

Abstract
Unlike wild-type mouse melanocytes, where melanosomes are concentrated in dendrites and dendritic tips, melanosomes in dilute (myosin Va−) melanocytes are concentrated in the cell center. Here we sought to define the role that myosin Va plays in melanosome transport and distribution. Actin filaments that comprise a cortical shell running the length of the dendrite were found to exhibit a random orientation, suggesting that myosin Va could drive the outward spreading of melanosomes by catalyzing random walks. In contrast to this mechanism, time lapse video microscopy revealed that melanosomes undergo rapid (∼1.5 μm/s) microtubule-dependent movements to the periphery and back again. This bidirectional traffic occurs in both wild-type and dilute melanocytes, but it is more obvious in dilute melanocytes because the only melanosomes in their periphery are those undergoing this movement. While providing an efficient means to transport melanosomes to the periphery, this component does not by itself result in their net accumulation there. These observations, together with previous studies showing extensive colocalization of myosin Va and melanosomes in the actin-rich periphery, suggest a mechanism in which a myosin Va–dependent interaction of melanosomes with F-actin in the periphery prevents these organelles from returning on microtubules to the cell center, causing their distal accumulation. This “capture” model is supported by the demonstration that (a) expression of the myosin Va tail domain within wild-type cells creates a dilute-like phenotype via a process involving initial colocalization of tail domains with melanosomes in the periphery, followed by an ∼120-min, microtubule-based redistribution of melanosomes to the cell center; (b) microtubule-dependent melanosome movement appears to be damped by myosin Va; (c) intermittent, microtubule-independent, ∼0.14 μm/s melanosome movements are seen only in wild-type melanocytes; and (d) these movements do not drive obvious spreading of melanosomes over 90 min. We conclude that long-range, bidirectional, microtubule-dependent melanosome movements, coupled with actomyosin Va–dependent capture of melanosomes in the periphery, is the predominant mechanism responsible for the centrifugal transport and peripheral accumulation of melanosomes in mouse melanocytes. This mechanism represents an alternative to straightforward transport models when interpreting other myosin V mutant phenotypes.
Keywords: myosin V, dilute, melanosomes, microtubule motors, organelle motility
Mice with mutations at the dilute locus (Jenkins et al., 1981; Strobel et al., 1990), which encodes the heavy chain of myosin Va (Mercer et al., 1991), exhibit a reduction or “dilution” in the intensity of hair coloration (for reviews see Silvers, 1979; Jackson, 1994). The pigments (melanins) that are responsible for the coloration of mammalian hair and skin are produced within melanocytes, which are dendritic cells of ectodermal origin that reside near the base of the hair follicle and within the bottom-most layer of epidermis (for reviews see Hearing and King, 1993; Jimbow et al., 1993). These cells are responsible for delivering pigment, via their extensive dendritic arbor, to numerous keratinocytes, the principal cell type of hair and skin, as these keratinocytes migrate into the hair shaft proper and to the skin surface. The synthesis of melanins occurs entirely within a specialized organelle of the melanocyte, the melanosome. These organelles form in the central cytoplasm and migrate out dendritic extensions to their sites of intercellular transfer at dendritic tips. Fully melanized melanosomes, which appear completely black in both light and electron micrographs, are the end product that melanocytes provide keratinocytes. Key to the biology of mammalian pigmentation, therefore, is that melanosomes are concentrated at the distal ends of the melanocyte's dendrites, and that the melanosomes accumulated at these sites are transferred to keratinocytes. This mechanism provides an efficient way to disperse the pigment so that it becomes visible in hair and skin. Mammalian pigmentation differs significantly from pigmentation in fish and amphibians, where rapid, highly coordinated aggregations and dispersions of pigment granules occurring entirely within the melanocyte itself lead to rapid changes in surface color (Haimo and Thaler, 1994).
Recently, we and others have shown that dilute melanocytes exhibit an abnormal intracellular distribution of melanosomes (Koyama and Takeuchi, 1981; Provance et al., 1996; Wei et al., 1997). Unlike wild-type mouse melanocytes, in which melanosomes are concentrated in dendrites and dendritic tips, melanosomes in dilute melanocytes, both in vitro and in situ, are highly concentrated in the center of the cell. While this shift in melanosome distribution is dramatic, it is not absolute, since some melanosomes are usually seen in dendrites and dendritic tips even in melanocytes from mice homozygous for a true null allele at dilute (d l20J) (Wei et al., 1997). Nevertheless, this phenotype, together with the facts that myosin Va colocalizes with melanosomes, resides on the limiting membrane of the melanosome, and is associated with purified melanosomes (Provance et al., 1996; Nascimento et al., 1997; Wu et al., 1997), supports the idea that the long-range centrifugal transport of melanosomes in mammalian melanocytes is a myosin Va–dependent process. This conclusion is also consistent with the fact that a microtubule-dependent component of melanosome translocation in mammalian melanocytes is generally thought not to exist (Hearing and King, 1993; Jimbow et al., 1993; Rogers and Gelfand, 1998).
In this paper, we sought to further define the role that myosin Va plays in the centrifugal transport and peripheral accumulation of melanosomes in mouse melanocytes. In contrast to our previous thinking, the experiments we performed here—which include the direct observation and quantitation of melanosome dynamics in wild-type and dilute melanocytes, the expression of dominant-negative myosin Va–tail domain fusion proteins in wild-type melanocytes, the determination of actin filament distribution and orientation, and the analysis of melanosome spreading in microtubule-depleted cells—implicate a cooperative mechanism involving long-range, bidirectional, microtubule-dependent melanosome transport, coupled with myosin Va–dependent capture of melanosomes in the actin-rich periphery, as the predominant mechanism responsible for the centrifugal transport and peripheral accumulation of melanosomes. These results, like the recent work in fish melanophores (Rodionov et al., 1998) showing that an intact actin cytoskeleton is required for the centrifugal, microtubule-dependent component of pigment granule motility to generate even granule distribution, and in frog melanophores (Rogers et al., 1997; Rogers and Gelfand, 1998) showing that pigment granules isolated from these cells move on both actin and microtubules, add to the growing evidence that microtubule and actin filament systems cooperate in organelle motility and distribution (Bearer et al., 1993; Evans et al., 1997; Fath et al., 1994; Kuznetsov et al., 1992; Langford, 1995; Lillie and Brown, 1992; Morris and Hollenbeck, 1995). Unlike melanophores, however, the cooperation occurring in mouse melanocytes results in the polarized distribution of the melanosome and operates in the context of ongoing bidirectional, microtubule-dependent melanosome transport, which may be closer to the situation for other organelles in other differentiated cell types. Moreover, because the nature of our study directly implicates myosin Va in this cooperative process, we suggest that our capture model may provide further insight into the mechanistic basis of other myosin V mutant phenotypes, especially those involving polarized organelle distribution.
Materials and Methods
Primary and Immortal Mouse Melanocyte Cultures
Control primary melanocytes were prepared from the skin of C57BL/6J (D/D) pups, while dilute melanocytes were prepared from the skin of C57BL/6J pups that were homozygous for the dilute lethal allele d l20J, which represents a true null allele at dilute (see Strobel et al., 1990 and Fig. 1 in Wu et al., 1997). These latter pups were obtained by crossing heterozygous parents of the genotype d l20J/d v se (short ear [se]1) (a generous gift of Dr. Neal Copeland and Dr. Nancy Jenkins, NCI, NIH), and were distinguished from their d l20J/d v se and d v se/d v se littermates based on Western blot analysis, as described previously (Wei et al., 1997; Wu et al., 1997). Only homozygous d l20J pups are devoid of detectable myosin Va heavy chain protein (Wu et al., 1997). Cultures of primary melanocytes were prepared from the epidermis of individual mice by a modification of the method of Tamura et al. (1987), as described previously (Wei et al., 1997), and were grown on gelatin-coated 24.5-mm round No. 1 1/2 coverslips in Ham's F-10 medium supplemented with 10% horse serum, 2% fetal calf serum, 1% (vol/vol) penn strep, 0.1 μM dbcAMP, and 85 nM tetradecanoyl phorbol acetate (TPA) (No. P8139; Sigma Chemical Co., St. Louis, MO). The nontransformed but immortal mouse melanocyte cell line melan-a (D/D) was a generous gift of Dr. Dorothy Bennett (St. George's Hospital Medical School, London, UK) and was maintained as described previously (Wu et al., 1997).
Figure 1.
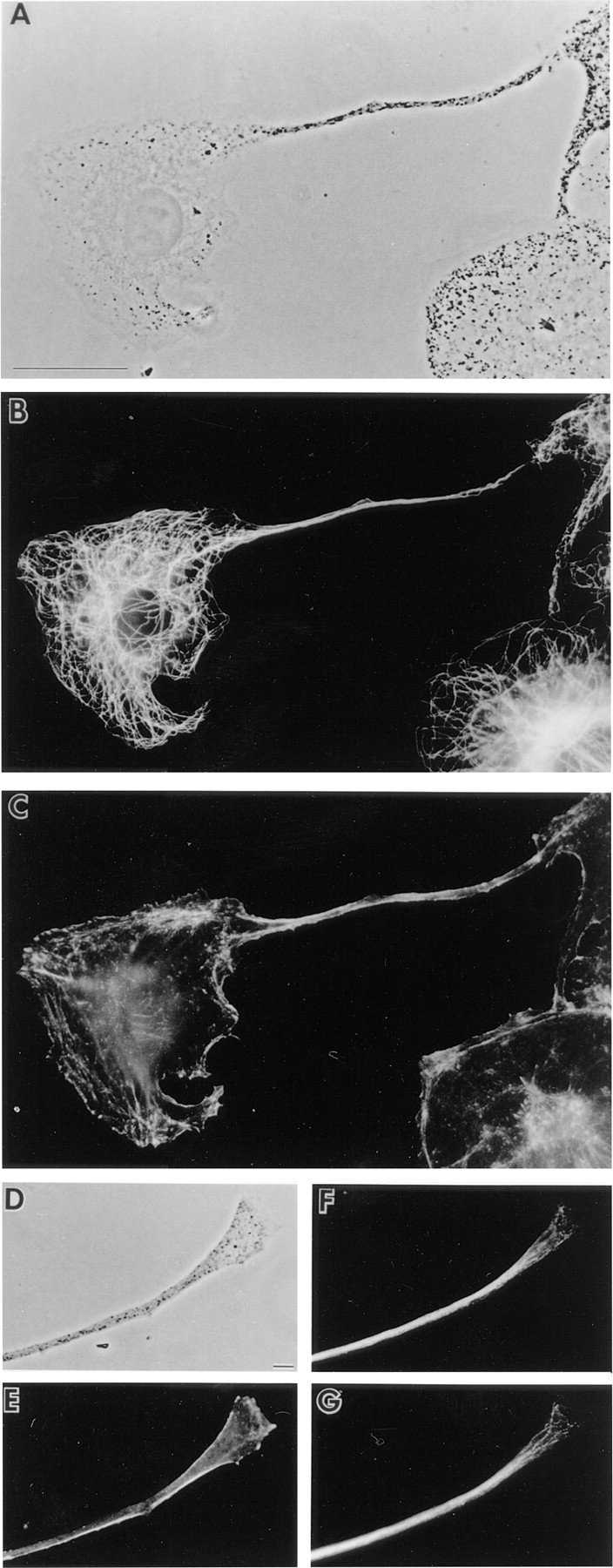
Organization of microtubules and F-actin in melanocytes. Shown are phase contrast images of a melan-a melanocyte exhibiting a single, long dendritic extension (A) and the distal portion of another dendrite (D), and the distributions of microtubules (B, F, and G; F and G are two different focal planes) and F-actin (C and E) within them. Bars: (A) 10 μm; (B) 2 μm.
Live Cell Imaging
The dynamics of melanosomes in melanocytes was monitored using a Zeiss Axiovert 135 microscope (Carl Zeiss, Thornwood, NY) equipped with differential interference contrast (DIC) optics, a Plan Apochromat 100× 1.25 NA oil immersion lens, and illumination provided by a halogen lamp through a green interference filter. Bright field or DIC images were recorded using a Newvicon camera (model C2400 Newvicon; Hamamatsu, Hamamatsu City, Japan) and were captured and stored at a rate of one frame (1/30 s) every second using an optical disc recorder (model TQ303IF; Panasonic) controlled through a PC. Background subtraction and contrast enhancement were performed in real time before recording images using an Argus-10 image processor (Hamamatsu).
Cells grown on 24.5-mm round coverslips were introduced into a Dvorak-Stotler perfusion chamber (Nicholson Precision Instruments, Gaithersburg, MD) that was fitted with a custom-made elliptical chamber spacer (∼35 μl internal volume), and with a stainless steel, two-way inlet manifold to allow for changes in the perfusion media. Perfusion medium was identical to the tissue culture medium used for culturing the cells and was delivered to the chamber by gravity feed at a flow rate of 0.2 ml/min, which allowed complete exchange of the chamber solution every ∼10 s. Perfusion medium was warmed to 37°C and equilibrated with humidified 5% CO2 using specially designed perfusion bottles (gift of Dr. Ken Springs, NHLBI, NIH). Solenoid-actuated pinch valves (Custom Electronics, Gaithersburg, MD) were used to switch between perfusion media of different composition. To maintain the chamber at 37°C, the entire microscope was placed inside a custom made enclosure connected to a forced air heater (Air Therm; World Precision, Inc., Sarasota, FL). The stage temperature was monitored using a temperature probe (model YSI 400; Coparma) placed adjacent to the perfusion chamber. Using this arrangement, the cells could be maintained for many hours in standard culture medium at 37°C. To depolymerize microtubules, cells were perfused at 37°C with medium containing 9.6 μg/ml (16 μM) nocodazole (No. M1404; Sigma Chemical Co.) diluted from a freshly prepared 5 mg/ml stock dissolved in DMSO. To drive depolymerization further, cells that had been perfused with nocodazole at 37°C for at least 10 min were placed on ice for 30 min in the presence of nocodazole, and then perfused at 37°C in the presence of nocodazole.
DNA Constructs and Cell Transfection
To prepare plasmid EGFP-MC-ST, a cDNA clone encoding the COOH-terminal ∼75% of mouse myosin Va was obtained from a B16 mouse melanoma cDNA library (gift of Dr. Murray Brilliant, University of Pennsylvania, Philadelphia, PA) and digested with SstI and EcoRI, and the ∼2.2-kb SstI/EcoRI fragment that begins at residue 1258 (numbering according to Mercer et al., 1991) and ends at the first poly A+ site was cloned into plasmid EGFP Cl (No. 6094-1; CLONTECH Labs, Palo Alto, CA). The fusion protein generated by this plasmid contains the COOH-terminal 619 amino acids of the predominant spliced isoform found in melanocytes (including both melanocyte-specific exons—exons D [27 residues] and F [25 residues]—identified in Seperack et al., 1995) fused to the COOH terminus of an enhanced version of green fluorescent protein (EGFP; 238 amino acids). To prepare plasmid FLAG-MC-LT, the cDNA clone described above was digested with Eco47III and converted to an EcoRI site by linker addition, and the ∼2.7-kb EcoRI fragment that begins at residue 1091 and ends at the first poly A+ site was cloned into plasmid pCMV2-FLAG (IBI Eastman Kodak, Rochester, NY). The fusion protein generated by this plasmid contains the COOH-terminal 786 amino acids of the predominant spliced isoform found in melanocytes fused to the COOH terminus of the FLAG epitope tag (nine amino acids). For transient transfections, melan-a melanocytes were grown to 70% confluence on 12-mm round No. 1 1/2 coverslips (for fixation/immunostaining), 24.5-mm round No. 1 1/2 coverslips (for live cell imaging in the Dvorak-Stotler Chamber), or chamber slides with No. 1 coverslip bottoms (No. P136307; Lab-tek, Naperville, IL) (for short-term viewing of live cells on the confocal microscope) and transfected with 1 μg of CsCl-purified plasmid DNA per 2.5 ml of culture media using FuGENE 6 transfection reagent (No. 1-814-443; Boehringer Mannheim, Indianapolis, IN), as described by the manufacturer. Cells were imaged or fixed/stained at various intervals between 24 and 60 h after transfection.
Immunostaining and Fluorescence Microscopy
Cells transfected with plasmid EGFP-MC-ST were fixed for 30 min at room temperature in freshly prepared 4% paraformaldehyde (No. 00380; Poly Sciences, Warrington, PA) in serum-free culture medium prepared using MEM powder (No. 41600-016, GIBCO BRL; Life Technologies, Gaithersburg, MD), washed briefly in PBS, and mounted on glass slides using Slow Fade (No. S-2828; Molecular Probes, Eugene, OR) and rubber cement. Cells transfected with plasmid FLAG-MC-LT were fixed as above, washed with PBS, blocked for 15 min in three changes of PBS containing 10% (vol/vol) fetal calf serum, incubated for 1 h at room temperature with anti-FLAG monoclonal antibody M5 (No. IB13091; Eastman Kodak) diluted 1:250 in blocking buffer supplemented with 0.2% saponin (No. S790; Sigma Chemical Co.), washed with PBS, incubated for 1 h with Texas red–labeled goat anti–mouse IgG (No. 115-025-146; Jackson ImmunoResearch Labs, Inc., West Grove, PA) diluted 1:100 in saponin-containing blocking buffer, washed with PBS, and mounted using Slow Fade and nail polish. To stain for F-actin and microtubules, cells were fixed in 4% paraformaldehyde dissolved in cytoskeletal stabilization buffer (Conrad et al., 1993) and processed as described above using Oregon green 488-labeled phalloidin (No. 0-7466; Molecular Probes) at a final concentration of 30 nM, a rat monoclonal antibody to tubulin (No. MAS 077P; Accurate Chemical) at a dilution of 1:100, and Texas red–labeled donkey anti–rat IgG (No. 712-075-153; Jackson ImmunoResearch Labs). For conventional light microscopy, samples were viewed with a Zeiss Axioplan microscope using either a 63× Plan-Apochromat Phase 3 objective or a 100× Plan Neofluar objective, and were recorded using Kodak TMAX film (400 ASA). For confocal microscopy, samples were viewed with a Zeiss LSM 410 confocal microscope using either a 63× Plan Neofluar objective for fixed cells or a 63× water-immersion objective for living cells in chamber slides. Images, which are either single, 1-μm-thick optical sections, or superimposed stacked images representing the full thickness of the cell (see figure legends), were transferred to Adobe Photoshop and printed using a Fuji (Tokyo, Japan) digital color printer. The fluorescent image in Fig. 7 H was obtained using an intensified CCD camera (model C2400; Hamamatsu) attached to the microscope for live cell imaging.
Figure 7.

Colocalization of GFP-tagged myosin Va tail domains and melanosomes in the periphery of transfected melanocytes before the generation of the dilute-like phenotype. (A) Untransfected cells (nonfluorescent) and a single cell transfected with plasmid EGFP-MC-ST before the appearance of the dilute-like phenotype (24 h after transfection). (B) GFP fluorescence in this transfected cell (1-μm section). Note the colocalization of fluorescence and black pigment (arrows). Bar, 7.5 μm.
Electron Microscopy and S1 Labeling
Melan-a melanocytes were permeabilized for 5 min at 37°C in PHEM buffer (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2, pH 6.9) supplemented with 0.02% saponin and 80 nM phalloidin (No. P-3457; Molecular Probes). The cells were then incubated for 30 min at room temperature in PHEM buffer containing 0.5 mg/ml rabbit skeletal muscle subfragment 1 (a generous gift of Dr. James R. Sellers, NHLBI, NIH), rinsed twice (5 min each) in PHEM buffer, and rinsed once (3 min) in 0.1 M sodium phosphate buffer, pH 6.8 (PB) (Lewis and Bridgman, 1992). The cells were then fixed for 30 min at room temperature in PB containing 2% glutaraldehyde, rinsed briefly with PB, and treated as described by McDonald (1984). In brief, cells were postfixed for 15 min on ice in PB containing 0.5% OsO4, 0.8% K3Fe (CN)6, and 1 mM MgCl2, rinsed three times (5 min each) in distilled water, mordanted for 1 min in PB containing 0.15% tannic acid, rinsed in PB, and stained en bloc for 2 h at room temperature in 2% aqueous uranyl acetate. The cells were then dehydrated in graded alcohols and embedded in Epon. Thin sections were examined using a Phillips 410 electron microscope (Mahwah, NJ). Actin polarity was scored as described in the text. Actin filaments within 2 μm of distal dendrite membrane were used to determine actin orientation at dendritic tips.
Image Analysis
Individual video frames acquired at 1-s intervals were transferred from the OMDR to a Macintosh Quadra 950 computer, and melanosome centroid positions in successive frames were hand digitized. Path plots were assembled using Dias software (version 1.2; Solltech, Inc., Iowa City, IA). The speed of each 1 s step was calculated using the direct method as follows:
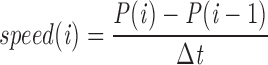 |
where i is the i th frame, Δt is the time interval (1 s), and P is the centroid position. The length of each step was the distance between consecutive centroid positions. The speed and path lengths of microtubule-dependent movements reported in Table I were calculated based on individual path plots representing one number. The maximum speed of each path plot is the average of the fastest step and any additional steps that were at least 70% the speed of the fastest step. The average speed for each path plot is the average of all the steps that were faster than 0.35 μm/s. Path lengths are the sum of all the individual steps in the path. Averages, standard deviations, and significance values (performed using the Student's t test) were calculated using Excel.
Table I.
Quantification of Microtubule-dependent Melanosome Movements in Wild-Type and Dilute Melanocytes
| Wild-type | Dilute | |||||||
|---|---|---|---|---|---|---|---|---|
| CF* | CP‡ | CF | CP | |||||
| Max. speed§ (n) | 0.95 ± 0.41‖ (38) | 1.03 ± 0.33‖ (34) | 1.67 ± 0.64‖ (34) | 1.86 ± 0.67‖ (34) | ||||
| Aver. speed§ (n) | 0.68 ± 0.17‖ (38) | 0.76 ± 0.19‖ (34) | 1.04 ± 0.29‖ (34) | 1.13 ± 0.35‖ (34) | ||||
| Path length (μm) | 4.6 ± 2.5‖ (38) | 5.4 ± 2.7‖ (34) | 13.3 ± 6.9‖ (34) | 11.9 ± 8.5‖ (34) | ||||
Centrifugal direction.
Centripetal direction.
For definition of maximum and average speeds (in μm/s), see Materials and Methods.
Each pair of values (wild-type CF/dilute CF and wild-type CP/dilute CP) are significantly different (P < 0.001).
Results
The Orientation of Actin Filaments within Dendrites Is Essentially Random
Light immunofluorescence micrographs of melan-a melanocytes (D/D) stained with fluorescent phalloidin (Fig. 1, C and E) typically reveal sparse staining in the central cytoplasm, intense edge staining along the length of the dendrite, and prominent staining at dendritic tips (see also Lacour et al., 1992; Wu et al., 1997). Confocal microscopy revealed that the edge staining in dendrites is due to a cortical shell of F-actin under the plasma membrane (data not shown). To determine the orientation of actin filaments within this cortical shell, melan-a melanocytes were permeabilized with detergent, labeled with rabbit skeletal muscle myosin subfragment 1, fixed, processed for transmission electron microscopy, and sectioned such that the cell body and as much of the dendritic extension as possible were present in a single section (Fig. 2 A). As in the light micrographs, the electron micrographs show that actin filaments are scarce in the central cytoplasm and positioned in dendrites almost exclusively just under the plasma membrane, forming a cortical shell that surrounds a microtubule-rich, actin-poor central zone. Actin polarity in dendrites was scored in a series of 1-μm-wide cross-sectional bands that spanned the short axis of the dendrite and that ranged from the base of the dendrite to where the dendrite left the plane of the section. A total of 28 such bands from seven dendrites, each from a different melanocyte, and each spanning a distance of between 20–30 μm from the nucleus, were scored. Decorated filaments were grouped into one of three categories, as described in the schematic in Fig. 1 B. Those in which the arrowhead pointed within a 90° arc whose center points directly away from the nucleus were scored as pointed end–out. Those in which the arrowhead pointed within a 90° arc whose center points directly towards the nucleus were scored as barbed end–out. Those that fell into the two remaining 90° arcs were scored as “other.” Three main points summarize what was found. First, the number of barbed end–out filaments per 1-μm cross-sectional band (4 ± 2.3) was not significantly different from the number of pointed end–out filaments (3.3 ± 2.2) (n = 28). Second, in several instances where the number of barbed end–out filaments exceeded pointed end–out filaments by a factor of greater than 3:1, the filaments in the adjacent 1-μm cross-sectional areas always exhibited an inverse ratio (data not shown). Third, of the 367 filaments scored, 112 (31%) were barbed end–out, 92 (25%) were pointed end–out, and 163 (44%) fell into the “other” category. These three results indicate that actin filament orientation in dendrites is essentially random. Using other thin sections that contained a total of 19 melanosome-rich dendritic tips, we also found that the number of barbed end–out filaments per tip (5.3 ± 3.5) was not significantly different from the number of pointed end–out filaments per tip (4.2 ± 2.9) (n = 19). Of 227 filaments scored in tips, 100 (44%) were barbed end–out, 80 (35%) were pointed end–out, and 47 (21%) were “other.” Therefore, as in the dendrite proper, the actin in dendritic tips exhibits a largely random orientation. Together, these results indicate that if myosin Va is responsible for the long-range centrifugal transport of melanosomes, it would be via a mechanism wherein random walks of melanosomes catalyzed by myosin Va on actin filaments of random orientation drive the outward spreading or “facilitated diffusion” of the organelle.
Figure 2.
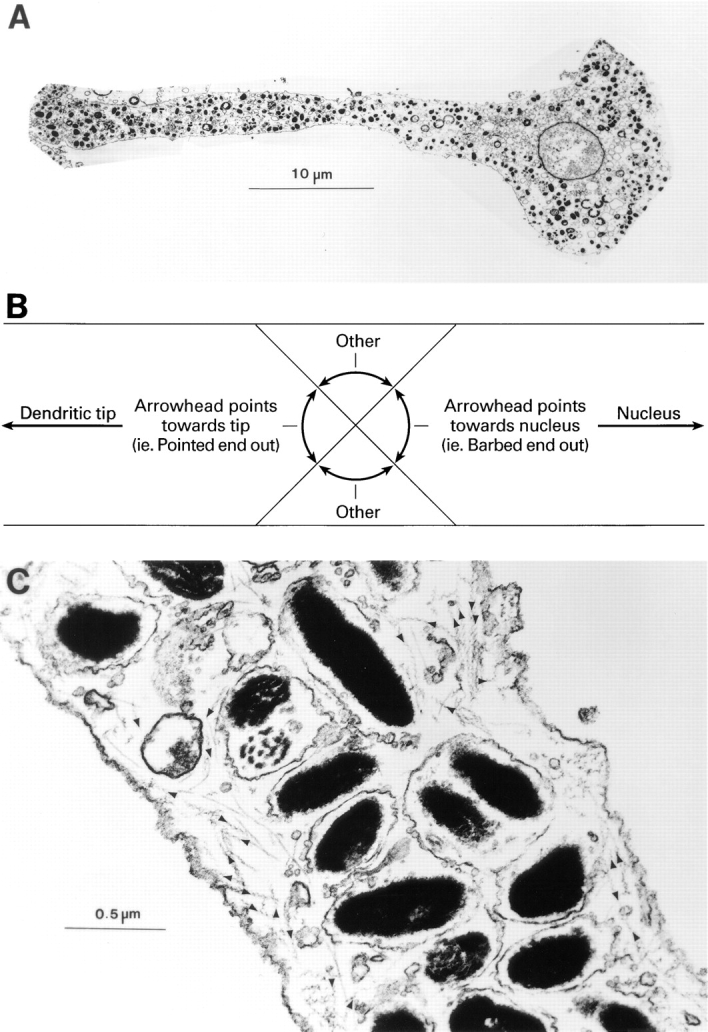
Orientation of F-actin within melanocyte dendrites. (A) Low-magnification transmission electron micrograph of the dendrite of a melan-a melanocyte labeled with subfragment 1. (B) Schematic indicating how F-actin orientation was scored in dendrites. (C) High-magnification micrograph of a section of dendrite from an S1-labeled melanocyte. Black arrowheads have been applied to aid in showing the orientation of filaments within this section.
Melanosomes in Dilute Melanocytes Move Rapidly and Bidirectionally between the Cell Center and the Periphery
In contrast to the mechanism described above, melanosomes in primary dilute null melanocytes were found to be undergoing rapid, bidirectional translocation between the cell center and the periphery. Fig. 3 A shows a still image of one such melanocyte (d l20J/d l20J) possessing a broad, well-spread cell extension projecting to the upper right. As is typical of heavily melanized dilute melanocytes (Wei et al., 1997), the majority of melanosomes are perinuclear, although some are clearly present in the broad flat cell extension. Time lapse video microscopy revealed that many of these latter melanosomes are moving rapidly (>1 μm/s) and bidirectionally between the cell center and the periphery. Two examples of these melanosome movements, which are very obvious when viewed at ∼15–30 times real speed, are shown in successive video stills (1-s time intervals) in Fig. 3, B1–B10 (centrifugal) and C1–C10 (centripetal). D and E show the paths (1-s time intervals) for some of the melanosomes whose centrifugal (D) and centripetal (E) movements within the boxed area in A were obvious to the eye over a 3-min time span (see video segment No. 1).2 While not all melanosomes traverse the entire cell extension in one continuous movement, some do (for example, see melanosome numbers 5 and 11 in D and E, respectively). Such long-range persistent movements are even more obvious in the lightly melanized mutant cell shown in Fig. 3 F, where melanosome numbers 2–4 (G, closed circles) move from near the nucleus to near the margin of the cell without significant pause, while melanosome numbers 7 and 13 (G, open circles) move from near the cell margin to near the nucleus with only a few short pauses. In typical fashion, essentially all of the melanosomes in the periphery of this cell are undergoing rapid bidirectional movement (or are temporarily pausing; see video segment No. 2), and individual melanosomes can be seen to move by this fast component in rapid succession from the center to the periphery and back again. One of these melanosomes is shown in successive video stills in Fig. 3, I1–I11, while H shows the paths for four different melanosomes undergoing such bidirectional movements. Fig. 3, J–L, shows that this bidirectional melanosome movement is also evident within longer cell extensions that are more typical of melanocyte dendrites in vivo (see video segment No. 3). Finally, Fig. 3, M and N, shows very striking bidirectional melanosome movements within a large dendritic extension emanating from a mutant cell (see video segment No. 4). These movements are notable in that they persist for long distances (melanosome 1, 48 μm; melanosome 6, 24 μm).
Figure 3.


Rapid, bidirectional melanosome translocation in the periphery of dilute melanocytes. (A) A typical dilute null melanocyte in primary culture. (B1–B10 and C1–C10) Successive video stills (1-s intervals) of typical centrifugal (B1–B10) and centripetal (C1–C10) melanosome movements (white arrows). (D and E) The paths for a portion of the centrifugal (D) and centripetal (E) melanosome movements that occurred over a 3-min time span within the boxed area in A. The path diagrams for this and some subsequent figures are reoriented relative to the image of the cell so that up is towards the periphery (P) and down is towards the nucleus (N). As in all subsequent path diagrams, the positions of melanosomes are shown at 1-s intervals. (F) A lightly melanized dilute melanocyte. (G) The paths for a portion of the centrifugal (closed circles) and centripetal (open circles) movements that occurred over a 3-min period within the boxed area in F. (H) The paths for four individual melanosomes that moved centrifugally and then centripetally. (I1–I11) Video stills of one such melanosome (white arrows). (J) A more dendritic dilute melanocyte. (K and L) The paths for the most obvious centrifugal and centripetal movements that occurred over a 40-s period within the boxed area in J. (M) A large dendritic extension from a dilute melanocyte. (N) Several highly persistent bidirectional melanosome movements that occurred over a 3-min interval within the boxed area in M. The identification of these organelles as melanosomes is certain because they can also be seen to move in brightfield images, where the black melanin within them is the only thing visible, and because these black objects match the match the size and shape of authentic melanosomes. Bars, 2.88 μm.
From analysis of these and other dilute melanocytes, we obtained the following information regarding this bidirectional component of melanosome motility. First, individual melanosomes move on average 13.3 ± 6.9 μm (n = 34) and 11.9 ± 8.5 μm (n = 34) between stops for centrifugal and centripetal movements,3 respectively (Table I). Second, stops may be short pauses on the order of seconds or longer pauses lasting for more than a minute, and pauses may or may not be associated with reversals in direction. Third, melanosomes that reverse direction sometimes appear to follow the same track in reverse (for example, see the path in the upper left corner of the Fig. 3 H). Fourth, while long (i.e., 20–30 μm) movements can occur without significant pauses, it is more common to see such movements interrupted by several pauses lasting on the order of a few seconds. Fifth, the average and maximum speeds of melanosomes undergoing this movement are 1.04 ± 0.29 and 1.67 ± 0.64 μm/s (n = 34), respectively, for centrifugal movements, and 1.13 ± 0.35 and 1.86 ± 0.67 μm/s (n = 34), respectively, for centripetal movements (Table I). These values for centrifugal and centripetal movements are not significantly different. Finally, while we did not attempt to quantitate the numbers of centrifugal and centripetal movements per unit time, their frequencies appear to be essentially the same. Indeed, they must be, given the phenotype of dilute null melanocytes, where net accumulation of melanosomes in the cell's periphery does not occur. Therefore, while this component of melanosome motility provides an efficient means to move melanosomes to the periphery, it does not by itself generate the net peripheral accumulation of melanosomes characteristic of wild-type melanocytes.
The Bidirectional Translocation of Melanosomes in the Periphery of Dilute Melanocytes Is Microtubule Dependent
The fact that the component of melanosome motility described above runs parallel to the long axis of cell extensions, exhibits linear trajectories often exceeding 15 μm in length without pausing, and possesses speeds characteristic of microtubule-dependent motor proteins (Hirokawa, 1998), together with the fact that in melanocytes stained with an α tubulin antibody, microtubules were found to run the length of dendritic extensions (Fig. 1 B), stopping just shy of the dendritic tip (Fig. 1, F and G), all suggest that this component of melanosome motility is microtubule dependent. To verify this, melanosome dynamics within dilute melanocytes were recorded before and after the addition of nocodazole. Fig. 4 B shows the paths for a portion of the fast centrifugal (left) and centripetal (right) melanosome movements that occurred over a 2-min time span within the dendritic extension of the dilute melanocyte in A just before the addition of nocodazole. C shows this same cell 5 min after the addition of 16 μM nocodazole to the media (37°C), while D shows the paths for every melanosome in this dendrite over the ensuing 1-min time interval (see video sequences Nos. 5A and 5B). Clearly, most if not all fast melanosome movements have disappeared (the slow displacements of a few melanosomes are due to local contractions of the cytoplasm). Occasional fast movements that were sometimes seen in dendrites of other similarly treated melanocytes completely disappeared when nocodazole treatment was supplemented with a 30-min incubation at 4°C, and fast bidirectional melanosome movements resumed when cells were returned to 37°C in the absence of nocodazole (see below). These observations, together with immunofluorescence staining of melanocytes incubated at 37°C in 16 μM nocodazole for 5 min, which indicated that this treatment largely eliminated microtubules from the periphery of melanocytes (data not shown), support the conclusion that the fast bidirectional component of melanosome movement is microtubule dependent. In addition, the nearly complete abrogation of this movement within cell extensions produced by nocodazole addition alone (i.e., without a 4°C incubation) suggests that microtubules within dendritic extensions are highly dynamic.
Figure 4.

Microtubule dependence of the fast peripheral and central melanosome movements. (A) A dilute melanocyte before the addition of nocodazole. (B) Some of the fast centrifugal (left) and centripetal (right) movements that occurred over a 2-min period within the boxed area in A. (C) The same cell as in A, but 5 min after the addition of 16 μM nocodazole to the perfusion medium. (D) The paths for all of the melanosomes within the boxed area in C over the ensuing 1 min. (E) The lightly melanized cell also described in Fig. 3, before the addition of nocodazole. (F) Some of the fast central movements that occurred within the boxed area in E over a 1-min span. (G) The same cell as in E, but after treatment with nocodazole and incubation at 4°C, as described in the text. (H) The complete paths for all of the melanosomes within the boxed area in G over a 1-min time span (i.e., all 60 positions for each melanosome are shown). (I) The same cell as in G, but following perfusion for 30 min without nocodazole. (J) A portion of the fast centrifugal (left) and centripetal (right) movements that occurred over a 2-min period within the boxed area in I. Bars, 2.88 μm.
Melanosomes in the Center of Dilute Melanocytes Are Also Undergoing Rapid, Microtubule-dependent Movements
Time lapse video microscopy of untreated and nocodazole-treated dilute melanocytes revealed that essentially all of the melanosomes in the central cytoplasm of these cells are also moving rapidly on microtubules. Fig. 4 E shows a still image of the same lightly melanized dilute melanocyte described in Fig. 3, F–I, while Fig. 4 F shows the paths for a portion of the fast melanosome movements seen in the central cytoplasm of this cell (boxed area in E) over a 2-min time span before the addition of 16 μM nocodazole. These fast movements exhibit nearly the same speed as fast movements in the periphery (1.05 ± 0.44 μm/s, n = 15 from this cell), but differ in that their average excursions are much shorter (3.1 ± 1.3 μm, n = 15 from this cell). G shows this same cell after incubation at 37°C for 10 min, at 4°C for 30 min, and at 37°C for 10 min, all in the presence of 16 μM nocodazole, while H shows the paths for all melanosomes in the boxed area over the ensuing 1 min. Clearly, most if not all of the short, fast movements have disappeared (see video segments Nos. 6A and 6B). We also found that these central, short-range movements reappeared first upon nocodazole washout, with peripheral movements following some time later once cell extensions have reformed. This latter point is demonstrated in I and J, which show this same cell 30 min after nocodazole wash out (see video segment No. 7). We conclude, therefore, that the majority of melanosomes in dilute melanocytes are undergoing microtubule-dependent translocation, with most moving on the large number of short microtubules (see Fig. 1) that terminate in the central cytoplasm, while the rest ride on the smaller number of long microtubules that extend to the periphery of the cell.
Bidirectional Microtubule-dependent Melanosome Traffic Also Exists in Wild-Type Melanocytes but Does Not Involve Melanosomes Concentrated along the Edge or at the Tip of Dendrites
Fig. 5 A shows a primary wild-type melanocyte, which exhibits the typical bias in peripheral distribution of melanosomes characteristic of myosin Va+ melanocytes, while B shows a portion of the fast centrifugal and centripetal melanosome movements that occurred within this cell over a 2-min time span. As in many wild-type melanocytes, these fast movements, which in well-spread cells like this one are most evident in the region between the nucleus and the melanosome-rich periphery, are considerably less obvious than in dilute melanocytes (see video sequence No. 8). This appears to be due in part to the fact that the peripheral cytoplasm of wild-type melanocytes, unlike dilute melanocytes, contains significant numbers of melanosomes that are not undergoing this fast movement and that block the view of those that are. Striking bidirectional melanosome movements in wild-type melanocytes are not uncommon, however, especially within dendritic extensions. D and G show the paths for a portion of the fast centrifugal (closed circles) and centripetal (open circles) melanosome movements that occurred over a 3-min time span within the boxed area of the dendritic extensions in C and F, respectively. In addition to the melanosomes that are undergoing these fast movements (which occur mostly in the central, microtubule-rich zone of the dendrite), wild-type cells possess large numbers of melanosomes that are at the tip (C) and along the edge (F) of their dendrites and that are undergoing slow, local motions (see video sequences Nos. 9 and 10 and Figs. 9 and 10). Importantly, while microtubule-dependent melanosome movements in these actin-rich zones are rare, melanosomes can be removed from the tip (E) and lateral edge (H) of the dendrite by the fast component, and new melanosomes can be delivered to the tip (E) and lateral edge (H) by the fast component. The extent to which the balance of these dynamics can change the degree of peripheral accumulation of melanosomes was evident in several instances where a surfeit of centrifugal or centripetal movements led to relatively rapid (on the order of minutes) increases or decreases, respectively, in the degree of peripherally accumulated melanosomes. For example, just before the capture of images used for the data in F–H, a surfeit of centrifugal melanosome movements spanning a period of ∼10 min lead to an approximate doubling of the number of melanosomes at the tip (I and J).
Figure 5.

Rapid, bidirectional melanosome translocation in wild-type melanocytes. (A) A well-spread wild-type melanocyte. (B) The paths for a portion of the fast centrifugal and centripetal (arrowheads) movements that occurred over a 2-min span within the boxed area in A. (C) The dendrite and melanosome-rich tip of the wild-type melanocyte shown in the inset. (D) The paths for a portion of the fast centrifugal and centripetal movements that occurred over a 3-min span within the boxed area in C. (E) The paths for two centrifugal movements (closed circles) that delivered melanosomes to, and two centripetal movements (open circles) that removed melanosomes from, the dendritic tip. (F) The dendritic extension of the cell shown in the inset, which had a prominent accumulation of melanosomes along the lateral edges of the dendrite. (G) Some of the fast centrifugal and centripetal movements that occurred over a 3-min period within the boxed area in F. (H) The paths for three centrifugal movements (closed circles) that delivered melanosomes to, and three centripetal movements (open circles) that removed melanosomes from, the lateral edge of this dendrite. (I and J) The tip of the cell shown in F before (I) and after (J) an ∼10-min period where pronounced centrifugal movements led to an approximately twofold increase in the numbers of melanosomes at the tip. Bars, 2.88 μm.
Figure 9.
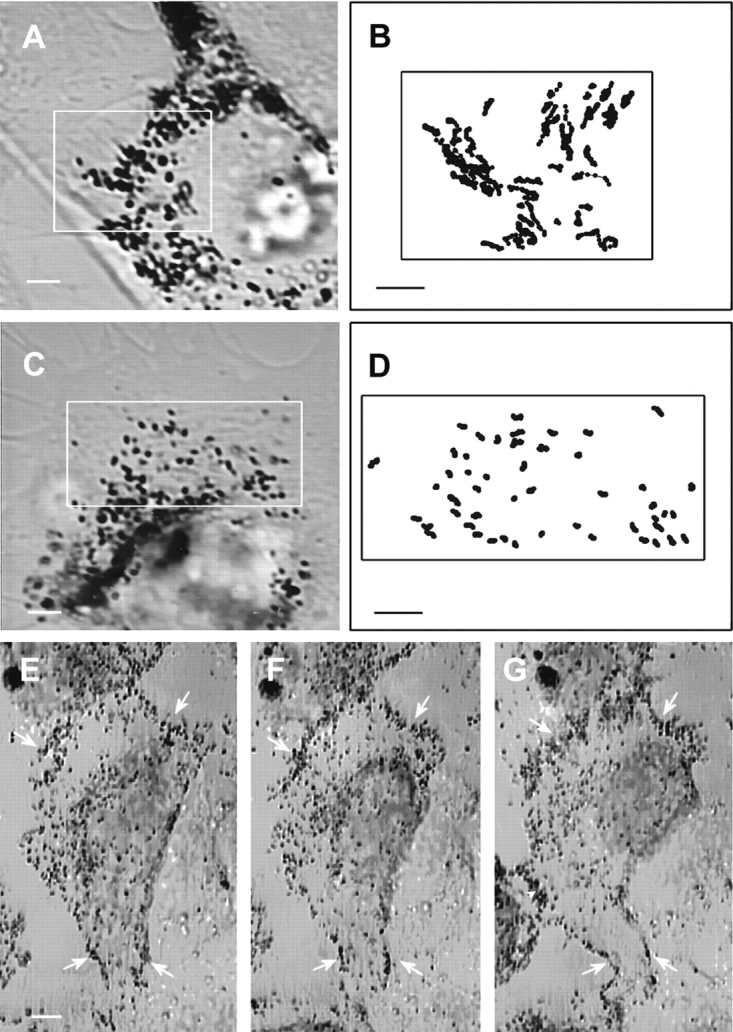
The dynamics of melanosomes in microtubule-depleted wild-type and dilute null melanocytes. (A and C) Microtubule-depleted wild-type and dilute null melanocytes, respectively. (B and D) The paths for all of the melanosomes present within the boxed areas in A and C, respectively, over an entire 100-s period (i.e., the positions for every melanosome were digitized over the entire 100 s, or until the melanosome was no longer visible because it moved out of the plane of focus or became indistinguishable from surrounding melanosomes). (E–G) A melan-a melanocyte before microtubule depletion (E), 5 min after being returned to 37°C (in the presence of nocodazole) after a 30-min incubation at 4°C with nocodazole (F) and 90 min later (G). Arrows in all three panels point to the prominent concentration of melanosomes in the cortex of this cell. The black area marked by an arrowhead in G is part of another cell. Bars: (A–D) 2.2 μm; (E–G) 4.5 μm.
Figure 10.

Histograms of the speeds of melanosomes in microtubule-depleted wild-type and dilute melanocytes. Speeds for wild-type (filled bars) and dilute (hashed bars) melanocytes were binned in 0.025-μm/s increments (except for the first bin, which represents true zero, i.e., not a range) and plotted as a percentage of total steps.
From analysis of these and other wild-type melanocytes (e.g., melan-a cells, which are essentially indistinguishable from primary wild-type melanocytes), we obtained the following information regarding this fast component in the context of wild type cells. First, as in dilute melanocytes, it is dependent on the presence of intact microtubules (see below). Second, the average and maximum speeds of melanosomes undergoing these movements in wild-type cells are 0.68 ± 0.17 and 0.95 ± 0.41 μm/s (n = 38), respectively, for centrifugal movements, and 0.76 ± 0.19 and 1.03 ± 0.33 μm/s (n = 34), respectively, for centripetal movements (Table I). While these values for centrifugal and centripetal movements are not significantly different from each other, they are all significantly slower than the values for dilute melanocytes (P < 0.001). Third, individual melanosomes move on average 4.6 ± 2.5 μm (n = 38) and 5.4 ± 2.7 μm (n = 34) between stops for centrifugal and centripetal movements, respectively (Table I). While these values are not significantly different from each other, they are both significantly shorter than the values obtained for dilute melanocytes (P < 0.001). These differences may be due to a myosin Va–dependent damping of both the rate and persistence of microtubule-dependent melanosome movements in vivo (see Discussion).
Expression of the Myosin Va Tail Domain in a Wild-Type Background Creates a Dilute-like Phenotype
The fact that the fast, bidirectional, microtubule-dependent component of melanosome transport identified above can move melanosomes to the periphery efficiently but cannot concentrate them there, together with previous studies showing extensive colocalization of myosin Va and melanosomes in the actin-rich periphery, suggested a mechanism in which a myosin Va–dependent interaction of melanosomes with F-actin in peripheral regions of the cell prevents these organelles from returning to the cell center via the centripetal aspect of this fast component, thereby causing their peripheral accumulation. To test this “capture” model, we expressed the myosin Va tail domain within wild-type melanocytes. If, as conjectured (Mooseker and Cheney, 1996), this portion of the myosin Va molecule contains the site that mediates myosin Va/ melanosome interaction, then it should compete with endogenous myosin Va for binding to the melanosome. This competition should, in turn, serve to uncouple the organelle from the peripheral actin cytoskeleton. Once uncoupled, the melanosome should redistribute to the cell center, the site of greatest microtubule density (see Fig. 1 B), by the action of bidirectional, microtubule-dependent traffic. Melan-a melanocytes (D/D) were transiently transfected with plasmid EGFP-MC-ST (see Materials and Methods), which drives the expression of a fusion protein containing EGFP fused to the COOH-terminal 619 amino acids of the melanocyte-specific isoform of myosin Va. This portion of the myosin Va heavy chain contains the last ∼40% of the central stalk domain (including both melanocyte-specific exons [exons D and F in Seperack et al., 1995]), and all of the COOH-terminal 410-residue globular tail domain. Fig. 6, A and C, shows the appearance of several such transfected cells, each surrounded by a number of untransfected cells. The transfected cells, with their striking perinuclear distribution of melanosomes, are very reminiscent of dilute melanocytes, and contrast sharply with the surrounding untransfected cells in which melanosomes are concentrated in the periphery. Similar results were obtained with plasmid FLAG-MC-LT, which drives the expression of a fusion protein containing the nine-residue FLAG epitope tag fused to the COOH-terminal 786 amino acids of the melanocyte-specific isoform of myosin Va (Fig. 6 E). Conversely, cells transfected with the GFP or FLAG vectors alone did not demonstrate this dilute-like phenotype (data not shown). In agreement with the mechanism proposed above, GFP and FLAG fluorescence in cells exhibiting the dilute-like phenotype was largely coincident with the mass of aggregated melanosomes in the cell center. This coincidence was seen not only in fixed cells (B and F), but in living cells expressing the GFP-tagged myosin Va tail chimera as well (D), indicating that the colocalization is not an artifact of fixation.
Figure 6.
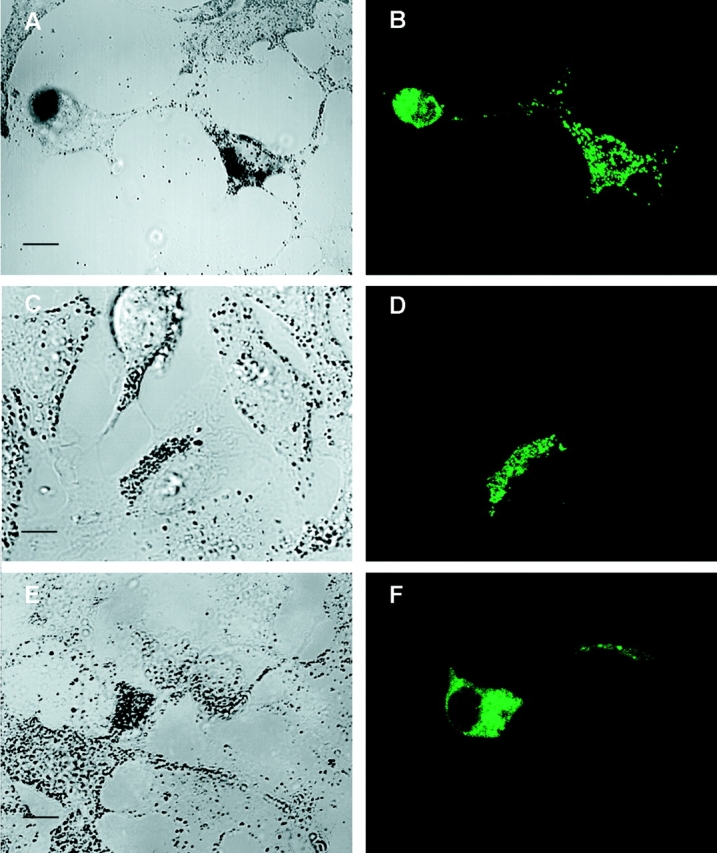
Expression of myosin Va tail domain fusion proteins in wild-type melanocytes creates a dilute-like phenotype. (A, C, and E) Fields of melan-a melanocytes showing untransfected cells (nonfluorescent) and cells transfected with plasmids EGFP-MC-ST (A and C) and FLAG-MC-LT (E) (48 h after transfection). (B, D, and F) Corresponding confocal micrographs of GFP fluorescence in fixed (B; stacked images) and living (D; 1-μm section) cells, and FLAG fluorescence (using rhodamine-labeled anti-FLAG monoclonal antibody) in fixed cells (F; 1-μm section). Bars: (A) 16.7 μm; (C and E) 7.5 μm.
Evidence that tail expression actually generates the dilute-like phenotype, and further insight into the mechanism by which it does so, came from analysis of GFP fluorescence in transfected cells that do not show the dominant-negative phenotype. Fig. 7 shows one of these cells, which are common between ∼24 and 36 h of transfection but rare after ∼50 h of transfection, and which are characterized by having peripherally distributed melanosomes (Fig. 7 A) and GFP fluorescence that largely colocalizes with these melanosomes (B, arrows). Given that this pattern is precisely what one would predict must precede the generation of the dilute-like phenotype if the mechanism is as described above, we sought to demonstrate by time lapse microscopy that transfected cells like the one in Fig. 7 do indeed turn into cells exhibiting the dilute-like phenotype. Fig. 8 A shows a transfected melan-a cell that was identified by eye as having peripheral, melanosome-associated fluorescence, while B–F show this cell at five successive 30-min intervals. Over this 150-min time span, melanosomes can be seen to undergo a dramatic redistribution, beginning predominantly at the margin of the cell (Fig. 8 A), and ending predominantly in the cell center (F) (see video sequence No. 11). The distribution of melanosomes and GFP fluorescence in this living cell ∼5 min after the time point corresponding to F are shown in G and H, respectively. Once again, GFP fluorescence is largely coincident with the mass of melanosomes in the cell center. These results, which were replicated in each of three similar transfected cells (all of which took ∼120 min to generate this dominant-negative phenotype), lend strong support to the idea that tail expression initiates the generation of the dilute-like phenotype by displacing endogenous myosin Va from melanosomes in the periphery. The microtubule-based melanosome traffic that drives the redistribution of these melanosomes, which are now uncoupled from the peripheral actin cytoskeleton, is evident in high-magnification time lapse images of transfected cells (data not shown), where melanosomes are seen to undergo rapid, bidirectional movements that gradually result in their accumulation in central regions of the cell, where the density of microtubules is greatest (Fig. 1).
Figure 8.

Time lapse video micrographs of a transfected melan-a melanocyte during the generation of the dilute-like phenotype. (A–F) Video stills at 30-min intervals of a single melan-a melanotype transfected with plasmid EGFP-MC-ST (center). At time zero, this cell had peripherally distributed fluorescence that localized with the melanosomes accumulated under the plasma membrane. (G) This same cell 5 min after the image in F. (H) The distribution of fluorescence ∼90 s after capturing the image in G (the time required to change cameras). The intense spot of fluorescence probably represents myosin Va tails accumulated at the microtubule-organizing center (see Wu et al., 1998). Bars, 6.4 μm.
Intermittent, Microtubule-independent, 0.14 μm/s Melanosome Movements Are Seen Only in Wild-Type Melanocytes
The capture mechanism described above requires a myosin Va–dependent interaction of melanosomes with the actin cytoskeleton. We sought to identify such interactions indirectly by comparing the dynamics of melanosomes in wild-type and dilute melanocytes. To focus on these interactions, the microtubule-dependent component of melanosome transport was eliminated by treating cells with nocodazole at 4°C for 30 min followed by incubation at 37°C in the presence of nocodazole. This treatment was also necessary to get reasonable numbers of melanosomes in dilute melanocytes that were not microtubule associated. To quantify melanosome dynamics, the positions of every melanosome within ∼120 μm2 areas were digitized over a period of 100 s (at 1-s intervals). Care was taken to choose cells in which there were no obvious changes in cell shape or position over this 100-s time span, and where melanosomes were well spread (a prerequisite for quantitating individual melanosome movements). Fig. 9 shows video stills of the wild-type (A) and dilute null (C) melanocytes used in this analysis, while B and D show the entire paths for all of the melanosomes present in the boxed areas. The difference between these paths, which is reasonably obvious from visual inspection (also see video sequences Nos. 12A and 12B), was borne out by quantitative analysis. Fig. 10 shows histograms in which the speeds of these melanosomes (41 for wild-type, 52 for dilute) were binned in 0.025-μm/s increments and plotted as a percentage of total steps (2,432 for wild-type, 5,174 for dilute). Three main points can be made from this plot. First, the percentage of steps in which melanosomes did not move (zero bin) is slightly more than twice as big in the dilute sample (75.3%) as in the wild-type sample (33.4%). Second, both wild-type and dilute samples have a peak (peak 1) centered around ∼0.05 μm/s. While we do not know the precise nature of these movements, they clearly are not myosin Va dependent. Third, the wild-type sample, but not the dilute sample, has another peak (peak 2) that falls roughly between the 0.075–0.1-μm/s bin and the 0.175–0.2-μm/s bin. This portion of the wild-type histogram contains 17.4% of the total steps. By contrast, only 0.17% of the total steps for the dilute sample fall in these bins, suggesting that the melanosome movements corresponding to peak 2 are myosin Va dependent. The average speed of these movements is 0.14 μm/s. Analysis of individual path plots from the wild-type sample revealed that 24 of the 41 paths (∼60%) exhibited at least three contiguous 1-s steps with a speed exceeding 0.1 μm/s. Whether these events represent myosin Va–dependent movements of melanosomes on actin filaments or the movements of actin filaments to which melanosomes are tethered by myosin Va is unclear. Nevertheless, because both situations require a myosin Va– dependent interaction of melanosomes with F-actin, these results support the capture mechanism.
We also examined the fate of melanosomes in microtubule-depleted cells over a longer time interval to estimate the ability of these movements to drive longer-range translocations of melanosomes. Fig. 9 E shows a melan-a melanocyte in which melanosomes were concentrated in the peripheral cortex (see arrows), and were undergoing rapid, bidirectional movements in the zone between the cortex and the nucleus before microtubule depletion (data not shown). Fig. 9 F shows this cell 5 min after being returned to 37°C (in the presence of nocodazole) following a 30-min incubation at 4°C in the presence of nocodazole, while G shows this same cell 90 min later. Comparison of F and G shows that the melanosomes concentrated in the peripheral cortex of this cell immediately after microtubule depletion (arrows) did not spread back into the cell, but rather remained highly concentrated in the cortex for at least 90 min. These results indicate that myosin Va– dependent melanosome movements, whatever their exact nature, do not drive obvious spreading of the organelles over 90 min.
Discussion
Myosin Va, Melanosome Transport, and Melanosome Distribution
Our current model (Fig. 11) for the role that myosin Va plays in the centrifugal transport and peripheral accumulation of melanosomes in mouse melanocytes stems from five observations: (a) the accumulation of melanosomes within dendrites and dendritic tips requires the expression of myosin Va (Koyama and Takeuchi, 1981; Provance et al., 1996; Wei et al., 1997); (b) the organization of F-actin in melanocytes is not optimal for efficient, long-range centrifugal transport of melanosomes by myosin Va (Figs. 1 and 2); (c) melanocytes possess a microtubule-dependent component of melanosome translocation that provides a very efficient means to transport melanosomes to the periphery (Figs. 3–5); (d) the characteristics of this component (e.g., bidirectionality) are such that it cannot (and does not) by itself generate a net peripheral accumulation of melanosomes (Figs. 3–5; and (e) myosin Va and melanosomes colocalize extensively in the actin-rich periphery (Nascimento et al., 1997; Wu et al., 1997). Together, these observations point to a cooperative mechanism in which a myosin Va–dependent interaction of melanosomes with F-actin in peripheral regions of the cell prevents melanosomes delivered there by centrifugal microtubule-dependent transport from being returned to the cell center by centripetal microtubule-dependent transport, thereby causing their distal accumulation. Three additional lines of evidence support this cooperative/capture mechanism. First, by expressing myosin Va tail domain fusion proteins in wild-type melanocytes, we demonstrated this cooperative/capture mechanism operating in reverse, where melanosomes, displaced from actin in the periphery by myosin Va tail domains, redistribute on microtubules to the cell center, creating a dilute-like phenotype (Figs. 6–8). While the bidirectional nature of microtubule-dependent melanosome transport might appear at first to preclude its ability to drive this redistribution, the much higher density of microtubules present in the cell body relative to cell extensions (Fig. 1) serve as a sink, causing the melanosomes to accumulate there over time. Indeed, this would be why melanosomes remain largely perinuclear in dilute melanocytes, despite the fact that most appear to be moving most of the time on microtubules.
Figure 11.

Model depicting how microtubule- and actomyosin Va–dependent melanosome traffic cooperate to drive the centrifugal transport and peripheral accumulation of melanosomes. Melanosomes undergo fast, bidirectional, long-range microtubule-dependent translocations within dendritic extensions (1). Myosin Va serves to capture melanosomes delivered to the periphery via the centrifugal component of microtubule-dependent movement by mediating their interaction with F-actin (2). The myosin Va molecules involved in this capture can be associated with peripheral F-actin (3), the soluble pool (4), and/or may be present as passengers on melanosomes as they are carried to the periphery on microtubules (5). This latter possibility is supported by the distributions of the GFP–myosin Va chimera reported here, and would be consistent with the landmark observation in squid axoplasm that individual organelles can move on both actin and microtubules (Kuznetsov et al., 1992). Melanosome capture occurs primarily at the actin-rich dendritic tip (2), but also along the lateral edge of dendrites (especially in less polarized melanocytes) (6). While a large percentage of captured melanosomes do not exhibit obvious motion (7), short-range, myosin Va–dependent melanosome movements in the periphery do occur. These movements may be due to the myosin Va–dependent translocation of these organelles on actin (8), and/ or to the movement of actin to which melanosomes are tethered by myosin Va (not shown). The myosin Va–dependent movement of melanosomes on actin will tend to be restricted by the physical barriers inherent in isotropic actin networks (9), by the association of individual melanosomes with filaments of opposite polarity (10), and by incomplete anchorage of actin filaments. Actin is sparse in the central cytoplasm (11), except for lamellae, which exclude melanosomes and do not contain myosin Va (12). Melanosomes also move on the large mass of microtubules in the center of melanocytes (13). These microtubules provide the “sink” that drives the accumulation of melanosomes in the center of myosin Va− cells and of wild-type cells transfected with dominant-negative constructs.
Second, by analyzing the characteristics of microtubule-dependent movements in wild-type and dilute melanocytes, we uncovered differences that may further reflect the functioning of the cooperative/capture mechanism (Table I). Specifically, the average and maximum speeds of microtubule-dependent melanosome movements in wild-type melanocytes (both centrifugal and centripetal) are ∼60% of that in dilute melanocytes. In addition, the average path lengths (both centrifugal and centripetal) in wild-type melanocytes are ∼50% of that in dilute melanocytes. We think that these differences are probably due to myosin Va–dependent interactions of the melanosome with F-actin as the organelle is being translocated on microtubules. By acting as a drag, these interactions serve to damp, or in other cases stop, the microtubule-dependent movements in wild-type cells. Moreover, we suggest that transient interactions that slow the movement predominate in the microtubule-rich, actin-poor central cytoplasm and core of the dendrite (Fig. 11), while strong interactions that stop the movement tend to predominate in the actin-rich, microtubule-poor zone under the plasma membrane of dendrites and dendritic tips. Inherent in this mechanism, therefore, is the concept (see Langford, 1995; Kelleher and Titus, 1998) that the coordination of the microtubule- and actin-dependent components is driven to a significant extent simply by differences in the spatial distributions of these polymers.
Third, by comparing the dynamics of melanosomes in microtubule-depleted wild-type and dilute melanocytes, we identified a class of intermittent, ∼0.14-μm/s melanosome movements that appear to be myosin Va dependent (Figs. 9 and 10). At present it is impossible to tell whether these movements represent myosin Va moving melanosomes on actin, or the movement of actin to which melanosomes are tethered by myosin Va. What we can reasonably conclude, however, is that since both situations require myosin Va–dependent interactions of melanosomes with actin, this data supports the cooperative/capture mechanism.
The data in Figs. 9 and 10, together with the fact that a random orientation of actin does not preclude long-range myosin Va–dependent melanosome transport (via facilitated diffusion), raises the issue of the relative significance of myosin Va–dependent melanosome motility versus the cooperative/capture mechanism identified here in driving the long-range centrifugal transport and peripheral accumulation of melanosomes. Our observation that melanosomes accumulated in the periphery of wild-type cells do not exhibit obvious, microtubule-independent spreading over 90 min (Fig. 9) suggests that the net outward displacement of centrally located melanosomes by the action of myosin Va alone would also be minimal over 90 min. By contrast, melanosomes are seen to undergo microtubule-dependent transport from the center to the periphery in as little as ∼20 s. With regard to peripheral accumulation, while a myosin-only mechanism working in the context of randomly oriented actin can facilitate melanosome diffusion, it cannot generate concentration. By contrast, the actomyosin Va–dependent capture of melanosomes delivered to the periphery by microtubule-dependent movement provides for not only efficient, long-range transport, but also a concentrating mechanism whose kinetics should easily exceed the ability of myosin-driven diffusion to dissipate the accumulated organelles. Evidence that mouse melanocytes actually use this mechanism was provided by forcing the cooperative/capture mechanism to run in the absence of capture (Figs. 6–8), where long-range melanosome movements are seen to be microtubule based. Furthermore, the ∼2-h time interval required for this redistribution to occur, where the sink driving redistribution is simply the difference in microtubule density between the periphery and the cell center, probably underestimates the ability of the cooperative/capture mechanism to generate a peripheral accumulation when working in the correct direction, where the sink is the myosin Va–dependent capture of melanosomes. Based on these and other observations (see below), we conclude that the cooperative/capture mechanism is the predominant mechanism responsible for the long-range centrifugal transport and peripheral accumulation of melanosomes in mouse melanocytes. We say predominant because we cannot rule out some contribution to long-range transport by the action of a slow myosin Va–dependent component. Furthermore, such myosin V–only organelle movements may be very important in other cellular contexts. Having said this, we note that the ability of myosin V to drive long-range displacements of organelles in the context of randomly oriented actin is thwarted not only by the inefficiency inherent in a random orientation of tracks, but also by problems involving (a) anchorage of actin filaments (if not anchored, actin may move over the surface of the organelle and not vice versa), (b) cancellation of forces (caused by myosin V molecules on the organelle's surface interacting simultaneously with actin filaments of opposing orientation), and (c) lack of tensile strength (individual, i.e., not bundled, actin filaments have little tensile strength, so may tend to bend when myosin V–dependent forces are applied). In the case of the melanosome, these problems are probably magnified, relative to 150-nm transport vesicles, by its large size and physical rigidity.
The cooperative/capture mechanism identified here not only overturns existing ideas about the mechanism of melanosome motility in mammalian melanocytes (Hearing and King, 1993; Provance et al., 1996; Wei et al., 1997; Wu et al., 1997; Mermall et al., 1998; Rogers and Gelfand, 1998) but provides the foundation for future efforts directed at understanding how the microtubule- and myosin V–dependent components are regulated to maintain an accumulation of melanosomes at dendritic tips in the context of constitutive, and up-regulatable, transfer to keratinocytes. Success in this area could lead to the identification of agents that augment this transfer, which in turn could have a significant impact on the prevention and treatment of melanoma.
In addition to providing strong support for our cooperative/capture mechanism, the results of expressing GFP- and FLAG-tagged myosin Va tail domain fusion proteins in wild-type melanocytes also provide strong confirmation of previous efforts to demonstrate that myosin Va associates with melanosomes (Nascimento et al., 1997; Wu et al., 1997), and indicate that the portion of myosin Va mediating this interaction resides within the COOH-terminal ∼33% of the molecule. Efforts to further define this domain, which may include two melanocyte-specific exons (Seperack et al., 1995), should aid in identifying the melanosomal protein that serves as the myosin V “receptor.”
Microtubule-dependent Melanosome Transport
This paper represents the first clear demonstration that melanosomes in mammalian melanocytes undergo microtubule-dependent translocation. Our initial appreciation of these movements was facilitated by examination of dilute melanocytes, where essentially all melanosomes are seen to be riding on microtubules most of the time, and where melanosomes moving on microtubules in the periphery stand out against the melanosome-free background (Figs. 3 and 4). While these movements are harder to see in wild-type cells, examples of striking, bidirectional, microtubule-dependent melanosome translocations are not uncommon (see video sequence No. 10) and can cause relatively rapid changes in the degree of peripheral accumulation of melanosomes (Fig. 5). The identity of the microtubule motor(s) responsible for these bidirectional movements, and the polarity of microtubules within melanocyte dendrites, are unknown. In neurons, microtubules are uniformly plus end–out in axons, but almost equally split between plus end–out and minus end–out in dendrites (Baas and Yu, 1998). If the former situation is the case for the melanocyte dendrite, then the melanosome must associate with both plus end– and minus end–directed motors. If the microtubules are of mixed polarity, then either type of motor alone could support bidirectional melanosome traffic. Identification of the motor(s) involved should be possible using function-blocking antibodies and/ or the expression of dominant-negative constructs, and may be facilitated by the characterization of melanosome dynamics in dilute suppressor (dsu) melanocytes. Dsu is an extragenic suppressor of dilute that acts in a semidominant, cell autonomous manner to reverse the effect of dilute mutations on coat color (Moore et al., 1988, 1994). Based on the example in yeast, where overexpression of a kinesin-related protein (smy1p) partially suppresses the phenotype of a myosin V mutant (myo2 66) (Lillie and Brown, 1992), it has been speculated that dsu might encode a kinesin family member that is either turned on or overexpressed in dsu melanocytes (Moore et al., 1994). Our results here make this hypothesis all the more attractive.
Mammalian Melanosome Motility and the Motility of Pigment Granules in Fish and Amphibian Melanophores
Fish melanophores generate very rapid changes in surface color through highly coordinated, microtubule-dependent, intracellular dispersion (dark) and aggregation (light) of their pigment granules (Haimo and Thaler, 1994). Recently, Rodoinov et al. (1998) reported that when dispersion was induced in these cells after destruction of the actin cytoskeleton, pigment granules were seen to move all the way to the margins of the cell. This phenotype, which contrasts with the even cytoplasmic distribution of pigment granules normally found in the fully dispersed state, and which is consistent with the unopposed action of a plus end–directed microtubule motor, suggested that an interaction of these granules with the actin cytoskeleton (presumably via a myosin) serves to limit plus end–directed pigment granule traffic by shifting granules from fast, anterograde movement to local movement on short actin filaments of random orientation that were shown to exist between the microtubules. We suggest that this cooperative mechanism, and the cooperative/capture mechanism identified here in mouse melanocytes, are related phenomena tuned to achieve different biological ends. In the case of fish melanophores, the cooperation between plus end– directed granule movements and local, actomyosin-dependent granule movements serves to position granules on actin at points all along the microtubule, thereby generating the even cytoplasmic distribution required for darkening. In mouse melanocytes, the cooperation between fast, bidirectional, microtubule-dependent melanosome transport and actomyosin V–dependent capture of melanosomes at dendritic tips serves to generate the peripheral accumulation of melanosomes required for efficient transfer to keratinocytes. Importantly, the actomyosin-dependent component in both these systems appears to be primarily short-range in nature (Fig. 9; see references in Haimo and Thaler [1994] and Discussion in Rogers and Gelfand [1998]), and, as such, is only apparent when working in cooperation with the long-range, microtubule-dependent component. This may not be the case, however, for frog melanophores, where Rogers and Gelfand (1998) argue that an actomyosin (possibly myosin V)–dependent component, operating in the context of a barbed end–out bias in actin filament organization, functions along with kinesin II (Rogers et al., 1997) to drive the long-range centrifugal transport of pigment granules.
Generality of the Cooperative/Capture Mechanism
The results obtained in this study add to the growing consensus that microtubule- and actin-based motors cooperate in organelle motility and distribution (Kuznetsov et al., 1992; Lillie and Brown, 1992; Bearer et al., 1993; Fath et al., 1994; Langford, 1995; Morris and Hollenbeck, 1995; Evans et al., 1997; Kelleher and Titus, 1998; Rogers and Gelfand, 1998; Rodionov et al., 1998), agree with the view that microtubules support long-range movements, while actin filaments support short-range movements in the periphery (Bray, 1992), and extend these concepts in several ways. Specifically, the cooperative/capture mechanism identified here generates a polarized (not even) distribution of an organelle and operates in the context of ongoing, bidirectional, microtubule-dependent traffic. This probably represents a common context in which a myosin would seek, in cooperation with microtubules, to determine organelle distribution. This would be the case, for example, in neuronal dendrites, where only a single type of microtubule motor would drive bidirectional transport (Baas and Yu, 1998), or in the case where microtubules are of uniform polarity, and where the organelle possesses both plus and minus end–directed motors, but where retrograde and anterograde movements are ongoing, not reciprocally controlled by hormones as in melanophores. Importantly, because the nature of our study directly implicates myosin Va in the cooperative/capture mechanism, this mechanism may provide insight into the function(s) of myosin V in other cell types. For example, this mechanism may explain the defect in distribution of smooth endoplasmic reticulum (SER) in cerebellar Purkinje cells from dilute rats and mice (Dekker-Ohno et al., 1996; Takagashi et al., 1996), where SER is present in the trunks of the dendrite proper but never penetrates the dendritic spines. By analogy with melanosomes, SER in the dendrite proper may be undergoing bidirectional, microtubule-dependent transport, while capture of SER tubules in the dendritic spines may be actomyosin Va dependent. Myosin Va may also have a general role in influencing the distribution of secretory lysosomes, given that (a) melanosomes are secretory lysosomes (Orlow, 1995); (b) humans with Griscellis' disease, the equivalent of mouse dilute (Pastural et al., 1997), have reported defects in natural killer cell function (Klein et al., 1994); (c) the lytic granules used by natural killer cells to kill target cells are also secretory lysosomes (Griffiths, 1996); and (d) lytic granule secretion is a targeted event involving microtubules (Griffiths, 1995). Finally, our results may provide further insight into the phenotypes of yeast cells lacking either of the two class V yeast myosins (myo2p and myo4p), which are usually classified solely as deficits in the transport of various cargo from mother cell to bud (for reviews see Brown, 1997; Mermall et al., 1998; Titus, 1998). While the steady-state concentration of these myosins at the bud tip (Lillie and Brown, 1994; Govidan et al., 1995) may only represent the pool of myosin V molecules that have completed this transport task, the likelihood that the diffusion of vesicles across the yeast cell is quite rapid (see Bloom and Goldstein, 1998) raises the possibility that the myosin V–dependent capture of vesicles that diffuse to the bud tip represents a significant mechanism driving the accumulation of at least certain cargo there.
Acknowledgments
We thank Brian Jankowitz for assistance in preparing the manuscript, Dr. Ken Springs and Dr. James Sellers for help with live cell imaging and video microscopy, Dr. Leah Haimo for advice and discussion, Dr. Neal Copeland and Dr. Nancy Jenkins for mouse strains, and Dr. Edward D. Korn for comments and support of this work.
Abbreviations used in this paper: dsu
dilute suppressor
- se
short ear
- SER
smooth endoplasmic reticulum
- EGFP
enhanced green fluorescent protein
Footnotes
2. The 12 video sequences referred to in the text can be viewed as Quicktime movies available at our website (www.nhlbi.nih.gov/nhlbi/relab/dir/ lcb/mcb.htm). Please note that because these time lapse video sequences were generated by recording one video frame per second instead of 30 frames per second, melanosomes appear to jump when moving rapidly (i.e., >1 μm/s).
3. We attribute the microtubule-dependent melanosome movements reported here to motor proteins, as opposed to microtubule dynamics, because the maximum rates of change in microtubule length (∼10–30 μm/ min for elongation velocity, and ∼20–50 μm/min for shortening velocity; Parsons and Salmon, 1997) are too slow to account for the maximum rates of movement we saw here (∼120 μm/min in both directions, based on the average of the single fastest steps in the paths analyzed in the text).
References
- Baas PW, Yu W. A composite model for establishing the microtubule arrays of the neuron. Mol Neurobiol. 1998;12:145–161. doi: 10.1007/BF02740651. [DOI] [PubMed] [Google Scholar]
- Bearer EL, DeGiorgis JA, Bodner RA, Kao AW, Reese TS. Evidence for myosin motors on organelles in squid axoplasm. Proc Natl Acad Sci USA. 1993;90:11252–11256. doi: 10.1073/pnas.90.23.11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS, Goldstein LSB. Cruising along microtubule highways: how membranes move through the secretory pathway. J Cell Biol. 1998;140:1277–1280. doi: 10.1083/jcb.140.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, D. 1992. In Cell Movements. Garland Publishing, Inc., New York. 243–264.
- Brown SS. Myosins in yeast. Curr Opin Cell Biol. 1997;9:44–48. doi: 10.1016/s0955-0674(97)80150-0. [DOI] [PubMed] [Google Scholar]
- Conrad PA, Giuliano KA, Fisher G, Collins K, Matsudaira PT, Taylor DL. Relative distributions of actin, myosin I, and myosin II during the wound healing response of fibroblasts. J Cell Biol. 1993;120:1381–1391. doi: 10.1083/jcb.120.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker-Ohno K, Hayasaka S, Takagishi Y, Oda S, Wakasugi N, Mikoshiba K, Inouye M, Yamamura H. Endoplasmic reticulum is missing in dendritic spines of Purkinje cells of the ataxic mutant rat. Brain Res. 1996;714:226–230. doi: 10.1016/0006-8993(95)01560-4. [DOI] [PubMed] [Google Scholar]
- Evans LL, Hammer JA, III, Bridgman PC. Subcellular localization of myosin V in nerve growth cones and outgrowth from dilute-lethal neurons. J Cell Sci. 1997;110:439–449. doi: 10.1242/jcs.110.4.439. [DOI] [PubMed] [Google Scholar]
- Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J Cell Biol. 1994;126:661–675. doi: 10.1083/jcb.126.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govidan B, Bowser R, Novick P. The role of myo2, a yeast class V myosin, in vesicular transport. J Cell Biol. 1995;128:1055–1068. doi: 10.1083/jcb.128.6.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths GM. The cell biology of CTL killing. Curr Opin Immunol. 1995;7:343–348. doi: 10.1016/0952-7915(95)80108-1. [DOI] [PubMed] [Google Scholar]
- Griffiths GM. Secretory lysosomes—a special mechanism of regulated secretion in haematopoietic cells. Trends Cell Biol. 1996;6:329–332. doi: 10.1016/0962-8924(96)20031-5. [DOI] [PubMed] [Google Scholar]
- Haimo LT, Thaler CD. Regulation of organelle transport: lessons from color change in fish. BioEssays. 1994;16:727–733. [Google Scholar]
- Hearing, V.J., and R.A. King. 1993. Determinants of skin color: melanocytes and melanization. In Pigmentation and Pigmentary Disorders. N. Levine, editor. CRC Press, Inc., London. 4–18.
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Jackson IJ. Molecular and developmental genetics of mouse coat color. Annu Rev Genet. 1994;28:189–217. doi: 10.1146/annurev.ge.28.120194.001201. [DOI] [PubMed] [Google Scholar]
- Jenkins NA, Copeland NG, Taylor BA, Lee B K. Dilute (d) coat colour mutation of DBA/2J mice is associated with the site of integration of an ecotropic MuLV genome. Nature. 1981;293:370–374. doi: 10.1038/293370a0. [DOI] [PubMed] [Google Scholar]
- Jimbow, K., W.C.J. Quevedo, T.B. Fitzpatrick, and G. Szabo. 1993. Biology of melanocytes. In Dermatology in General Medicine. T.B. Fitzpatrick et al., editors. McGraw-Hill, Inc., New York. 261–288.
- Kelleher JF, Titus MA. Intracellular motility: how can we all work together? . Curr Biol. 1998;8:R394–R397. doi: 10.1016/s0960-9822(98)70246-5. [DOI] [PubMed] [Google Scholar]
- Klein C, Philippe N, Le Deist F, Fraitag S, Prost C, Durandy A, Fischer A, Griscelli C. Partial albinism with immunodeficiency (Griscelli syndrome) J Pediatr. 1994;125:886–895. doi: 10.1016/s0022-3476(05)82003-7. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Takeuchi T. Ultrastructural observations on melanosome aggregation in genetically defective melanocytes of the mouse. Anat Rec. 1981;201:599–611. doi: 10.1002/ar.1092010404. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Langford GM, Weiss DG. Actin-dependent organelle movement in squid axoplasm. Nature. 1992;356:722–725. doi: 10.1038/356722a0. [DOI] [PubMed] [Google Scholar]
- Lacour JP, Gordot PR, Eller M, Bhawan J, Gilchrest BA. Cytoskeletal events underlying dendrite formation by cultured pigment cells. J Cell Physiol. 1992;151:287–299. doi: 10.1002/jcp.1041510210. [DOI] [PubMed] [Google Scholar]
- Langford GM. Actin-and microtubule-dependent organelle motors: interrelationships between the two motility systems. Curr Opin Cell Biol. 1995;7:82–88. doi: 10.1016/0955-0674(95)80048-4. [DOI] [PubMed] [Google Scholar]
- Lewis AK, Bridgman PC. Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J Cell Biol. 1992;119:1219–1243. doi: 10.1083/jcb.119.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie SH, Brown SS. Suppression of a myosin defect by a kinesin-related gene. Nature. 1992;356:358–361. doi: 10.1038/356358a0. [DOI] [PubMed] [Google Scholar]
- Lillie SH, Brown SS. Immunofluorescence localization of the unconventional myosin, myo2p, and the putative kinesin-related protein, smylp, to the same regions of polarized growth in Saccharomyces cerevisiae. . J Cell Biol. 1994;125:825–842. doi: 10.1083/jcb.125.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald K. Osmium ferricyanide fixation improved microfilament preservation and membrane visualization in a variety of animal cell types. J Ultrastruct Res. 1984;86:107–118. doi: 10.1016/s0022-5320(84)80051-9. [DOI] [PubMed] [Google Scholar]
- Mercer JA, Seperack PK, Strobel MC, Copeland NG, Jenkins NA. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature. 1991;349:709–713. doi: 10.1038/349709a0. [DOI] [PubMed] [Google Scholar]
- Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:533–537. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Seperack PK, Strobel MC, Swing DA, Copeland NG, Jenkins NA. Dilute suppressor dsu acts semidominantly to suppress the coat color phenotype of a deletion mutation, dl20J, of the murine dilute locus. Proc Natl Acad Sci USA. 1988;85:8131–8135. doi: 10.1073/pnas.85.21.8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Swing DA, Copeland NG, Jenkins NA. The murine dilute suppressor gene encodes a cell autonomous suppressor. Genetics. 1994;138:491–497. doi: 10.1093/genetics/138.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooseker MS, Cheney RE. Unconventional myosins. Annu Rev Cell Dev Biol. 1996;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate cells. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento AAC, Amaral RG, Bizario JCS, Larson RE, Espreafico EM. Subcellular localization of myosin-V in the B16 melanoma cells, a wild-type cell line for the dilute gene. Mol Biol Cell. 1997;8:1971–1988. doi: 10.1091/mbc.8.10.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlow SJ. Melanosomes are specialized members of the lysosomal lineage of organelles. J Invest Dermatol. 1995;105:3–7. doi: 10.1111/1523-1747.ep12312291. [DOI] [PubMed] [Google Scholar]
- Parsons SF, Salmon ED. Microtubule assembly in clarified Xenopusegg extracts. Cell Motil Cytoskel. 1997;36:1–11. doi: 10.1002/(SICI)1097-0169(1997)36:1<1::AID-CM1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Pastural E, Barrat F, Dufourcq-Lagelouse R, Certain S, Sanal O, Jabado N, Seger R, Griscelli C, Fischer A, Saint G, Basile Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet. 1997;16:289–291. doi: 10.1038/ng0797-289. [DOI] [PubMed] [Google Scholar]
- Provance DWJ, Wei W, Ipe V, Mercer JA. Cultured melanocytes from dilute mutant mice exhibit dendritic morphology and altered melanosome distribution. Proc Natl Acad Sci USA. 1996;93:14554–14558. doi: 10.1073/pnas.93.25.14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov VI, Hope AJ, Svitkina TM, Borisy GG. Functional coordination of microtubule-based and actin-based motility in melanophores. Curr Biol. 1998;8:165–168. doi: 10.1016/s0960-9822(98)70064-8. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Gelfand VI. Myosin cooperates with microtubule motors during organelle transport in melanophores. Curr Biol. 1998;8:161–163. doi: 10.1016/s0960-9822(98)70063-6. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Tint IS, Fanapour PC, Gelfand VI. Regulated bidirectional motility of melanophore pigment granules along microtubules in vitro. Proc Natl Acad Sci USA. 1997;94:3720–3725. doi: 10.1073/pnas.94.8.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seperack PK, Mercer JA, Strobel MC, Copeland NG, Jenkins NA. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO (Eur Mol Biol Organ) J. 1995;14:2326–2332. doi: 10.1002/j.1460-2075.1995.tb07227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers, W.K. 1979. Dilute and Leaden, the p-locus, rub-eye and ruby-eye 2. In A Model for Mammalian Gene Action and Interaction. W.K. Silvers, editor. Springer-Verag, New York. 83–104.
- Strobel MC, Seperack PK, Copeland NG, Jenkins NA. Molecular analysis of two mouse dilute locus deletion mutations: spontaneous dilute lethal20Jand radiation-induced dilute prenatal lethal Aa2 alleles. Mol Cell Biol. 1990;10:501–509. doi: 10.1128/mcb.10.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagishi Y, Oda S, Hayaska S, Dekker-Ohno K, Shikata T, Inouye M, Yamamura H. The dilute-lethal (dl) gene attacks a Ca2+store in the dendritic spine of Purkinje cells in mice. Neurosci Lett. 1996;215:169–172. doi: 10.1016/0304-3940(96)12967-0. [DOI] [PubMed] [Google Scholar]
- Tamura A, Halaban R, Moellmann G, Cowan JM, Lerner MR, Lerner AB. Normal murine melanocytes in culture. In Vitro Cell Dev Biol. 1987;23:519–522. doi: 10.1007/BF02628423. [DOI] [PubMed] [Google Scholar]
- Titus MA. Motor proteins: myosin V—the multi-purpose transport motor. Curr Biol. 1998;7:R301–R304. doi: 10.1016/s0960-9822(06)00143-6. [DOI] [PubMed] [Google Scholar]
- Wei Q, Wu X, Hammer JA., III The predominant defect in dilute melanocytes is in melanosome distribution and not cell shape, supporting a role for myosin V in melanosome transport. J Muscle Res Cell Motil. 1997;18:517–527. doi: 10.1023/a:1018659117569. [DOI] [PubMed] [Google Scholar]
- Wu X, Bowers B, Wei Q, Kocher B, Hammer JA., III Myosin V associates with melanosomes in mouse melanocytes: evidence that myosin V is an organelle motor. J Cell Sci. 1997;110:847–859. doi: 10.1242/jcs.110.7.847. [DOI] [PubMed] [Google Scholar]
- Wu X, Kocher B, Wei Q, Hammer JA., III Myosin Va associates with microtubule-rich domains in both interphase and dividing cells. Cell Motil Cytoskel. 1998;40:286–303. doi: 10.1002/(SICI)1097-0169(1998)40:3<286::AID-CM7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]