 9 E shows that no significant tyrosine phosphorylation of Shc was detected under these conditions although tyrosine phosphorylation of Shc was detected in PDGF-stimulated cells (Fig. 9 E, lane 9). Western blotting of the immunoprecipitates with anti-Shc confirmed presence of similar amount of Shc in all samples (Fig. 9 F). Taken together, these results demonstrated that Shc was not phosphorylated in cell adhesion to FN under our experimental conditions, where Erk activation is observed (Fig. 9). Therefore it is unlikely that ΔC14 inhibited Erk activation by interfering with Shc's ability to couple integrins to Erk.
To further evaluate our hypothesis that ΔC14 functioned as a dominant-negative mutant by reducing FAK-associated Src and/or Fyn in focal contacts rather than by interfering with Shc or other signaling pathways, we examined the effect of FAK COOH-terminal fragment FRNK and its corresponding mutant lacking the last 14 amino acids (FRNKΔC14) on BrdU incorporation and Erk activation. Consistent with a previous report (Gilmore and Romer, 1996), we found that overexpression of FRNK resulted in a decrease in BrdU incorporation in comparison with the control HFF cells whereas FRNKΔC14 had little effect (Fig. 10 A). Because overexpression of FRNK (but not FRNKΔC14) can compete with FAK for focal contacts localization (Gilmore and Romer, 1996; Richardson and Parsons, 1996) and FRNK is not expected to bind Shc directly, these results further support our hypothesis that interaction of FAK with signaling molecules such as Src and Fyn in focal contacts is critical for cell cycle progression.
Figure 10.
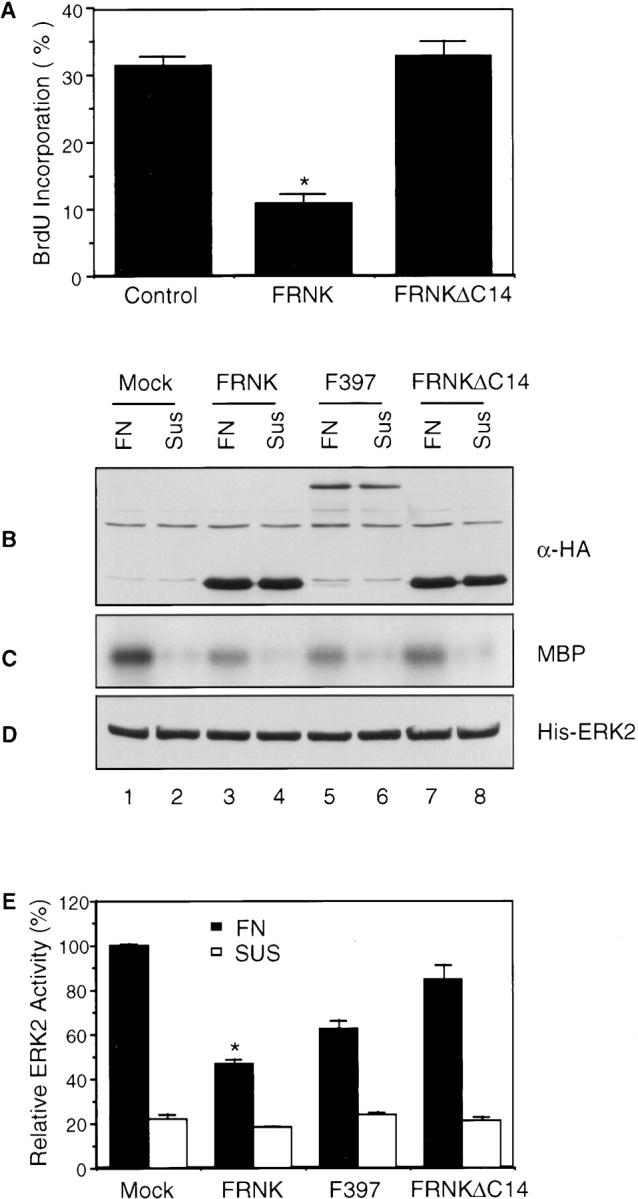
Effects of FAK constructs on BrdU incorporation and Erk activation in HFF cells. (A) HFF cells were transiently transfected with expression vectors encoding FRNK, FRNKΔC14, or pKH3 vector alone (Control). They were then analyzed for BrdU incorporation as described in Materials and Methods. The percentage of BrdU (+)/positively transfected cells was determined by analyzing 40–50 positively transfected cells for each transfection in multiple fields. The results show mean + SE for at least three independent experiments. *P = 0.001 in comparison to value from control cells. (B–E) HFF cells were transiently transfected with expression vectors encoding FRNK, FRNKΔC14, Y397F (i.e., F397), or pKH3 vector alone (Mock). They were serum starved, detached by trypsinization and then either maintained in suspension (Sus) or replated on fibronectin (FN), as described in Materials and Methods. Whole cell lysates were Western blotted with α-HA (B) or α-His (D), or immunoprecipitated by α-His followed by in vitro kinase assay with MBP as substrate (C). (E) The average and standard deviation of relative Erk activites were obtained from four independent experiments. The relative activities were normalized to MOCK cells plated on FN. *P = 0.015 and 0.052 in comparison to value from MOCK cells on FN for cells transfected with FRNK and Y397F on FN, respectively.
We then examined the effect of these constructs on Erk activation in HFF cells. Cells were transiently transfected with these FAK constructs along with a His-tagged Erk2. As shown in Fig. 10 C, overexpression of FRNK significantly reduced the increase of Erk activation in cell adhesion when compared with mock-transfected cells (Fig. 10 C, compare lanes 3 and 1) whereas overexpression of FRNKΔC14 had little effect (lane 7). Overexpression of Y397F also reduced Erk activation, although to a lesser extent (lane 5). Western blotting with α-HA shows the expression levels of these FAK constructs (Fig. 10 B). Blotting with anti-His antibody confirmed similar expression levels of Erks under all conditions (Fig. 10 D). Quantitation of Erk activity from four independent experiments indicated an ∼50% and 35% reduction in Erk activation by FRNK and Y397F, respectively (Fig. 10 E). The inhibition of Erk activation by these FAK constructs correlated with their effects on BrdU incorporation (see Figs. 1 and 10 A). Taken together, these results suggested strongly that activation of Erk in cell adhesion is mediated at least in part by FAK and that inhibition of Erk activation by ΔC14 and FRNK may play a role in inhibition of cell cycle progression.
Regulation of Cyclin D1 and p21 by FAK
Integrin-mediated cell adhesion has been found to regulate several cyclins and cyclin-dependent kinases (CDKs) (Fang et al., 1996; Zhu et al., 1996; Assoian, 1997). Therefore the effects of induced exogenous FAK expression on these cell cycle regulators are examined. Fig. 11 A shows that induction of ΔC14 expression increased p21 expression and reduced cyclin D1 expression (a and b, compare lanes 4 and 3). Conversely, overexpression of WT FAK decreased p21 expression and enhanced cyclin D1 expression (Fig. 11 A, a and b, compare lanes 8 and 7). The expression levels of p21 and cyclin D1 were not altered by induction of ΔC14F expression (Fig. 11 A, a and b, compare lanes 6 and 5). The expression levels of Cdk2, cyclin E, cyclin A, and p27 remained the same in all cells under either induced or uninduced conditions (Fig. 11 A, c–f).
Figure 11.

Regulation of the expression levels of components of G1/S cyclin-Cdk complexes by exogenous FAK and mutants. (A) α-p21, α-cyclin D1, α-Cdk2, α-cyclin E, α-cyclin A, or α-p27 Western blot of whole cell lysates prepared from cells expressing the indicated proteins under uninduced (lanes U) and induced (lanes I) conditions. (B) Serum-starved cells expressing ΔC14 were incubated in media containing 10% CS in the presence (U) or absence (I) of tetracycline. At the indicated times, lysates were prepared and analyzed by Western blot with α-p21, α-cyclin D1, α-Cdk2, α-cyclin E, or α-cyclin A.
Regulation of p21 and cyclin D1 by ΔC14 was then examined more closely by the following time course experiments. Lysates were prepared from cells with or without induced ΔC14 expression at various times after serum stimulation. They were subjected to Western blot analysis as shown in Fig. 11 B. Expression of ΔC14 was detected 4–8 h after induction and it reached a maximal level 12 h after induction (data not shown and see Fig. 2). An increase of p21 expression was observed in the induced cells 8–12 h after induction (Fig. 11 B, b) relative to that in uninduced cells (Fig. 11 B, a). The reduction of cyclin D1 expression by ΔC14 was observed 12 h after tetracycline removal (Fig. 11 B, c and d). Induction of ΔC14 expression did not affect expression of Cdk2, cyclin E or cyclin A (Fig. 11 B, e–j). These results suggested that the altered expression of p21 and cyclin D1 was a consequence of ΔC14 expression and that ΔC14 inhibited cell cycle progression by inducing p21 expression and/or decreasing cyclin D1 expression in G1 phase.
Discussion
In this paper, we examine the possible roles and mechanisms of FAK in cell cycle regulation by integrins. We established stable cell lines with tetracycline-regulated inducible overexpression of exogenous FAK and mutants. Using this inducible system as well as transient transfections, we showed that overexpression of WT FAK accelerated G1 to S phase transition in both primary HFF cells and NIH3T3 cells. Conversely, overexpression of a dominant negative FAK mutant ΔC14 inhibited cell cycle progression at G1 phase and this inhibition required the Y397 in ΔC14. Biochemical analyses using the inducible expression system revealed that ΔC14 was mislocalized but still associated with Src and Fyn. They also showed that ΔC14 reduced Erk activation in cell adhesion, blocked cyclin D1 upregulation, and induced p21 expression, whereas WT FAK increased cyclin D1 expression and decreased p21 expression. Together these results demonstrate that FAK serves as a mediator of cell cycle regulation by integrins. They also suggest that FAK/Src complex formation in the focal contacts, Erk activation, and cyclin D1 and/or p21 are some of the important FAK downstream events in the signaling pathway leading to cell cycle regulation.
Results presented here are consistent with a previous observation showing that inhibition of FAK by microinjection of the COOH-terminal fragment reduced BrdU incorporation in HUVEC and Balb/c 3T3 cells (Gilmore and Romer, 1996). Interestingly, FAK−/− cells have been derived from FAK knockout mice, suggesting that FAK is not required for cell proliferation under some conditions (Ilic et al., 1995). However, these FAK−/− cells do not contain a functional p53 because a p53 mutation was introduced into the embryo from which the FAK−/− cells were derived. Since it is a critical cell cycle regulator, the lack of p53 might account for cell cycle progression without FAK function in these cells. Alternatively, the properties of the FAK−/− cells could be due to the increased expression of FAK-related kinases such as Pyk2, which may serve to compensate some of the functions of FAK.
The use of an inducible expression system in stable cell lines has allowed us to begin to investigate the mechanisms of cell cycle regulation by FAK. Analyses of FAK mutant ΔC14 showed that it was mislocalized but still capable of association with Src and Fyn. Furthermore, cell cycle progression was not affected by the double mutant ΔC14F that did not associate with Src family kinases as expected. These results suggested that ΔC14 functioned as a dominant negative mutant by competing with endogenous FAK in focal contacts for binding Src and/or Fyn. Consistent with this, we also observed cell cycle inhibition by Y397F mutant and FRNK, but not FRNKΔC14, which are expected to compete with endogenous FAK for focal contacts localization. Src family kinases have been shown to play a critical role in cell cycle regulation (Thomas and Brugge, 1997). Inhibition of their activities blocked DNA synthesis induced by several growth factors (Twamley-Stein et al., 1993; Roche et al., 1995). However, our results provide first evidence suggesting that the FAK complex formation with Src family kinases in focal contacts (but not necessarily the total Src family kinase activities, see Fig. 8, G and H) is necessary for cell cycle regulation in cell adhesion.
Recent studies have shown that integrin-mediated cell adhesion can regulate several components of cell cycle regulation machinery (Fang et al., 1996; Zhu et al., 1996; Assoian, 1997; Day et al., 1997), although the intracellular signaling pathways in cell cycle regulation by integrins are largely unknown. Results in this report revealed that cyclin D1 and p21 are two major targets of FAK signaling pathways in cell cycle regulation by integrins. Induced overexpression of WT FAK increased cyclin D1 expression and decreased p21 expression whereas induction of ΔC14 expression blocked cyclin D1 upregulation and increased p21 expression. Wu and Schonthal (1997) recently reported that disruption of cell–matrix interactions caused rapid p21 upregulation via p53. It will be interesting to determine whether regulation of p21 by FAK involves a similar mechanism. If so, these results will further support the notion that ΔC14 functions as a dominant-negative FAK (inhibiting endogenous FAK function that results in p53 activation and then p21 upregulation). This will also be consistent with the previous observation that loss of FAK function did not cause cell cycle arrest in p53-deficient cells (Ilic et al., 1995).
Integrin-mediated cell adhesion has been shown to regulate a number of cyclins including cyclin D1, cyclin E, and cyclin A (Assoian, 1997). However, a recent report showed that overexpression of cyclin D1 but not cyclin E–induced anchorage-independent cell cycle progression, suggesting that adhesion-dependent cell cycle regulation is mediated through cyclin D1 (Resnitzky, 1997). Experiments are in progress to determine whether cyclin D1 or p21 or both are necessary for the effects of FAK and ΔC14 overexpression on cell cycle regulation. Nevertheless, these results are consistent with our conclusion that FAK is a mediator of cell cycle regulation by integrins.
Integrin-mediated cell adhesion has also been shown to regulate expression levels of cyclin A (Assoian, 1997). Furthermore, cyclin A expression is induced in a cyclin D/ Cdk4/6-dependent manner at G1/S transition. It is therefore surprising that we did not observe any change in cyclin A expression in spite of the evident alterations in cyclin D expression upon induction of ΔC14 expression (Fig. 11). It is possible that the effects of FAK on cell cycle regulation may be more complex than its effect on cyclin D expression. Future studies using the inducible expression systems will be directed toward identifying additional factors involved in cell cycle regulation by FAK.
Two other intracellular signaling molecules Shc and ILK have also been implicated in cell cycle regulation in cell adhesion (Wary et al., 1996; Radeva et al., 1997). The signaling pathways associated with ILK in cell cycle regulation is unclear at present, although phosphorylation of integrin cytoplasmic domain by ILK may be involved (Hannigan et al., 1996). In contrast, Shc has also been shown to link some integrins to the MAP kinase pathways. Interestingly, we found that cell adhesion to FN induced Erk activation in a FAK-dependent manner under the experimental conditions where Shc was not tyrosine phosphorylated (Fig. 9). Taken together these results suggest that multiple pathways could be involved in cell cycle regulation by integrins. For example, overexpression of WT FAK and ΔC14 affected expression levels of p21, but not p27 (Fig. 11) whereas both p21 and p27 have been found to be upregulated by disruption of cell adhesion (Assoian, 1997). This possibility may also explain the partial inhibition of new DNA synthesis by the ΔC14 mutant (Fig. 4). Alternatively, the partial inhibition could be due to heterogeneity of expression in the clone. Co-staining with α-BrdU and α-FAK indicated that the ΔC14 cells with BrdU incorporation contained less or almost undetectable level of the exogenous ΔC14 (Fig. 4 B, a and b).
The mechanisms of regulation of cyclin D1 and/or p21 by FAK and its associated signaling molecules such as Src and Fyn are not clear at present. Cell spreading has been correlated with cell cycle progression at G1/S transition (Chen et al., 1997). However, we did not detect any differences in cell spreading or adhesion to fibronectin among the different cell lines upon induction of FAK or mutants (data not shown). Apparently normal focal contacts were also detected in cells expressing ΔC14, although we can not exclude completely any subtle differences in these structures among these cells (Fig. 8). In contrast we have observed inhibition of Erk activity by ΔC14 (Fig. 9) and its activation by inducible overexpression of WT FAK (data not shown). These results are consistent with a previous report showing Erk activation by transient transfection of FAK into HEK293 cells (Schlaefper et al., 1997). However, FAK phosphorylation and Erk activation have been dissociated under some other conditions (Lin et al., 1997). Furthermore, Wary et al. (1996) have shown that some integrins activate Erk by coupling to Shc without affecting FAK phosphorylation. It is not clear how to explain these conflicting data at present, although it is possible that this is due to the use of different cells and possibly subtle differences in various approaches (e.g., stable expression vs transient and inducible expression). In any case, the effects on the Erk activation by WT FAK and ΔC14 mutants might account for the regulation of cyclin D1 expression and cell cycle progression in our systems, as activation of Erk has been shown to induce cyclin D1 expression (Lavoie et al., 1996).
One surprising finding here is that the mislocalized FAK mutant ΔC14 was tyrosine phosphorylated in attached cells (Fig. 8). Furthermore, tyrosine phosphorylation of ΔC14 was regulated by cell adhesion in the same manner as the WT FAK with more phosphorylation detected in attached cells than in detached cells (data not shown). Although the mechanism of FAK activation by integrins is still unclear, it has been proposed that oligomerization of FAK with integrins and other cytoskeletal proteins at the focal contacts (i.e., focal contacts localization) is responsible for its activation (Giancotti, 1997; Guan, 1997). However, a FAK mutant lacking the focal adhesion targeting sequence has been shown to be highly tyrosine phosphorylated in attached cells (Hildebrand et al., 1993), suggesting that FAK activation and tyrosine phosphorylation may be independent of its focal contacts localization. Consistent with this, Lyman et al. (1997) have recently found that integrin-activated FAK phosphorylation was independent of focal contacts formation in attached cells. These results suggest the interesting possibility that FAK phosphorylation is regulated by proteins not present in focal contacts such as the recently described protein tyrosine phosphatase PTEN (Li et al., 1997; Steck et al., 1997; Tamura et al., 1998).
Although we have provided evidence suggesting the importance of FAK/Src complex formation and Erk activation, other downstream effectors may also be involved in cell cycle regulation by FAK. It has been shown that PI3 kinase binds to FAK also at Y397 (Chen et al., 1996) and that Grb2 binding to FAK at Y925 depends on Y397 phosphorylation and Src binding (Schlaepfer et al., 1994). Future studies with additional mutants that selectively disrupt these interactions will be necessary to determine if recruitment of these other signaling molecules to focal contacts plays a role in cell cycle regulation by FAK.
Integrin signaling through FAK has been demonstrated to regulate cell migration (Ilic et al., 1995; Cary et al., 1996; Gilmore and Romer, 1996). Furthermore, phosphorylation of Y397 was necessary for FAK functions in both cell migration and cell cycle regulation (Cary et al., 1996; this study). Cell proliferation and migration are often coupled under many physiological conditions such as wound healing and development. Thus FAK may play a role in the coordinate regulation of these two cellular functions in response to integrin-mediated cell adhesion.
Acknowledgments
We are grateful to Dr. Y. Xiong for antibodies against cyclin D1, p21, and p27; Dr. R. Hynes for antibody against integrin α5; and Dr. M. Roberson for an expression vector encoding His-Erk2. We thank Drs. A. Yen and R. A. Hemendinger for help in FACS® analysis, and N. Winand, C. Zheng, D.-C. Han, L. Cary, and S. Abbi for critical reading of the manuscript.
Abbreviations used in this paper
- BrdU
5-bromodeoxyuridine
- CDK
cyclin-dependent kinase
- CS
calf serum
- E4Y1
poly(Glu,Tyr)
- Erk
extracellular signal–regulated kinase
- FAK
focal adhesion kinase
- FN
fibronectin
- MBP
myelin basic protein
- PI 3-kinase
phosphatidylinositol 3-kinase
- SH2
Src homology 2
- WT
wild type
Footnotes
This research was supported by National Institutes of Health grant GM52890 to J.-L. Guan. J.-L. Guan is an Established Investigator of American Heart Association.
References
- Assoian RK. Anchorage-dependent cell cycle progression. J Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L, Chang J, Guan J-L. Stimulation of cell migration by overexpression of focal adhesion kinase and its assocation with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Cary LA, Han DC, Polte T, Hanks S, Guan J-L. Identification of p130cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P-Y, Kanner SB, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK . J Biol Chem. 1994;269:20567–20574. [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Chen H-C, Guan J-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-C, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan J-L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- Chen H-C, Appeddu PA, Isoda H, Guan J-L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Schaller MD, Leu T-H, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein kinase, pp125FAK . Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day ML, Foster RG, Day KC, Zhao X, Humphrey P, Swanson P, Postigo AA, Zhang SH, Dean DC. Cell anchorage regulates apoptosis through the retinoblastoma tumor suppressor/E2F pathway. J Biol Chem. 1997;272:8125–8128. doi: 10.1074/jbc.272.13.8125. [DOI] [PubMed] [Google Scholar]
- Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E. Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science. 1996;271:499–502. doi: 10.1126/science.271.5248.499. [DOI] [PubMed] [Google Scholar]
- Freundlieb S, Baron U, Bonin AL, Gossen M, Bujard H. Use of tetracycline-controlled gene expression systems to study mammalian cell cycle. Methods Enzymol. 1997;283:159–173. doi: 10.1016/s0076-6879(97)83014-5. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG. Integrin signaling: specificity and control of cell survival and cell cycle progression. Curr Opin Cell Biol. 1997;9:691–700. doi: 10.1016/s0955-0674(97)80123-8. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J-L. The role of focal adhesion kinse in integrin signaling. Int J Biochem Cell Biol. 1997;29:1085–1096. doi: 10.1016/s1357-2725(97)00051-4. [DOI] [PubMed] [Google Scholar]
- Guan J-L, Trevithick JE, Hynes RO. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul. 1991;2:951–964. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAKin cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;272:13937–13944. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Lin TH, Aplin AE, Shen Y, Chen Q, Schaller M, Romer L, Aukhil I, Juliano RL. Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J Cell Biol. 1997;136:1385–1395. doi: 10.1083/jcb.136.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman S, Gilmore A, Burridge K, Gidwitz S, White GC., II Integrin-mediated activation of focal adhesion kinase is independent of focal adhesion formation or integrin activation. J Biol Chem. 1997;272:22538–22547. doi: 10.1074/jbc.272.36.22538. [DOI] [PubMed] [Google Scholar]
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Integrin-mediated signalling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- Radeva G, Petrocelli T, Behrend E, Leung-Hagesteijn C, Filmus J, Slingerland J, Dedhar S. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J Biol Chem. 1997;272:13937–13944. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- Resnitzky D. Ectopic expression of cyclin D1 but not cyclin E induces anchorage-independent cell cycle progression. Mol Cell Biol. 1997;17:5640–5647. doi: 10.1128/mcb.17.9.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK . Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Roche S, Koegl M, Barone MV, Roussel MF, Courtneidge SA. DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol Cell Biol. 1995;15:1102–1109. doi: 10.1128/mcb.15.2.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JX, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src . Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances Ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interaction with and activation of c-Src. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to ras pathway by Grb2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130Cas, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;4:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Sato T, D'Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal-adhesion-targeting mechanism of pp125FAK. J Exp Med. 1995;182:1089–1100. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Turner CE, Miller J. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motif and multiple LIM domains: identification of a vinculin and p125FAKbinding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- Twamley-Stein GM, Pepperkok R, Ansorge W, Courtneidge SA. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc Natl Acad Sci USA. 1993;90:7696–7700. doi: 10.1073/pnas.90.16.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori K, Hirai H, Aizawa S, Ruoslahti E. Introduction of p130Cassignaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- Wu RC, Schonthal AH. Activation of p53-p21(waf1) pathway in response to disruption of cell-matrix interactions. J Biol Chem. 1997;272:29091–29098. doi: 10.1074/jbc.272.46.29091. [DOI] [PubMed] [Google Scholar]
- Xing Z, Chen H-C, Nowlen JK, Taylor SJ, Shalloway D, Guan J-L. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- Zhu X, Assoian RK. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Ohtsubo M, Bohmer RM, Roberts JM, Assoian RK. Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2, and phosphorylation of the retinoblastoma protein. J Cell Biol. 1996;133:391–403. doi: 10.1083/jcb.133.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]