

Abstract
Desmosomes first assemble in the E3.5 mouse trophectoderm, concomitant with establishment of epithelial polarity and appearance of a blastocoel cavity. Throughout development, they increase in size and number and are especially abundant in epidermis and heart muscle. Desmosomes mediate cell–cell adhesion through desmosomal cadherins, which differ from classical cadherins in their attachments to intermediate filaments (IFs), rather than actin filaments. Of the proteins implicated in making this IF connection, only desmoplakin (DP) is both exclusive to and ubiquitous among desmosomes. To explore its function and importance to tissue integrity, we ablated the desmoplakin gene. Homozygous −/− mutant embryos proceeded through implantation, but did not survive beyond E6.5. Mutant embryos proceeded through implantation, but did not survive beyond E6.5. Surprisingly, analysis of these embryos revealed a critical role for desmoplakin not only in anchoring IFs to desmosomes, but also in desmosome assembly and/or stabilization. This finding not only unveiled a new function for desmoplakin, but also provided the first opportunity to explore desmosome function during embryogenesis. While a blastocoel cavity formed and epithelial cell polarity was at least partially established in the DP (−/−) embryos, the paucity of desmosomal cell–cell junctions severely affected the modeling of tissue architecture and shaping of the early embryo.
Keywords: desmoplakin, desmosomes, cadherins, embryonic lethal, knockout
Cadherins are transmembrane receptors implicated in cell–cell adhesion, and in transducing signals that regulate such diverse processes as morphogenesis, tissue polarity and intercellular communication (for review, see Garrod, 1993; Koch and Franke, 1994; Marrs and Nelson, 1996; Yap et al., 1997). Cadherins form calcium-mediated, homotypic associations that enable cells expressing the same cadherins to recognize and attach to each other during development and differentiation. Intracellularly, cadherins associate dynamically with the cytoskeleton to transmit cues that a stable cell–cell contact has been achieved. Desmosomes are specialized junctions characteristic of tissues where resilient, stable intercellular associations are required. They are especially abundant in heart muscle and in skin epidermis. At the core of the desmosome are the desmoglein (Dsg)1 and desmocollin (Dsc) cadherins, each encoded by three distinct genes that are differentially expressed as pairs in a tissue-specific fashion (Garrod, 1993; Koch and Franke, 1994).
At the intracellular surface, these desmosomal cadherins associate with one or more of three ubiquitously expressed members of the armadillo family of cadherin binding proteins (Barth et al., 1997). Plakophilin 1a localizes to desmosomes in stratified and complex epithelia (Schmidt et al., 1997), plakophilin 2 localizes to desmosomes in simple and complex epithelia (Mertens et al., 1996), and plakoglobin localizes broadly not only to desmosomes, but also adherens junctions (Cowin et al., 1986). Besides plakoglobin, desmoplakin is the only other protein characteristic of all desmosomes. A member of the small plakin family of coiled-coil proteins, desmoplakin is similar to plakoglobin in that it lacks a transmembrane domain and resides on the cytoplasmic surface of the desmosome (Green et al., 1992; Ruhrberg and Watt, 1997). In vitro experiments suggest that the NH2-terminal head segment of desmoplakin associates with itself and with other desmosomal components (Stappenbeck et al., 1993; Smith and Fuchs, 1998), whereas the nonhelical tail domain can bind to IFs (Stappenbeck and Green, 1992; Kouklis et al., 1994; Kowalczyk et al., 1997). These studies suggest that desmoplakin may function to anchor the IF network to desmosomes, thereby imparting to desmosomes those characteristics that distinguish them from classical adherens junctions.
Cadherin-mediated cell–cell junctions form early in embryonic development. Intercellular adhesion begins with E-cadherin expression at the 8 cell stage (for review see Fleming, 1994). These classical adherens junctions associate with β- and α-catenins and form stable linkages to the actin cytoskeleton (for review see Barth et al., 1997). E-cadherin null embryos survive through compaction, a feature attributed to residual maternal E-cadherin (Larue et al., 1994). However, E-cadherin null embryos fail to form a trophectoderm and a blastocoel cavity. In contrast to adherens junctions, which are ubiquitous at these early stages, desmosomes do not appear until E3.5 and are restricted to the developing trophectoderm (Ducibella et al., 1975; Jackson et al., 1980; Fleming et al., 1991). The timing and location of nascent desmosome formation has led to the speculation that desmosomes may be required for cell type diversification, establishment of epithelial polarity and/or for fortifying adhesion to allow blastocoel fluid accumulation.
Recently, targeting of two desmosomal genes in mice have provided insights into the functions of different desmosomal proteins and of the robust desmosomes of stratified epithelia and heart muscle. Ablation of Dsg3, a desmosomal cadherin, produces the balding mutant mouse displaying hair fragility and oral lesions due to splitting of epithelial desmosomes (Koch et al., 1997). In contrast, targeted mutation of plakoglobin leads to bursting of the heart ventricles in E12.5 embryos due to a paucity of desmosomes (Bierkamp et al., 1996; Ruiz et al., 1996). Epidermal and gut desmosomes are formed in these embryos, but they appear mechanically fragile, as judged by the epidermal degeneration observed in a few embryos that survive to birth (Bierkamp et al., 1996). Taken together, both studies suggest that desmosomal cadherins and plakoglobin are important in desmosome formation and/or stability. It has been postulated that functional redundancy accounts for why some desmosomes are more affected than others in these knockouts (see also McGrath et al., 1997).
An interesting and important issue presently unresolved is the possibility that desmosomes that are assembled from different components may have different structural properties and functions. Related to this issue is the tremendous diversity that exists in desmosome size in different tissues and at various stages of development. In the early embryo, desmosomes begin as very small structures, referred to as nascent desmosomes, and often only a fraction of the size of the robust desmosomes present in heart muscle and in epidermis (Jackson et al., 1980, 1981). Thus far, the potential caveat of redundancy has precluded the use of mouse knockouts to determine when desmosomes acquire essential functions in embryonic development, and what these functions might be.
Another issue unaddressed is the role of cytoskeletal linkages to desmosomes. In E-cadherin–mediated cell–cell adhesion, in vitro studies argue that association with the actin cytoskeleton is required for stabilization of these junctions (Adams et al., 1996; Angres et al., 1996). In contrast, based on gene targeting studies with hemidesmosomal members of the plakin family, namely BPAG1e and plectin, severing the linkage between IFs and the α6β4 integrins can occur without grossly affecting either hemidesmosome structure (BPAG1e and plectin) or number (BPAG1e; Guo et al., 1995; Andra et al., 1997).Therefore, it seems that either (a) actin and IFs differ in their ability to stabilize the junctions to which they adhere, or (b) cadherins and integrins differ in the role of the cytoskeleton in stabilizing their respective junctions.
To begin to address the answers to these questions, we have used embryonic stem cell technology to ablate the desmoplakin gene in mouse embryos. In striking contrast to other desmosomal genes thus far studied in this way, desmoplakin is critical to the development of the early embryo, and null embryos do not survive beyond E6.5. We show that desmoplakin plays a central role not only in establishing IF cytoskeletal architecture, but also in assembly and or stabilization of the desmosome. Moreover, we provide evidence that these two functions are separable. Finally, we show that embryonic desmosomes are important in providing mechanical integrity to the endoderm at a stage when the embryo begins to diversify and develop shape and form.
Materials and Methods
Screening the 129/sv Genomic Library
A mouse DP cDNA (Kouklis, P., V. Vasioukhin, and E. Fuchs, unpublished data) was used to screen a mouse 129/sv genomic DNA library in phage λ (Stratagene, Palo Alto, CA), and a 17.5-kb NotI restriction endonuclease fragment of mDP sequence was subcloned into bluescript KS+ and partially sequenced. This clone, mDP1, was subjected to extensive restriction map analyses and segments were subsequently selected for preparation of the targeting vector. The partial sequence of mDP1 was used to design primers for PCR and probes for Southern blot analysis. Primer sets used for Fig. 2 are: no. 1, left, 5′CTGTGGTTATCCTAACGCC3′, right, 5′GCAGGGATCTTACGAGAAGG3′; no. 2, left, same as no. 1, right, 5′TAGGGGAGGAGTAGAAGGTG3′; no. 3, left, 5′GACAAGAACACCAACATCGC3′, right, 5′CCTACATCACTGAACAGTGTACC3′.
Figure 2.

Absence of desmoplakin in a portion of E3.5 blastocysts that still undergo blastocoel cavity formation. DP (+/−) heterozygous matings were conducted, and embryos at E3.5 were removed before implantation from pregnant females. (A and B) Blastocysts were analyzed by indirect immunofluorescence using the antibodies indicated in the boxes, and embryos were visualized by whole mount confocal microscopy. Anti-desmoplakin (αDP, green), anti-E-cadherin (αEcad, red), and DAPI (DNA, blue); ICM, inner cell mass; TE, trophectoderm; BC, blastocoel cavity. Note yellow dots, reflective of αDP/αEcad double-labeling, in endoderm of wt (w.t.), but not mutant (mut) trophectoderm (A, arrow). (C) DNAs were isolated from blastocysts, and genotyping was done by PCR and Southern blot analysis (necessary due to the small size of the embryos). Two different primer sets were used to delineate between wild type and targeted allele (see Fig. 1 A). Primer set no. 1 amplified a 215-bp fragment of the wt allele (w.t.) while primer set no. 2 amplified a 300-bp fragment in the targeted allele (mut). After resolution by agarose gel electrophoresis, bands were transferred to Hybond paper, which was then hybridized with a ECL-labeled probe specific for the sequences present in both mutant and wt alleles. Note that the embryo DNA in first and last lanes scored negative for the wt allele. Shown are data from mouse line 11-10. 1 cm = 20 μm.
Electroporation and Analysis of Embryonic Stem Cells and Knockout Mice
Embryonic stem (ES) cells (R1 strain from Andreas Nagy and Janet Rossant, University of Toronto) were transfected by electroporation, followed by positive and negative selection in tissue culture as described previously (Guo et al., 1995). DNAs were assayed by Southern blot analysis. ES cells harboring the desired targeting event were injected into mouse 129/sv blastocysts, which were then transferred to C57BL/6 mothers. After breeding, heterozygous mice were identified by Southern blot analysis of tail DNAs.
Confocal Microscopy on Frozen Tissue Sections
Embryos were analyzed by either (a) whole mount immunofluorescence after dissection from their decidua; or (b) ±dissection from their decidua, freezing in OCT compound, sectioning (10 μm), and conventional indirect immunofluorescence. Blastocysts, E5.0, E6.0, E6.5, and E7.0 embryos were isolated and fixed with either methanol (−20°C) for 10 min or 4.0% formaldehyde for 10 min, followed by washing 2× with PBS. Embryos were then transferred to blocking (1.0% BSA, 0.1% Triton X-100, and 1.0% gelatin in PBS) for 1–12 h before incubating for 12 h in 100 μl fresh solution containing primary antibody at 4°C. After transferring embryos into wash buffer 3× for 10 min, embryos were transferred to a 100-μl droplet blocking buffer containing secondary Texas red– or FITC-conjugated antibodies (1:250 dilution) for 1 h before washing and mounting. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI).
αDP antibodies used were: (a) DP2.15, a mouse monoclonal antibody against the rod/tail of DP (5 μg/ml; ICN Biochemicals, Costa Mesa, CA), or (b) three affinity-purified rabbit polyclonal αDP antibodies directed against either the tail (1:50; Kouklis, P., and E. Fuchs, unpublished data), the rod (1:100; Kouklis et al., 1994), or the head (1:10; Smith and Fuchs, 1998). Other antibodies used were: rat monoclonal αEcadherin (1:100; Zymed, San Francisco, CA), rat monoclonal αK8 (TROMA-1, 1:100; a kind gift from Dr. Robert Oshima, Burnham Institute, La Jolla, CA), rabbit αplakoglobin (αPg; 1:100, a kind gift of from Dr. Jackie Papkoff, Megabios Corp., Burlingame, CA), αDSCII 7G6 (αDsc2; 1:200, a kind gift from Dr. Margaret Wheelock, University of Toledo, OH), and αDSGII (αDsg2; 1:250) and guinea pig αplakophilin-2 (1:200, both kind gifts from Dr. Werner Franke, German Cancer Research Center, Heidelberg, Germany).
For sectioning, embryos or decidua were frozen in OCT compound. 10-μm sections were fixed with methanol (−20°C) for 10 min or with 4.0% formaldehyde for 10 min and then washed 2× with PBS. Blocking, primary, and secondary antibody incubations were as above. Specimens were examined using a confocal microscope (model LSM 410; Carl Zeiss Inc., Thornwood, NY).
Histology and Ultrastructural Analyses
Dissected embryos were embedded in LX-112 medium, sectioned (0.75 μm), stained with toluidine blue, and visualized by light microscopy. Embryos not dissected from their decidua were prepared for paraffin sectioning as described by Kaufman (1992), and sections were stained with hematoxylin and eosin.
For regular EM, embryos were fixed at room temperature for >1 h in 0.2 M sodium cacodylate buffer, pH 7.4, containing 2.5% glutaraldehyde and 4% formaldehyde. After washing 3× in the same buffer, embryos were postfixed with 1% aqueous osmium tetroxide for 1 h at room temperature. Embryos were washed in sodium cacodylate buffer, followed by maleate buffer, pH 5.1, and then dehydrated with ascending grades of ethanol and propylene oxide. Samples were embedded in LX-112 medium, polymerized at 70°C for 48 h, trimmed, sectioned at 70–90 nm, poststained in 50% saturated uranyl acetate and 0.2% lead citrate, and examined with a transmission electron microscope (Philips CM120). Immunoelectron microscopy with anti-plakoglobin antibodies was performed as previously described (Allen et al., 1996).
Results
Isolation, Characterization, and Targeting of the Murine Desmoplakin Gene
A mouse desmoplakin genomic clone, mDP1, was isolated and partially characterized for construction of the targeting vector. Fig. 1 A shows a restriction map of the mDP1 sequence, with approximate positioning of exons. Exons were not assigned numbers since the complete gene structure of DP remains undetermined. A 4.5-kb XhoI-AvrII fragment was targeted for deletion and replaced by the neomycin resistance gene, under the control of the phosphoglycerate kinase 1 (pgk1) promoter. This fragment begins in an exon and ends in an intron, and contains coding sequence for amino acids (aa) 281–474 of the ∼350-kD DP protein. In the targeting vector, a stop codon resides 30 bp 3′ from the encoded aa 281. We chose this strategy to ensure against expression of a truncated DP protein from the mutant allele: DP head segments less than 387 aa residues are not stable in transfected keratinocytes unless coupled to a reporter protein (Smith and Fuchs, 1998; unpublished data). We later verified this with NH2-terminal-specific DP antibodies and Northern blot analyses (see below).
Figure 1.

Targeting of the desmoplakin allele. (A) Stick diagrams illustrate restriction map of mDP1 genomic locus (WT), targeting vector used, and resulting genomic locus following successful homologous recombination with targeted construct (mutant allele). A 4.5-kb XhoI-AvrII fragment beginning at aa 281 (843 bp downstream from the ATG translation start codon) and ending in an intron 3′ from encoded aa 473 (1,419 bp downstream of the ATG) was targeted for removal and replaced with the pgk1-neomycin resistance gene (neo). The pgk1-Herpes thymidine kinase gene (TK) was inserted 5′ for negative selection. 5′ and 3′ probes used for Southern blot analyses are shown in bold; thin lines correspond to the predicted sizes of the restriction fragments that will hybridize to these probes. C, Cla 1; Nc, Nco 1; B, Bam H1; N, Nhe 1; R, EcoRI; X, XhoI; A, AvrII. Primer sets correspond to those used in PCR analysis for identifying the mutant and wt alleles. (B–D) Southern blot analysis. Restriction endonucleases used for digestions are indicated beneath each blot; sizes of hybridizing bands are indicated in kb. (B) Tail DNAs were from brown mice that were candidates for germline transmission of the targeted allele. These mice were derived from ES clones 11-10 and 71. (C) Tail DNAs from an established mouse line of 11-10, showing 5′ and 3′ analysis as well as verification that the recombination was a single event. Note: Both ES clones went germline; however our study focuses on clone 11-10, since clone 71 showed inappropriate recombination at the 3′ end of the locus, even though 5′ recombination and deletion of the 4.5-kb DP sequence was successful (data not shown). (D) E9.5 DNAs from heterozygous (+/−) matings. Only those embryos that scored positive in a PCR prescreen were subjected to Southern analysis. As shown, all of these were genotypically heterozygous (wt at right was a control). This was true for all matings >E6.5.
The targeting vector shown in Fig. 1 A was used to generate ES cells with a disrupted DP allele. Of a total of >1,000 clones analyzed, three scored positive for homologous recombination, as judged by Southern blot analysis. Clones 71 and 11-10 were then injected separately into C57BL/6 blastocysts to produce chimeric animals, which were mated with C57BL/6 females to generate heterozygous offspring. Southern blot analyses of tail DNAs confirmed the mutant genotype and the homologous recombination event (Fig. 1, B and C).
Offspring analyzed from matings of heterozygous DP (+/−) mice from 71 and 11-10 lines were either (+/−) or (+/+) in genotype and appeared indistinguishable from wild-type (wt) mice (Table I). The ratio of (+/−) to (+/+) mice was ∼2:1, suggesting that mice heterozygous for the DP mutant allele develop normally. In support of these statistics, histological analysis of heterozygous newborn mice revealed no obvious defects.
Table I.
Summary of Phenotype and Genotype Analyses of Embryos from DP (+/−) Matings
| Age | Phenotype | Genotype* | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | Abnormal | Resorbed | +/+ | +/− | −/− | |||||||
| E3.5 | 55 | 0 | — | 10 | 19 | 8 | ||||||
| E5.5 | 43 | 0 | 0 | 4 | 10 | 3 | ||||||
| E6.0 | 151 | 35 | 5 | 6 | 12 | 5 | ||||||
| E6.5 | 82 | 10 | 15 | 6 | 10 | 5 | ||||||
| E7.5 | 42 | 1 | 10 | 4 | 5 | 0 | ||||||
| E9.5 | 31 | 0 | — | 8 | 11 | 0 | ||||||
| E12.5 | 9 | 0 | — | 4 | 5 | 0 | ||||||
| Post | 350 | 0 | — | 93 | 184 | 0 | ||||||
Comparable phenotypes were found with either 129/C57BI/6J or 129/Black/CD-1 backgrounds.
Not all embryos were genotyped.
The data also suggested that (−/−) embryos die before birth. In investigating (−/−) embryonic lethality further, we learned that heterozygous (+/−) matings produced no (−/−) embryos at any stage after gastrulation (Fig. 1 D, examples shown from E9.5). These data indicated that targeting of both DP alleles gave rise to early embryonic lethality in mice.
Mutant E3.5 DP Embryos Can Undergo Blastocoel Cavity Formation
DP is first expressed in the trophectoderm of developing wt E3.5 embryos, concomitant with blastocoel cavity formation, and it has been suggested that DP may be essential for the trophectoderm to withstand the mechanical stress involved in blastocoel cavity formation (Fleming et al., 1991; Collins et al., 1995). Therefore, in exploring the DP mutant phenotype in greater detail, we began by examining E3.5 blastocysts from heterozygous DP (+/−) matings.
In contrast to wt matings that produced E3.5 blastocysts that were unanimously positive for DP antibody (αDP) staining, heterozygous DP (+/−) matings produced a portion of E3.5 embryos that did not label with αDP (Fig. 2, compare frame A with B). These embryos also failed to stain with an antibody against the NH2-terminal portion of DP, whose encoded sequence exists in the targeted allele. Taken together, these data suggested that the αDP negative embryos were homozygous for the targeted DP allele, and that a truncated form of DP was not produced by the targeting event.
To confirm that a portion of the E3.5 blastocysts from heterozygous (+/−) DP matings were homozygous for the targeting event, we developed a PCR–Southern blot strategy for DNAs isolated from individual blastocysts of heterozygous matings. Primer sets and PCR resulted in fragments that were diagnostic for mutant and wt alleles, and that could be detected by Southern blot analysis. As shown in Fig. 2 C, (−/−) blastocyst DNAs were present in a portion of the embryos. Further genotyping showed that E3.5 embryos from heterozygous matings gave the expected Mendelian ratio with ∼25% homozygous for the DP mutation (Table I).
The most remarkable feature of the αDP negative E3.5 blastocysts was that they formed a trophectoderm and a blastocoel cavity (Fig. 2, frame B). Additionally, anti-E-cadherin (αEcad) antibody staining appeared normal and was located at cell–cell borders of both the inner cell mass and the trophectoderm of αDP-negative embryos. Thus, despite the correlation between DP expression and blastocoel cavity formation in wt embryos, DP did not appear to be required for this preimplantation morphologic process.
DP Mutant Embryos Progress to Early Postimplantation
Thus far, our observations suggested that DP mutant embryos die sometime between blastocyst formation and gastrulation. Since most studies on desmosome and adherens junction formation have focused either on the preimplantation embryo (Jackson et al., 1980; Douglas and King, 1990; Fleming et al., 1991; Collins et al., 1995) or on stages after gastrulation (Jackson et al., 1981), our first task was to examine desmoplakin and E-cadherin expression in early postimplantation embryos from wt matings. By E5.0, embryos have implanted and are separated from maternal decidua (Dec) by Reichert's membrane (Fig. 3 A; Kaufman, 1992). At this stage, the embryo consists of three basic tissue types: primitive ectoderm (Ect), primitive endoderm (PN; and its derivative, parietal endoderm, PE), and trophectodermally derived ectoplacental cone (EPC; and its derivative mural trophectoderm). Of these, only the ectoderm at the embryo base that opposes the EPC, will give rise to the mouse; remaining embryonic tissues contribute to formation of placenta, yolk sac, and chorion.
Figure 3.
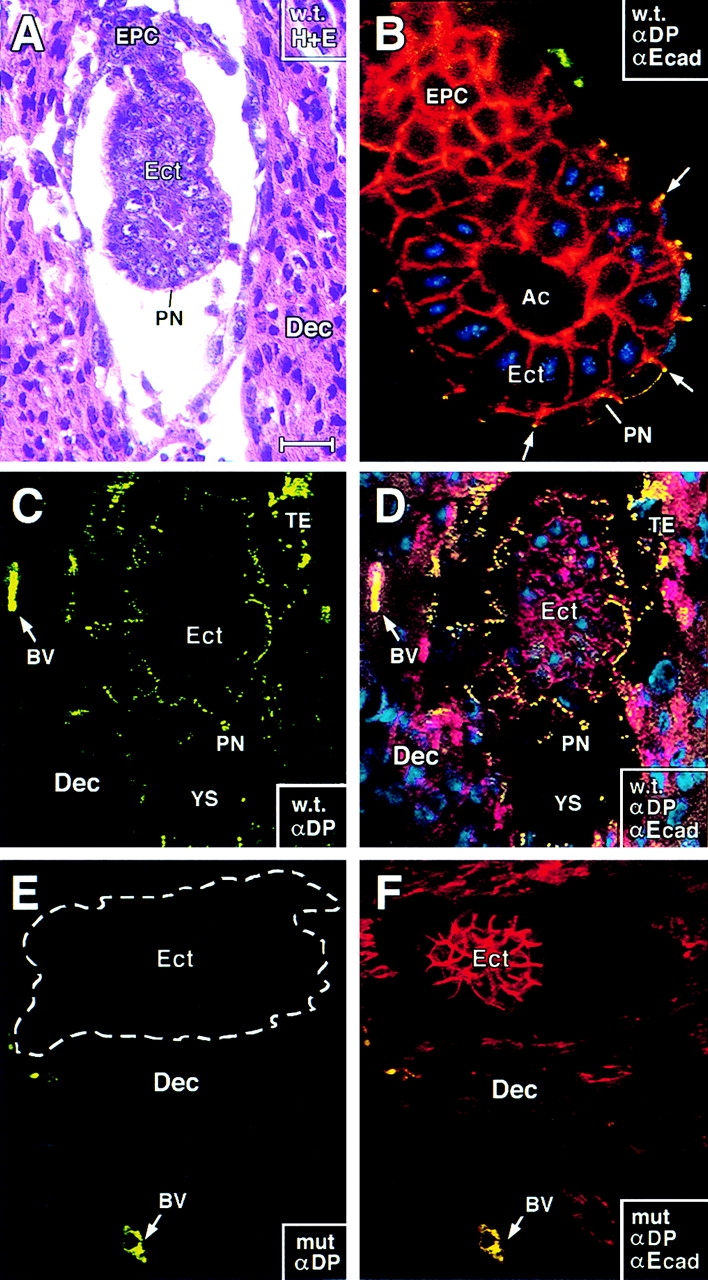
E5.0 DP mutant embryos form and appear similar in size and in anti-E-cadherin staining to wt embryos. E5.0 embryos were either dissected from their decidua and processed for whole mount immunofluorescence (B) or left intact and subjected to sectioning and either histology (hematoxylin and eosin staining; A) or immunofluorescence (C–F). Antibodies used for immunofluorescence are: anti-desmoplakin (αDP, green), anti-E-cadherin (αEcad, red), and DAPI (DNA, blue). Corner boxes denote wt (w.t.) or DP null (mut) and also where relevant, which of the immunofluorescence images are captured in each frame. All frames shown are of longitudinal views through the central region of the embryos. Ect, primitive ectoderm; PN, primitive endoderm; EPC, ectoplacental cone; TE, giant trophectoderm cells (visible only in undissected embryos); BV, maternally derived blood vessels in decidua; Ac, proamniotic cavity; YS, yolk sac. Note yellow dots in PN, EPC, and TE of wt embryos and in maternal BV of wt or mutant embryos, indicative of double-labeling with αDP and αEcad. Note: At E5.0, mutant and wt embryos are still similar in size; the section shown in E and F was slightly off center, giving it a smaller appearance than in C and D. Bar, (A, E, and F) 75 μm; (B) 20 μm; (C and D) 50 μm.
Confocal imaging of E5.0 embryos subjected to whole mount indirect immunofluorescence revealed αEcad staining at all cell–cell borders as expected (Fig. 3 B). This also confirmed antibody penetration and the validity of the whole mount technology. Staining with αDP was not seen in the columnar cells of Ect, but it was detected at cell–cell borders of the single layer of PN cells that encase the embryo (Fig. 3 B; yellow dots at surface). In frozen sections of intact E5 decidua, αDP-labeled cell borders not only of PN, but also of the Ect and the TE attached to Reichert's membrane on the other side of the developing yolk sac cavity (Figs. 3, C and D, YS). Overall, these staining patterns correlated well with the temporal appearance and location of nascent desmosomes (αDP) and adherens junctions (αEcad) in these cell-types (see below).
Approximately 25% of the E5.0 embryos from heterozygous matings failed to stain with αDP (Fig. 3, E). The only αDP staining seen in these embryos was that of the maternal blood vessels (BV) in the decidua, which served as a control to monitor αDP efficacy. These embryos stained normally with αEcad (Fig. 3, F). In addition, when dissected from their decidua so that embryos could be examined by size and shape, the embryos within the litters of heterozygous matings appeared morphologically similar (not shown). Taken together, these data indicated that DP mutant embryos can progress through implantation.
Desmoplakin (−/−) Embryos >E6.0 Are Significantly Smaller Than Normal
In contrast to E5.0 embryo batches from heterozygous matings, ∼25% of E6.0 embryos were significantly smaller than normal (Fig. 4 A). The larger embryos had a length of 500 ± 30 μm and a diameter of 200 ± 20 μm; the small embryos were of similar diameter, but with a length of only 150 ± 30 μm. Greater than 95% of small embryos harvested between E6.0 and E6.5 had DNA that scored positive by PCR for the mutant DP allele and negative for the wt allele (Fig. 5). Occasionally, heterozygous crossings gave rise to small E6 embryos that were genotypically wt; however, since such embryos were also produced from wt crossings, we attribute these rare situations to spontaneous abortions that occur naturally at low frequency (<5%). Generally, it was easy to distinguish the DP mutant embryos from these rare aborted embryos.
Figure 4.
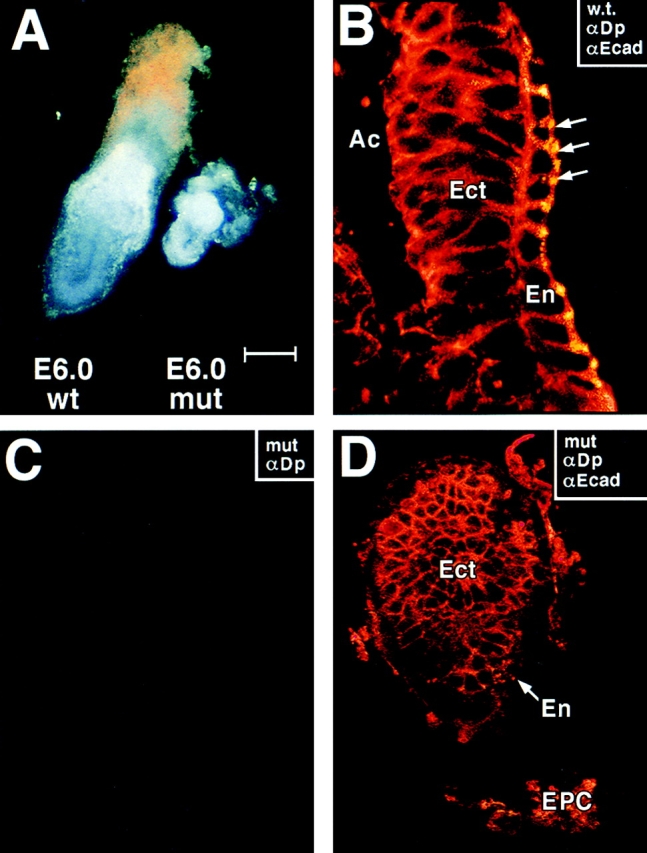
E6.0 mutant embryos that lack desmoplakin are abnormally small. DP (+/−) heterozygous matings were conducted, and E6.0 embryos were removed from pregnant females and dissected from their decidua. Embryos were either photographed directly (A) or analyzed by indirect immunofluorescence using the antibodies indicated in the boxes (all other panels). Immunofluorescence was visualized by whole mount confocal microscopy. Approximately 25% of the embryos were small for their age and these were negative for αDP staining. Anti-desmoplakin (αDP, green), anti-E-cadherin (αEcad, red), and DAPI (DNA, blue); Ac, proamniotic cavity; Ect, embryonic ectoderm; En, visceral endoderm; Epc, ectoplacental cone. Note yellow dots, reflective of αDP/αEcad double-labeling, in endoderm of wt (w.t.) E6.0 embryo (B, arrows). Bar, (A) 125 μm; (B) 25 μm; (C and D) 50 μm.
Figure 5.

Identification of desmoplakin null embryos from E6.5 heterozygous matings. E6.5 embryos of heterozygous DP matings were dissected from their decidua, and their DNAs were isolated, and subjected to PCR analysis. (A) Two different primer sets were used to delineate between wt and targeted allele, as described in the legend to Fig. 2. Note that the embryo DNA in the left-most lane scored negative for the wt allele. Shown are data from mouse line 11-10. (B) To verify that the 4.5-kb fragment targeted for removal was missing from the genome of (−/−) E6.5 embryos, primer set no. 3 was engineered from sequences only contained within the targeted fragment. Production of a 150-bp fragment (targeted exon) was indicative for the presence of a wt allele. A 500-bp actin fragment was amplified as a control in this experiment. Shown are data from mouse line 71.
In the wt E6.0 embryo, αDP staining was still restricted to the surface endoderm (En) of wt embryos, and was not present in developing ectoderm (Fig. 4, B). The genetically targeted E6.0 embryos were not only smaller, but were also negative for αDP staining with either NH2-terminal, rod, or COOH-terminal antibodies, providing a second means of distinguishing DP null from (+/−) or (+/+) wt embryos. That the targeted allele did not produce a stable transcript was also confirmed by Northern blot analysis of DP (+/−) mouse skin (not shown).
Despite the smaller size of these embryos, αEcad staining was still normal, and was present in both the developing endoderm and ectoderm (Fig. 4, D). Thus, despite the lack of cellular expansion, adherens junctions were still maintained, suggesting that the embryos had retained their cellular integrity. We did detect a reduction in adherens junctions and morphological signs of cell degeneration in older embryos (>E6.5). Therefore, we restricted our analysis to E6.0 and earlier.
The strong correlation between small embryo size at E6 and (−/−) genotype was interesting in that wt embryos normally undergo a dramatic increase in proliferation between E5.0 and E7.0 of embryogenesis. Based on our data, the E5.5 DP (−/−) embryos seemed unable to cope with the massive wave of proliferation and cellular reorganization necessary to form the egg cylinder, mesoderm, and amniotic and coelomic cavities (see also Kaufman, 1992). The disorganization appeared to prevent polarization of the embryo.
BrdU labeling of embryos (intraperitoneal injections 30 min before killing pregnant female mice) confirmed a failure of the mutant embryos to undergo cellular expansion, and TUNEL assays revealed that this failure was not due to an increase in apoptosis within mutant embryos (data not shown). The ability of E6.0 mutant embryos to incorporate BrdU and subsequent histological and EM analysis (see below) provided further evidence that the embryos were still alive at this age. Since expression of desmoplakin and desmosome formation only begin in wt embryonic ectoderm at >E7.0 (data not shown; see Jackson et al., 1981 b), it seemed most likely that this DP null-mediated defect was rooted in some fundamental failure occurring in extraembryonic tissues.
A Collapse of the Keratin Network Is an Early Defect in the Endoderm and Ectoplacental Cone Cells of the Desmoplakin Null Embryos
Since desmoplakin can directly associate with IFs in vitro and in tissue culture (Kouklis et al., 1994; Kowalczyk et al., 1997), we tested whether the keratin network might be affected in DP null mutant embryos. K8 and its partner K18 are the primary keratins expressed in E7 and E8 embryos, where they are seen in the visceral layer of the En, throughout the EPC, and to a lesser extent, in the extraembryonic and Ect (Jackson et al., 1981; Oshima, 1982). Therefore, we used antibodies directed against K8 (αK8) to detect the keratin network in whole E5.0 and E6.0 DP null and wt embryos isolated from their decidua and analyzed by confocal imaging.
In wt embryos, αK8 staining was observed in the En and EPC but not in Ect (Fig. 6, A and B). The filamentous nature of the keratin network present in the visceral endoderm was best visualized through higher magnification of αK8 images of the embryo surface (Fig. 6, C–E). Double labeling with αDP revealed desmosomes at the borders of cells that also stained positive for αK8 (D and E). From these views, it appeared that a cortical network of keratin existed at the endodermal cell periphery, and a network of keratin fiber bundles spanned the cytoplasm. This was true of both E5.0 and E6.0 embryos.
Figure 6.
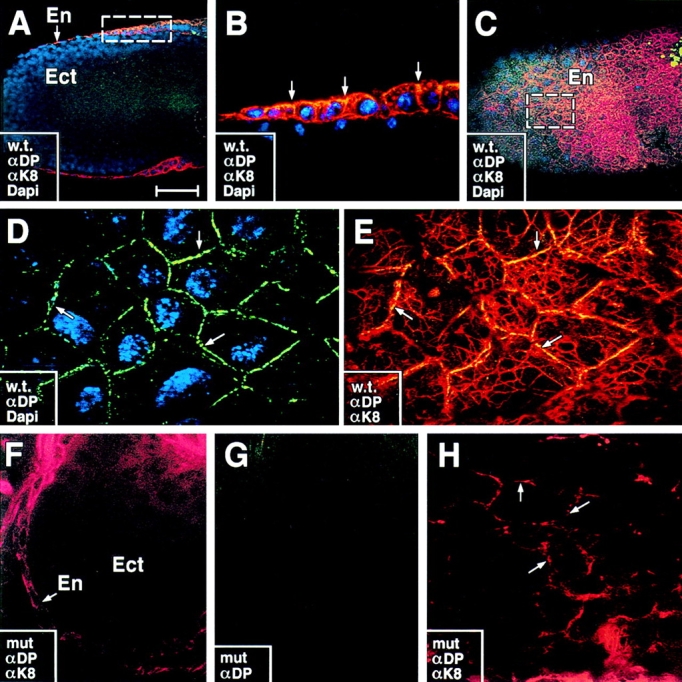
The keratin network is perturbed in DP mutant embryos. E6.0 embryos were taken from DP (+/−) heterozygous matings, dissected from their decidua and analyzed by whole mount indirect immunofluorescence microscopy using the antibodies indicated in the boxes. Embryos were visualized by confocal microscopy. Shown are either photographs of the embryo surface (C–E, G, and H) or cross sections (A, B, and F). αK8, TROMA-1, a K8-specific antibody (red); anti-desmoplakin (αDP, green), DAPI (DNA, blue). Wt embryos are shown in A–E; mutant embryos from the same litter are shown in F–H. En, visceral endoderm; Ect, embryonic ectoderm. Note: In wt embryos, αDP and αK8 staining is visible at intercellular borders and throughout the cytoplasm of En cells, but not Ect cells. Labeling at intercellular borders is best visualized in cross sections; arrows in B denote double-labeling (yellow) at junction sites where αDP also stains. Boxes in A and C denote areas that are presented at higher magnification in B and D, respectively. Labeling of intracellular keratin network is best visualized in surface views (e.g., E and H). Many keratin bundles were seen in close contact with DP positive sites at cell–cell borders (D and E, arrows). Surface area of mutant embryo in F and G is identical. Note absence of αDP staining and disorganization of keratin filament bundles in F–H. Arrows in H denote concentration of anti-K8 staining at cell borders; cytoplasmic staining was either collapsed (cells in lower right in H) or absent. Bar: (A and C) 75 μm; (B) 25 μm; (D and E) 10 μm; (F and G) 50 μm; (H) 10 μm.
In contrast to their wt siblings, E5.0 and E6.0 embryos that lacked αDP staining displayed marked abnormalities in the organization of keratin IF networks (Fig. 6, F–H). Instead of an extensive cytoskeleton of fibers, keratin was localized primarily at or near the cell periphery. Occasionally, aggregates of fiber bundles were detected within the cytoplasm (Fig. 6 H), but the network did not span the cytoplasm as it did in wt embryos. The appearance of IF defects by E5.0 provided evidence that biochemical perturbations in the DP null cells preceded overt morphological aberrations visible by E6.0.
Antibody Staining Reveals a Perturbation in Desmosomal Cadherin Localization in DP Null Embryos
To further pursue the structural consequences of desmoplakin ablation, we examined the localization of desmosomal cadherins. Coimmunofluorescence with αDP was conducted to confirm the identity of embryos as either wt (+/−) or (+/+) or mutant, and αK8 staining was used to mark the aberrant cells within mutant embryos. Antibodies against desmoglein 2 (αDsg2) strongly stained the ectoplacental cone and endoderm of wt embryos (Fig. 7, A and B). Markedly reduced staining was obtained in these cells within DP (−/−) embryos (Fig. 7, C). Similar reductions were seen in labeling patterns obtained with anti-desmocollin 2 (αDsc2; shown) and anti-plakophilin 2 (not shown; Fig. 7, D and E). Interestingly, the low level of α-desmosomal cadherin staining was still punctate in mutant embryos, suggesting that nascent desmosomes may be reduced in number and/or otherwise affected in DP mutant embryos. This alteration in desmosomal cadherin localization did not seem to affect E-cadherin localization, as judged by our earlier αEcad immunofluorescence studies.
Figure 7.

Reduction but not absence of anti-desmosomal cadherin staining in E6.0 DP mutant embryos. E6.0 embryos were taken from DP (+/−) heterozygous matings, dissected from their decidua and analyzed by whole mount indirect immunofluorescence microscopy using the antibodies indicated in the boxes. Embryos were visualized by confocal microscopy. Shown in A and B are sectional views, with the embryonic ectoderm in the center and endoderm at the surface. C–G are surface views, showing only the endodermal cells, positive for desmoplakin in the wt. Antibodies are: (A and B) anti-desmoglein 2 (αDsg2, red) ±anti-desmoplakin (αDP, green); (C) αDsg2 (green), αDP (blue, absent), αK8 (red); (D and E) anti-desmocollin2 (Dsc2, green). Note: αDsg2 and αDsc2 did not label the embryonic ectoderm, consistent with the similar restricted residence of desmoplakin and keratin. Embryos were identified as mutant (mut) or wt (w.t.) based upon αDP labeling, which when absent was accompanied by perturbations in the K8/K18 network. Arrows in C denote presence of a few punctate dots of αDsg2 staining in the mutant embryo. Bar: (A and B) 30 μm; (C–E) 20 μm.
Morphological Abnormalities in Desmoplakin Null Embryos
Since E6 mutant embryos could be distinguished from wt littermates by size, we used this age to dissect embryos from their decidua and process them for detailed morphological studies. Mutant embryos had an additional distinguishing feature: they were extremely fragile to mechanical dissection. Toluidine blue-stained sections of fixed and Epon-embedded sections revealed that upon careful handling, morphologies could be preserved. Approximately 75% of the mutant E6.0 embryos were very small (Fig. 8 A, inset). The larger wt embryos displayed a highly organized, columnar layer of ectoderm, surrounded by a flat layer of endodermal cells (Fig. 8, B and C). This was confirmed not only by our prior immunofluorescence studies with keratin and desmosomal markers, but also by additional studies using in situ hybridizations with a mouse antisense probe to HNF4α, an endodermal marker (data not shown; see Duncan et al., 1994; Levinson-Dushnik and Benvenisty, 1997). Additional ultrastructural evidence revealed that microvilli were formed on the visceral endoderm (see below). Finally, a proamniotic cavity developed, albeit poorly (Fig. 8 D). When taken together, these data further implied that the mutant embryos could at least partially establish epithelial polarity and progress through proamniotic cavity formation in the absence of desmoplakin.
Figure 8.
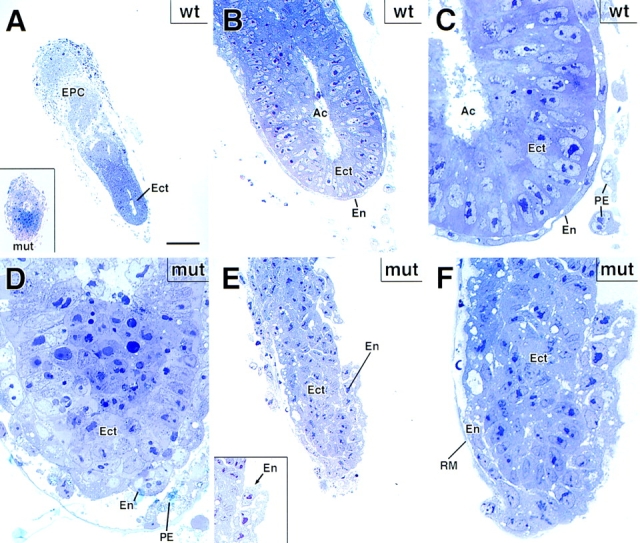
Mutant DP embryos are defective in egg cylinder elongation. Wt and mutant E6 embryos were processed for semithin sectioning, toluidine blue staining and microscopy. Shown are: (A–C) representative example of a wt E6 embryo, depicting a single layer of flattened visceral endoderm cells (En) encasing a single layer of columnar embryonic ectoderm (Ect). Inset to A shows an example of a small mutant E6 embryo, which was quite typical and accounted for ∼75% of the mutant DP E6 embryos within heterozygous matings. Ac, proamniotic cavity; EPC, ectoplacental cone; RM, Reichert's membrane; PE, parietal endoderm. D shows higher magnification of the mutant embryo in inset to A, revealing a very small proamniotic cavity (light blue area at top center of panel). E, E inset, and F provide example of a larger mutant DP embryo, which was still markedly smaller than its wt counterparts, but which did show some signs of egg cylinder formation, albeit aberrant. Note defects in cell–cell adhesion within the embryonic endoderm. These larger embryos most likely had additional secondary defects, and thus were not used for most analyses. Bar: (A and inset) 100 μm; (B and E inset) 35 μm; (C, D, and F) 20 μm; (E) 50 μm.
Those embryos that progressed further through development were larger and displayed major morphological abnormalities (embryo in Fig. 8, E and F). Even at the light microscopy level, defects in cell–cell adhesion were apparent. Adhesive defects were largely restricted to cells that normally express desmoplakin. In some regions, endodermal cells detached from each other as well as from ectoderm, leading to gaps in the endodermal layer (Fig. 8 E; inset shows higher magnification). In these larger embryos, the proamniotic cavity was still poorly developed, although the ectodermal cells still appeared adhesive (examples shown). In some embryos, cell–cell adhesion and cell organization was disrupted in the ectoderm, but presumably this effect was secondary and late-stage, as only the largest mutant embryos displayed this abnormality. When taken together with the difficulties we encountered in handling the larger mutant embryos, it appeared that the endoderm (and ectoplacental cone) of these mutant embryos was mechanically fragile due to a weakening of cell–cell junctions, and this may have subsequently led to secondary defects such as disorganization in the ectoderm.
Since ultrastructural analysis has not yet been reported for the early embryonic stages post-implantation, it was important to examine intercellular junctions of both wt and DP null embryos (Fig. 9). Desmosomes in wt E6.0 embryos could be distinguished from adherens junctions on the basis of their attachment to intermediate filaments (Fig. 9 A). Desmosomes were more abundant in the ectoplacental cone than elsewhere at this stage (Fig. 9, A and B). In the EPC, an occasional desmosome was quite large, as shown in the example in Fig. 9 B. These few large desmosomes in the EPC resembled those found in epidermis, where desmosomes are prominent (Fig. 9 C). Desmosomes were also found in developing endoderm (Fig. 9, D and D′), although they tended to be less abundant than in the EPC. Overall, the number and size of desmosomes were significantly smaller in both the E6.0 ectoplacental cone and endoderm than in postnatal epidermis. In addition, the number of IFs present and in association with smaller (nascent) desmosomes was typically less than in epidermis. As expected from our immunofluorescence studies, no desmosomes were detected in the embryonic ectoderm.
Figure 9.
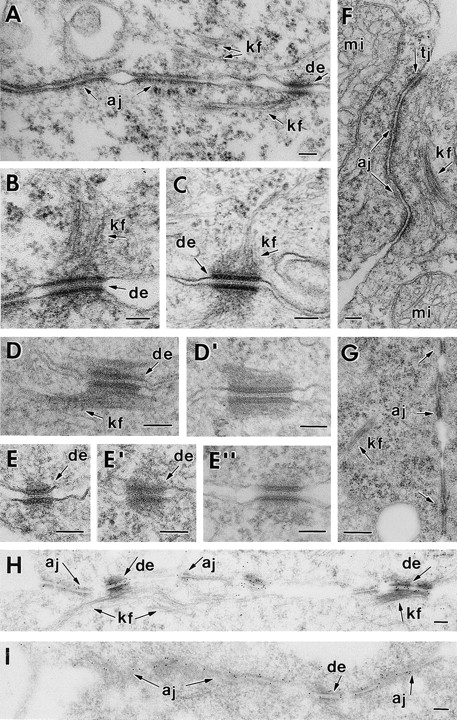
Mutant DP embryos display a dramatic reduction in desmosomes with loss of keratin filament attachment, but with no major changes in adherens or tight junctions. Wt and mutant E6.0 embryos, and newborn backskin epidermis from a wt embryo, were dissected from their decidua and processed for conventional electron microscopy or for immunoelectron microscopy using αPg antibodies. Shown are examples of cell–cell junctions present in the three cell types of the embryo. Note: No desmosomes were detected in wt or mutant embryonic ectoderm, consistent with immunofluorescence data. (from A to B) E6.0 wt ectoplacental cone, depicting desmosomes (de), with keratin filaments (kf) attached, and adherens junctions (aj); (C) desmosome in first suprabasal layer of newborn epidermis; these junctions were many fold more numerous in epidermis than in E6.0 embryos. (D and D′) Desmosomes from wt embryonic endoderm. (E and E′′) desmosomes in mutant embryonic endoderm (E and E′) and ectoplacental cone (E′′). Note: these examples were rare, particularly the one in E′′. Overall very few discernible desmosomes were observed. (F and G) Adherens junctions (aj) and tight junctions (tj) from DP mutant endoderm cells (F) and EPC (G). Note: Despite the paucity of desmosomes in these cells, these junctions appeared normal. (H and I) Immunoelectron microscopy of wt and mutant ectoplacental cone tissue, respectively, labeled with αPg antibodies. Mi, mitochondria; mv, microvilli at the embryo surface; gj, gap junction. Bars: (A–I) 100 nm.
In marked contrast to wt embryos, mutant E6.0 embryos displayed very few structures that resembled desmosomes, irrespective of the cell type or position within the embryo. This was true for both the ectoplacental cone and the endoderm. Upon quantitation of wt and mutant embryo sections, we estimated a >10× reduction in overall desmosome number and a >2× reduction in their size. This was the case whether comparing wt versus mutant EPC or comparing wt versus mutant endoderm. We did not take into consideration in our calculations the formation of cell–cell junctions that we could not definitely identify as desmosomes. The paucity of desmosomes could not be attributed to the arrest in embryonic development, as we did not detect a dramatic difference in either the overall number or the size of desmosomes per cell between E5.0 and E6.0 (not shown). Rather, the impressive reduction in desmosomes was consistent with our immunofluorescence data suggesting that the absence of DP results in a marked defect in desmosome assembly and/or stabilization.
In searching through many sections of DP mutant EPC and endoderm, we occasionally came across a structure that was recognizable as a desmosome (examples in Fig. 9, E and E′′). The largest desmosome we found was that shown in Fig. 9 E′′, stemming from E6.0 ectoplacental cone. While few in number, these examples enabled us to see that the overall structure of the plaque was largely normal at least in the larger desmosomes, although we could not identify discernible keratin filaments attached to these plaques. In contrast, most wt embryonic desmosomes displayed easily visible keratin filaments attached to their plaques (Fig. 9, A–D′). When taken together with our immunofluorescence data, we conclude that keratin filaments within the cells of DP mutant embryos were disorganized for two reasons: (a) the number of desmosomes was reduced dramatically, and (b) what desmosomes remained did not attach IFs efficiently.
Throughout E6.0 DP (−/−) embryos adherens junctions, tight junctions and gap junctions were found at cell–cell borders. Most importantly, in both the DP mutant embryonic endoderm and ectoplacental cone, the ultrastructure, size and numbers of these other junction types appeared to be largely normal (Fig. 9, F and G, respectively). In this way, E6.0 DP null embryonic cells seemed to differ from E12.5 plakoglobin null muscle cells, where adherens junctions were significantly larger and more prominent than normal, and where desmosomes were largely absent (Ruiz et al., 1996; Bierkamp et al., 1996). There are a number of possible reasons for this difference, ranging from functional differences in plakoglobin versus desmoplakin to tissue-specific differences in muscle versus embryonic endoderm. Further studies will be needed before this distinction is fully understood.
To explore whether the paucity of desmosomes might affect the overall distribution and/or partitioning of plakoglobin in the EPC and/or endoderm, we conducted immunoelectron microscopy using an anti-plakoglobin antibody. As shown in Fig. 9 H, anti-plakoglobin-labeled wt desmosomes more strongly than adherens junctions. In contrast, while anti-plakoglobin labeled the occasional desmosome remaining in DP (−/−) cells, this labeling was not markedly stronger than that of adherens junctions (Fig. 9 I). It seems likely that this difference in anti-plakoglobin labeling patterns between wt and mutant is a reflection of the smaller overall size of the desmosomes in the DP (−/−) cells.
Discussion
Two Major Functions for Desmoplakin
Desmoplakin Is Required for Linkage Between IFs and Desmosomes.
Numerous biochemical studies have suggested that members of the plakin family act as intermediate filament linker proteins, binding directly to IFs and to components of the hemidesmosome or desmosome. The function of two hemidesmosomal plakins, bullous pemphigoid antigen 1e (BPAG1e) and plectin, has recently been explored through the use of gene targeting. Indeed, ablation of these proteins in mice resulted in the severing of the connection between the IF network and the hemidesmosomal junctions to which these proteins localize (Guo et al., 1995; Andra et al., 1997).
The disruption of the keratin filament network and lack of discernible IFs attached to the few desmosomes remaining in our DP null embryos now enable us to include desmoplakin as a bona fide IF linker protein for desmosomes. This finding confirms and extends the previous in vitro data showing that desmoplakin's tail segment can bind to IFs and its head segment can associate with desmosomes (Stappenbeck and Green, 1992; Stappenbeck et al., 1993; Kouklis et al., 1994; Kowalcyzk et al., 1997; Smith and Fuchs, 1998). The results of the DP knockout further suggest that if plakophilins and plakoglobin also associate with desmosomal cadherins and IFs, as suggested by in vitro studies (Troyanovsky et al., 1993, 1996; Smith and Fuchs, 1998), these interactions are not sufficient to anchor IFs to desmosomes in the absence of desmoplakin.
Desmoplakin Is Required for Assembly and/or Stabilization of Desmosomes.
In the BPAG1e knockout, ablation severed the keratin cytoskeleton from hemidesmosomes, without affecting hemidesmosome number, structure or attachment to the basement membrane (Guo et al., 1995). The plectin knockout resulted in a reduction in hemidesmosomes, but this could be a consequence of other cytoskeletal perturbations arising from plectin's multifaceted ability not only to interact with IFs at hemidesmosomes, but also to interact with the cytoskeleton throughout the cell (Andra et al., 1998). Given the prevailing notion that desmoplakin's role is limited to desmosome-IF connections, it was surprising that the loss of desmoplakin resulted not only in disorganization of IFs, but also in dramatic reduction (∼10×) in the number of desmosomal-like junctions within the developing DP null embryo. Thus, in vivo, desmoplakin seems to perform a dual function in linking IFs and in providing stabilization to the junction to which it associates.
A priori, the substantial degree of sequence similarity, secondary structure and in vitro behavior of plakins might suggest that this new role of desmoplakin, not shared by BPAG1e, may be rooted in fundamental differences in the reliance of hemidesmosomes and desmosomes upon their cytoskeletal interactions. Certainly, the dependence of adherens junctions on the actin cytoskeleton has been strongly inferred from many studies (Hirano et al., 1992; McNeill et al., 1993; Kofron et al., 1997; Torres et al., 1997), and one might be tempted to argue that cadherins in general require cytoskeletal associations for stabilization. However, in striking contrast to our DP null embryos, ablation of K8 or K18 leads to a complete loss of the keratin network in early postimplantation embryos, with no discernible defects in desmosome formation, structure or stabilization (Baribault and Oshima, 1991; Baribault et al., 1993; Magin et al., 1998). Thus, it seems that desmoplakin's function must go beyond simply desmosome stabilization by virtue of cytoskeletal linkage.
Our studies provide compelling evidence that desmoplakin plays an integral role in the assembly and/or stabilization of desmosomes in a fashion that cannot be explained by the current literature derived from in vitro studies. Although a few in vitro studies have suggested that desmoplakin may be able to bind directly to desmosomal cadherins (Troyanovsky et al., 1993; Smith and Fuchs, 1998), most explorations have led primarily to a linear model of protein-protein interactions, where desmosomal cadherins cluster through association with plakoglobin and/or plakophilins, which then interact with desmoplakins, which then link to the IF cytoskeleton (for example see Kowalczyk et al., 1997 and references therein). Our findings here argue against a linear model, and suggest a more complex and multifunctional role for DP in desmosome assembly/stability. Whether desmoplakin performs this essential role for most types of desmosomes seems likely, but must await generation of conditional knockouts to selectively ablate the DP gene in specific somatic tissues.
The Role of Desmosomes in Early Embryogenesis
The essential role of adherens junctions in early embryogenesis has been well documented in a variety of vertebrate systems (Heasman et al., 1994a ,b; Larue et al., 1994; Haegel et al., 1995; Riethmacher et al., 1995). Adherens junctions are required for preimplantation development, blastocoel cavity formation and/or epithelial polarization, findings that are consistent with cell culture experiments suggesting that the conversion of unpolarized precursor cells to a polarized epithelium involves cadherin-mediated cell–cell junction assembly (Balcarova-Standler et al., 1984; Vega-Salas et al., 1987; Wollner et al., 1992).
The targeting of the desmoplakin gene has provided the first opportunity to directly explore the functions of desmosomes in early development. The timing of desmosome formation has led investigators to speculate that desmosomes might be required to withstand the mechanical stress induced by blastocoel cavity formation and fluid accumulation (Fleming et al., 1991, 1994). Our findings show that DP null embryos progress well beyond the blastocyst stage and indeed even make it through implantation. The explanation does not seem to reside in the carryover of residual maternal DP mRNAs, since αDP staining was not measurable in DP null blastocysts from heterozygous DP (+/−) matings. Rather, it seems more likely that the few and very small desmosomes that exist in wt trophectoderm at this stage of development (Jackson et al., 1980) are not essential for blastocoel cavity formation.
Similarly, we can say that the establishment of a bilayered embryo of endoderm and ectoderm occurred in the DP mutant embryos. Despite some cellular disorganization within these DP null embryos, the surface microvilli, tight junctions and adherens junctions were still formed, and the biochemistry (desmosomal, keratin, and HNF4α markers) distinguishing ectoderm and endoderm was still established. In addition, a proamniotic cavity still formed as DP null embryos progressed from E5 to E6, although it was significantly smaller than in control E6 embryos. Taken together, our findings suggested that DP null embryos proceed up to, but not beyond the stage at which the egg cylinder is elongated.
The defect in the elongation of the egg cylinder in DP null embryos was both striking and consistent and appeared to be directly due to abnormalities in desmosome assembly/stabilization. Perturbations in the keratin network could exacerbate the situation, although ablation of the network altogether is without affect to this process (Baribault et al., 1993; Magin et al., 1998). Egg cylinder formation involves a dramatic increase in cell proliferation accompanied by major changes in cellular organization that must occur while simultaneously maintaining very tight occluding barriers and desmosomes. This burst of proliferation is impaired in DP null embryos, and as the ectoderm increased in size, intercellular gaps and cell degeneration became prevalent in the surface endoderm. Thus, whereas nearly all extraembryonic tissues were affected at this stage, it seems most likely that the inhibition in egg cylinder formation originated at least in part from a failure of the surface endoderm to withstand mechanical and/or organizational stress.
Our findings that early postimplantation DP null embryos undergo a dramatic reduction in cell proliferation are interesting in light of recent keratinocyte culture studies revealing that disruption of intercellular junctions by a dominant negative E-cadherin mutant inhibits proliferation and promotes terminal differentiation (Zhu and Watt, 1996). An intriguing possibility is that cell signaling pathways might be perturbed in embryos that lack proper desmosomes. Whereas direct links between desmosomes and signal transduction pathways have not yet been established, stimulation of adherens junction formation is accompanied by a marked increase in tyrosine phosphorylation (Levenberg et al., 1998 and references therein), and it seems likely that control of all types of intercellular adhesion must in some fashion be linked to the mechanisms that govern cell cycle control, intercellular organization and differentiation.
Finally, it is also important to consider that eventually, the DP null embryonic ectoderm also became disorganized, a process that must be secondary, given that these cells did not possess desmosomes at this stage of development. One possibility is that alterations in the intercellular organization and/or signal transduction pathways within extraembryonic tissues led to defective intercellular signaling between visceral endoderm and embryonic ectoderm. Unequivocal evidence awaits the use of tetraploid morulae aggregation to generate embryos derived solely from DP (−/−) ES cells. However, such studies have been done recently for several genes encoding visceral endoderm-specific signaling factors (Duncan et al., 1997; Waldrip et al., 1998). Interestingly, the phenotypes of (−/−) embryos involving visceral endoderm signaling genes and our DP (−/−) embryos were quite similar, and showed ectodermal defects leading to a failure of the early mouse embryo to establish its polarity and/or body plan (Duncan et al., 1997; Waldrip et al., 1998). As future studies are conducted, the possible relation between desmosome assembly and signaling pathways between endoderm and ectoderm should become clearer.
In summary, despite their appearance as static structures, desmosomes must be dynamic, undergoing rapid changes and reorganization to accompany the molding of an expanding egg cylinder and yet still imparting to the developing endoderm an ability to withstand and sustain mechanical pressures imposed by an increase in growth and the formation of a proamniotic cavity. The failure of the egg cylinder and proamniotic cavity to form properly in DP null embryos appears to be a reflection of a functional importance of these structures during dynamic processes of tissue remodeling.
Differences Between Plakoglobin and Desmoplakin Null Embryos: What Can A Comparison Tell Us About Desmosome Structure/Assembly?
A number of key differences distinguish the recently described plakoglobin knockout embryos (Bierkamp et al., 1996; Ruiz et al., 1996) from our desmoplakin knockout embryos. Most obvious is the timing at which perturbations are detected: while the loss of plakoglobin was not felt until E12.5, the aftermath of desmoplakin's ablation resulted in a failure to progress to egg cylinder formation and mesoderm development. It has been surmised, but not demonstrated directly, that the late onset of plakoglobin null perturbations stems from redundancy, presumably by a plakophilin, at earlier stages (Ruiz et al., 1996; Bierkamp et al., 1996).
Another difference worth considering is that in the plakoglobin knockout, a new “mixed type” adhering junction arose with Dsg2 diffusely spread over the cell surface. In contrast, Dsg2, Dsc2, and plakophilin 2 still appeared to be clustered and colocalized in DP null embryos, although the amount of overall staining was low and the number of sites that labeled were significantly reduced. A final difference between Pg and DP null embryos seemed to be in adherens junctions, which were increased in size in Pg null muscle cells, but seemed largely unaffected in the DP mutant embryos. The differences in the two knockout phenotypes suggest that plakoglobin and desmoplakin may play different roles in desmosome formation and stabilization. Direct testing of this notion now awaits conditional knockout studies enabling the functions of these two proteins to be assessed in the same cell type.
Acknowledgments
We thank Dr. Andreas Nagy and Dr. Janet Rossant (University of Toronto) for the R1 strain of ES cells, and the following for antibodies, as described in the text: Dr. Werner W. Franke, Dr. Jackie Papkoff, Dr. Robert Oshima, Dr. Margaret Wheelock. We thank Tyler Jacks (Massachusetts Institute of Technology) for the method on BrdU labeling. Finally, we thank Chuck Wellek for his assistance in the preparation of the computer generated figures.
Abbreviations used in this paper
- aa
amino acids
- BV
blood vessels
- DAPI
4, 6-diamidino-2-phenylindole
- Dec
maternal decidua
- DP
desmoplakin
- Dsc
desmocollin
- Dsg
desmoglein
- Ect
primitive ectoderm
- En
embryonic endoderm
- EPC
ectoplacental cone
- ES
embryonic stem
- IF
intermediate filaments
- PN
parietal endoderm
- PN primitive endoderm
wt, wild-type
- YS
yolk sac
Footnotes
E. Fuchs is an Investigator of the Howard Hughes Medical Institute. G.I. Gallicano is a postdoctoral fellow funded by the National Institutes of Health.
Drs. Gallicano and Kouklis contributed equally to this manuscript.
References
- Adams CL, Nelson WJ, Smith SJ. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell–cell adhesion. J Cell Biol. 1996;135:1899–1911. doi: 10.1083/jcb.135.6.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen E, Yu QC, Fuchs E. Mice expressing a mutant desmosomal cadherin exhibit abnormalities in desmosomes, proliferation, and epidermal differentiation. J Cell Biol. 1996;133:1367–1382. doi: 10.1083/jcb.133.6.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andra K, Lassmann H, Bittner R, Shorny S, Fassler R, Propst F, Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andra K, Nikolic B, Stocher M, Drenckhahn D, Wiche G. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev. 1998;12:3442–3451. doi: 10.1101/gad.12.21.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angres B, Barth A, Nelson WJ. Mechanism for transition from initial to stable cell–cell adhesion: kinetic analysis of E-cadherin-mediated adhesion using a quantitative adhesion assay. J Cell Biol. 1996;134:549–557. doi: 10.1083/jcb.134.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcarova-Stander J, Pfeiffer SE, Fuller SD, Simons K. Development of cell surface polarity in the epithelial MadinDarby canine kidney (MDCK) cell line. EMBO (Eur Mol Biol Organ) J. 1984;3:2687–2694. doi: 10.1002/j.1460-2075.1984.tb02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribault H, Price J, Miyai K, Oshima RG. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 1993;7:1191–1201. doi: 10.1101/gad.7.7a.1191. [DOI] [PubMed] [Google Scholar]
- Baribault H, Oshima RG. Polarized and functional epithelia can form after the targeted inactivation of both mouse keratin 8 alleles. J Cell Biol. 1991;115:1675–1684. doi: 10.1083/jcb.115.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AI, Nathke IS, Nelson WJ. Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 1996;180:780–785. doi: 10.1006/dbio.1996.0346. [DOI] [PubMed] [Google Scholar]
- Bornslaeger EA, Corcoran CM, Stappenbeck TS, Green KJ. Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell–cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J Cell Biol. 1996;134:985–1001. doi: 10.1083/jcb.134.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JE, Lorimer JE, Garrod DR, Pidsley SC, Buxton RS, Fleming TP. Regulation of desmocollin transcription in mouse preimplantation embryo. Development. 1995;121:743–753. doi: 10.1242/dev.121.3.743. [DOI] [PubMed] [Google Scholar]
- Cowin, P., H.-P. Kapprell, W.W. Franke, J. Tamkun, and R.O. Hynes. 1986. Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 1063–1073. [DOI] [PubMed]
- Douglas GC, King BF. Differentiation of human trophoblast cells in vitro as revealed by immunocytochemical staining of desmoplakin and nuclei. J Cell Sci. 1990;96:131–141. doi: 10.1242/jcs.96.1.131. [DOI] [PubMed] [Google Scholar]
- Ducibella T, Albertini DF, Anderson E, Biggers JD. The preimplantation mammalian embryo: characterization of intercellular junctions and their appearance during development. Dev Biol. 1975;45:231–250. doi: 10.1016/0012-1606(75)90063-9. [DOI] [PubMed] [Google Scholar]
- Duncan SA, Manova K, Chen WS, Hoodless P, Weinstein DC, Bachvarova RF, Darnell JE., Jr Expression of transcription factor HNF4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: HNF4 is a marker for primary endoderm in the implanting blastocyst. Proc Natl Acad Sci USA. 1994;91:7598–7602. doi: 10.1073/pnas.91.16.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SA, Nagy A, Chan W. Murine gastrulation requires HNF4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf4(−/−) embryos. Development. 1997;124:279–287. doi: 10.1242/dev.124.2.279. [DOI] [PubMed] [Google Scholar]
- Fleming TP, Butler L, Lei X, Collins J, Javed Q, Sheth B, Stoddart N, Wild A, Hay M. Molecular maturation of cell adhesion systems during mouse early development. Histochemistry. 1994;101:1–7. doi: 10.1007/BF00315824. [DOI] [PubMed] [Google Scholar]
- Fleming TP, Garrod DR, Elsmore AJ. Desmosome biogenesis in the mouse preimplantation embryo. Development. 1991;112:527–539. doi: 10.1242/dev.112.2.527. [DOI] [PubMed] [Google Scholar]
- Garrod DR. Desmosomes and hemidesmosomes. Curr Opin Cell Biol. 1993;5:30–40. doi: 10.1016/s0955-0674(05)80005-5. [DOI] [PubMed] [Google Scholar]
- Green KJ, Virata ML, Elgart GW, Stanley JR, Parry D. Comparative structural analysis of desmoplakin, bullous pemphigoid antigen and plectin: members of a new gene family involved in organization of intermediate filaments. Int J Biol Macromol. 1992;14:145–153. doi: 10.1016/s0141-8130(05)80004-2. [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu Q-C, Wollmann R, Perman B, Fuchs E. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified squamous epithelia and severe neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Heasman J, Ginsberg D, Geiger B, Goldstone K, Pratt T, Yoshida-Noro C, Wylie C. A functional test for maternally inherited cadherin in Xenopusshows its importance in cell adhesion at the blastula stage. Development. 1994a;120:49–57. doi: 10.1242/dev.120.1.49. [DOI] [PubMed] [Google Scholar]
- Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, Kintner C, Noro CY, Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopusembryos. Cell. 1994b;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeichi M. Identification of a neural alpha-catenin as a key regulator of cadherin function and multicellular organization. Cell. 1992;70:293–301. doi: 10.1016/0092-8674(92)90103-j. [DOI] [PubMed] [Google Scholar]
- Jackson BW, Grund C, Winter S, Franke WW, Illmensee K. Formation of cytoskeletal elements during mouse embryogenesis. II. Epithelial differentiation and intermediate-sized filaments in early postimplantation embryos. Differentiation. 1981;20:203–216. doi: 10.1111/j.1432-0436.1981.tb01177.x. [DOI] [PubMed] [Google Scholar]
- Jackson BW, Grund C, Schmid E, Burki K, Franke WW, Illmensee K. Formation of cytoskeletal elements during mouse embryogenesis. Intermediate filaments of the cytokeratin type and desmosomes in preimplantation embryos. Differentiation. 1980;17:161–179. doi: 10.1111/j.1432-0436.1980.tb01093.x. [DOI] [PubMed] [Google Scholar]
- Kaufman, M.H. 1992. The Atlas of Mouse Development. Academy Press, San Diego, CA. pp. 2–8.
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PJ, Franke WW. Desmosomal cadherins: another growing multigene family of adhesion molecules. Curr Opin Cell Biol. 1994;6:682–687. doi: 10.1016/0955-0674(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Kofron M, Spagnuolo A, Klymkowsky M, Wylie C, Heasman J. The roles of maternal alpha-catenin and plakoglobin in the early Xenopusembryo. Development. 1997;124:1553–1560. doi: 10.1242/dev.124.8.1553. [DOI] [PubMed] [Google Scholar]
- Kouklis P, Hutton E, Fuchs E. Making the connection: keratin intermediate filaments and desmosomes proteins. J Cell Biol. 1994;127:1049–1060. doi: 10.1083/jcb.127.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, Denning MF, Green KJ. The NH2-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol. 1997;139:773–784. doi: 10.1083/jcb.139.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci USA. 1994;91:8263–8267. doi: 10.1073/pnas.91.17.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenberg S, Katz BZ, Yamada KM, Geiger B. Long-range and selective autoregulation of cell–cell or cell-matrix adhesions by cadherin or integrin ligands. J Cell Sci. 1998;111:347–357. doi: 10.1242/jcs.111.3.347. [DOI] [PubMed] [Google Scholar]
- Levinson-Dushnik M, Benvenisty N. Involvement of hepatocyte nuclear factor 3 in endoderm differentiation of embryonic stem cells. Mol Cell Biol. 1997;17:3817–3822. doi: 10.1128/mcb.17.7.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magin TM, Schroder R, Leitgeb S, Wanninger F, Zatloukal K, Grund C, Melton DW. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J Cell Biol. 1998;140:1441–1451. doi: 10.1083/jcb.140.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs JA, Nelson WJ. Cadherin cell adhesion molecules in differentiation and embryogenesis. Int Rev Cytol. 1996;165:159–205. doi: 10.1016/s0074-7696(08)62222-6. [DOI] [PubMed] [Google Scholar]
- McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RA. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet. 1997;17:240–244. doi: 10.1038/ng1097-240. [DOI] [PubMed] [Google Scholar]
- McNeill H, Ryan TA, Smith SJ, Nelson WJ. Spatial and temporal dissection of immediate and early events following cadherin-mediated epithelial cell adhesion. J Cell Biol. 1993;120:1217–1226. doi: 10.1083/jcb.120.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol. 1996;135:1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Oshima RG. Developmental expression of murine extra-embryonic endodermal cytoskeletal proteins. J Biol Chem. 1982;257:3414–3421. [PubMed] [Google Scholar]
- Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E- cadherin gene results in defective preimplantation development. Proc Natl Acad Sci USA. 1995;92:855–859. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhrberg C, Watt FM. The plakin family: versatile organizers of cytoskeletal architecture. Curr Opin Genet Dev. 1997;7:392–397. doi: 10.1016/s0959-437x(97)80154-2. [DOI] [PubMed] [Google Scholar]
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Langbein L, Rode M, Pratzel S, Zimbelmann R, Franke WW. Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque component. Cell Tissue Res. 1997;290:481–499. doi: 10.1007/s004410050956. [DOI] [PubMed] [Google Scholar]
- Smith E, Fuchs E. Defining desmoplakin's interactions with desmosomes. J Cell Biol. 1998;141:1229–1241. doi: 10.1083/jcb.141.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, Bornslaeger EA, Corcoran CM, Luu HH, Virata ML, Green KJ. Functional analysis of desmoplakin domains: specification of the interaction with keratin versus vimentin intermediate filament networks. J Cell Biol. 1993;123:691–705. doi: 10.1083/jcb.123.3.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, Green KJ. The desmoplakin carboxyl terminus coaligns with and specifically disrupts intermediate filament networks when expressed in cultured cells. J Cell Biol. 1992;116:1197–1209. doi: 10.1083/jcb.116.5.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Stoykova A, Huber O, Chowdhury K, Bonaldo P, Mansouri A, Butz S, Kemler R, Gruss P. An alpha-E-catenin gene trap mutation defines its function in preimplantation development. Proc Natl Acad Sci USA. 1997;94:901–906. doi: 10.1073/pnas.94.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky RB, Chitaev NA, Troyanovsky SM. Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci. 1996;109:3069–3078. doi: 10.1242/jcs.109.13.3069. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM, Eshkind LG, Troyanovsky RB, Leube RE, Franke WW. Contributions of cytoplasmic domains of desmosomal cadherins to desmosome assembly and intermediate filament anchorage. Cell. 1993;72:561–574. doi: 10.1016/0092-8674(93)90075-2. [DOI] [PubMed] [Google Scholar]
- Vega-Salas DE, Salas PJ, Gundersen D, Rodriguez-Boulan E. Formation of the apical pole of epithelial (MadinDarby canine kidney) cells: polarity of an apical protein is independent of tight junctions while segregation of a basolateral marker requires cell–cell interactions. J Cell Biol. 1987;104:905–916. doi: 10.1083/jcb.104.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ. Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell. 1998;92:797–808. doi: 10.1016/s0092-8674(00)81407-5. [DOI] [PubMed] [Google Scholar]
- Wollner DA, Krzeminski KA, Nelson WJ. Remodeling the cell surface distribution of membrane proteins during the development of epithelial cell polarity. J Cell Biol. 1992;116:889–899. doi: 10.1083/jcb.116.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7:308–315. doi: 10.1016/s0960-9822(06)00154-0. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM. Expression of a dominant negative cadherin mutant inhibits proliferation and stimulates terminal differentiation of human epidermal keratinocytes. J Cell Sci. 1996;109:3013–3023. doi: 10.1242/jcs.109.13.3013. [DOI] [PubMed] [Google Scholar]